

Abstract
Current cancer treatment strategies have been advanced by chimeric antigen receptor (CAR) cell therapy, a rapidly emerging cellular immunotherapy. The numerous revolutionary achievements of CAR T cells against hematological malignancies initiated an upsurge in research on translating this therapy into a treatment for solid tumors. Unfortunately, no equivalent success has yet been achieved in treating solid tumors. The main challenges posed by solid tumors have gradually been recognized and include a lack of unique antigen targets, antigen heterogeneity, limited infiltration into the tumor, and an immunosuppressive tumor microenvironment. Surmounting the limitations of solid tumors remains critical in popularizing CAR T cell applications. Various approaches to augmenting the efficiency of CAR T cells through directly optimizing CAR constructs or through innovative combination strategies such as vaccines, biomaterials, and oncolytic virus have arisen. In addition to describing the main obstacles that restrict the promotion of CAR T cells, this paper focuses on reviewing new ongoing strategies to circumvent these limitations.
Keywords: Chimeric antigen receptor, solid tumors, vaccine, oncolytic virus, cancer immunotherapy
Introduction
Cancer immunotherapy, including immune checkpoint inhibitors, adoptive cell therapy (ACT), vaccines, and monoclonal antibodies, has achieved great success over the past few decades, with genetically engineered CAR T cells being the most exciting achievement. The addition of specific CARs enables T cells to identify specific tumor cells in an MHC-independent manner. CARs typically consist of extracellular regions that contain a single-chain variable fragment (scFv) and recognize specific antigens and intracellular signal transduction or activation regions that are linked by the transmembrane domain and hinge regions and transmit signals [1]. Unlike first-generation CARs, which contained only one activation signal and possessed limited functionality, subsequent constructs have been continuously optimized. Second-generation and third-generation CARs can be combined with multiple costimulatory molecules, while fourth-generation CARs are engineered to release transgenic products, accompanied by enhanced proliferative capacities and antitumor efficacies [1]. Remarkable results in eliminating hematological malignancies by adoptive infusion of CAR T cells have been obtained, particularly in achieving persistent disease regression in B-cell lymphoma and leukemia [2-4]. The ensuing approval by the FDA of two CD19 CAR T cell products targeting B cell tumor antigens to struggle against relapsed or refractory B-cell malignant tumors has set off a climax of CAR T cell research [5,6]. These achievements have stimulated further exploration of applying CAR T cell therapy to combat solid tumors and initiated numerous preclinical or clinical studies to evaluate the effect of CAR T cells [7-10]. However, the current results of CAR T cell treatment of solid tumors has remained unsatisfactory.
Several elements are hypothesized to contribute to the significant discrepancies in the clinical effects in hematological malignancies and solid tumors. The lack of unique tumor antigens and heterogeneous antigen expression are pivotal factors that result in side effects and antigen escape [11]. Moreover, transporting CAR T cells from the bloodstream to the tumor site after infusion is difficult due to the disordered vasculature and dense matrix within solid tumors [12]. In addition, the hostile tumor microenvironment (TME) is another obvious obstacle that significantly impedes the function and persistence of CAR T cells [13]. To overcome the challenges associated with solid tumors, various strategies have been developed involving optimized CAR structures and innovative combination therapy aimed at enhancing the specificity, infiltration, and efficacy of CAR T cells and reprogramming the inhibitory conditions. In this review, the main challenges presented by solid tumors and feasible strategies to support CAR T cell therapy will be elaborated in detail.
Selection of CAR T cell targets
Choosing an appropriate target on tumor cells is extremely crucial and affects not only the accurate identification and removal of tumor cells by CAR T cells but also the safety of the treatment. In fact, as an obvious obstacle, the lack of tumor specific antigens in solid tumors remains unconquered. To date, some overexpressed endogenous molecules in tumor tissues, especially those that promote tumor proliferation and persistence, such as GD2, interleukin 13 receptor α (IL13Rα), mesothelin, and human epidermal growth factor receptor 2 (HER2), have been selected as targets for CAR T therapy [8,9,14,15]. However, there is the potential of damage to normal tissue triggered by on-target and off-tumor toxicity because some normal tissues may also express nonspecific tumor antigens [16]. To enhance safety and reduce off-target toxicity, much attention has focused on optimizing CAR constructs with improved tumor antigen selectivity and specificity.
One way to improve specificity is to design bispecific CAR T cells with split signaling pathways connected to a costimulatory signal and an activation signal (Figure 1A). Only when CAR T cells simultaneously encounter two antigens expressed on tumors, can T cells become activated to produce powerful effects [17]. Similarly, improving the ability to control CAR T cells to turn them on or off under specific conditions has also been studied (Figure 1A). On-switch CAR T cells, which are designed to be conditionally activated only in the presence of inducible foreign molecules, provide a strategy to accurately control CAR T cell activation. This effect is achieved by dividing the key recognition and activation signaling of the CAR into different modules that can only be combined with the application of heterodimerizing small molecules [18]. Using a bifunctional small molecule “switch”, which is composed of folate and fluorescein isothiocyanate (folate-FITC), CAR T cells can specifically identify tumor cells overexpressing folate receptors [19].
Figure 1.
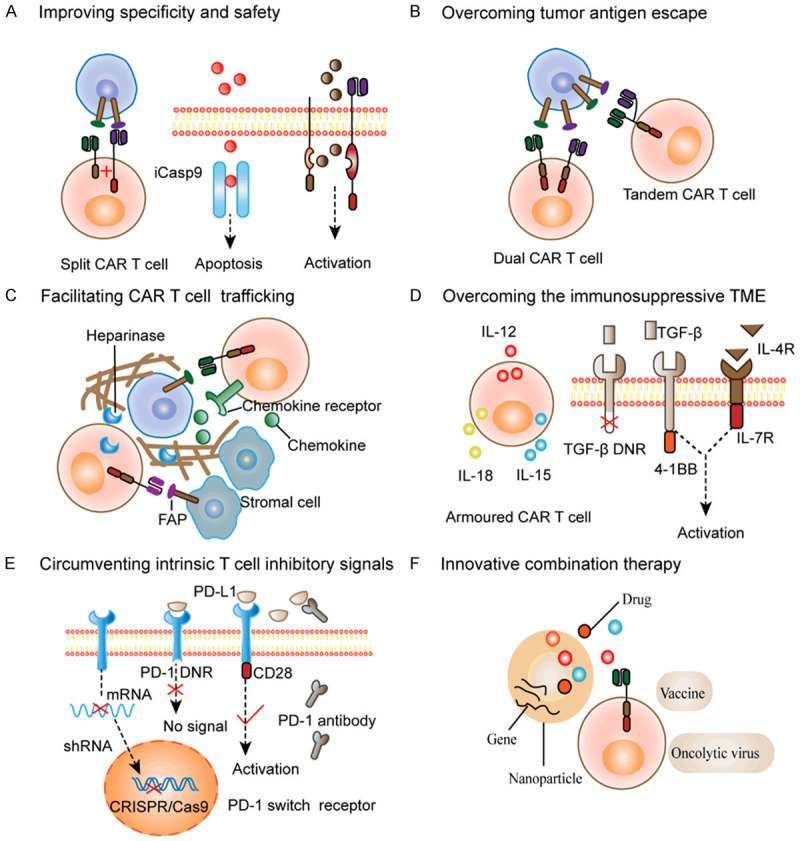
Innovative strategies to circumvent the challenges of CAR T cell therapy. A. Improving specificity and safety: Split CAR T cells contain separate costimulatory signals and activation signals, which require the presence of two different tumor antigens for full activation. On-switch CAR T cells or off-switch CAR T cells are induced to activate or undergo apoptosis by small molecule dimerizers. B. Overcoming tumor antigen escape: Multitarget CAR T cells are designed to target different tumor antigens, including dual CAR T cells with two CARs or tandem CAR T cells with multiple scFvs in one CAR. C. Facilitating CAR T cell trafficking: This can be achieved by optimizing CAR T cells to express appropriate CCRs, including CXCR2, CCR2b, and CCR4, which bind to chemokine ligands secreted by tumors. Alternatively, targeting CAR T cells to FAP to remove stromal cells, and engineering CAR T cells secreting the HPSE enzyme to degrade the tumor matrix will further overcome physical barriers. D. Overcoming the immunosuppressive tumor microenvironment (TME): Armoured CAR T cells secreting immunostimulatory factors such as IL12, IL18, and IL15 can modulate the local cytokine microenvironment to benefit CAR T cell survival and recruit endogenous immune cells. Another strategy is to modify CAR T cells to withstand inhibitory factors through TGF-β dominant negative receptors (DNRs) that cannot transmit immunosuppressive signals, TGF-β receptors that convert TGF-β signals into 4-1BB or IL-12 stimulatory signals, and inverted cytokine receptors infused with the extracellular domain of the IL-4 receptor and the endodomain of the IL-7 receptor. E. Circumventing intrinsic T cell inhibitory signals: One approach is to directly block the production of PD-1, using CRISPR/Cas9 to knock out the PD-1 gene in CAR T cells or using shRNA to degrade mRNA in CAR T cells. In addition, PD-1 DNRs lacking the intracellular domain are unable to transmit inhibitory signals. PD-1 switch receptors with intracellular immunostimulatory signals can convert inhibitory signals into stimulatory signals. PD-1 blocking antibodies secreted by CAR T cells can competitively bind PD-L1. F. Innovative combination therapy to synergistically enhance CAR T cell functions: Vaccines including viruses or dendritic cells provide a way to activate CAR T cells in vivo. Nanoparticles or oncolytic viruses can be modified to carry drugs or genes or stimulatory cytokines to enhance the efficacy of CAR T cells.
Subsequently, some studies have proposed another approach to suppress T cells function if toxic reactions arise by engineering suicide genes or using antibody-mediated killing, also known as off-switch CAR T cells [20,21]. Incorporating the inducible caspase 9 (iCasp9) system into T cells engineers the T cells to be killed by induction. When administered iCasp9 induction-dependent small molecule dimerizers that primarily damage activated cells that highly express certain genes, T cells are rapidly induced to undergo apoptosis, shutting down the effect of CAR T cells [22].
Tumor antigen heterogeneity and tumor antigen escape
Solid tumors are composed of molecularly heterogeneous subgroups, and antigen expression in different tumors or in individuals is different. Heterogenous target antigen expression restricts the outcome of CAR T cell therapy and is related to subsequent clonal escape, tumor resistance and recurrence after removing the most immunogenic epitopes [23]. Tumor resistance caused by tumor antigen loss has been shown in patients with recurrent glioblastoma after epidermal growth factor receptor (EGFR)-targeted CAR T cell therapy [24]. Therefore, addressing tumor heterogeneity and overcoming tumor escape are imperative for achieving long-term remission.
Recently, investigators have been dedicated to designing multitarget CAR T cells, aiming to restrain tumor antigen escape by discerning multiple antigens. Tandem CAR T cells have two scFVs that recognize different antigens connected to one CAR construct (Figure 1B). One group compared the effect of tandem CAR T cells co-expressing HER2 and IL13Rα to single-specific CAR T cells in mouse models of glioblastoma and found that the former exhibited reduced antigen escape and increased tumor clearance [25]. CAR T cells simultaneously expressing multiple CAR constructs have also been developed. Similarly, another study found that trivalent CAR T cells specific for three different glioblastoma targets overcame antigen heterogeneity between different patients and promoted tumor clearance in an autologous mouse model [26]. Targeting mutant antigens or non-tumor cell antigens in the tumor environment, such as fibroblasts or the tumor vasculature, may also overcome antigen escape [27]. As shown by one group, EGFRvIII CAR T cells, which recognize EGFRvIII, suppressed tumor growth in glioblastoma models [28].
Other attempts to surmount antigen escape include the use of modified T cells expressing bispecific T cell engagers (BiTEs) and immunomodulatory agents capable of inducing an endogenous immune response to synergistically eliminate tumors. The main mechanism of BiTEs is to connect T cells with tumor cells utilizing two scFvs targeting CD3 and the tumor antigen, thereby recruiting and activating endogenous T cells to kill tumors [29]. One study found that CAR T cells expressing EGFR-targeted BiTEs were able to recruit endogenous bystander T cells in a glioblastoma model, effectively attenuating antigen expression heterogeneity and bypassing antigen escape [30]. Furthermore, CAR T cells expressing CD40 ligand (CD40L) have also been proven to provide a CD40 activation signal and activate endogenous antigen-presenting cells (APCs) to secrete stimulating cytokines, thus enhancing antitumor efficacy and overcoming tumor escape [31,32].
Limited CAR T cell trafficking
Successful transport from the bloodstream to the tumor site is the prerequisite for CAR T cells to attack tumor cells. However, limited trafficking to solid tumors presents another major obstacle. Unlike easy access to hematological malignancies, there are some factors in solid tumors that result in inefficient T cell homing and limited infiltration of the tumor site. One factor is mismatched chemokines expressed by tumor tissues, which are usually incompatible with chemokine receptors (CCRs) on CAR T cells [33]. In addition, infiltration is further inhibited by physical barriers constituted with dense extracellular matrix (ECM), as well as abnormal tumor blood vessels in solid tumors [12]. Therefore, many strategies have been proposed to overcome limited T cell trafficking. Local infusion to deliver CAR T cells to the tumor sites or cranial cavity seems to be a potential strategy that can also avoid the toxicity of systemic injection and off-target effects. Intracranial administration and intraperitoneal delivery of CAR T cells were evaluated in breast cancer brain metastases and ovarian cancer, respectively [34-36]. However, these strategies are not feasible in the case of extensive tumor metastasis.
Exploring other strategies to potentiate T cell infiltration and achieve accurate cell homing are necessary. By directly optimizing CAR T cells to express favorably matched CCRs, CAR T cells can better interact with chemokine ligands expressed by tumor cells (Figure 1C). Moon et al. utilized lentiviral vectors to obtain anti-mesothelin CAR T cells expressing CCR2b, which interacted with chemokine ligand 2 (CCL2) secreted by cancer cells, and showed improved T cell infiltration in the mesothelioma xenograft model, accompanied by improved antitumor activity [37]. Recently, another study found that CAR T cells expressing CXCR2 could facilitate T cell homing and augment the antitumor response [38]. Alternatively, additional CCRs such as CCR4 have also been proven to be effective to varying degrees [39].
To enhance migration and infiltration capacity, another potential approach includes disrupting physical barriers in solid tumors, either by designing CAR T cells to recognize stromal cell-associated antigens or secrete matrix-degrading enzymes (Figure 1C). One of the attractive targets seems to be the protease fibroblast activation protein (FAP), which is expressed on fibroblasts in many solid tumors [40]. Furthermore, evaluation of FAP CAR T cells in multiple tumor models showed effective tumor suppression and decreased numbers of FAP positive stromal cells [41]. In another study, heparinase (HPSE)-modified CAR T cells aimed to supplement the HPSE insufficiency in the matrix have also been shown to promote matrix degradation and T cell infiltration [42].
The immunosuppressive tumor microenvironment
Even when CAR T cells are successfully transported, another major roadblock is the highly immunosuppressive TME that can render them anergic [43]. Solid tumors are typically characterized by a suppressive TME composed of multiple immunosuppressive molecules and inhibitory immune cells. Suppressive immune cells such as regulatory T cells can hinder CAR T cell proliferation and cytotoxic abilities through contact-dependent methods, while some inhibitory molecules, such as transforming growth factor-β (TGF-β) and IL-10, can promote T cell anergy by indirect contact [13]. Recently, various strategies have emerged to overcome the immunosuppressive TME and make it beneficial for T cell survival and proliferation (Figure 1D).
Some research has endeavored to modify CAR T cells to overexpress cytokines that promote inflammation such as IL-12, IL-15, and IL-18, and these cells are known as armoured CAR T cells [44-46]. In contrast to the toxicity induced by systemic injection of stimulating molecules, this approach safely modulates the local microenvironment. Engineering T cells to secrete IL-12 was confirmed to induce increased proliferative capacities, survivability and cytotoxicity, as well as resistance to apoptosis and PD-L1-induced functional inhibition, in ovarian cancer [47]. IL-15 expressing anti-CD19 CAR T cells were also found to achieve persistence and induce sustained remission in a model of leukaemia, and the mechanism was possibly related to the formation of a memory cell subset [48]. Similarly, in a neuroblastoma metastasis model, optimizing GD2 CAR T cells with IL-15 resulted in improved antitumor abilities [49]. Furthermore, related researches on CAR T cells secreting IL-18 showed that these cells had enhanced proliferation and infiltration capacities, and could recruit endogenous immune cells to regulate the TME [50,51].
Other strategies to shelter CAR T cells from inhibitory molecules, such as blocking immunosuppressive signals in CAR T cells and the TME, are also worth considering (Figure 1D). Since TGF-β signaling is an important inhibitory pathway, a feasible alternative to promote antitumor responses is to block TGF-β signaling. One group found that a TGF-β dominant negative receptor (DNR) could effectively protect CAR T cells from immunosuppressive cytokines with reduced inhibitory signal transduction in preclinical prostate cancer models [52]. Alternatively, CAR T cells engineered with receptors that convert TGF-β signals into 4-1BB or IL-12 stimulation signals exerted powerful antitumor effects and were rendered resistant to immunosuppressive signals [53]. Another group also found that TGF-β CAR T cells had reinforced cytotoxicity and strengthened antitumor immune functions, protecting adjacent immune cells from TGF-β-mediated immunosuppression [54].
Similarly, research in models of pancreatic cancer showed that CAR T cells expressing inverted cytokine receptors could promote T cell survival. The extracellular segment of the IL-4 receptor and the intracellular segment of the IL-7 receptor were fused to convert immunosuppressive cytokine signals into activation signals [55]. Inducing the endogenous antitumor immune response and enhancing costimulatory signals have also been explored [56]. In addition, metabolic disorders such as nutrient deficiency, low pH and hypoxia are another feature of the TME, and metabolic competition further restricts the activity of CAR T cells [57]. Reforming the metabolic pathway of CAR T cells might be another strategy. For example, under hypoxic conditions, the transcription factor hypoxia-inducible factor 1-α (HIF-1α) can be stably expressed. By modifying CAR T cells to express HIF-1α, the cells gained the ability to tolerate hypoxia, which resulted in increased CAR expression and oncolysis to improve the antitumor effect [58].
Intrinsic T cell inhibitory signals
T cells have intrinsic inhibitory mechanisms, and upregulation of important inhibitory receptors CTLA-4/PD-1 results in T cell exhaustion and restrains T cell persistence by interacting with ligands that are overexpressed on tumor cells [59,60]. Thus, overcoming endogenous suppressive signals to reduce CAR T cell depletion is also a promising approach. Some novel strategies, including the use of gene silencing, PD-1 switch receptors, DNRs and the secretion of PD-1 antibodies to circumvent the internal and external inhibitory mechanism of T cells, have been developed (Figure 1E).
Recent research has focused on the use of gene silencing techniques such as CRISPR/Cas9 or short hairpin RNAs (shRNAs) to directly knockout the genes that encode the inhibitory receptor PD-1 in CAR T cells. For example, one group silenced PD-1 in anti-CD19 CAR T cells via CRISPR/Cas9 and showed rescued T cell activity and improved antitumor efficacy in the PD-L1-positive tumor models [61]. Similarly, transduction of shRNA into anti-MSLN CAR T cells to silence the PD-1 gene led to downregulation of endogenous PD-1 levels, enhanced T cell expansion and tumor cell lysis [62].
In addition, modifying CAR T cells to express switch receptors seems to be an attractive strategy to overcome immune checkpoints or inhibitory molecules, which allows the conversion of inhibitory signals to stimulatory signals [63]. PD-1 switch receptors (Figure 1E) were created by fusing the extracellular region of the immunosuppressive signal with the activated intracellular domain and achieved conversion of the PD-L1 signal into a stimulatory signal, thereby resisting T cell dysfunction and promoting tumor regression [64]. Attempts to engineer CAR T cells with dominant negative PD-1 receptors have also been explored (Figure 1E). Due to the lack of transmembrane segments and intracellular signal transduction domains, T cell with DNRs are endowed with the ability to compete with endogenous cell receptors to bind PD-1, but no inhibitory signal can be transduced [65]. In addition, engineered CAR T cells that can release PD-1 blocking antibodies have been demonstrated to boost the antitumor effects of CAR T cells [66].
Innovative combination therapy
Although high remission rates have been obtained in hematological malignancies, patients lack sustained remission after CAR T cell therapy and ultimately experience disease relapse. The function of CAR T cells is intimately associated with the clinical outcomes of patients, and the activation, killing functions and persistence of these cells in vivo are dynamic processes that are affected by diverse factors, such as tumor factors, T cell factors, and individual factors [67]. Each aspect may impact the application of CAR T cells, and so it seems insufficient to settle these intricate factors only through a monotonous approach. In addition to direct genetic modifications of T cells, alternative strategies to overcome insufficient CAR T cell expansion and enhance persistence in vivo should be explored [68-71]. The recent advances in combined vaccines, biomaterials, and oncolytic viruses have good application prospects to achieve desired outcomes, either by directly enhancing the function of T cells or by recruiting endogenous immune cells, as well as remodeling the TME.
CAR T cell therapy combined with vaccines
Therapeutic cancer vaccines have been a key breakthrough in cancer therapy by specifically inducing T cells to attack tumors. Some studies found that dual-specific T cells expressing tumor-specific CARs and endogenous T cell receptors (TCRs) against strong immunogens, such as influenza viruses showed strong amplification and antitumor activity after immunization with an immunogen vaccine [72-75]. Preparing CAR T cells using virus-specific cytotoxic T lymphocytes (CTLs) has shown great potential in many early studies. Notably, Epstein-Barr virus (EBV)-specific CTLs expressing GD2 CARs lasted longer in patients with neuroblastoma than activated non-virus-specific T cells [76].
Vaccination provides an alternative strategy to further facilitate the amplification, activation and cytotoxicity of these CAR T cells in vivo (Figure 1F). One of the common vaccines is a virus-based vaccine: for example, a cytomegalovirus (CMV)-based vaccine combined with adoptive infusion of T cells synergistically promoted tumor clearance [75]. Inoculation with a viral vaccine expressing gp100 enhanced T cell expansion and tumor regression in multiple mouse models [72]. A recent study in B cell acute lymphoblastic leukaemia (B-ALL) patients also showed that using viral vaccines to stimulate natural TCRs, even without lymphodepletion, could enable CD19-modified virus-specific T cells to successfully expand and increase sustainability [77]. However, there are risks of viral reactivation and viremia, and the safety of viral vaccines needs to be further evaluated.
Other cancer vaccines containing soluble tumor-associated antigens (TAAs) and dendritic cell (DC) adjuvants have been shown to activate TAA-specific effector cells and stimulate the production of antibodies in preclinical and clinical trials [78]. DCs are full-time APCs that regulate natural and acquired immunity and are vital in the process of immunotherapy. One study revealed the enhanced T cell activation, expansion, and antitumor effect of ACT by a DC vaccine in vivo [79]. In a clinical trial of melanoma, tumor infiltrating T cells were infused after the patients were vaccinated with tumor antigen-bearing DC vaccines, resulting in one person achieving complete remission and two people having stable disease [80].
There are also parallel results in a study of DC vaccine-mediated stimulation of CAR T cells. One group discovered that GD2 CAR T cells prepared from virus-specific CTLs killed target cells when cultured with neuroblastoma for the first time in vitro, but these T cells failed to control the tumor after the second exposure. However, virus-specific TCRs can be reactivated by utilizing DCs or DC supernatant alone and rescue CAR T cell functions [73]. Inoculation with DC cell vaccines shocked with tumor-specific antigen peptide enhanced the amplification and function of intracellular oncoprotein WT1 CAR T cells and promoted tumor clearance in xenograft mouse models [81]. Although DC vaccines may be a safe way to enhance CAR T cells, patients receiving cancer vaccine treatment have demonstrated moderate antitumor immune responses, and the clinical treatment effect has been very limited. These approaches need to be improved in the future.
CAR T cell therapy combined with biomaterials
Some shortcomings exist when directly modifying CAR T cells to express supporting factors, such as complicated operations and uncontrollable systemic toxicity. Hence, it is imperative to develop new strategies, and biomaterials offer a potential avenue for supplementation. Some methods based on nanomedicine to improve the functions of adoptively infused T cells are being continuously explored by more researchers.
Synthesized nanoparticles can be designed to direct larger gene cargoes or drugs to target cell subpopulations in vivo through chemically conjugated antibodies or ligand binding to receptors on the target cell surface (Figure 1F) [82]. Some studies have explored the potential of liposomes to deliver stimulatory molecules in ACT: Zhang et al. used pegylated liposomes modified with cytokines or cell-specific antibodies to transfer cargoes and simulate T cell expansion and observed successful labeling and significant expansion of T cells in vivo [83]. Recently, backpacking nanoparticles have also been developed to connect supporting drugs to immune cells and en-hance cell function in an autocrine manner [84]. Protein nanogels are another kind of biological material that can carry therapeutic substances and release cargoes depending on antigen recognition. Li Tang et al. applied protein nanogels to transport IL-15 super-agonists to the TME and observed markedly increased effectiveness of EGFR CAR T cells and improved survival of tumor loaded mice [85]. Furthermore, nanoparticles have broken through the barrier of long-term preparation of CAR T cells in vitro. One group utilizing DNA nanocarriers to transfer CAR genes that recognize leukaemia cells found that circulating T cells could be rapidly and accurately reprogrammed into antigen-specific T cells in situ and achieve equal effects [86].
Using nanoparticles loaded with immunosuppressive inhibitors is another promising approach to reverse the suppressive TME. Zhang et al. found that tumor-targeted liposomes encapsulating the immunomodulator PI-3065 and 7DW8-5 suppressed monocytic myeloid-derived suppressor cells while simultaneously increasing endogenous antitumor cells and provided a two-week window for CAR T cell treatment. During this period of infusion, CAR T cells could effectively penetrate, robustly expand and promote tumor clearance in a mouse model [87]. Similarly, PEGylated immunoliposomes transported small molecule inhibitors of TGF to ACT cells and maintained T cell proliferation in vitro [88].
Nanoparticles can also transport antigens to lymphoid tissues and stimulate endogenous APCs to present antigens, assisting CAR T cell therapy. Leyuan Ma et al. designed an albumin-bound phospholipid polymer by connecting a small molecule or peptide ligand of CAR to the lipid tail to achieve lymph node targeting. As a result, CAR ligand was effectively delivered to the lymph nodes by albumin and inserted into the APC membrane through the lipid tail to modify macrophages and DCs, and subsequent endogenous cell activation further boosted CAR T cell proliferation and enhanced antitumor effects [89]. Recently, another study revealed that a nanoparticle RNA vaccine can directly enhance the cytotoxic effect of CAR T cells and overcome insufficient stimulation and low sustainability. This nanoparticle vaccine can transport CAR antigen to APCs in lymphoid tissues and simultaneously initiate a Toll-like receptor-dependent type I interferon-driven immune stimulation program [90].
CAR T cell therapy combined with oncolytic viruses
As a kind of virus that selectively infects tumor cells, oncolytic viruses not only directly lyse cells but also induce endogenous antitumor immunity by recruiting immune cells and releasing proinflammatory molecules; moreover, oncolytic viruses can also be modified as platforms to deliver cargoes [91]. Hence, combined treatment with oncolytic viruses is a promising combination strategy for reverting tumor immunosuppression and enhancing the capabilities of CAR T cells, especially the use of oncolytic viruses to transport therapeutic drugs or genes to the TME [91].
On the one hand, oncolytic viruses can present multiple tumor antigens to CAR T cells and contribute to overcoming antigen escape. Recently, CAR T cells combined with oncolytic viruses expressing BiTEs specific for EGFR were investigated in vivo and showed a synergistic effect in overcoming tumor heterogeneity [92]. On the other hand, genetically modified oncolytic viruses that encode chemokines can attract CAR T cells to the tumor site and promote infiltration. For example, the combination of CAR T cells with oncolytic viruses expressing the chemokine RANTES and the cytokine IL15 have been shown to improve infiltration and promote obvious tumor regression in mouse models [93]. Similar encouraging results were found in another study of oncolytic viruses expressing the chemokine CXCL11 [94]. Investigators have also attempted to overcome immunosuppression by modifying oncolytic viruses to express immunosuppressive agents and promote inflammatory factors. Oncolytic adenoviruses expressing the cytokines IL-15, IL-2, and TNF have been proven to reverse the immunosuppressive TME, promote CAR T cell infiltration and enhance persistence [95]. Another study demonstrated that combining intratumoral injection of an oncolytic adenovirus secreting a PD-L1 blocking mini-body had the potential to improve the antitumor efficacy of HER2 CAR T cells [96].
Conclusions
As demonstrated by the inspiring efficacy in a variety of hematological malignancies, CAR T cell therapy remains a potential strategy for cancer treatment. However, no parallel success has been achieved in the treatment of solid tumors to date. Challenges posed by solid tumors, including the lack of specific tumor targets, heterogeneous antigen expression, limited infiltration, and an inhibitory TME are the main factors that hamper the success of CAR T cell therapy. More studies are urgently needed to elucidate the mechanisms that affect treatment efficacy and overcoming these obstacles in solid tumors will further expand the application scope of CAR T cells. A variety of precise and controllable schemes are currently underway to modify CAR structure. Moreover, the combinatorial treatment strategies in preclinical models have achieved success, indicating the potential for translation into clinical applications, and more clinical studies to evaluate the effects of combination therapy are needed in the future.
Acknowledgements
We thank the research group for their contributions and work. This project was supported by the funding from the National Natural Science Foundation of China (No. 81700126).
Disclosure of conflict of interest
None.
References
- 1.Abreu TR, Fonseca NA, Goncalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Control Release. 2020;319:246–261. doi: 10.1016/j.jconrel.2019.12.047. [DOI] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng ZH, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen XY, Soma L, Wood B, Li D, Heimfeld S, Riddell SR, Maloney DG. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8(+) and CD4(+) CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DA, Morton KE, Toomey MA, Rosenberg SA. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, Chester KA, Westwood NB, Halford SER, Nabarro S, Wan S, Austin E, Hawkins RE. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: driving T cells to solid tumors. Cancer Discov. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, Grossman RG, Heslop HE, Gottschalk S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagley SJ, O’Rourke DM. Clinical investigation of CAR T cells for solid tumors: lessons learned and future directions. Pharmacol Ther. 2020;205:107419. doi: 10.1016/j.pharmthera.2019.107419. [DOI] [PubMed] [Google Scholar]
- 11.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 12.Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. 2015;33:55–63. doi: 10.1016/j.coi.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 14.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142–148. doi: 10.1038/cgt.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ Jr. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1:43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, Gonzalez J, Schultz PG, Young TS, Kim CH. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137:2832–2835. doi: 10.1021/jacs.5b00106. [DOI] [PubMed] [Google Scholar]
- 20.Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 24.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot JM, Zheng ZH, Levine BL, Okada H, June CH, Brogdon JL, Maus MV. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK, Brawley VS, Byrd TT, Krebs S, Gottschalk S, Wels WS, Baker ML, Dotti G, Mamonkin M, Brenner MK, Orange JS, Ahmed N. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu MF, Orange JS, Sumazin P, Man TK, Joseph SK, Hegde M, Ahmed N. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20:506–518. doi: 10.1093/neuonc/nox182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chinnasamy D, Yu ZY, Theoret MR, Zhao YB, Shrimali RK, Morgan RA, Feldman SA, Restifo NP, Rosenberg SA. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010;120:3953–3968. doi: 10.1172/JCI43490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, Fraietta JA, Kloss CC, Posey AD, Engels B, Singh R, Ezell T, Idamakanti N, Ramones MH, Li N, Zhou L, Plesa G, Seykora JT, Okada H, June CH, Brogdon JL, Maus MV. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velasquez MP, Torres D, Iwahori K, Kakarla S, Arber C, Rodriguez-Cruz T, Szoor A, Bonifant CL, Gerken C, Cooper LJ, Song XT, Gottschalk S. T cells expressing CD19-specific engager molecules for the immunotherapy of CD19-positive malignancies. Sci Rep. 2016;6:27130. doi: 10.1038/srep27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick MB, Scarfo I, Cetrulo CL, Demehri S, Nahed BV, Cahill DP, Wakimoto H, Curry WT, Carter BS, Maus MV. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37:1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 31.Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, Brentjens RJ. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell. 2019;35:473–488. e6. doi: 10.1016/j.ccell.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ols M, Schebesta M, Brideau E, Elpek K, Fleury M, Gori J, Heller S, Li DJ, Primack B, Reardon C, Sethi D, Storer A, Sun D, Tran K, Weisman E, Briskin M, Richardson C, Suri V, Shamah S. CAR-Ts armored with small molecule-regulated 1L12 or CD40L cassettes for enhanced activity against solid tumors. Cancer Res. 2019:79. [Google Scholar]
- 33.Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. 2017;28:xii18–xii32. doi: 10.1093/annonc/mdx238. [DOI] [PubMed] [Google Scholar]
- 34.Hardaway JC, Prince E, Arepally A, Katz SC. Regional infusion of chimeric antigen receptor T cells to overcome barriers for solid tumor immunotherapy. J Vasc Interv Radiol. 2018;29:1017–1021. e1. doi: 10.1016/j.jvir.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Murad JP, Kozlowska AK, Lee HJ, Ramamurthy M, Chang WC, Yazaki P, Colcher D, Shively J, Cristea M, Forman SJ, Priceman SJ. Effective targeting of TAG72(+) peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front Immunol. 2018;9:2268. doi: 10.3389/fimmu.2018.02268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, Chang WC, Ostberg JR, Neman J, Jandial R, Portnow J, Forman SJ, Brown CE. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2 thorn breast cancer metastasis to the brain. Clin Cancer Res. 2018;24:95–105. doi: 10.1158/1078-0432.CCR-17-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ Jr, Riley JL, June CH, Albelda SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Idorn M, Skadborg SK, Kellermann L, Halldorsdottir HR, Holmen Olofsson G, Met O, Thor Straten P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology. 2018;7:e1450715. doi: 10.1080/2162402X.2018.1450715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S, Rataj F, Janssen KP, Endres S, Anz D, Kobold S. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology. 2016;5:e1105428. doi: 10.1080/2162402X.2015.1105428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mondino A, Vella G, Icardi L. Targeting the tumor and its associated stroma: one and one can make three in adoptive T cell therapy of solid tumors. Cytokine Growth Factor Rev. 2017;36:57–65. doi: 10.1016/j.cytogfr.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E, Albelda SM. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, June CH, Riley JL, Wherry EJ, Albelda SM. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, Sandalcioglu IE, Neyazi B, Moritz T, Meyer J, Rossig C, Altvater B, Eiz-Vesper B, Morgan MA, Abken H, Schambach A. Design and characterization of an “all-in-one” lentiviral vector system combining constitutive anti-GD2 CAR expression and inducible cytokines. Cancers (Basel) 2020;12:375. doi: 10.3390/cancers12020375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257:83–90. doi: 10.1111/imr.12125. [DOI] [PubMed] [Google Scholar]
- 46.Huang Y, Li D, Qin DY, Gou HF, Wei W, Wang YS, Wei YQ, Wang W. Interleukin-armed chimeric antigen receptor-modified T cells for cancer immunotherapy. Gene Therapy. 2018;25:192–197. doi: 10.1038/gt.2017.81. [DOI] [PubMed] [Google Scholar]
- 47.Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7:10541. doi: 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, Olivares S, Rabinovich B, Huls H, Forget MA, Datar V, Kebriaei P, Lee DA, Champlin RE, Cooper LJ. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113:E7788–E7797. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. 2019;25:2915–2924. doi: 10.1158/1078-0432.CCR-18-1811. [DOI] [PubMed] [Google Scholar]
- 50.Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, Park H, Purdon TJ, Daniyan AF, Spitzer MH, Brentjens RJ. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23:2130–2141. doi: 10.1016/j.celrep.2018.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu BL, Ren JT, Luo YP, Keith B, Young RM, Scholler J, Zhao YB, June CH. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20:3025–3033. doi: 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, Brenner MK, Leen AM, Vera JF. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 2018;8:972–987. doi: 10.1158/2159-8290.CD-17-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-beta-responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med. 2018;3:75–86. doi: 10.1002/btm2.10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, Brenner MK, Fisher WE, Leen AM, Vera JF. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25:249–258. doi: 10.1016/j.ymthe.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 2017;7:1306–1319. doi: 10.1158/2159-8290.CD-17-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu X, Gnanaprakasam JNR, Sherman J, Wang R. A metabolism toolbox for CAR T therapy. Front Oncol. 2019;9:322. doi: 10.3389/fonc.2019.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, Poirot L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833. doi: 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leick M, Maus MV. Wishing on a CAR: understanding the scope of intrinsic T-cell deficits in patients with cancer. Cancer Discov. 2019;9:466–468. doi: 10.1158/2159-8290.CD-19-0073. [DOI] [PubMed] [Google Scholar]
- 61.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immun. 2012;51:263–272. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 64.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76:1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology. 2017;6:e1273302. doi: 10.1080/2162402X.2016.1273302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, Yan S, Xiang J, Ma X, Seshan VE, Hendrickson RC, Liu C, Brentjens RJ. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25:1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 68.Chruściel E, Urban-Wójciuk Z, Arcimowicz L, Kurkowiak M, Kowalski J, Gliwiński M, Marjański T, Rzyman W, Biernat W, Dziadziuszko R, Montesano C, Bernardini R, Marek-Trzonkowska N. Adoptive cell therapy harnessing antigen-specific T cells to target solid tumours. Cancers. 2020;12:683. doi: 10.3390/cancers12030683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lanitis E, Coukos G, Irving M. All systems go: converging synthetic biology and combinatorial treatment for CAR-T cell therapy. Curr Opin Biotechnol. 2020;65:75–87. doi: 10.1016/j.copbio.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Rafiq S, Hackett CS, Brentjens RJ, Ramello MC, Haura EB, Abate-Daga D. Engineering strategies to overcome the current roadblocks in CAR T cell therapy CAR-T cells and combination therapies: what s next in the immunotherapy revolution? Nat Rev Clin Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramello MC, Haura EB, Abate-Daga D. CAR-T cells and combination therapies: what’s next in the immunotherapy revolution? Pharmacol Res. 2018;129:194–203. doi: 10.1016/j.phrs.2017.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Slaney CY, von Scheidt B, Davenport AJ, Beavis PA, Westwood JA, Mardiana S, Tscharke DC, Ellis S, Prince HM, Trapani JA, Johnstone RW, Smyth MJ, Teng MW, Ali A, Yu Z, Rosenberg SA, Restifo NP, Neeson P, Darcy PK, Kershaw MH. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res. 2017;23:2478–2490. doi: 10.1158/1078-0432.CCR-16-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, Mehta B, Dotti G, Kinchington PR, Leen AM, Rossig C, Rooney CM. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res. 2017;23:3499–3509. doi: 10.1158/1078-0432.CCR-16-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xin G, Schauder DM, Jing W, Jiang A, Joshi NS, Johnson B, Cui W. Pathogen boosted adoptive cell transfer immunotherapy to treat solid tumors. Proc Natl Acad Sci U S A. 2017;114:740–745. doi: 10.1073/pnas.1614315114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grenier JM, Yeung ST, Qiu Z, Jellison ER, Khanna KM. Combining adoptive cell therapy with cytomegalovirus-based vaccine is protective against solid skin tumors. Front Immunol. 2017;8:1993. doi: 10.3389/fimmu.2017.01993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lapteva N, Gilbert M, Diaconu I, Rollins LA, Al-Sabbagh M, Naik S, Krance RA, Tripic T, Hiregange M, Raghavan D, Dakhova O, Rouce RH, Liu H, Omer B, Savoldo B, Dotti G, Cruz CR, Sharpe K, Gates M, Orozco A, Durett A, Pacheco E, Gee AP, Ramos CA, Heslop HE, Brenner MK, Rooney CM. T-cell receptor stimulation enhances the expansion and function of CD19 chimeric antigen receptor-expressing T cells. Clin Cancer Res. 2019;25:7340–7350. doi: 10.1158/1078-0432.CCR-18-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lou YY, Wang G, Lizee G, Kim GJ, Finkelstein SE, Feng CG, Restifo NP, Hwu P. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer Res. 2004;64:6783–6790. doi: 10.1158/0008-5472.CAN-04-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poschke I, Lovgren T, Adamson L, Nystrom M, Andersson E, Hansson J, Tell R, Masucci GV, Kiessling R. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol Immunother. 2014;63:1061–1071. doi: 10.1007/s00262-014-1575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y, Amaishi Y, Okamoto S, Mineno J, Ikeda H, Maki T, Fujiwara H, Akatsuka Y, Kato T, Shiku H. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132:1134–1145. doi: 10.1182/blood-2017-08-802926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20:321–334. doi: 10.1038/s41577-019-0269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release. 2013;172:426–435. doi: 10.1016/j.jconrel.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siriwon N, Kim YJ, Siegler E, Chen X, Rohrs JA, Liu Y, Wang P. CAR-T Cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol Res. 2018;6:812–824. doi: 10.1158/2326-6066.CIR-17-0502. [DOI] [PubMed] [Google Scholar]
- 85.Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ, Li N, Kudchodkar SB, Wong HC, Jeng EK, Maus MV, Irvine DJ. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707–716. doi: 10.1038/nbt.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12:813–820. doi: 10.1038/nnano.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang F, Stephan SB, Ene CI, Smith TT, Holland EC, Stephan MT. Nanoparticles that reshape the tumor milieu create a therapeutic window for effective T-cell therapy in solid malignancies. Cancer Res. 2018;78:3718–3730. doi: 10.1158/0008-5472.CAN-18-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng Y, Tang L, Mabardi L, Kumari S, Irvine DJ. Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or noninternalizing receptors. Acs Nano. 2017;11:3089–3100. doi: 10.1021/acsnano.7b00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, Fichter M, Wang C, Liang S, Silva M, Kumari S, Mehta NK, Abraham W, Thai N, Li N, Wittrup KD, Irvine DJ. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–168. doi: 10.1126/science.aav8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, Klein O, Kuna K, Ouchan Y, Woll S, Christ E, Weber D, Suchan M, Bukur T, Birtel M, Jahndel V, Mroz K, Hobohm K, Kranz L, Diken M, Kuhlcke K, Tureci O, Sahin U. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–453. doi: 10.1126/science.aay5967. [DOI] [PubMed] [Google Scholar]
- 91.Guedan S, Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol. 2018;9:2460. doi: 10.3389/fimmu.2018.02460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T, Young RM, Alemany R, June CH, Guedan S. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6:605–616. doi: 10.1158/2326-6066.CIR-17-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195–5205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Lo A, Thorne SH, Albelda SM. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7:e1395997. doi: 10.1080/2162402X.2017.1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, Keith B, Young RM, Engels B, Sorsa S, Siurala M, Havunen R, Tahtinen S, Hemminki A, June CH. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018;3:e99573. doi: 10.1172/jci.insight.99573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77:2040–2051. doi: 10.1158/0008-5472.CAN-16-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]