

Abstract
Epigenetic mechanisms comprising DNA methylation, histone modifications, and noncoding RNAs affect chromatin structure and regulate gene expression. These mechanisms control normal embryonic development and adult life and their deregulation contributes to several diseases including cancer. The process of tumorigenesis is complex and results from the evolution of different “hallmarks of cancer”. Hanahan and Weinberg presented in 2000 and 2011 seminal contributions in the cancer field, first the six hallmarks of cancer and a decade later two additional hallmarks and two enabling characteristics were added. Here, we surmise that epigenetic mechanisms regulate and contribute to every single hallmark in cancer, and thus represent the hallmark of hallmarks in tumorigenesis. Focusing on epigenetics as a major hallmark in cancer formation has profound preventive, therapeutic, and clinical implications.
Keywords: Epigenetics, cancer, hallmarks
Introduction
Cancer is one of the most prominent causes of mortality. It is estimated that 8.2 million deaths occurred worldwide in 2012 [1] and more than six hundred thousand cancer deaths are predicted to follow just in the United States in 2020 [2]. Cancer is a multifaceted and diverse disease in which cancer cells share common mechanisms of tumor formation, progression, and ability to grow beyond their natural environment. Scientists proposed several theories throughout the years to explain the origins of cancer [3]. However, no theories or features of cancer cells were more discussed and adopted then the “hallmarks of cancer” as proposed by Hanahan and Weinberg [4,5]. Hanahan and Weinberg have defined the hallmarks of cancer “as acquired functional capabilities that allow cancer cells to survive, proliferate, and disseminate” [6]. Within a decade, eight hallmarks were characterized namely “the ability of cancer cells to sustaining proliferative signaling, evading growth suppressors, resisting cell death, inducing angiogenesis, activating invasion and metastasis, enabling replicative immortality, deregulating cellular energetics, and avoiding immune destruction”. In addition, “genome instability and mutation and tumor-promoting inflammation” were added as two enabling characteristics in the achievement of hallmarks [5].
All the different hallmarks and enabling characteristics describe functional properties of normal cells that are acquired during the process of tumor formation and are “the driving forces of tumorigenesis” [7]. Horne at al. reevaluated the hallmarks and unified them under the umbrella of cancer evolution rather than being separated into discrete categories [8]. Some of these hallmarks may have overlapping properties such as avoiding growth suppressors and resisting cell death which will in turn sustain the proliferative signaling of tumor cells. Other hallmarks may be needed at different phases of tumor development, for instance activating invasion and metastasis occurs later in tumorigenesis. Furthermore, the contribution and chronology of the hallmarks may vary according to the different cancers, tissue of origin, composition of the tumor, and the contributions of the microenvironment and immune system.
Historically cancer was mostly considered a genetic disease, however the “somatic mutation theory” did not explain the origin of most cancers [9]. Tumor cells are mostly characterized by abnormalities in responding to internal and external signals, cellular identity, and deregulation of gene expression [10-12]. In fact, epigenetic mechanisms are tightly controlled and regulate embryonic development and adult life and their deregulation has been involved in many disorders including cancer [13,14]. Large-scale cancer genome sequencing efforts have indicated that almost half of human cancers bear mutations in chromatin proteins [15,16]. In addition, malignant cells show CpG islands hypermethylation, mostly in tumor suppressor genes, a reduction of total DNA methylation, and progressive histone modifications changes [17]. Recently, noncoding RNAs (ncRNAs) whether small ncRNA (< 200 nt) namely microRNAs (miRNAs) and long ncRNAs (lncRNAs) were observed to be deregulated in cancer and to majorly contribute to tumorigenesis [18-20]. Recently, Flavahan et al. concluded that abnormal epigenetic mechanisms and chromatin structures give rise to oncogenic properties that manifest themselves in all the hallmarks of cancer [21]. The focus of this review is to present evidence showing that epigenetic mechanisms regulate and contribute to every single hallmark in cancer and that perhaps present themselves as hallmark of hallmarks. The role of mutations cannot be overlooked in tumor formation as there is a major crosstalk between genetic and epigenetic modifications but will not be the focus of this is review [22].
Epigenetic mechanisms and the hallmarks of cancer
Epigenetics is “defined as the study of mitotically and meiotically heritable changes in gene function that are not dependent on DNA sequence” [23]. It was recently postulated that epigenetic aberrations and chromatin states may cause extensive oncogenic gain of function properties and may satisfy all of the hallmarks of cancer [21]. External and internal signals may enhance or reduce chromatin resistance leading to a “restrictive state that blocks differentiation programs” or epigenetic plasticity, respectively [21]. Epigenetic plasticity in turn provides a permissive environment for premalignant and malignant cells to stimulate different gene regulatory pathways resulting in abnormal cell fates. Some of these acquired changes may be “passengers” or “drivers” to the process of tumorigenesis. These driver epigenetic conditions may be fixed during cell proliferation by various mechanisms including DNA methylation, histone modifications, and ncRNA contributions resulting in tumor suppressor gene inhibition and oncogene activation.
Here we will focus on the most investigated epigenetic mechanisms in tumorigenesis namely DNA methylation, histone modifications, miRNA, and lncRNA deregulation in cancer and their contributions to the different hallmarks. We will select few examples that justify our selection of epigenetic mechanism contributions to all of cancer’s hallmarks. For further examples and details, the reader is referred to excellent comprehensive reviews [18-22].
Sustaining proliferative signaling
Sustaining proliferative signaling is considered as one of the utmost crucial properties of tumor cells [6]. Several players and signaling pathways ensure the coordinated regulation of cell division during normal embryonic development and adult life. Cancer cells obstruct these signals by a variety of mechanisms ranging from secreting their own growth factors that function in autocrine fashion. These growth factors in turn bind to receptors commonly endowed with tyrosine kinase activities that regulate cell cycle progression. Cancer cell proliferation may be also fueled by signals received from cells in the stroma that may in turn be affected by the tumor cells themselves. Alternatively, cancer cells may upregulate the receptors that receive the mitogenic signal or may acquire mutations in receptors that result in their ligand-independent activation.
Proto-oncogenes are commonly hypomethylated in several cancers [24]. It is well established that the epidermal growth factor (EGF) and c-myc are hypermethylated in liver cancer [25]. In addition, negative feedback signals that inhibit cell proliferation or cell cycle progression may be impaired. For instance, PTEN counteracts the oncogenic PI3K signaling by degrading its product PIP. PTEN promoter is methylated in some cancers resulting in shut down of its transcription and loss of PTEN expression [26].
Genetic and epigenetic mechanisms are interrelated. For instance in gliomas and other tumors isocitrate dehydrogenase (IDH) mutations are common initiating events [27,28]. This results in the formation of the 2-hydroxyglutarate oncometabolite which obstructs hydroxylases which are involved in DNA demethylation, resulting in hypermethylated DNA. The DNA binding protein CTCF is involved in insulating chromatin loops from excessive enhancer stimulation and thus its function is reduced in IDH mutants [21]. Consequently, in gliomas, the oncogene PDGFRA, that encodes the platelet-derived growth factor receptor A, is due to loss of CTCF gene insulation and results in hyperproliferation of glioma cells. The loss of insulator function is conserved through cell proliferation due to the stability of DNA methylation.
The histone code is commonly deregulated in cancer [17]. Several proto-oncogenes are abnormally expressed by aberrations in histone modifications. For instance, fibroblast growth factor receptor 2 (FGFR2) is implicated in breast cancer susceptibility. High levels of FGFR2 in cancer cells have been linked to polymorphic sequences with constitutively acetylated histones [29]. Other patterns of histone post-translational modifications such as methylation, phosphorylation, and ubiquitination are commonly deregulated in cancer in several genes impacting cell proliferation [30].
Cell proliferation is tightly controlled by proto-oncogene products as well as by ncRNAs. The expression of coding and noncoding genes is affected by ncRNAs. Recently, ncRNAs, epigenetics, and cancer were shown to be regulating each other [18]. ncRNAs affect epigenetic mechanisms which in turn control ncRNAs [31] and these mechanisms are aberrant in cancer [18]. miRNAs control gene expression by abrogating transcript translation or reducing its stability. miRNAs are crucial for the control of cell growth and survival [32]. These miRNAs can be amplified at certain loci or lost in deleted chromosomal segments [33]. miRNAs are deregulated in most cancers and affect proto-oncogenes or tumor suppressor genes and, therefore, function as tumor suppressors or oncomiRs [34]. miR-17 cluster encodes six miRNAs that function as oncomiRs due to their upregulated expression by c-myc, resulting in cell cycle progression and regulation of E2F [35]. miR-124a represses the oncogene CDK6 and is commonly silenced in colon cancer, resulting in retinoblastoma (Rb) phosphorylation and inactivation of [36].
lncRNAs are a heterogeneous group of ncRNAs that exceed 200 nucleotides in length and encode less than 100 amino acids in their open reading frame [37]. lncRNAs are deregulated in cancer and affect most of its hallmarks [20]. For instance, lncRNAs sustain proliferative signaling through the sex steroid hormones. Sex steroidal hormones in female and males control female mammary glands, uterus, and ovary or prostate gland and testis, respectively. They function by binding to their intracellular receptors activating or repressing gene expression through co-activators and co-repressors, respectively. SRA, the steroid receptor RNA activator, functions as a lncRNA [38]. SRA with SRC-1 is a constituent of an RNA and protein complex that regulates the nuclear receptors through their AF-1 domain. SRA levels are elevated in breast tumors which might result in altered ER/PR action observed in breast tumorigenesis [39]. The situation is even more complex as SRA was found to produce a protein that functions as a corepressor and activator of nuclear receptors [40]. There are also other examples of lncRNAs than SRA that play a role in cell proliferation. More than hundred differentially expressed lncRNAs were found by RNA-Sequencing to be differentially regulated in different prostate benign and malignant tissues [41]. PCAT-1 (prostate cancer associated transcript-1) was one characterized lncRNA to be overexpressed in aggressive and advanced prostate tumors. PCAT silencing in prostate cell lines reduced cell growth and resulted in more than two hundred upregulated genes. Gene ontology showed the enrichment of these latter genes in cell cycle control and mitosis. Interestingly, the promoter region of cell cycle genes were shown to encode more than 200 lncRNAs, underscoring the importance of these RNA molecules in cell cycle and cell proliferation control [42].
Evading growth suppressors
In addition to sustaining proliferation in tumor formation, cancer cells need to evade negative growth regulators. The most prominent brakes to cell division and cell cycle progression are the Rb protein, the p53 pathway, and the cyclin-dependent kinase inhibitors (CDKIs) [43,44]. The Rb protein is responsive to external and internal cues to dictate whether cells should continue cell division or halt cell cycle progression. The p53 protein on the other hand is mostly responsive to internal cues and would halt cell cycle progression if the amount of stress or DNA damage can be repaired. If the damage is excessive and irreparable then p53 will activate cell death signaling pathways, mostly apoptotic ones.
In cancer the tumor suppressor genes have their promoter regions commonly hypermethylated [45]. The Rb promoter is regulated by CTCF which is in turn controls promoter stability. CTCF binding to promoter sequences is methylation-dependent and in case of hypermethylated DNA, commonly observed in human cancers, the Rb promoter is silenced [46]. p53 is the most prevalently mutated and inactivated gene in cancer [47]. p53 promoter methylation is frequent in various cancers including neuroblastomas and melanomas [48,49]. p53 gene expression is tightly regulated by the activator p14ARF and the repressor Mdm2. p14ARF promoter region is commonly methylated in many cancers resulting in reduced levels of p53. Finally, the CDKIs are also tumor suppressor genes commonly inactivated or reduced in cancer. A prominent and first example is the promoter methylation of p16 [50].
Histone code aberrations are also identified in tumor suppressor genes that negatively control cell division. The transcriptional factor Rb inhibits proliferation by downregulating the expression of several genes needed for cell cycle progression through the assembly of a multi-protein complex including repressors such as histone deacetylases (HDACs). The fine-tuning of HDACs and histone acetyltransferases (HATs) is tightly regulated in normal cells but unchecked in cancer cells. HDAC type I levels are upregulated in some tumors that are of high grades and may result in transcriptional repression of some genes needed for proliferation and cell cycle control [51].
p53 and signaling network are regulated by miRNAs at various levels. miRNAs can directly target p53 or indirectly through its regulators [52]. So far more than 20 miRNAs were listed to directly target p53 and to reduce its expression. Many of these latter miRNAs are elevated in human tumors resulting in decreased p53 levels [53]. p53 repressors such as Mdm2 and Mdm4 can be targeted by other miRNAs. More than nine miRNAs can directly target MDM2, reducing its expression, thus activating p53. Interestingly, some of these miRNAs are transcriptionally regulated by p53 and, therefore, constitute a positive feedback loop. In conclusion, these findings indicate that p53 expression and activity are controlled by miRNAs and aberration of these miRNAs in cancer may result in the loss of p53.
Several lncRNAs were also demonstrated to control p53 levels and activity [54]. 7SL is a lncRNA that is overexpressed in several cancers and silencing of 7SL reduces cell proliferation, induces senescence, and autophagy [55]. On the other hand, overexpressing 7SL enhances DNA replication and cell growth. RNA pulldown experiments showed that 7SL interacts with p53 transcripts and reduces its translation [55]. Tumor cells have also utilized lncRNAs in avoiding growth suppressors such as CDKIs. For instance, the lncRNA ANRIL induces the proliferation of non-small cell lung cancer (NSCLC) and cervical cancer cells via the silencing of the p15 and p16 genes [56].
Resisting cell death
Aberrant cells are eliminated in multicellular organisms by cell death, such as apoptosis, to preserve the organism. It is therefore crucial for tumor cells to reduce or eliminate this mechanism during their development [57]. Apoptosis is a regulated process and is divided into extrinsic or intrinsic programs according to external or internal cell death signals, respectively. Deregulation of intrinsic apoptosis is more commonly involved in cancer formation [6].
The Bcl-2 family members are major regulators of intrinsic apoptosis transferring signals between apoptotic stimuli and effector players [58]. They are functionally divided as prosurvival members such as Bcl-2 and its close members, such as Bcl-w, Bcl-xL, A1, Mcl-1, and apoptotic ones namely Bak and Bax and the BH3 only members (Bim, Bid, Puma, Bad, Noxa…). Bax and Bak interrupt the mitochondrial membrane potential and result in the burst of cytochrome c and Apaf-1 and formation of the apoptosome complex that activates caspase 9 [59]. Apoptosis is tightly controlled by the balance between the pro-and anti-apoptotic members. Although Bcl-2 chromosomal translocations are commonly observed in hematological malignancies, however other overexpression mechanisms have been observed. In melanomas, EZH2, the histone methyltransferase enhancer of zeste homolog 2, shows aberrant activity resulting in its reduced binding to the promoter region of Bcl-2, resulting in its activation and contributing to apoptosis resistance in melanoma aggressiveness [60]. The Bcl-2 gene was observed to be hypomethylated in B-cell chronic lymphocytic leukemia, resulting in high expression levels [61]. Deregulated methylation of Bcl-2 and bax is observed in glioblastoma multiform resulting in apoptosis disruption [62]. Interestingly, HDAC inhibitors result in reduced Bcl-2 levels and apoptosis induction in t(14;18) lymphomas [63].
The Bcl-2 family members are also under post-transcriptional control by miRNAs [64]. More than 35 miRNAs reduce the levels of the survival Bcl-2 members of which 12 were demonstrated to target directly Bcl-2 including miR-15/16. This latter cluster is one of the first discovered tumor suppressor miRNA to be silenced or excised in chronic lymphocytic leukemia (CLL) [65]. miR-206 directly targets Bcl-2 and its reduced expression enhanced Bcl-2 levels in glioblastoma tissues and correlated with disease progression [66]. Also lncRNA contributed to the regulation of Bcl-2 expression levels. The lncRNA HOTTIP was shown to upregulate Bcl-2 levels and to associate with chemoresistance in SCLC [67].
A key sensor in DNA damage is the “guardian of the genome” p53. Several epigenetic mechanisms were previously listed to reduce p53 levels and to result in reduced DNA repair checkpoints [68]. However, other mechanisms may contribute to the inactivation of the DNA damage response such as dysfunction of the DNA methyltransferase DNMT3A in acute myeloid leukemia and mutations in DNMT3A are linked to poor disease prognosis [69].
Enabling replicative immortality
Cells acquire replicative immortality to form tumors, in contrast to normal cells that have a limited replicative capability. Unlimited proliferation is linked to the property of cancer cells to protect and keep elongated the ends of telomeric DNA [70]. Nonimmortalized cells have limited telomerase activity as opposed to elevated levels detected in immortalized and cancer cells. Telomeric shortening is recognized as a tumor suppressive mechanism in normal cells and leads to senescence and/or apoptosis [71].
Evidence indicates that the catalytic subunit of human telomerase hTERT (human telomerase transcriptase) can be epigenetically regulated. Expression of hTERT is regulated by DNA methylation, histone modifications, and ncRNAs [72]. Usually hypermethylation at promoter sequences is correlated with gene silencing, however the situation differs on the hTERT promoter. The hypermethylated hTERT promoter inhibits repressors such as CTCF from binding [73]. On the other hand, hTERT promoter is stimulated when the activators SP1 and c-myc bind to unmethylated CpG sites at positions 11, 12, 19, and 27 [74]. The hTERT promoter displays elevated DNA methylation levels, and lower methylation correlates with enhanced telomerase expression and activity [75]. In hepatocellular carcinoma, histone modification and DNA methylation regulate hTERT.
Histones organize chromatin in the cell nucleus and modification of their charges affects their affinity to DNA. Histone tail post-translational modifications include acetylation, methylation, ubiquitination, and phosphorylation [76]. Active gene transcription is usually associated with hyperacetylated histones and H3K4 (lysine 4 on histone 3) methylation. However, inactive gene expression is linked to hypoacetylated histones and methylation at both lysine 9 and 27 of histone 3. The highest levels of acetylated histones H3 and H4 and methylation at lysine 4 of histone 3 correlate with elevated levels of hTERT expression in cancer cells [77].
The hTERT promoter is also regulated by miRNAs [72]. For instance, miR-491-5p inhibits hTERT and its expression is reduced in cervical cancer [78]. It is considered a tumor suppressor miRNA that targets hTERT in cervical cancer as its enforced expression inhibits the growth of tumor cells. In several cancers, specific miRNAs have been detected to regulate hTERT by a diversity of mechanisms. The most common mechanism of these hTERT-specific miRNAs is their interaction with hTERT-3’UTR to downregulate its expression. In gastric cancer, miR-1207-5p and miR-1266 levels are reduced resulting in elevated levels of hTERT [79]. Other miRNAs bind to the ORF of hTERT transcripts and inhibit their translation as observed for miR-1182 [80]. These latter miRNAs were shown to be downregulated in gastric cancer resulting in elevated levels of hTERT. Interestingly, a new mechanism of hTERT regulation by lncRNA was noted through its sponging effect of specific miRNAs that target hTERT. The lncRNA BC032469 is elevated in gastric cells and upregulates hTERT by buffering its specific miR-1207-5p [81]. In addition, the lncRNA H19 was observed to target hTERT and to negatively correlate with hTERT expression levels in acute promyelocytic leukemia [82].
Recently, the nucleoprotein structure of telomeres was noted to be aberrant in neoplastic transformation [83]. Tumors can rely on alternative mechanism for telomere lengthening (ALT) than telomerase activation to elongate their telomeres. Telomeric DNA is in a heterochromatin hypoacetylated form due to the inclusion of histones H3.3 and the function of sirtuin deacetylases. ALT mechanism and telomeric nucleoprotein are deregulated in cancer due to H3.3 mutations resulting in an oncohistone and sirtuin deacetylase functions destabilizing the chromatin landscape [84].
Inducing angiogenesis
Tumors are not able to grow beyond 1-2 mm in size without inducing angiogenesis. These new blood vessels, sprouting from preexisting ones, are needed by tumor cells for the supply of oxygen and nutrients and for the disposal of carbon dioxide and waste. The angiogenic process is under strict control within the tumor microenvironment due to the opposing actions of angiogenic and anti-angiogenic factors such as VEGF and their receptors (VEGFR) and thrombospondin-1, respectively [85].
The epigenome methylation status has been implicated recently in tumor angiogenesis [86]. VEGF-A represents one of the strongest inducers of angiogenesis and its expression is upregulated in several tumors [87]. Recurrent bladder transitional cell carcinoma (TCC) with muscle invasive properties is characterized by aggressive angiogenesis and bad prognosis. VEGF-A levels are elevated in TCC and methylation status of VEGFA differed between low-grade TCC and high-grade TCC where promoter hypermethylation characterizes the former one [88]. VEGFRs are also elevated in tumors and methylation status of their promoters regulates their expression levels [89].
The von Hippel-Lindau (VHL) is a tumor suppressor gene that reduces tumor growth and down-regulates many angiogenic factors [90]. The loss of VHL in several tumors causes extensive tumor vascularization [91]. The VHL gene encodes pVHL which is a constituent of E3 ubiquitin ligase that adds ubiquitin residues and targets the hypoxia-inducible factor (HIF) for proteasomal-mediated degradation under normoxic conditions. HIF is a transcription factor that controls the expression of VEGF. Loss of pVHL by genetic or epigenetic mechanisms causes accumulation of HIF and consequently VEGF overproduction resulting in marked angiogenesis. Hypermethylation of VHL promoter commonly occurs in several cancers [92].
Histone-methylating enzymes such as EZH2 constitute the catalytic subunit of the Polycomb Repressive Complex 2. EZH2 causes histone H3 lysine 27 trimethylation which is a marker of gene silencing. EZH2 regulates angiogenesis and its silencing abrogates capillary tube formation in cultured cells. In vivo, EZH2 promotes tumor growth, metastasis, and angiogenesis as it silences anti-angiogenic genes [93].
Recently, miRNAs and lncRNAs were shown to regulate angiogenesis and to contribute to neoplastic transformation [94]. Several miRNAs such as “miR-17-92 cluster, miR-378, miR-296, let-7f, miR-27b, miR-130, and miR-126” were demonstrated to have angiogenic activities [95]. More than 40 miRNAs have been implicated in VEGF regulation [19]. The miR-17-92 induces angiogenesis in several solid tumors [96]. c-myc is a powerful inducer of angiogenesis by reducing the expression of Tsp1 which is also down-regulated by miR-17-92 [96]. miR-378 is elevated in several cancers and regulates VEGF expression [97]. miR-125a binds to VEGF-3’UTR to degrade it, miR-378 binds and competes to the same region, thus inhibiting VEGF silencing. Another study demonstrated that miR-378 is an oncogene, not only through induction of angiogenesis but also by promoting growth and tumor survival through targeting Fus-1 and Sufu tumor suppressor genes [98].
LncRNAs impact tumor formation through angiogenesis and have been quoted as “angio-LncRNAs” [99]. LncRNAs can function as natural antisense transcripts that can impact the proteins or can be located between protein-coding genes [100]. Hepatocellular carcinomas overexpress lncRNA MVIH which promotes angiogenesis and microvessel formation through abrogation of phosphoglycerate kinase 1 secretion [101]. Studies have indicated that lncRNAs can regulate VEGF signaling pathway [102]. MALATI “Metastasis-associated lung adenocarcinoma transcript 1” is a lncRNA that impacts tumor angiogenesis by enhancing endothelial cell proliferation [103]. MALATI is overexpressed in several tumors and stimulates invasion and metastasis [104]. The loss of MALAT1 reduces the proliferation of tumor cells and their ability to undergo angiogenesis as this lncRNA was shown to regulate cellcycle related endothelial factors [104]. Other lncRNAs were also demonstrated to regulate angiogenesis such as HOXD-AS1, HIF-1A-AS2, and MEG3 among others by regulating angiogenic factors or hypoxia inducible factor 1-α (HIF-1α) or through other unknown mechanisms [94].
Activating invasion and metastasis
Activating invasion and metastasis is the property of all hallmarks that contributes the most to malignancy and cancer-related death. Invasion and metastasis lead to an intertwined cascade that starts with tumor cell invasion, intravasation, extravasation, and successful growth of metastatic colonies [105]. Several listed epigenetic players in the regulation of angiogenesis also impact invasion and metastasis. Cell-cell and cell to extracellular matrix adhesion molecules are altered in invasion and metastasis such as the loss of E-cadherin and replacement by N-cadherin. However, the “epithelial-to-mesenchymal transition” (EMT) mostly regulates invasion and metastasis of carcinoma cells [106]. Genetic and most recently epigenetic mechanisms have been involved in the activation of EMT [107]. EMT is a normal process of embryogenesis and organogenesis, however this process is majorly deregulated in invasion and metastasis. EMT is a dedifferentiation program that involves a wide array of transcriptional factors that impact broadly epigenetic mechanisms and gene regulation.
ATP-chromatin dependent remodeling complexes play major roles in chromatin relaxation and compaction and impact gene regulation, DNA replication, DNA repair, chromosome segregation, and recombination. Chromatin remodeling complexes of which there are four major families: SWI/SNIF, CHD, ISWI, and INO80, contain an ATPase domain that hydrolyzes ATP to modify histones and affect nucleosome structure [108]. These chromatin remodeling complexes are deranged during malignant progression [109]. MTA1/2 is a member of the CHD family and its overexpression is involved with the invasiveness of several cancers. However, MTA3 is another member of CHD family that inhibits transcription of SNAI1 and inhibits EMT, invasion, and metastasis in breast cancer [110]. In addition, mutations are commonly observed in the SWI/SNIF remodeling complex where at least one member is mutated in 20% of human tumors [111]. DNA methylation also contributes to EMT where the DNA binding proteins MeCP2 and MBD1/2 recognize and attach to the methylated CpG in E-cadherin promoter resulting in gene silencing in tumor cells [112].
The EMT results in wide range transcriptional silencing of epithelial genes and activation of mesenchymal ones which is directed by histone modifications. In breast cancer cells, the HAT hMOF catalyzes H4K16 acetylation to preserve the expression of EMT tumor suppressors, namely E-cadherin and TSM1 [113]. Furthermore, the nuclear factor HNF3 collaborates with p300/CBP on E-cadherin promoter to antagonize EMT and metastasis of breast cancer cells [114]. Histone deacetylases are also implicated in the regulation of E-cadherin. The EMT transcriptional factor ZEB1 recruits HDAC1/2 to the E-cadherin promoter to decrease its expression and to induce EMT and invasion in pancreatic cancer [115]. However, ZEB1 recruits SIRT1 for the silencing of E-cadherin and to induce EMT and metastasis in prostate cancer cells [116]. Other epithelial genes, such as EPCAM and ESRP1, are involved in ZEB1-induced repression of EMT and induction of metastasis. Histone methylation may play repressive functions in EMT. Symmetric demethylation on H3K9 mainly by G9a results in transcriptionally repressed genes. For instance, G9a dimethylates H3K9 in the promoter of the cell adhesion EPCAM and results in EMT and metastasis in lung cancer cells [117].
Regulators RNAs such as miRNAs and lncRNAs are also involved in the regulation of EMT and their deregulation was observed in numerous cancers [107]. Several of the previously listed miRNAs and lncRNAs that affect cell proliferation and angiogenesis also impact invasion and metastasis. For instance, miR-133a inhibits cell growth and invasion in NSCLC [118]. miR-223 directly silences PARP1 and enhances migration and invasion in esophageal adenocarcinoma [119].
ZEB1/2 transcripts are targeted at their 3’UTR by a dozen of miRNAs in several types of cancer cells [120] of which five miR-200 family members regulate differentiation and epithelial properties of several cell types and tissues [121]. ZEB1/2 and miR-200 and miR-205 reciprocally bind to suppress each other’s expression to retain epithelial properties. In particular, the latter miRNAs inhibit ZEB1/2 expression and induction of epithelial phenotype [122]. Therefore, the delicate balance between miR-200/205 and ZEB1/2 constitutes a “feedback regulatory loop” which controls EMT and metastasis [123].
Several lncRNAs were demonstrated to control invasion and metastasis by several mechanisms including transcriptional and translational control, scaffolding structure, decoy for other miRNAs, and miRNA sponging effect [124]. In metastatic breast cancer, the lncRNA treRNA is overexpressed. TreRNA inhibits the translation of the E-cadherin epithelial marker by sequestering the RNA-binding proteins hnRNPK, PUF60, RXR1, and FXR2 and, therefore, promoting EMT and metastasis [125]. The lncRNA HOTAIR is overexpressed in several cancers where it acts as a scaffold needed for instance for the assembly of c-myc transcription initiation complex. In breast cancer cells, the oncoprotein HBXIP recruits c-myc to HOTAIR resulting in c-myc transcriptional activation of genes involved in metastasis [126]. Furthermore, HOTAIR overexpression retargets the polycomb repressive complex 2 (PRC2) to several genomic regions impacting colorectal and breast cancer metastasis [127,128]. HOTAIR enhances invasion and migration and its silencing reduces cell growth and metastasis in colorectal cancer cells [129]. HOTAIR can also sponge miR-148a which reactivates Snail2 resulting in EMT in esophageal carcinoma cells [130]. The lncRNA ZFAS1 is an oncogene in hepatocellular carcinoma with elevated expression in tumor versus normal tissues [131]. The oncogenic function of ZFAS1 is due to the sponging of miR-150, resulting in the reactivation of the metalloproteinases (MMP) 14 and 16 and ZEB1 and subsequent metastasis [131]. The lncRNA HNF1A-AS1 is overexpressed in adenocarcinomas where it directly attaches to DNMT1 facilitating its association with the E-cadherin promoter and subsequent EMT suppression [132]. HNF1A-AS1 downregulation upregulates E-cadherin while downregulating N-cadherin and β-catenin expression.
Reprogramming energy metabolism
Tumor cells boost their cellular and energy metabolism in order to keep up with the sustained proliferation and to adjust to the challenging tumor microenvironment [133,134]. For almost a century, it has been noted that cancer cell metabolism differs from normal ones due to their rewiring of energy metabolism to keep up with the hyperproliferative state [135]. The “Warburg effect” ensures that tumor cells utilize glucose by aerobic glycolysis instead of oxidative phosphorylation to ensure rapid energy demand and provides intermediates for anabolic reactions. It was not until the last decade that cancer metabolism was added as one of the cancer hallmarks [5] and the benefits of its therapeutic targeting became obvious [136].
Pavlova and Thompson classified deregulated cancer metabolism into six hallmarks as listed by verbatim: “(1) deregulated uptake of glucose and amino acids, (2) use of opportunistic modes of nutrient acquisition, (3) use of glycolysis/TCA cycle intermediates for biosynthesis and NADPH production, (4) increased demand for nitrogen, (5) alterations in metabolite-driven gene regulation, and (6) metabolic interactions with the microenvironment” [137]. In general, tumor cells acquire most of these hallmarks while few gain all of them. Oncogenes and tumor suppressor genes regulate cell proliferation and may achieve this by redirecting tumor cell metabolism [138,139]. In particular, the effects of k-ras, c-myc, HIF1α, and mTOR on cancer metabolism may be counteracted by p53, pRB, and PTEN. Oncogenic mutations in PIK3CA, AKT, PTEN, and k-ras can substantially induce glycolysis by upregulating the mTOR-AKT signaling, the rate limiting steps in glycolysis as well as the GLUT1, glucose transporter [140,141]. On the other hand, p53 inhibits glycolysis and promotes oxidative phosphorylation while pRB reduces glutamine utilization in the TCA cycle and downregulates the glutamine transporter ASCT2 [142,143].
Epigenetic mechanisms link nutritional status to changes in gene expression. Tumor cells may express a proliferative gene expression profile by highjacking the epigenome. In fact, metabolic alterations and epigenetic mechanisms are intimately intertwined in cancer cells [144,145]. Cancer metabolism and epigenetics crosstalk resulting in the metabolic rewiring that provides cofactors needed for epigenetic enzymes and production of oncometabolites that regulate epigenetic mechanisms.
The Warburg effect may be observed due to the differential DNA methylation detected in the glycolytic phenotype in tumor cells. VHL hypermethylation results in constitutive expression of HIF1α and increased glycolysis in renal cell carcinoma [146]. The glucose transporter GLUT1 is downregulated by Derlin-3-proteasomal-mediated degradation. Derlin-3 is epigenetically silenced by DNA hypermethylation, resulting in increased transport of glucose to tumor cells and enhanced Warburg effect [147]. The rate limiting steps in gluconeogenesis, fructose 1,6-biphosphatase 1 and fructose 1,6-biphosphatase 2, that antagonize glycolysis are due to promoter hypermethylation in gastric cancer cells [148,149]. This results in enhanced glycolysis needed for anabolic metabolism and ATP production. Tumor progression and enhanced glycolysis have been observed by genetic silencing of BRCA1 by DNA hypermethylation in breast cancer cells [150]. Therefore, differential DNA methylation regulates the Warburg effect in tumor cells.
The Sirtuins (SIRTs) are family members of HDAC inhibitors that can either operate as oncogenes or tumor suppressor genes [151]. They are the most studied histone modifying enzymes in tumor metabolism. SIRT6 can function as a tumor suppressor by deacetylating and reducing HIF-induced glycolysis and c-myc-related glutaminolysis [152]. Frequent deletions of SIRT6 are observed in several human cancers. In addition, SIRT7 directly interacts with c-myc and represses its metabolic aberrations [152]. Further studies are required to study the effect of other histone modifications on the metabolic alterations in tumor cells.
ncRNAs can regulate metabolism of cancer cells at different metabolic pathways. miRNAs control gene expression of several players in cancer metabolism [153]. They can contribute to the Warburg effect by enhancing glycolysis and downregulating oxidative phosphorylation through the TCA cycle. miRNAs also regulate common tumor oncogenic pathways such as c-myc, HIF1α, and k-ras that may impact cancer metabolism [154]. miRNAs also impact the genes involved in glucose uptake, namely miR-1291 for GLUT1 [155], miR-195-5p for GLUT3 [156], and miR-93 for GLUT4 [157]. miRNAs also affect key glycolytic enzymes such as miR-143 and miR-155 for hexokinase 2 [157] and miR-326 for pyruvate kinase M2 [158]. Glutaminase targeting by miR23a reduces glutaminolysis in cancer cells [159].
LncRNAs regulate glucose, lipid, and amino acid metabolism [160]. LncRNAs were shown to regulate glucose metabolism by binding to their GLUTs transporters. ANRIL is a large antisense lncRNA that transcribes an RNA in antisense INK4B-ARF-INK4A cluster [161]. ANRIL is increased and promotes nasopharyngeal carcinoma development where it activates the AKT/mTOR pathway resulting in elevated GLUT1 and lactate dehydrogenase expression [162]. LncRNAs also regulate glycolysis by binding to vital enzymes such as 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in cancer-associated fibroblasts to promote invasion in ovarian cancer [163]. The lncRNA ceruloplasmin upregulates the key glycolytic enzyme glucose-6-phosphate isomerase causing glycolysis and tumor progression [164]. The lncRNA UCA1 is elevated in bladder carcinoma and enhances glycolysis by upregulating its first regulatory enzyme hexokinase [165]. A variety of these lncRNAs are deregulated in a several solid and hematological malignancies [160] and understanding of their metabolic roles will shed light on cancer prognostics and therapeutics.
Evading immune destruction
The immune response plays a double-edge sword in cancer as it can fight tumor cells or can generate an inflammatory tumor microenvironment that enhances tumor progression. Immune checkpoints are mechanisms that keep in check the immune response and control autoimmunity. Tumor cells have managed to avoid tumor surveillance and evading immune destruction by “hijacking” immune checkpoints [166]. Recent successes in generating drugs for targeting and silencing these immune checkpoints and subsequent activation of tumor immunity provided hope for previously untreatable advanced malignancies [167]. The most investigated immune checkpoint inhibitors are focused on inhibitors of programmed death protein-1 (PD-1) and its ligands PD-L1 and PD-L2, in addition to inhibitors of cytotoxic T lymphocyte associated antigen 4 (CTLA-4). PD-1 is present on B cells, activated T cells (CD4 and CD8), natural killer (NK) cells, and dendritic cells. While CTLA-4 controls the T-cell activity in the initial phase of immune reaction, PD-1 regulates the late activity of the immune phase in the tumor microenvironment.
The epigenetic modulation of immune checkpoints has revealed crucial mechanisms [168]. An elevated risk of gastric cancer was correlated with enhanced hypermethylation of CTLA-4 promoter [169]. The cytotoxic activity of NK cells is epigenetically regulated by methylation of the killer cell immunoglobulin-like receptors and the DNMT inhibitor azacytidine restores the function of NK cells [170]. PD-1, PD-L1, and PD-L2 expression are also regulated by promoter methylation where the use of the demethylating agent decitabine results in their upregulation in myelodysplastic syndromes [171]. Clinical trials are studying the efficacy of combining immune checkpoint inhibitors and DNA demethylating agents [172].
The CTLA-4 promoter is controlled by histone acetylation and nuclear factor of activated T-cell binding (NFAT) [173]. PD-1 expression on CD8 T cells is rich with enhancer sequences containing acetylation of H3K27 and monomethylation of H3K4 which are markers of activated transcription [174]. In response to several cytokines, the T helper (TH)-1 genes TBET and INFγ show elevated expression due to promoter acetylation [175]. Several clinical trials are evaluating the promise of combining immune checkpoint inhibitors and HDAC inhibitors [168].
Numerous studies point out to the link between miRNAs and immune checkpoints regulation. PD-1 is regulated in cancer by miR-138-5p [176]. Several miRNAs target PD-L1 such as miR-34a-5p, miR-200, miR-513a-5p, and miR-570-3p [19]. CTLA-4 is directly targeted by miR-138 and has shown efficacy in anti-glioma therapy [177]. A great number of miRNAs loci display CpG rich islands indicating that DNA methylation is a key regulatory mechanism [178]. In lung cancer, p53 was demonstrated to regulate tumor evasion through miR-34 which targets PD-L1 [179]. Several miRNAs regulate epigenetically immune checkpoints and, therefore, present potential therapeutic strategies in targeting these miRNAs either alone or in combination with checkpoint inhibitors.
Several lncRNAs were recently shown to regulate cancer immunity [180]. The lncRNA APOC1P1-3 is overexpressed in breast cancer, due to hypomethylation of its promoter region, and is related to tumor size [181]. APOC1P1-3 binds directly to tubulin resulting in its decreased acetylation, inactivation of caspase-3, and apoptosis inhibition. T cell activity in the tumor microenvironment affects tumor development. Several reports point out to the crucial role of lncRNA in mediating T cell functions. Some lncRNAs regulate Treg differentiation and others abrogate cytotoxic functions [182]. The lncRNA HOTAIR induces retinoic acid-mediated differentiation of myeloid cells to granulocytes [183]. The lncRNA lnc-DC controls dentritic cells differentiation by binding directly to STAT3 and activating it [184]. These few listed examples indicate that lncRNAs can be used as targets for anticancer therapies.
Hanahan and Weinberg have defined two enabling characteristics which facilitate the acquisition of the different hallmarks [5]. In particular, the contributions of the genome instability that lead to random mutations and gross chromosomal abnormalities leading to the different hallmarks. In addition, the role of tumor-promoting inflammation in tumorigenesis starting from the pre-tumorigenic to the benign then malignant phases is well-established [185]. Epigenetic mechanisms and these two enabling characteristics crosstalk and impact each other as discussed next.
How epigenetic mechanisms contribute to genome instability and mutations
Genome instability leads to the buildup of point-mutations and gross chromosomal changes throughout the genome. These in turn lead to the formation of oncogenes and the loss of tumor suppressor genes that favor tumor development. Although genetic mutations are crucial for tumor formation, however, epigenetic mechanisms are as essential. Epigenetic mechanisms modify chromatin structure and can lead to global DNA mutations. Also, epigenetic mechanisms can affect concomitantly the expression of hundreds of genes that affect tumor progression.
The DNA methylation pattern of the genome of cancer cells differs from that of normal ones. Cancer genomes are typically hypermethylated at CpG islands at specific genes and hypomethylated at repetitive DNA elements [186]. For instance the hypermethylation of CpG islands occurs early in approximately 25% of lung and colorectal cancer tissues [187,188]. Several DNA repair genes are silenced by DNA hypermethylation in many tumor types leading to further accumulation of DNA mutations and genome instability. The DNA repair gene MGMT is hypermethylated and silenced in more than 40% of colorectal and brain tumor tissues [189]. The MLH1 gene is hypermethylated and silenced in colorectal, ovarian, and endometrial cancers which results in malfunctioning of the DNA mismatch repair complex and aberrations in microsatellite repeat stability [190-192]. The BRAC1 gene is hypermethylated in medullary breast carcinomas, sporadic mucinous breast carcinomas, and sporadic ovarian carcinomas by 67%, 55%, and 31%, respectively [193,194]. Alternatively global DNA hypomethylation leads to the activation of oncogenes and of chromosomal and centromeric instability [195,196]. Global DNA hypomethylated was suggested to cause chromosomal instability in the early steps of hepatocellular carcinoma [197].
Chromatin modifiers are crucial for the maintenance of chromatin, packing into nucleosomes, and regulated gene expression and their dysregulation is observed in early and advanced phases of neoplastic transformation. This balance is carefully maintained by enzymes that add or remove acetyl groups (HATs versus HDACs), methyl groups (HMTs versus HDMs), and phosphoryl groups (histone kinases versus histone phosphatases). Mutations are detected in cancers in genes encoding HATs, HMTs, and HDMs in particular on lysine residues [198]. For instance, NSD1 and NSD3, two lysine methyl transferases, are subject to chromosomal translocation in acute myeloid leukemia while the lysine HDMs 5A is mutated in this leukemia [199]. HAT mutations are commonly detected in SCLC and B cell lymphomas [200,201]. Several HDACs are overexpressed in cancer and result in silencing of tumor suppressor genes. The following HDACs are upregulated in cancers such HDAC1 in breast, prostate, colorectal, esophageal, and gastric cancers, HDAC2 in gastric, cervical, and colorectal cancer, HDAC3 in colorectal, gastric, and prostate cancer, and HDAC6 in breast cancer [202,203]. The overexpression of HDACs in several cancers is due to the interplay between transcription factors and epigenetic modulators on their promoter regions. HDAC1 and HDAC2 are overexpressed in colorectal cancer due to the binding of the transcriptional factors, Sp1/Sp3, HAT p300, and histone H3K4 methyltransferase SET1 [204]. These results show that the use of HDAC inhibitors may be a therapeutic strategy in reactivating tumor suppressor genes in HDACs overexpressing cancers.
miRNAs are also controlled by epigenetic mechanisms and are commonly altered in human cancers [205]. Several miRNAs, miR-9, miR-124, miR-137, miR-148, and miR-512 are silenced by CpG hypermethylation in some cancers. Some specific miRNAs called “epi-miRNAs” were demonstrated to target epigenetic regulators [206] and their deregulation contributes to genomic instability [207]. miRNA-induced genomic instability may be due to loss of control of the cell cycle checkpoints, DNA repair mechanism, and mitotic separation. miR-372 is an oncomiR that targets p53 and deregulates p53-mediated CDKI and cell cycle regulation through its inhibition of LATS2 [208]. The stability of the genome was also shown to be guarded by p53 and LATS2 to prevent tetraploidization [209]. Therefore, deregulation of miR-372 promotes oncogenic transformation and genomic instability. The oncomiR miR-155 is overexpressed in several cancers and can result in lymphoblastic leukemia [210]. Studies indicated that DNA mismatch genes (MLH1, MSH2, and MSH6) are controlled by miR-155. Colorectal cancer cells overexpressing miR-155 reduced the levels of MLH1, MSH2, and MSH6 and showed microsatellite instability [211]. In addition, miR-155 was shown to damage telomeres by reducing the levels of the telomere binding factor TERF1 leading to genomic instability and telomere fragility in breast cancer [212]. LncRNA can affect chromatin remodeling and gene expression and, therefore, regulate genome stability [213]. It remains to be determined whether lncRNA aberrations by epigenetic mechanisms lead to genome instability.
Tumor-promoting inflammation and epigenetic mechanisms
It is well-established from epidemiological studies that inflammatory conditions enhance cancer incidence [214]. Epidemiological studies estimate that chronic inflammation is linked to 15% of cancer incidence [215]. In particular, the following chronic inflammatory conditions make individuals susceptible to cancer namely infection by H. pylori is linked to high incidence of gastric cancer while hepatitis C virus is associated with liver cancer; inflammatory bowel condition with colorectal cancer; and inflammation due to chronic ultraviolet exposure with skin cancer. Inflammation and cancer are closely linked. While chronic inflammation increases the incidence of cancer and consumption of drugs that reduce inflammation decreases cancer incidence and death [216].
Although acute inflammation may have beneficial effects in fighting infections and repairing tissue damage, however chronic infection by viruses, environmental exposures to irritating agents such as asbestos, ultraviolet radiation, and air pollutants cause chronic inflammation that predisposes individuals to certain cancers. Several signaling pathways are activated due to reactive oxygen species (ROS), inflammatory interleukins (ILs), and cytokine secretions (IL-6, IL-1β, TNF-α), and survival signaling pathways (NF-κB, STAT3, MAPK). In turn, the inflammatory microenvironment accentuates tumor progression by the release of protumorigenic cytokines and growth factors by inflammatory and immune cells.
Epidemiological, in vivo, and in vitro studies show a connection between chronic inflammation and deregulated epigenetic mechanisms in cancer formation [214]. H. pylori infection causes gastric cancer. It was also demonstrated that epigenetic alterations are involved in gastric cancer [217]. Patients infected with H. pylori display a high incidence of E-cadherin promoter methylation and silencing in comparison with non-infected individuals [218]. E-cadherin silencing was detected by more than 50% as early as in intestinal metaplasia and was further increased in tumor invasion and metastasis. In addition, several CpG islands containing genes were hypermethylated in H. pylori-infected mucosa [219]. Epigenetic alterations are also detected in other inflammatory conditions such as ulcerative colitis, that predispose individuals to colorectal cancer, have shown increased methylation of the CDKI p16 [220].
The association studies between inflammatory conditions and epigenetic mechanisms were causally linked in animal studies. Suppression of H. pylori infection in animal model of gastric cancer resulted in reversal of infection-causing methylation changes in gastric mucosa of infected animals [221]. In precancerous colorectal tissues of mice treated with azoxymethane- and dextran sulfate sodium show elevated levels and activity of HDAC and depletion of acetylated H3K27 [222]. Histone hypoacetylation was reversed with aspirin treatment, a nonsteroidal inflammatory drug. The literature is rich in providing more examples relating chronic inflammation and epigenetic mechanisms in early and late phases of tumor formation in vivo and in vitro models [214].
Some of the listed molecular mechanisms that link cancer-causing chronic inflammation and epigenetic mechanisms are alterations of cellular metabolism and cofactors namely S-adenosyl methionine (SAM) needed for several epigenetic enzymes such as DNMTs and HMTs and the cofactor acetyl CoA needed for HAT activity [223]. Also inflammation may affect epigenetic mechanisms through DNA damage. ROS generation in the tumor microenvironment by inflammatory cells can damage DNA. In colorectal cancer, the tumor suppressor gene caudal type homeobox-1 (CDX1) is hypermethylated and silenced due to hydrogen peroxide [224]. DNA is susceptible to ROS-induced DNA damage particularly at guanine bases [225]. Also ROS hinder the ability of DNMTs to bind to hemimethylated DNA resulting in global DNA hypomethylation and genomic instability [226].
Several reports have indicated that inflammation alters miRNA profile in epithelial cells of the colon and, therefore, linking miRNAs to tumor development [227]. There are 11 differentially regulated miRNAs that affected inflammatory chemokine production such as macrophage inflammatory peptide (MIP)-2 alpha that is secreted by the epithelial cells. miRNA profile in epithelial cells can be modified by various mechanisms during inflammation including NF-κB activation, IL-6 induced STAT3 phosphorylation, or cytokine secretion [227,228]. One of these modified during inflammation is the tumor-suppressor miR-7 which targets EGFR. In a mouse model of inflammation-induced gastric cancer, 40 miRNAs were regulated during the different phases of inflammation-induced cancer [229]. Activated macrophages where shown to inhibit miR-7 and contribute to gastric cancer formation. The Let-7 family of miRNAs consists of 12 members which target c-myc and ras [230,231] and their genomic positions are commonly deleted in colorectal cancer and other tumors [232]. miRNA genes are commonly observed at fragile sites of chromosomes and common chromosomal breakpoints. More than 50% of 186 investigated miRNAs were demonstrated to map at fragile sites and in cancer-linked chromosomal regions [232]. miR-31 expression was shown to increase in inflammatory bowel to cancer which targets the negative regulator of HIF-1α and consequently upregulating HIF-1α activity [233]. The miRNAs profile observed in cancer inflammatory tissues can be present in epithelial or immune cells.
Recently, lncRNA profile was shown through a genome profiling to be modified during different phases of tumor progression in cholangiocarcinoma [234]. Several of these lncRNAs regulate genes involved in inflammation and oxidative stress signaling. Another lncRNA lincRNA-Cox2 was shown to control the body inflammatory reactions to injury and infections. In lincRNA-Cox2 knockout animal models, macrophages and mouse tissues had deregulated expression of inflammatory genes [235]. Deletion of lincRNA-Cox2 reduced the expression of cyclooxygenase-2 which is a major enzyme in prostaglandin synthesis. LincRNA-Cox2 functions as an enhancer RNA for the gene Ptgs2 that encodes cyclooxygenase-2.
Conclusions
The hallmarks of cancer by Hanahan and Weinberg have shaped and focused research in cancer biology for the last two decades [4,5]. Although we agree with most of the hallmarks in shaping the chronic process of neoplastic transformation, however, we propose that aberrations of epigenetic mechanisms lie at their hearts or so called “hallmark of hallmarks” (Figure 1). Some of these hallmarks may have overlapping functions such as supporting proliferative signaling, escaping growth suppressors, and counteracting cell death, however each one individually contributes to cancer formation.
Figure 1.
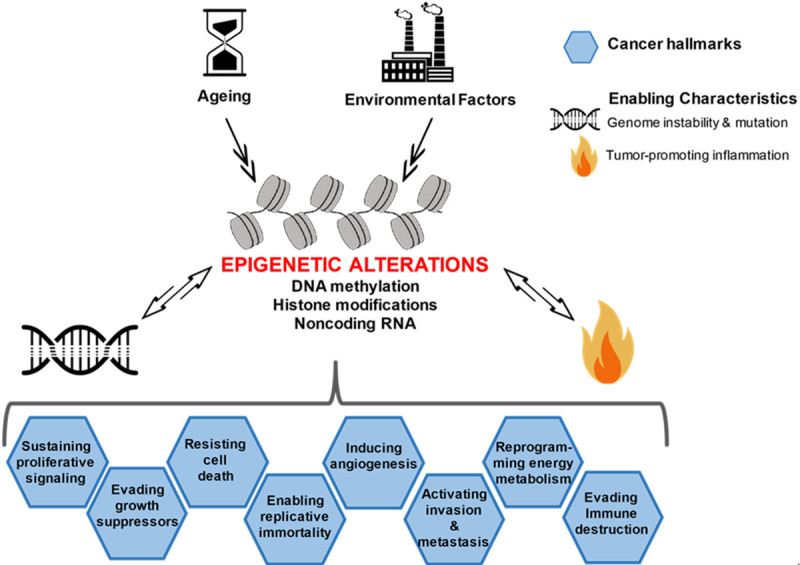
The central role of epigenetic mechanisms in the hallmarks of cancer. Epigenetic alterations comprise DNA methylation, histone modifications, and noncoding RNAs. Ageing and environmental factors collaborate with epigenetic mechanisms to contribute to the different hallmarks. The hallmarks of cancer are shaped by the enabling characteristics: “genomic instability and mutation and tumor-promoting inflammation” that in turn lead to abnormalities in epigenetic mechanisms.
The chronology of the different hallmarks is not crucial in orchestrating the transformation process but what matters is that epigenetic mechanisms are early events in the transformation process and that genomic instability and mutation and tumor promoting inflammation assist in the acquisition of these hallmarks. Previous reports have perceived the different hallmarks as continuous evolutionary events in cancer formation [8,236]. The hallmarks of cancer are shaped by genomic instability and tumor-promoting inflammation that in turn contribute to abnormalities in epigenetic mechanisms. A couple of years before Hanahan and Weinberg suggested that tumor-promoting inflammation is an enabling characteristic of cancer hallmarks, Colotta et al. proposed that “cancer-related inflammation, is the seventh hallmark of cancer and it links to genetic instability” [185].
Ageing and environmental factors collaborate with epigenetic mechanisms to contribute to the different hallmarks. It was observed that hypomethylation-associated ageing may be a contributing factor of cancer and chronic inflammation as it increases the immunogenicity of DNA [237]. Epigenetic damage results from ageing where genome methylation status of individuals resembles general genome hypomethylation observed in cancer genomes. Environmental factors encompass toxicants, diets, and alcohol intake that affect the epigenome [238]. The effects of diets such as energy intake, folate consumption, and polyphenol content may contribute to changes in the epigenome [239]. Some of these alterations may be subtle and occurring earlier than any carcinogenic effect detection. In fact epigenetic deregulation occurs very early such as DNA methylation changes are identified in normal tissues and increase steadily with age [240]. For instance, the EN1 gene expresses engrailed-1 homeobox protein which controls pattern formation during embryogenesis and is widely expressed in the cerebellum [241]. EN1 promoter hypermethylation has been detected in several solid tumors including colorectal [242] and prostate cancers [243]. Hypermethylation was associated with tumor grading and patient clinical outcome as 30% reduction in the 5-year survival rate was observed in colorectal cancer patients compared to those without EN1 hypermethylation [242]. These findings stipulate that aberrant methylation of certain biomarkers such as EN1 gene can be used for early cancer detection.
Loss of imprinting (LOI) of insulin-growth factor 2 (IGF2) may happen early in colorectal cancer cell transformation and may be used as an early marker of cancer. IGF2 LOI results in increased cell proliferation due to activation of the second silent allele resulting in doubling of gene expression of this mitogen [244]. In Barrett’s esophagus, chronic acid reflux results in inflammatory reactions and epigenetic changes well before cancer formation [245]. Epigenetic modifications and DNA methylation changes are common pre-tumorigenic events for decades earlier than cancer appearance. This underscores the usefulness of detecting epigenetic changes early in the carcinogenic process and thus may have chemopreventive and therapeutic implications.
The significance of epigenetic mechanisms in the hallmarks of cancer is their reversibility and, therefore, therapeutic implications. The exciting aspect of epigenetics model of cancer is its central role in the cancer hallmarks and that its dysregulation may act as a “common driver through cancer progression” [22]. Some of these epigenetic mechanisms directly impact the hallmarks of cancer and others may have indirect effects through genomic instability, mutations, and tumor promoting environment as listed in this review. The original hallmarks of cancer are very much designed by the mutation theory, and thus reversibility and therapeutic targeting may not be easily accomplished, hence the crucial role of epigenetic mechanisms in chemoprevention and therapeutic tumor targeting.
Finally, by targeting epigenetic mechanisms we will be aiming at several hallmarks of cancer. This represents a great advantage of “epidrugs” as they aim reversible processes. Unfortunately, attempts to use “epidrugs” has been linked to several genome-wide effects. The use of general DNA methylase inhibitors generated genome instability [246] and the use of general HDAC inhibitors sometimes promoted tumor growth [247]. We have not deciphered yet most functions of ncRNAs. Further research will uncover the crucial role ncRNAs play in the various steps of tumor formation and may contribute substantially to understanding the role of epigenetic mechanisms as biomarkers and therapeutic targets of cancer [18,20].
Acknowledgements
This manuscript was an assignment in the Cancer Biology and Therapeutics, High Impact Cancer Research at Harvard Medical School, class of 2018-2019. The author greatly acknowledges the enthusiasm, dedication, and help of the course directors and staff throughout the program. The author would like to thank Chirine El-Baba for her help in designing Figure 1 and for editing the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Paduch R. Theories of cancer origin. Eur J Cancer Prev. 2015;24:57–67. doi: 10.1097/CEJ.0000000000000024. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer. An organizing principle for cancer medicine. Cancer: Principles & Practice of Oncology. 2015 [Google Scholar]
- 7.Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:e1007362. doi: 10.1371/journal.pgen.1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horne SD, Pollick SA, Heng HH. Evolutionary mechanism unifies the hallmarks of cancer. Int J Cancer. 2015;136:2012–2021. doi: 10.1002/ijc.29031. [DOI] [PubMed] [Google Scholar]
- 9.Brucher BL, Jamall IS. Somatic mutation theory - why it’s wrong for most cancers. Cell Physiol Biochem. 2016;38:1663–1680. doi: 10.1159/000443106. [DOI] [PubMed] [Google Scholar]
- 10.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16:803–810. doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- 13.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 14.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 15.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira HJ, Esteller M. Non-coding RNAs, epigenetics, and cancer: tying it all together. Cancer Metastasis Rev. 2018;37:55–73. doi: 10.1007/s10555-017-9715-8. [DOI] [PubMed] [Google Scholar]
- 19.Van Roosbroeck K, Calin GA. Cancer hallmarks and microRNAs: the therapeutic connection. Adv Cancer Res. 2017;135:119–149. doi: 10.1016/bs.acr.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703–719. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497. doi: 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 24.Blair LP, Yan Q. Epigenetic mechanisms in commonly occurring cancers. DNA Cell Biol. 2012;31(Suppl 1):S49–61. doi: 10.1089/dna.2012.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaneko Y, Shibuya M, Nakayama T, Hayashida N, Toda G, Endo Y, Oka H, Oda T. Hypomethylation of c-myc and epidermal growth factor receptor genes in human hepatocellular carcinoma and fetal liver. Jpn J Cancer Res. 1985;76:1136–1140. [PubMed] [Google Scholar]
- 26.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730–741. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- 29.Zhu X, Asa SL, Ezzat S. Histone-acetylated control of fibroblast growth factor receptor 2 intron 2 polymorphisms and isoform splicing in breast cancer. Mol Endocrinol. 2009;23:1397–1405. doi: 10.1210/me.2009-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muntean AG, Hess JL. Epigenetic dysregulation in cancer. Am J Pathol. 2009;175:1353–1361. doi: 10.2353/ajpath.2009.081142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9:3–12. doi: 10.4161/epi.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, O’Brien-Jenkins A, Katsaros D, Hatzigeorgiou A, Gimotty PA, Weber BL, Coukos G. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svoronos AA, Engelman DM, Slack FJ. OncomiR or tumor suppressor? The duplicity of microRNAs in cancer. Cancer Res. 2016;76:3666–3670. doi: 10.1158/0008-5472.CAN-16-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 36.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Git A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 37.Chen LL, Carmichael GG. Long noncoding RNAs in mammalian cells: what, where, and why? Wiley Interdiscip Rev RNA. 2010;1:2–21. doi: 10.1002/wrna.5. [DOI] [PubMed] [Google Scholar]
- 38.Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O’Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 39.Leygue E, Dotzlaw H, Watson PH, Murphy LC. Expression of the steroid receptor RNA activator in human breast tumors. Cancer Res. 1999;59:4190–4193. [PubMed] [Google Scholar]
- 40.Chooniedass-Kothari S, Hamedani MK, Troup S, Hube F, Leygue E. The steroid receptor RNA activator protein is expressed in breast tumor tissues. Int J Cancer. 2006;118:1054–1059. doi: 10.1002/ijc.21425. [DOI] [PubMed] [Google Scholar]
- 41.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, Cao X, Jing X, Wang X, Siddiqui J, Wei JT, Robinson D, Iyer HK, Palanisamy N, Maher CA, Chinnaiyan AM. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong B, Langerod A, Borresen-Dale AL, Kim SK, van de Vijver M, Sukumar S, Whitfield ML, Kellis M, Xiong Y, Wong DJ, Chang HY. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 44.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci. 2018;19:1166. doi: 10.3390/ijms19041166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De La Rosa-Velazquez IA, Rincon-Arano H, Benitez-Bribiesca L, Recillas-Targa F. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. 2007;67:2577–2585. doi: 10.1158/0008-5472.CAN-06-2024. [DOI] [PubMed] [Google Scholar]
- 47.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin SG, Xiong W, Wu X, Yang L, Pfeifer GP. The DNA methylation landscape of human melanoma. Genomics. 2015;106:322–330. doi: 10.1016/j.ygeno.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiss NB, Kogner P, Johnsen JI, Martinsson T, Larsson C, Geli J. Quantitative global and gene-specific promoter methylation in relation to biological properties of neuroblastomas. BMC Med Genet. 2012;13:83. doi: 10.1186/1471-2350-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 51.Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6:579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Zhang C, Zhao Y, Feng Z. MicroRNA control of p53. J Cell Biochem. 2017;118:7–14. doi: 10.1002/jcb.25609. [DOI] [PubMed] [Google Scholar]
- 53.Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12:613–626. doi: 10.1038/nrc3318. [DOI] [PubMed] [Google Scholar]
- 54.Chaudhary R, Lal A. Long noncoding RNAs in the p53 network. Wiley Interdiscip Rev RNA. 2017;8 doi: 10.1002/wrna.1410. 10.1002/wrna.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdelmohsen K, Panda AC, Kang MJ, Guo R, Kim J, Grammatikakis I, Yoon JH, Dudekula DB, Noh JH, Yang X, Martindale JL, Gorospe M. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014;42:10099–10111. doi: 10.1093/nar/gku686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naemura M, Murasaki C, Inoue Y, Okamoto H, Kotake Y. Long noncoding RNA ANRIL regulates proliferation of non-small cell lung cancer and cervical cancer cells. Anticancer Res. 2015;35:5377–5382. [PubMed] [Google Scholar]
- 57.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 58.Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorstyn L, Akey CW, Kumar S. New insights into apoptosome structure and function. Cell Death Differ. 2018;25:1194–1208. doi: 10.1038/s41418-017-0025-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sengupta D, Avaritt N, Tackett A. Epigenetic deregulation of Bcl-2 leads to apoptosis resistance in melanoma (942.2) FASEB J. 2014;28:942. [Google Scholar]
- 61.Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–1828. [PubMed] [Google Scholar]
- 62.Hervouet E, Vallette FM, Cartron PF. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010;1:e8. doi: 10.1038/cddis.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cui J, Placzek WJ. Post-transcriptional regulation of anti-apoptotic BCL2 family members. Int J Mol Sci. 2018;19:308. doi: 10.3390/ijms19010308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hao W, Luo W, Bai M, Li J, Bai X, Guo J, Wu J, Wang M. MicroRNA-206 inhibited the progression of glioblastoma through BCL-2. J Mol Neurosci. 2016;60:531–538. doi: 10.1007/s12031-016-0824-6. [DOI] [PubMed] [Google Scholar]
- 67.Sun Y, Hu B, Wang Q, Ye M, Qiu Q, Zhou Y, Zeng F, Zhang X, Guo Y, Guo L. Long non-coding RNA HOTTIP promotes BCL-2 expression and induces chemoresistance in small cell lung cancer by sponging miR-216a. Cell Death Dis. 2018;9:85. doi: 10.1038/s41419-017-0113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6:a026070. doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, Tien HF. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119:559–568. doi: 10.1182/blood-2011-07-369934. [DOI] [PubMed] [Google Scholar]
- 70.Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Semin Cancer Biol. 2011;21:349–353. doi: 10.1016/j.semcancer.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 72.Lewis KA, Tollefsbol TO. Regulation of the telomerase reverse transcriptase subunit through epigenetic mechanisms. Front Genet. 2016;7:83. doi: 10.3389/fgene.2016.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Renaud S, Loukinov D, Abdullaev Z, Guilleret I, Bosman FT, Lobanenkov V, Benhattar J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007;35:1245–1256. doi: 10.1093/nar/gkl1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi JH, Min NY, Park J, Kim JH, Park SH, Ko YJ, Kang Y, Moon YJ, Rhee S, Ham SW, Park AJ, Lee KH. TSA-induced DNMT1 down-regulation represses hTERT expression via recruiting CTCF into demethylated core promoter region of hTERT in HCT116. Biochem Biophys Res Commun. 2010;391:449–454. doi: 10.1016/j.bbrc.2009.11.078. [DOI] [PubMed] [Google Scholar]
- 75.Valls-Bautista C, Bougel S, Pinol-Felis C, Vinas-Salas J, Benhattar J. hTERT methylation is necessary but not sufficient for telomerase activity in colorectal cells. Oncol Lett. 2011;2:1257–1260. doi: 10.3892/ol.2011.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 77.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao Q, Zhai YX, Liu HQ, Shi YA, Li XB. MicroRNA-491-5p suppresses cervical cancer cell growth by targeting hTERT. Oncol Rep. 2015;34:979–986. doi: 10.3892/or.2015.4013. [DOI] [PubMed] [Google Scholar]
- 79.Chen L, Lu MH, Zhang D, Hao NB, Fan YH, Wu YY, Wang SM, Xie R, Fang DC, Zhang H, Hu CJ, Yang SM. miR-1207-5p and miR-1266 suppress gastric cancer growth and invasion by targeting telomerase reverse transcriptase. Cell Death Dis. 2014;5:e1034. doi: 10.1038/cddis.2013.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang D, Xiao YF, Zhang JW, Xie R, Hu CJ, Tang B, Wang SM, Wu YY, Hao NB, Yang SM. miR-1182 attenuates gastric cancer proliferation and metastasis by targeting the open reading frame of hTERT. Cancer Lett. 2015;360:151–159. doi: 10.1016/j.canlet.2015.01.044. [DOI] [PubMed] [Google Scholar]
- 81.Lu MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, Wang SM, He FT, Yang SM. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016;35:3524–3534. doi: 10.1038/onc.2015.413. [DOI] [PubMed] [Google Scholar]
- 82.El Hajj J, Nguyen E, Liu Q, Bouyer C, Adriaenssens E, Hilal G, Segal-Bendirdjian E. Telomerase regulation by the long non-coding RNA H19 in human acute promyelocytic leukemia cells. Mol Cancer. 2018;17:85. doi: 10.1186/s12943-018-0835-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cacchione S, Biroccio A, Rizzo A. Emerging roles of telomeric chromatin alterations in cancer. J Exp Clin Cancer Res. 2019;38:21. doi: 10.1186/s13046-019-1030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohammad F, Helin K. Oncohistones: drivers of pediatric cancers. Genes Dev. 2017;31:2313–2324. doi: 10.1101/gad.309013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19:329–337. doi: 10.1016/j.semcancer.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Pirola L, Ciesielski O, Balcerczyk A. The methylation status of the epigenome: its emerging role in the regulation of tumor angiogenesis and tumor growth, and potential for drug targeting. Cancers (Basel) 2018;10:268. doi: 10.3390/cancers10080268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 88.Ping SY, Shen KH, Yu DS. Epigenetic regulation of vascular endothelial growth factor a dynamic expression in transitional cell carcinoma. Mol Carcinog. 2013;52:568–579. doi: 10.1002/mc.21892. [DOI] [PubMed] [Google Scholar]
- 89.Quentmeier H, Eberth S, Romani J, Weich HA, Zaborski M, Drexler HG. DNA methylation regulates expression of VEGF-R2 (KDR) and VEGF-R3 (FLT4) BMC Cancer. 2012;12:19. doi: 10.1186/1471-2407-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shiao YH. The von Hippel-Lindau gene and protein in tumorigenesis and angiogenesis: a potential target for therapeutic designs. Curr Med Chem. 2003;10:2461–2470. doi: 10.2174/0929867033456639. [DOI] [PubMed] [Google Scholar]
- 91.Kondo K, Kaelin WG Jr. The von Hippel-Lindau tumor suppressor gene. Exp Cell Res. 2001;264:117–125. doi: 10.1006/excr.2000.5139. [DOI] [PubMed] [Google Scholar]
- 92.Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep. 2009;11:94–101. doi: 10.1007/s11912-009-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, Muller-Tidow C, Burdach S. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–5329. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khorshidi A, Dhaliwal P, Yang BB. Noncoding RNAs in tumor angiogenesis. Adv Exp Med Biol. 2016;927:217–241. doi: 10.1007/978-981-10-1498-7_8. [DOI] [PubMed] [Google Scholar]
- 95.Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett. 2009;285:116–126. doi: 10.1016/j.canlet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 96.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1:e116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A. 2007;104:20350–20355. doi: 10.1073/pnas.0706901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yu B, Wang S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics. 2018;8:3654–3675. doi: 10.7150/thno.26024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 101.Yuan SX, Yang F, Yang Y, Tao QF, Zhang J, Huang G, Yang Y, Wang RY, Yang S, Huo XS, Zhang L, Wang F, Sun SH, Zhou WP. Long noncoding RNA associated with microvascular invasion in hepatocellular carcinoma promotes angiogenesis and serves as a predictor for hepatocellular carcinoma patients’ poor recurrence-free survival after hepatectomy. Hepatology. 2012;56:2231–2241. doi: 10.1002/hep.25895. [DOI] [PubMed] [Google Scholar]
- 102.Huang JL, Zheng L, Hu YW, Wang Q. Characteristics of long non-coding RNA and its relation to hepatocellular carcinoma. Carcinogenesis. 2014;35:507–514. doi: 10.1093/carcin/bgt405. [DOI] [PubMed] [Google Scholar]
- 103.Thum T, Fiedler J. LINCing MALAT1 and angiogenesis. Circ Res. 2014;114:1366–1368. doi: 10.1161/CIRCRESAHA.114.303896. [DOI] [PubMed] [Google Scholar]
- 104.Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 105.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 107.Sun L, Fang J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol Life Sci. 2016;73:4493–4515. doi: 10.1007/s00018-016-2303-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 109.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13:373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez-Perez A, Jene-Sanz A, Lopez-Bigas N. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013;14:r106. doi: 10.1186/gb-2013-14-9-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koizume S, Tachibana K, Sekiya T, Hirohashi S, Shiraishi M. Heterogeneity in the modification and involvement of chromatin components of the CpG island of the silenced human CDH1 gene in cancer cells. Nucleic Acids Res. 2002;30:4770–4780. doi: 10.1093/nar/gkf593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kapoor-Vazirani P, Kagey JD, Powell DR, Vertino PM. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008;68:6810–6821. doi: 10.1158/0008-5472.CAN-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24:8277–8290. doi: 10.1038/sj.onc.1208991. [DOI] [PubMed] [Google Scholar]
- 115.Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, Friess H, Buchler M, Evert M, Lerch MM, Weiss FU. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 116.Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, Dai Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. 2012;31:4619. doi: 10.1038/onc.2011.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, Shiah SG, Chen PS, Jeng YM, Cheng TY, Lai TC, Chang JS, Jan YH, Chien MH, Yang CJ, Huang MS, Hsiao M, Kuo ML. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010;70:7830–7840. doi: 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- 118.Wang LK, Hsiao TH, Hong TM, Chen HY, Kao SH, Wang WL, Yu SL, Lin CW, Yang PC. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PLoS One. 2014;9:e96765. doi: 10.1371/journal.pone.0096765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Streppel MM, Pai S, Campbell NR, Hu C, Yabuuchi S, Canto MI, Wang JS, Montgomery EA, Maitra A. MicroRNA 223 is upregulated in the multistep progression of Barrett’s esophagus and modulates sensitivity to chemotherapy by targeting PARP1. Clin Cancer Res. 2013;19:4067–4078. doi: 10.1158/1078-0432.CCR-13-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abba ML, Patil N, Leupold JH, Allgayer H. MicroRNA regulation of epithelial to mesenchymal transition. J Clin Med. 2016;5:8. doi: 10.3390/jcm5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 124.Huang Q, Yan J, Agami R. Long non-coding RNAs in metastasis. Cancer Metastasis Rev. 2018;37:75–81. doi: 10.1007/s10555-017-9713-x. [DOI] [PubMed] [Google Scholar]
- 125.Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, Tchou J, Liu Q, Zhang L, Speicher DW, Calin GA, Huang Q. Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. EMBO J. 2013;32:2672–2684. doi: 10.1038/emboj.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li Y, Wang Z, Shi H, Li H, Li L, Fang R, Cai X, Liu B, Zhang X, Ye L. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Res. 2016;76:293–304. doi: 10.1158/0008-5472.CAN-14-3607. [DOI] [PubMed] [Google Scholar]
- 127.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, Miyano S, Mori M. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 129.Wu ZH, Wang XL, Tang HM, Jiang T, Chen J, Lu S, Qiu GQ, Peng ZH, Yan DW. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol Rep. 2014;32:395–402. doi: 10.3892/or.2014.3186. [DOI] [PubMed] [Google Scholar]
- 130.Xu F, Zhang J. Long non-coding RNA HOTAIR functions as miRNA sponge to promote the epithelial to mesenchymal transition in esophageal cancer. Biomed Pharmacother. 2017;90:888–896. doi: 10.1016/j.biopha.2017.03.103. [DOI] [PubMed] [Google Scholar]
- 131.Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z, Deng X, Chen H, Shen B, Peng C, Li H, Zhan Q, Zhu Z. Amplification of long noncoding RNA ZFAS1 promotes metastasis in hepatocellular carcinoma. Cancer Res. 2015;75:3181–3191. doi: 10.1158/0008-5472.CAN-14-3721. [DOI] [PubMed] [Google Scholar]
- 132.Wu Y, Liu H, Shi X, Yao Y, Yang W, Song Y. The long non-coding RNA HNF1A-AS1 regulates proliferation and metastasis in lung adenocarcinoma. Oncotarget. 2015;6:9160–9172. doi: 10.18632/oncotarget.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kang YP, Ward NP, DeNicola GM. Recent advances in cancer metabolism: a technological perspective. Exp Mol Med. 2018;50:31. doi: 10.1038/s12276-018-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24:1161–1180. doi: 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iurlaro R, Leon-Annicchiarico CL, Munoz-Pinedo C. Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol. 2014;542:59–80. doi: 10.1016/B978-0-12-416618-9.00003-0. [DOI] [PubMed] [Google Scholar]
- 139.Min HY, Lee HY. Oncogene-driven metabolic alterations in cancer. Biomol Ther (Seoul) 2018;26:45–56. doi: 10.4062/biomolther.2017.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 141.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE. p53 and metabolism: from mechanism to therapeutics. Oncotarget. 2018;9:23780–23823. doi: 10.18632/oncotarget.25267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, Clem BF. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2013;33:556. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene. 2017;36:3359–3374. doi: 10.1038/onc.2016.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Miranda-Goncalves V, Lameirinhas A, Henrique R, Jeronimo C. Metabolism and epigenetic interplay in cancer: regulation and putative therapeutic targets. Front Genet. 2018;9:427. doi: 10.3389/fgene.2018.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39:231–234. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- 147.Lopez-Serra P, Marcilla M, Villanueva A, Ramos-Fernandez A, Palau A, Leal L, Wahi JE, Setien-Baranda F, Szczesna K, Moutinho C, Martinez-Cardus A, Heyn H, Sandoval J, Puertas S, Vidal A, Sanjuan X, Martinez-Balibrea E, Vinals F, Perales JC, Bramsem JB, Orntoft TF, Andersen CL, Tabernero J, McDermott U, Boxer MB, Vander Heiden MG, Albar JP, Esteller M. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat Commun. 2014;5:3608. doi: 10.1038/ncomms4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, Yu J, Chan FK, Sung JJ, Jin HC. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. 2010;29:442–450. doi: 10.1038/onc.2009.332. [DOI] [PubMed] [Google Scholar]
- 149.Li H, Wang J, Xu H, Xing R, Pan Y, Li W, Cui J, Zhang H, Lu Y. Decreased fructose-1,6-bisphosphatase-2 expression promotes glycolysis and growth in gastric cancer cells. Mol Cancer. 2013;12:110. doi: 10.1186/1476-4598-12-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, Sapin V, Bignon YJ, Morvan D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS One. 2014;9:e102438. doi: 10.1371/journal.pone.0102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Carafa V, Altucci L, Nebbioso A. Dual tumor suppressor and tumor promoter action of sirtuins in determining malignant phenotype. Front Pharmacol. 2019;10:38. doi: 10.3389/fphar.2019.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, McGlynn L, Maxwell F, Edwards J, Giacosa S, Guccione E, Weissleder R, Bernstein BE, Regev A, Shiels PG, Lombard DB, Mostoslavsky R. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ye J, Zou M, Li P, Liu H. MicroRNA regulation of energy metabolism to induce chemoresistance in cancers. Technol Cancer Res Treat. 2018;17:1533033818805997. doi: 10.1177/1533033818805997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Masliah-Planchon J, Garinet S, Pasmant E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget. 2016;7:38892–38907. doi: 10.18632/oncotarget.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yamasaki T, Seki N, Yoshino H, Itesako T, Yamada Y, Tatarano S, Hidaka H, Yonezawa T, Nakagawa M, Enokida H. Tumor-suppressive microRNA-1291 directly regulates glucose transporter 1 in renal cell carcinoma. Cancer Sci. 2013;104:1411–1419. doi: 10.1111/cas.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fei X, Qi M, Wu B, Song Y, Wang Y, Li T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012;586:392–397. doi: 10.1016/j.febslet.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 157.Chen YH, Heneidi S, Lee JM, Layman LC, Stepp DW, Gamboa GM, Chen BS, Chazenbalk G, Azziz R. miRNA-93 inhibits GLUT4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62:2278–2286. doi: 10.2337/db12-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kefas B, Comeau L, Erdle N, Montgomery E, Amos S, Purow B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncol. 2010;12:1102–1112. doi: 10.1093/neuonc/noq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rathore MG, Saumet A, Rossi JF, de Bettignies C, Tempe D, Lecellier CH, Villalba M. The NF-kappaB member p65 controls glutamine metabolism through miR-23a. Int J Biochem Cell Biol. 2012;44:1448–1456. doi: 10.1016/j.biocel.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 160.Sun H, Huang Z, Sheng W, Xu MD. Emerging roles of long non-coding RNAs in tumor metabolism. J Hematol Oncol. 2018;11:106. doi: 10.1186/s13045-018-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007;67:3963–3969. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 162.Zou ZW, Ma C, Medoro L, Chen L, Wang B, Gupta R, Liu T, Yang XZ, Chen TT, Wang RZ, Zhang WJ, Li PD. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing side-population stem-like cancer cells. Oncotarget. 2016;7:61741–61754. doi: 10.18632/oncotarget.11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao L, Ji G, Le X, Wang C, Xu L, Feng M, Zhang Y, Yang H, Xuan Y, Yang Y, Lei L, Yang Q, Lau WB, Lau B, Chen Y, Deng X, Yao S, Yi T, Zhao X, Wei Y, Zhou S. Long noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 2017;77:1369–1382. doi: 10.1158/0008-5472.CAN-16-1615. [DOI] [PubMed] [Google Scholar]
- 164.Rupaimoole R, Lee J, Haemmerle M, Ling H, Previs RA, Pradeep S, Wu SY, Ivan C, Ferracin M, Dennison JB, Millward NMZ, Nagaraja AS, Gharpure KM, McGuire M, Sam N, Armaiz-Pena GN, Sadaoui NC, Rodriguez-Aguayo C, Calin GA, Drapkin RI, Kovacs J, Mills GB, Zhang W, Lopez-Berestein G, Bhattacharya PK, Sood AK. Long noncoding RNA ceruloplasmin promotes cancer growth by altering glycolysis. Cell Rep. 2015;13:2395–2402. doi: 10.1016/j.celrep.2015.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li Z, Li X, Wu S, Xue M, Chen W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014;105:951–955. doi: 10.1111/cas.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 167.Fan L, Li Y, Chen JY, Zheng YF, Xu XM. Immune checkpoint modulators in cancer immunotherapy: recent advances and combination rationales. Cancer Lett. 2019;456:23–28. doi: 10.1016/j.canlet.2019.03.050. [DOI] [PubMed] [Google Scholar]
- 168.Ali MA, Matboli M, Tarek M, Reda M, Kamal KM, Nouh M, Ashry AM, El-Bab AF, Mesalam HA, Shafei AE, Abdel-Rahman O. Epigenetic regulation of immune checkpoints: another target for cancer immunotherapy? Immunotherapy. 2017;9:99–108. doi: 10.2217/imt-2016-0111. [DOI] [PubMed] [Google Scholar]
- 169.Kordi-Tamandani DM, Davani SK, Baranzehi T, Hemati S. Analysis of promoter methylation, polymorphism and expression profile of cytotoxic T-lymphocyte-associated antigen-4 in patients with gastric cancer. J Gastrointestin Liver Dis. 2014;23:249–253. doi: 10.15403/jgld.2014.1121.233.dmkt. [DOI] [PubMed] [Google Scholar]
- 170.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, Rodgers K, Yen RW, Zahnow CA, Taube JM, Brahmer JR, Tykodi SS, Easton K, Carvajal RD, Jones PA, Laird PW, Weisenberger DJ, Tsai S, Juergens RA, Topalian SL, Rudin CM, Brock MV, Pardoll D, Baylin SB. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4:2067–2079. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, Parmar S, Cortes J, Kantarjian H, Garcia-Manero G. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–1288. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fazio C, Covre A, Cutaia O, Lofiego MF, Tunici P, Chiarucci C, Cannito S, Giacobini G, Lowder JN, Ferraldeschi R, Taverna P, Di Giacomo AM, Coral S, Maio M. Immunomodulatory properties of DNA hypomethylating agents: selecting the optimal epigenetic partner for cancer immunotherapy. Front Pharmacol. 2018;9:1443. doi: 10.3389/fphar.2018.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gibson HM, Hedgcock CJ, Aufiero BM, Wilson AJ, Hafner MS, Tsokos GC, Wong HK. Induction of the CTLA-4 gene in human lymphocytes is dependent on NFAT binding the proximal promoter. J Immunol. 2007;179:3831–3840. doi: 10.4049/jimmunol.179.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, Boss JM. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211:515–527. doi: 10.1084/jem.20130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Morinobu A, Kanno Y, O’Shea JJ. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J Biol Chem. 2004;279:40640–40646. doi: 10.1074/jbc.M407576200. [DOI] [PubMed] [Google Scholar]
- 176.Smolle MA, Calin HN, Pichler M, Calin GA. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J. 2017;284:1952–1966. doi: 10.1111/febs.14030. [DOI] [PubMed] [Google Scholar]
- 177.Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, Ling X, Huang N, Qiao W, Zhou S, Ivan C, Fuller GN, Gilbert MR, Overwijk W, Calin GA, Heimberger AB. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016;18:639–648. doi: 10.1093/neuonc/nov292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–1005. doi: 10.4161/cc.6.9.4209. [DOI] [PubMed] [Google Scholar]
- 179.Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, Kelnar K, Martin D, Komaki R, Gomez DR, Krishnan S, Calin GA, Bader AG, Welsh JW. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303. doi: 10.1093/jnci/djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Denaro N, Merlano MC, Lo Nigro C. Long noncoding RNAs as regulators of cancer immunity. Mol Oncol. 2019;13:61–73. doi: 10.1002/1878-0261.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liao XH, Wang JG, Li LY, Zhou DM, Ren KH, Jin YT, Lv L, Yu JG, Yang JY, Lu Q, Zou Q, Yu J, Liu XP, Zhou P. Long intergenic non-coding RNA APOC1P1-3 inhibits apoptosis by decreasing alpha-tubulin acetylation in breast cancer. Cell Death Dis. 2016;7:e2236. doi: 10.1038/cddis.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hu G, Tang Q, Sharma S, Yu F, Escobar TM, Muljo SA, Zhu J, Zhao K. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol. 2013;14:1190–1198. doi: 10.1038/ni.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang X, Weissman SM, Newburger PE. Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle progression during myeloid maturation in NB4 human promyelocytic leukemia cells. RNA Biol. 2014;11:777–787. doi: 10.4161/rna.28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, Cao X. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–313. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 185.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 186.Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet. 2010;70:277–308. doi: 10.1016/B978-0-12-380866-0.60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998;95:11891–11896. doi: 10.1073/pnas.95.20.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zou HZ, Yu BM, Wang ZW, Sun JY, Cang H, Gao F, Li DH, Zhao R, Feng GG, Yi J. Detection of aberrant p16 methylation in the serum of colorectal cancer patients. Clin Cancer Res. 2002;8:188–191. [PubMed] [Google Scholar]
- 189.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 190.Menigatti M, Di Gregorio C, Borghi F, Sala E, Scarselli A, Pedroni M, Foroni M, Benatti P, Roncucci L, Ponz de Leon M, Percesepe A. Methylation pattern of different regions of the MLH1 promoter and silencing of gene expression in hereditary and sporadic colorectal cancer. Genes Chromosomes Cancer. 2001;31:357–361. doi: 10.1002/gcc.1154. [DOI] [PubMed] [Google Scholar]
- 191.Ozdemir F, Altinisik J, Karateke A, Coksuer H, Buyru N. Methylation of tumor suppressor genes in ovarian cancer. Exp Ther Med. 2012;4:1092–1096. doi: 10.3892/etm.2012.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bischoff J, Ignatov A, Semczuk A, Schwarzenau C, Ignatov T, Krebs T, Kuster D, Przadka-Rabaniuk D, Roessner A, Costa SD, Schneider-Stock R. hMLH1 promoter hypermethylation and MSI status in human endometrial carcinomas with and without metastases. Clin Exp Metastasis. 2012;29:889–900. doi: 10.1007/s10585-012-9478-0. [DOI] [PubMed] [Google Scholar]
- 193.Dobrovic A, Simpfendorfer D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997;57:3347–3350. [PubMed] [Google Scholar]
- 194.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 195.Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Fuchs CS. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2008;122:2767–2773. doi: 10.1002/ijc.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Martinez JG, Perez-Escuredo J, Castro-Santos P, Marcos CA, Pendas JL, Fraga MF, Hermsen MA. Hypomethylation of LINE-1, and not centromeric SAT-alpha, is associated with centromeric instability in head and neck squamous cell carcinoma. Cell Oncol (Dordr) 2012;35:259–267. doi: 10.1007/s13402-012-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nishida N, Kudo M, Nishimura T, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, Minami Y, Ueshima K, Sakurai T, Yokomichi N, Nagasaka T, Goel A. Unique association between global DNA hypomethylation and chromosomal alterations in human hepatocellular carcinoma. PLoS One. 2013;8:e72312. doi: 10.1371/journal.pone.0072312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 199.Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NC, Cheng JF, Aplan PD, Kearney L, Boultwood J, Wainscoat JS. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98:1264–1267. doi: 10.1182/blood.v98.4.1264. [DOI] [PubMed] [Google Scholar]
- 200.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, Altmuller J, Baessmann I, Becker C, de Wilde B, Vandesompele J, Bohm D, Ansen S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grutter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Lund-Iversen M, Sanger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Buttner R, Wolf J, Nurnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, Mullighan CG, Gaidano G, Rabadan R, Brindle PK, Dalla-Favera R. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 203.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 204.Yang H, Salz T, Zajac-Kaye M, Liao D, Huang S, Qiu Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 2014;28:4265–4279. doi: 10.1096/fj.14-250654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kunej T, Godnic I, Ferdin J, Horvat S, Dovc P, Calin GA. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res. 2011;717:77–84. doi: 10.1016/j.mrfmmm.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 206.Liu X, Chen X, Yu X, Tao Y, Bode AM, Dong Z, Cao Y. Regulation of microRNAs by epigenetics and their interplay involved in cancer. J Exp Clin Cancer Res. 2013;32:96. doi: 10.1186/1756-9966-32-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Vincent K, Pichler M, Lee GW, Ling H. MicroRNAs, genomic instability and cancer. Int J Mol Sci. 2014;15:14475–14491. doi: 10.3390/ijms150814475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga LH, Agami R. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 209.Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, Adair B, Vannini I, Fanini F, Bottoni A, Costinean S, Sandhu SK, Nuovo GJ, Alder H, Gafa R, Calore F, Ferracin M, Lanza G, Volinia S, Negrini M, McIlhatton MA, Amadori D, Fishel R, Croce CM. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107:6982–6987. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dinami R, Ercolani C, Petti E, Piazza S, Ciani Y, Sestito R, Sacconi A, Biagioni F, le Sage C, Agami R, Benetti R, Mottolese M, Schneider C, Blandino G, Schoeftner S. miR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res. 2014;74:4145–4156. doi: 10.1158/0008-5472.CAN-13-2038. [DOI] [PubMed] [Google Scholar]
- 213.D’Alessandro G, di Fagagna FdA. Long non-coding RNA in the control of genome stability and cancer phenotypes. Non-coding RNA Investigation. 2018:2. [Google Scholar]
- 214.Maiuri AR, O’Hagan HM. Interplay between inflammation and epigenetic changes in cancer. Prog Mol Biol Transl Sci. 2016;144:69–117. doi: 10.1016/bs.pmbts.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 215.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 217.Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell. 2004;5:121–125. doi: 10.1016/s1535-6108(04)00033-9. [DOI] [PubMed] [Google Scholar]
- 218.Chan AO, Lam SK, Wong BC, Wong WM, Yuen MF, Yeung YH, Hui WM, Rashid A, Kwong YL. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, Tamura G, Saito D, Sugimura T, Ichinose M, Ushijima T. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 220.Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942–3945. [PubMed] [Google Scholar]
- 221.Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 222.Guo Y, Liu Y, Zhang C, Su ZY, Li W, Huang MT, Kong AN. The epigenetic effects of aspirin: the modification of histone H3 lysine 27 acetylation in the prevention of colon carcinogenesis in azoxymethane- and dextran sulfate sodium-treated CF-1 mice. Carcinogenesis. 2016;37:616–624. doi: 10.1093/carcin/bgw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett. 2015;356:309–314. doi: 10.1016/j.canlet.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang R, Kang KA, Kim KC, Na SY, Chang WY, Kim GY, Kim HS, Hyun JW. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene. 2013;524:214–219. doi: 10.1016/j.gene.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 225.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. Journal of The American Chemical Society. 1997;119:617–618. [Google Scholar]
- 226.Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008;266:6–11. doi: 10.1016/j.canlet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 227.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, Brant SR, Chakravarti S, Kwon JH. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135:1624–1635. e24. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 228.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31:37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kong D, Piao YS, Yamashita S, Oshima H, Oguma K, Fushida S, Fujimura T, Minamoto T, Seno H, Yamada Y, Satou K, Ushijima T, Ishikawa TO, Oshima M. Inflammation-induced repression of tumor suppressor miR-7 in gastric tumor cells. Oncogene. 2012;31:3949–3960. doi: 10.1038/onc.2011.558. [DOI] [PubMed] [Google Scholar]
- 230.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 231.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 232.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, Cheng Y, Jin Z, Yang J, Agarwal R, Abraham JM, Dassopoulos T, Harris M, Bayless TM, Kwon J, Harpaz N, Livak F, Meltzer SJ. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis. 2011;17:221–231. doi: 10.1002/ibd.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Han BW, Ye H, Wei PP, He B, Han C, Chen ZH, Chen YQ, Wang WT. Global identification and characterization of lncRNAs that control inflammation in malignant cholangiocytes. BMC Genomics. 2018;19:735. doi: 10.1186/s12864-018-5133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Elling R, Robinson EK, Shapleigh B, Liapis SC, Covarrubias S, Katzman S, Groff AF, Jiang Z, Agarwal S, Motwani M, Chan J, Sharma S, Hennessy EJ, FitzGerald GA, McManus MT, Rinn JL, Fitzgerald KA, Carpenter S. Genetic models reveal cis and trans immune-regulatory activities for lincRNA-Cox2. Cell Rep. 2018;25:1511–1524. e6. doi: 10.1016/j.celrep.2018.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Alkhazraji A, Elgamal M, Ang SH, Shivarov V. All cancer hallmarks lead to diversity. Int J Clin Exp Med. 2019;12:132–157. [Google Scholar]
- 237.Agrawal A, Tay J, Yang GE, Agrawal S, Gupta S. Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY) 2010;2:93–100. doi: 10.18632/aging.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bai ZT, Bai B, Zhu J, Di CX, Li X, Zhou WC. Epigenetic actions of environmental factors and promising drugs for cancer therapy. Oncol Lett. 2018;15:2049–2056. doi: 10.3892/ol.2017.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Morgan AE, Davies TJ, Mc Auley MT. The role of DNA methylation in ageing and cancer. Proc Nutr Soc. 2018;77:412–422. doi: 10.1017/S0029665118000150. [DOI] [PubMed] [Google Scholar]
- 240.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zec N, Rowitch DH, Bitgood MJ, Kinney HC. Expression of the homeobox-containing genes EN1 and EN2 in human fetal midgestational medulla and cerebellum. J Neuropathol Exp Neurol. 1997;56:236–242. doi: 10.1097/00005072-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 242.Mayor R, Casadome L, Azuara D, Moreno V, Clark SJ, Capella G, Peinado MA. Long-range epigenetic silencing at 2q14.2 affects most human colorectal cancers and may have application as a non-invasive biomarker of disease. Br J Cancer. 2009;100:1534–1539. doi: 10.1038/sj.bjc.6605045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Devaney J, Stirzaker C, Qu W, Song JZ, Statham AL, Patterson KI, Horvath LG, Tabor B, Coolen MW, Hulf T, Kench JG, Henshall SM, Pe Benito R, Haynes AM, Mayor R, Peinado MA, Sutherland RL, Clark SJ. Epigenetic deregulation across chromosome 2q14.2 differentiates normal from prostate cancer and provides a regional panel of novel DNA methylation cancer biomarkers. Cancer Epidemiol Biomarkers Prev. 2011;20:148–159. doi: 10.1158/1055-9965.EPI-10-0719. [DOI] [PubMed] [Google Scholar]
- 244.Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Powe NR, Feinberg AP. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- 245.Alvarez H, Opalinska J, Zhou L, Sohal D, Fazzari MJ, Yu Y, Montagna C, Montgomery EA, Canto M, Dunbar KB, Wang J, Roa JC, Mo Y, Bhagat T, Ramesh KH, Cannizzaro L, Mollenhauer J, Thompson RF, Suzuki M, Meltzer SJ, Melnick A, Greally JM, Maitra A, Verma A. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet. 2011;7:e1001356. doi: 10.1371/journal.pgen.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013;33:2989–2996. [PubMed] [Google Scholar]
- 247.Heng HH, Liu G, Stevens JB, Bremer SW, Ye KJ, Ye CJ. Genetic and epigenetic heterogeneity in cancer: the ultimate challenge for drug therapy. Curr Drug Targets. 2010;11:1304–1316. doi: 10.2174/1389450111007011304. [DOI] [PubMed] [Google Scholar]