

Abstract
Anoikis resistance is an important mechanism that mediates tumor metastasis. Studies have found that Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP1) promotes the occurrence, development, and metastasis of nasopharyngeal carcinoma (NPC). However, the related mechanism, especially whether LMP1 is involved in NPC metastasis through anoikis resistance, has not yet been elucidated. In present study, we showed that LMP1 enhanced the ability of NPC cells to resist anoikis by upregulating neurotrophic tyrosine kinase receptor type 2 (NTRK2 or TrkB) expression through NF-κB signaling and promoted the migration and invasion of NPC cells. After knockdown of NTRK2, the p-ERK and p-AKT in NPC cells were inhibited, and twist expression was further reduced, resulting in upregulation of E-cadherin expression and downregulation of vimentin expression. Subsequently, the results of a xenograft experiment showed that inhibiting NTRK2 could reduce LMP1-mediated NPC metastasis in vivo. In summary, these findings demonstrated that EBV-LMP1 upregulates twist expression to promote epithelial-mesenchymal transition (EMT) through the NTRK2-mediated AKT/ERK signaling pathway, thus mediating anoikis resistance and promoting NPC metastasis. These data will provide new molecular markers and potential targets for NPC metastasis.
Keywords: LMP1, NTRK2, anoikis, metastasis, nasopharyngeal carcinoma
Introduction
Nasopharyngeal carcinoma (NPC) is a malignant tumor derived from the nasopharyngeal epithelium. Clinically, 70%-80% of patients with NPC have cervical lymph node metastasis or distant metastasis at the time of diagnosis, which has become an important factor limiting the survival of patients [1]. Epstein-Barr virus (EBV) infection is closely related to the occurrence and development of NPC [2,3]. Latent membrane protein 1 (LMP1) is a tumorigenic protein encoded by EBV that can activate multiple signaling pathways including the NF-κB, MAPK, PI3K/AKT, IRF7, and STAT signals in a ligand-independent manner [4], and participate in tumor cell metabolism [5,6], angiogenesis [7], DNA damage repair [8], and telomerase activity [9]. Studies have found that LMP1-positive NPC cells have a stronger metastatic capacity than LMP1-negative NPC cells [10-13], but the mechanism has not been fully elucidated.
Anoikis is a programmed cell death process induced by the separation of cells from adjacent cells or the extracellular matrix (ECM) that can prevent cells from growing and attaching to inappropriate substrates, thereby avoiding the cell colonization in distant organs [14]. Resistance to anoikis is a key step in tumor invasion and metastasis [15,16]. Tumor cells can avoid anoikis by changing integrin expression, activating apoptotic signaling pathways, inducing epithelial-mesenchymal transition (EMT), regulating microRNA, inducing oxidative stress, or activating autophagy to promote distant metastasis. For example, integrin α2β1 protects melanoma cells against anoikis [17]. The activated PI3K/AKT pathway can help breast cancer cells prevent anoikis [18], and the activated MAPK signal promotes anoikis resistance of ovarian cancer cells by inducing EMT [19]. In esophageal cancer cells, high expression of miR-21 can promote anoikis resistance by regulating PDCD4 and PTEN [20]. Activation of the BLT2-ROS cascade promotes anoikis resistance when prostate cancer cells detach from the ECM [21]. Upregulation of protective autophagy in glioma stem cells can promote anoikis resistance [22]. These studies imply that multiple mechanisms are involved in regulating anoikis resistance.
The current study found that LMP1 could enhance the anoikis resistance, migration and invasion of NPC cells. Furthermore, neurotrophic tyrosine kinase receptor type 2 (NTRK2 or TrkB), a key molecule in the promotion of anoikis resistance, was selected in NPC. We found that LMP1 could upregulate NTRK2 expression, thereby activating AKT and ERK signaling that induced EMT through twist, and promoting anoikis resistance and metastasis in NPC.
Materials and methods
Cell cultures, transfection and reagents
CNE1 and HK1 are LMP1-negative NPC cell lines, CNE1-LMP1 (CM) and HK1-LMP1 (HM) are cell lines that stably express LMP1, C666-1 is an EBV-positive NPC cell line, C666-1-shLMP1 is an NPC cell line with LMP1 stably knocked down [5], CM-shNTRK2 and C666-1-shNTRK2 are NPC cell lines with NTRK2 stably knocked down, and the sequences of shRNAs are described in Table S1. 293T/17 [HEK 293T/17] (ATCC® CRL-11268) is a human renal epithelial cell line. All NPC cells were cultured in RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS, HyClone), and 293T/17 cells were cultured in Dulbecco’s modified Eagle medium (HyClone) supplemented with 10% FBS. Cells were transfected using LipofectamineTM 2000 (1888676, Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. LMP1-, p65- and control-siRNA were synthesized by Ribo Bio-technology (Guangzhou, China), and the siRNA sequences are listed in Table S2. The inhibitors used in this study were as follows: K252a (Abcam, Cambridge, MA, USA), MK-2206 (Topscience, Shanghai, China) and PD98059 (MedChem Express, Monmouth Junction, NJ, USA).
Cell viability
Cells were plated in ultralow-attachment 6-well plates (5×105 cells per well) and incubated for 24 h at 37°C. The cells were trypsinized and stained with a trypan blue solution (T10282, Thermo Fisher Scientific, Waltham, MA, USA), and the proportion of living cells was calculated as previously described [23].
Western blot analysis
Cells were lysed with IP lysis buffer containing a 10% cocktail (B14001, Bimake, Houston, TX, USA), the supernatants were collected by centrifugation at 13,000 rpm for 15 min, and the protein concentration was determined using a BCA kit (Pierce Chemical, Rockford, IL, USA). SDS-PAGE was performed using 30-50 µg of total cell protein. Then, the protein was transferred to a PVDF membrane, blocked with 5% skim milk at room temperature, and incubated with the indicated antibodies. The primary antibodies used were anti-LMP1 (1:500, M0897) purchased from DAKO (Glostrup, Denmark), anti-β actin (1:10,000, ab026) purchased from Abclonal (Wuhan, China); anti-p-JNK1/2/3 (Thr183+Tyr185) (1:1000, AF3318) purchased from Affinity Biosciences (Cincinnati, OH, USA); anti-vimentin (1:2000, NBP1-31327) purchased from Novus Biologicals (Littleton, CO, USA); anti-twist (1:1000, 25465-1-ap), anti-snail (1:1000, 13099-1-ap), anti-p-AKT (S473) (1:1000, 66444-1-IG), anti-AKT (1:1000, 60203-2-IG), anti-p38 (1:1000, 66234-1-IG), and anti-JNK (1:10,000, 66210-1-IG) purchased from Proteintech (Wuhan, China); anti-ERK (1:1000, D160317) purchased from Sangon Biotech (Shanghai, China); and anti-cleaved PARP (1:1000, 5625), anti-cleaved caspase3 (1:1000, 9664), anti-NTRK2 (1:1000, 4603S), anti-p-p65 (Ser536) (1:1000, 3033), anti-p65 (1:1000, 8242S), anti-E-cadherin (1:1000, 3195S), anti-LC3 (1:1000, 3868S), anti-p-p38 (Thr180/Tyr182) (1:1000, 4511T), and anti-p-ERK (Thr202/Tyr204) (1:1000, 4370T) purchased from Cell Signaling Technology (Danvers, MA, USA). Blots were analyzed using a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA).
Real-time quantitative PCR (qPCR)
Total RNA was isolated with TRIzol (15596026, Thermo Fisher Scientific) according to the manufacturer’s protocol. Reverse transcription was performed according to manufacturer instructions (K1621, Thermo Fisher Scientific). qPCR was performed using TaqManTM Gene Expression Master Mix (4369016, Thermo Fisher Scientific) on the ABI 7500 Real-Time PCR System (Foster City, CA, USA). qPCR primers are listed in Table S3.
Annexin-V FITC/PI analysis
Cells were plated in ultralow-attachment 6-well plates (5×105 cells per well) and incubated for 24 h at 37°C. The cells were trypsinized and treated according to the instructions of the Annexin-V FITC/PI Apoptosis Detection Kit (KGA107, KeyGEN BioTECH, Jiangsu, China). The proportion of apoptotic cells was observed and calculated under a fluorescence microscope (DMI3000B, Leica, Wetzlar, Germany), or apoptotic analysis was performed with a flow cytometer (FlowSight, Millipore, Billerica, MA, USA).
Soft agar assay
Six-well plates were precoated with 0.6% agar (A9414, Sigma-Aldrich, St. Louis, MO, USA) in growth medium. After solidification, cells (500-1000 cells per well) were suspended in 0.35% agar and added to the 6-well plates to form a double agar layer. The cells were fed by adding 0.2 mL of medium to the top of each well every 2 days. After 2 weeks, the cell clones were observed and counted under a light microscope (DMI3000B, Leica) as previously described [24].
Scratch assay
A cell monolayer was scraped to create a ‘scratch’ with a tip. Then, the debris was removed, serum-free medium was added, and an image was observed and acquired with a microscope (DMI3000B, Leica). After 8 h of culture, a second image was acquired. Photoshop software was used to analyze the narrowed distance, and SPSS statistical software was used for statistical analysis.
Transwell assay
Two hundred microliters of cell suspension (2×104) was added to a transwell chamber (353097, Corning, NY, USA) covered with Matrigel (356230, BD Biosciences, San Jose, CA, USA), and medium supplemented with 10% FBS was added to the lower chamber. After 24 h of incubation, the cells were fixed with methanol and stained with crystal violet for 15 min. The number of invaded cells was counted under a microscope (DMI3000B, Leica).
Immunohistochemistry (IHC)
Twenty-one NPC tissue samples were obtained from Xiangya Hospital. IHC was performed using a universal two-step immunohistochemistry kit (PV-9000, ZSGB-BIO, Beijing, China) and DAB kit (ZLI-9017, ZSGB-BIO, Beijing, China) according to the kit instructions. The primary antibodies used were anti-LMP1 (1:50, M0897, DAKO), anti-NTRK2 (1:100, 13129-1-AP, Proteintech), and anti-cleaved PARP (1:200, 5625, CST). Positive staining was identified according to the percentage and the staining intensity.
Luciferase reporter assay
To generate wild-type luciferase reporter plasmids, an NTRK2 promoter fragment amplified by PCR was cloned into the pGL3-Basic vector (Promega, Madison, WI, USA). Mutant-type plasmids with NF-κB p65 binding site mutations in the promoter of NTRK2 were generated using a mutagenesis kit (C214-02, Vazyme, Nanjing, China) according to the manufacturer’s instructions. The primers are listed in Table S4. After respectively transfecting wild-type and mutant-type plasmids into cells, luciferase activities were measured according to the instructions of the Dual-Luciferase Reporter System (E1910, Promega, Madison, WI, USA), and fluorescence values were detected with a multifunctional chemiluminescence detector (VICTOR X3, PerkinElmer, Waltham, MA, USA).
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using a ChIP assay kit (265157, Thermo) following the manufacturer’s instructions. The antibody used was anti-NF-κB p65 (1:100, 8242S, CST). The primers for the NTRK2 promoter domain containing the NF-κB p65-binding site used in the ChIP assays are shown in Table S5.
Immunofluorescence assay
Cells were washed with PBS, treated with 500 µl/well prechilled ice-cold methanol, fixed for 10 min, and then blocked with 10% donkey serum for 1 h. The primary antibody was incubated overnight at 4°C. The primary antibodies used were anti-p65 (1:400, 8242S, CST), anti-E-cadherin (1:100, Cusabio, Wuhan, China), anti-vimentin (1:400, NBP1-31327, NOVUS), anti-twist (1:50, 25465-1-ap, Proteintech), and anti-snail (1:50, 13099-1-ap, Proteintech), and the fluorescent secondary antibody (SAB4600042, anti-mouse or SAB4600234, anti-rabbit, Sigma) was incubated for 45 min at room temperature. A DAPI staining solution (E607303, BBI Life Sciences, Shanghai, China) was used to stain the nucleus. A reagent for preventing fluorescence quenching (E675001, BBI Life Sciences, Shanghai, China) was used to seal slides, and a confocal microscope (LSM 510 META, Carl Zeiss, Germany) was used to capture fluorescence images.
In vivo experiments
Animal studies were conducted in accordance with institutional ethical guidelines for the care and use of experimental animals. Nine 6-week-old female mice (BALB/c-nu) were randomly divided into 3 groups. CNE1, CM or CM-shNTRK2 cells (2×106) were suspended in 200 µl of PBS and injected via the tail veins to establish an animal metastasis model as previously described [25,26]. All mice were sacrificed 9 weeks after the injection, the lungs were isolated, and metastases on the lung surface were observed. Tissue embedding and H&E staining were performed, and the expression of LMP1, NTRK2 and cleaved PARP was detected by immunohistochemistry.
Statistical analysis
All data are represented as the average value and standard deviation obtained from three independent experiments. Comparisons between groups were performed using Student’s t-test. Statistical significance was defined as P<0.05. The relationship between LMP1 and NTRK2 was analyzed by the Pearson method.
Results
LMP1 promotes anoikis resistance and enhances the migration and invasion of NPC cells
To investigate whether LMP1 regulates anoikis, the NPC cell lines CNE1, CM, HK1, HM, C666-1 and C666-1-shLMP1, which have different LMP1 expression level, were selected for experiments (Figure 1A). The cells were suspended in ultralow-attachment plates, and total protein was extracted for western blot analysis after 48 h. The results showed that the LMP1-positive NPC cell lines CM, HM and C666-1 had lower expression of cleaved PARP and caspase3 than the LMP1-negative NPC cell lines CNE1, HK1 and C666-1-shLMP1, suggesting that LMP1-positive NPC cells are more resistant to anoikis (Figure 1B). Under suspension culture conditions for 24 h, the NPC cell lines CM, HM and C666-1 had a greater ability to form clusters than the LMP1 negative cell lines (Figure 1C), indicating that there was more communication among these cells, which is conducive for signal transmission among cells and promotes cell survival [27]. Then, Annexin-V/PI staining results showed that the apoptosis of HM and C666-1 cells were decreased compared with those of HK1 and C666-1-shLMP1 cells after suspension culture (P<0.01) (Figure 1D, 1E). After 24 h of suspension cell culture, trypan blue exclusion and flow cytometric analysis experiments found that the survival of LMP1-positive cells were higher than those of LMP1-negative cells, meanwhile, the cell apoptosis of C666-1 cells were lower than those of C666-1-shLMP1 cells, and this phenomenon was more obvious when the cell culture density was relatively low (Figure S1A, S1B). After transfection of siRNA-LMP1 into CM and HM cells and suspension culture for 24 h, trypan blue exclusion assay showed that the cell survival decreased (P<0.01) (Figure 1F). Furthermore, the ability to resist anoikis is necessary for cells to form clones in soft agar, and the ability to form soft agar clones predicts the metastatic potential of tumor cells [28]. As shown in Figure 1G, the data showed that CM and HM cells formed more clones than CNE1 and HK1 cells (P<0.05), indicating that LMP1-positive NPC cells are more resistant to anoikis and have greater metastatic potential than LMP1 negative cells. Scratch experiments showed that the wound healing ability of CM and HM cells was stronger than that of CNE1 and HK1 cells (P<0.05 and P<0.01, respectively), and transwell assay results also showed that CM and HM cells were more invasive than CNE1 and HK1 cells (P<0.01 and P<0.001, respectively) (Figure S2A, S2B). These results indicate that LMP1 can promote anoikis resistance and enhance the migration and invasion of NPC cells.
Figure 1.

LMP1 promotes anoikis resistance. (A) qPCR was performed to determine the expression of LMP1 in NPC cells (CNE1, CM, HK1, HM, C666-1, and C666-1-shLMP1 cells). (B) Western blot was used to analyze LMP1, cleaved PARP and caspase3 levels in NPC cells after suspension culture for 48 h. (C) NPC cells were grown under anchorage-free conditions (in ultralow-attachment cell culture plates) for 24 h. Scale bar: 200 μm. (D) Fluorescence microscopy and (E) flow cytometric analysis were performed to observe cell apoptosis after culture under suspension conditions for 24 h. Scale bar: 50 μm. (F) After transfection with siRNA-NC or siRNA-LMP1, the viability of nonadherent cells was assessed in a trypan blue rejection experiment. Scale bar: 100 μm. (G) The colony numbers of NPC cells in soft agar (2 weeks) are shown. Scale bar: 100 μm. CM (CNE1-LMP1), HM (HK1-LMP1). UN: untreated, NC: negative control. Columns: mean of three replicates; statistical significance was calculated using a t-test (ns: no significance, *P<0.05, **P<0.01, and ***P<0.001).
LMP1 upregulates NTRK2 expression
After determining that LMP1 can promote anoikis resistance in NPC cells, we analyzed the key molecules involved in LMP1 regulate anoikis. Ninety-eight genes related to anoikis were selected from the literature, and their mRNA levels were analyzed in CNE1, CM, HK1, and HM cells by a qPCR array. By using a twofold difference as a screening condition, five genes were identified to have upregulated expression in CM and HM cells: BCL-2, CXCR4, MMP9, FN1 and NTRK2 (Figure 2A). Among these genes, the alteration of NTRK2 was the most obvious (Figure 2B). In LMP1-positive CM, HM, and C666-1 cells, the mRNA expression of NTRK2 was significantly higher than that in LMP1-negative cells (P<0.001) (Figure 2C). Furthermore, western blot analysis showed that the protein level of NTRK2 was also high in LMP1-positive cells (Figure 2D). IHC results for 21 NPC tissue samples showed that NTRK2 was highly expressed in LMP1-positive NPC tissue samples, implying that LMP1 and NTRK2 were positively correlated (P=0.0262) (Figure 2E). These results confirm that LMP1 can upregulate NTRK2 expression in NPC.
Figure 2.

LMP1 upregulates NTRK2 expression. (A) A qPCR array was performed to analyze alteration of anoikis-related genes in LMP1-positive and LMP1-negative NPC cells. The red rectangle indicated the differentially expressed genes. (B) qPCR was performed to verify the expression alteration of BCL-2, CXCR4, FN1, MMP9 and NTRK2. (C) qPCR and (D) Western blot were performed to detect NTRK2 expression in NPC cells. (E) IHC staining for LMP1 and NTRK2 in clinical NPC tissue samples, and the correlation between the expression of these two proteins are shown. Scale bar: 50 μm. Columns: mean of three replicates; statistical significance was calculated using a t-test (*P<0.05, **P<0.01, and ***P<0.001).
NTRK2 promotes anoikis resistance in NPC cells
Further, we investigated whether LMP1-induced NTRK2 promotes anoikis resistance in NPC. The NTRK2 inhibitor K252a was used to knock down NTRK2 activity in HM cells (Figure S3A), and C666-1-shNTRK2 and CM-shNTRK2 cells with NTRK2 stably knocked down were established (Figures S3B, S4). After 48 h of suspension culture, western blot analysis showed that the protein expression of cleaved caspase3 and cleaved PARP was increased (Figure 3A). In addition, the formation of cell clusters was decreased when cells were cultured in ultralow-attachment plates (Figure 3B). Annexin-V/PI staining results indicated increased apoptosis after knocking down NTRK2 (P<0.01) (Figure 3C, 3D), whereas a trypan blue exclusion test showed that the cell survival was reduced (P<0.05 and P<0.01, respectively) (Figure 3E). Furthermore, the ability of cellular clone formation was weakened in two cell lines CM-shNTRK2 and C666-1-shNTRK2, which were NTRK2 stably knocked down (P<0.01) (Figure 3F). These results indicate that NTRK2 induced by LMP1 can promote anoikis resistance in NPC cells.
Figure 3.

NTRK2 induces anoikis resistance in NPC cells. The NTRK2 inhibitor K252a (200 nM) was used to knock down NTRK2 activity in HM cells, and C666-1-shNTRK2 and CM-shNTRK2 cells with NTRK2 stably knocked down were used. (A) The expression of NTRK2, cleaved PARP and cleaved caspase3 was analyzed by western blot after suspension culture for 48 h. (B) NPC cells were grown under anchorage-free conditions for 24 h. Scale bar: 50 µm. (C) Fluorescence microscopy and (D) flow cytometric analysis were performed to investigate cell apoptosis after culture under suspension conditions for 24 h. Scale bar: 50 µm. (E) The viability of nonadherent cells was assessed by a trypan blue rejection experiment. (F) The colony numbers of NPC cells in soft agar (2 weeks) are shown. Scale bar: 100 μm. Columns: mean of three replicates; statistical significance was calculated using a t-test (*P<0.05, **P<0.01, and ***P<0.001).
LMP1 upregulates NTRK2 expression through NF-κB
Studies have shown that LMP1 activates IKKβ through TRAF and TAK1, then IKKβ phosphorylates the NF-κB inhibitor IκBα and lets its rapid degradation, further the p65: p50 or cRel: p52 translocate into the nucleus to regulate target gene expression. In parallel with this, increasing phosphorylation at Ser536 of the NF-ĸB p65 by IKKβ and IKKα, plays a key role in the transcriptional competence of NF-ĸB and the nuclear translocation of p65. LMP1 also stimulates noncanonical NF-κB activation through TRAF and NIK. In this pathway, IKKα is activated by NIK and phosphorylates p100 to trigger its proteasomal processing into the mature p52, then enables the nuclear translocation of RelB: p52 [10,29,30]. Meanwhile, bioinformatics analysis of our study showed that in NTRK2 promoter, p65: p50 is the only binding molecular dimer of NF-κB pathway. So, we will study whether LMP1 regulates NTRK2 through p65. After transfection of siRNA-p65 into HM and C666-1 cells, the mRNA levels of NTRK2 were decreased (P<0.05) (Figure 4A). Western blot analysis showed that both p-p65 and NTRK2 were overexpressed in LMP1-positive NPC cells, while total p65 was slightly alteration. We also found that the expression of p-p65 and NTRK2 was reduced after LMP1 or NF-κB p65 was silenced (Figure 4B). Immunofluorescence observation showed that more NF-κB p65 translocated into nuclei, suggesting that more activated NF-κB signaling occurred in LMP1-positive CM and HM cells than in CNE1 and HK1 cells (Figure 4C). Genmatix software analysis predicted two NF-κB p65 binding sites in the promoter of NTRK2 (Figure 4D). The ChIP analysis showed that more NF-κB p65 was enriched at the site 2 of the NTRK2 promoter in LMP1-positive C666-1 cells than in C666-1-shLMP1 cells (P<0.01), but the enrichment at the site 1 of the NTRK2 promoter was not significantly changed (Figure 4E). By luciferase reporter assay, NF-κB p65 was proved to enhance the luciferase activity of wild-type NTRK2. The luciferase activity of mutant site 1 NTRK2-1 was not significantly different from that of wild-type NTRK2, however, the luciferase activity of mutant site 2 NTRK2-2 showed a lower alteration of promoter activity (P<0.01) (Figure 4F). These results indicate that LMP1-mediated NF-κB activity is capable of targeting the upregulation of NTRK2 expression.
Figure 4.

LMP1 upregulates NTRK2 expression through NF-κB p65. A. After transfection with siRNA-NC or siRNA-p65, the expression of p65 and NTRK2 in HM and C666-1 cells was detected by qPCR. B. The expression of p-p65, p65 and NTRK2 in HK1, HM, C666-1 and C666-1-shLMP1 cells was detected by western blot after siRNA-p65 treatment. C. The subcellular location of NF-κB p65 was analyzed by immunofluorescence staining in CNE1, CM, HK1 and HM cells. Cells were stained with a primary anti-p65 antibody (green), and the nucleus was counterstained with DAPI (blue). Scale bar: 50 μm. D. A schematic of the NTRK2 luciferase (Luc) construct is shown. The locations of the putative p65 binding elements and the mutations are indicated. E. ChIP analysis was carried out using a p65 Ab, and the enrichment of the NTRK2 promoter sequence was detected by qPCR. F. A luciferase assay was performed using the NTRK2 promoter-firefly luciferase reporter with a wild-type or mutated p65 binding site in 293T cells. UN: untreated, NC: negative control. Columns: mean of three replicates; statistical significance was calculated using a t-test (ns: no significance, *P<0.05, **P<0.01, and ***P<0.001).
LMP1-induced NTRK2 promotes anoikis resistance through EMT
Studies have shown that EMT [18], autophagy [22] and oxidative stress response [21] are important mechanisms of anoikis resistance. After 48 h of suspension culture, western blot analysis showed that NTRK2 expression was increased, E-cadherin expression was downregulated and Vimentin expression was upregulated in LMP1-positive HM cells compared with LMP1-negative HK1 cells, After knocking down of NTRK2, the expression of E-cadherin and Vimentin showed opposite alterations in HM-shNTRK2 cells (Figure 5A). The results showed that NTRK2 was the key molecule of LMP1 mediated EMT in NPC cells. Further, the NTRK2 inhibitor K252a was used to knock down NTRK2 activity in HM cells, and C666-1-shNTRK2 cells with NTRK2 stably knocked down were used for following study. After 48 h of suspension culture, the data showed that E-cadherin expression was significantly upregulated, and vimentin expression was downregulated, along with cleaved PARP and caspase3 level was increased (Figure 5B). At the same time, the cell morphology was changed significantly, showing a trend for a rounded morphology instead of a spindle shape (Figure 5C). However, autophagy-related LC3 did not change significantly after NTRK2 silencing (Figure S4), suggesting that NTRK2 participates in anoikis resistance through EMT rather than autophagy in NPC cells. Furthermore, the results of immunofluorescence experiments also showed that the expression of E-cadherin was enhanced and that of vimentin was weakened in NPC cells after NTRK2 knocking down (Figure 5D). These results indicate that LMP1-induced NTRK2 resists anoikis by promoting EMT.
Figure 5.
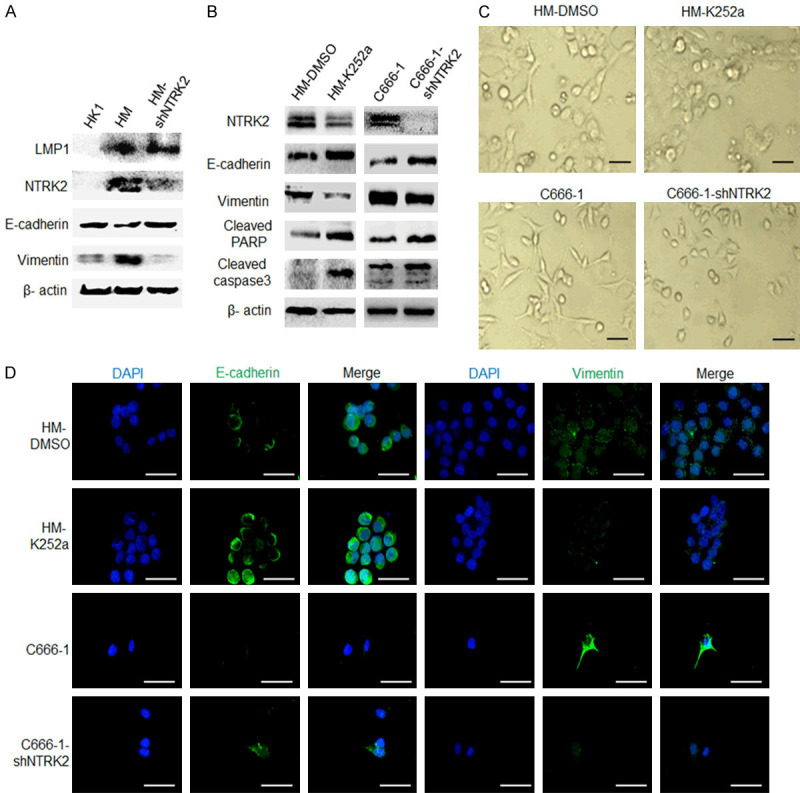
LMP1-induced NTRK2 promotes anoikis resistance through EMT. The NTRK2 inhibitor K252a (200 nM) was used to knock down NTRK2 activity in HM cells, and C666-1-shNTRK2 cells with NTRK2 stably knocked down were used. A. The expression of LMP1, NTRK2, E-cadherin and Vimentin was analyzed by western blot. B. The expression of E-cadherin, Vimentin, and cleaved PARP and caspase3 was analyzed by western blot. C. Cell morphology under a microscope is shown. D. The expression of E-cadherin and vimentin in HM and C666-1 cells was evaluated by immunofluorescence. Scale bar: 50 µm.
NTRK2 promotes EMT though twist by activating ERK/AKT in NPC cells
Studies have indicated that NTRK2 regulates EMT mainly through two transcription factors, twist and snail [12,13,19]. After knocking down NTRK2 activity, the data showed that twist expression was downregulated, while snail expression did not change significantly in HM and C666-1 NPC cells (Figure 6A). Immunofluorescence analysis showed the same results (Figure 6B). These data indicate that NTRK2 participates in EMT mainly through twist in NPC cells. Studies have also shown that NTRK2 is involved in the regulation of many signaling pathways, including the MAPK [31], AKT [32], and JAK/STAT3 [33] pathways. Western blot results showed that the expression of p-ERK and p-AKT in NPC cells was significantly downregulated after stably knocked down of NTRK2 expression and that there were no significant changes in the total protein levels of ERK and AKT, in addition, there were no significant changes in the total protein and phosphorylation levels of p38 and JNK (Figure 6C). After inhibiting AKT using MK-2206 or inhibiting ERK using PD98059 in HM and C666-1 cells, the data indicated that twist expression was reduced, vimentin expression was downregulated, and E-cadherin expression was upregulated and its alteration was more obvious after inhibiting both AKT and ERK (Figure 6D). Further, western blot results showed that the expression of NTRK2, p-ERK and p-AKT in NPC cells was significantly reduced after stably knocked down of LMP1 expression and that there were no significant changes of the total protein levels of ERK and AKT, at the same time, twist and vimentin expression was downregulated, and E-cadherin expression was increased (Figure 6E). These results indicated that LMP1-induced NTRK2 regulates twist through ERK and AKT.
Figure 6.
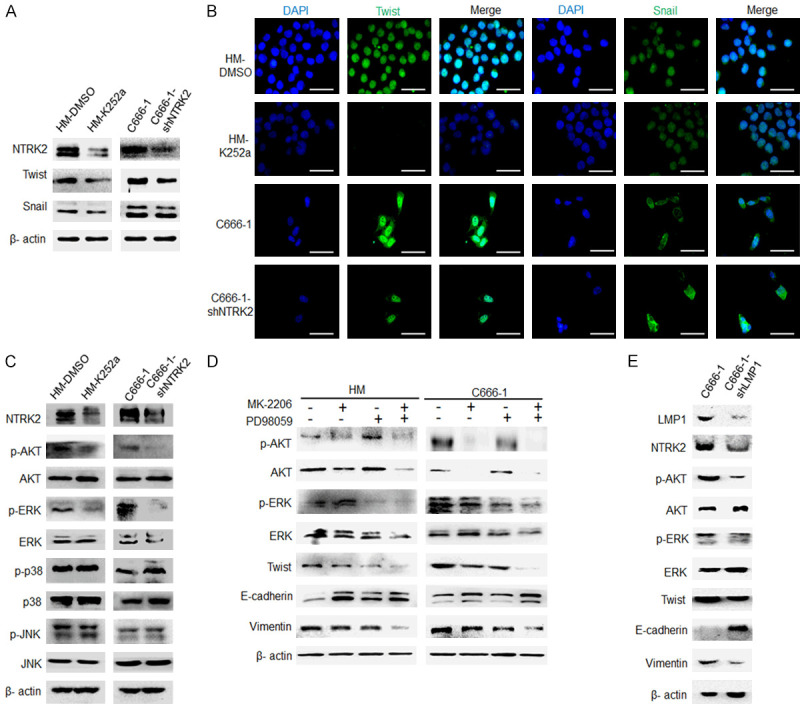
NTRK2 promotes EMT though twist by activating ERK/AKT in NPC cells. The NTRK2 inhibitor K252a (200 nM) was used to knock down NTRK2 activity in HM cells, and C666-1-shNTRK2 cells with NTRK2 stably knocked down were used. A. The expression of twist and snail was analyzed by western blot. B. The expression of twist and snail in HM and C666-1 cells was evaluated by immunofluorescence. Scale bar: 50 µm. C. Levels of AKT and MAPK signaling were determined by western blot analysis. D. The expression of twist, E-cadherin and vimentin was detected by western blot after inhibiting AKT and ERK. E. The expression of NTRK2, AKT, ERK, twist, E-cadherin and vimentin was detected by western blot after LMP1 stably knocked down in C666-1 cells.
Down-regulation of NTRK2 inhibits the metastasis of LMP1-positive NPC
CNE1, CM, or CM-shNTRK2 cells were injected into nude mice via the tail vein. After 9 weeks, the mice were sacrificed, and the lungs were removed. Figure 7A shows that there were no obvious metastases on the lung surface in the mice of the CNE1 group. However, a large number of metastases was observed in the CM group, and the number of metastases in the CM-shNTRK2 mice were significantly reduced. H&E staining showed that the area of tumor metastases in the CM group was significantly larger than that in both CNE1 and CM-shNTRK2 groups (Figure 7B). IHC results indicated that in the LMP1-positive CM group, NTRK2 was highly expressed and cleaved PARP expression was low, whereas the results were reversed in the LMP1-negative CNE1 group. In the CM-shNTRK2 group, both LMP1 and cleaved PARP were highly expressed (Figure 7C). These results verify that LMP1 enhances the ability of NPC to resist anoikis by up-regulating NTRK2 expression, thereby promoting NPC metastasis.
Figure 7.
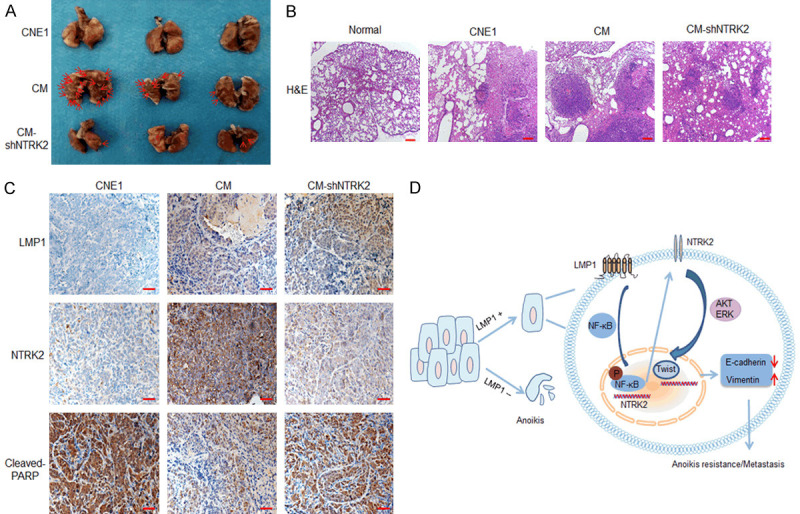
Down-regulation of NTRK2 expression inhibits the metastasis of LMP1-positive NPC. A. The NPC cells CNE, CM, or CM-shNTRK2 (2×106) were injected into nude mice via the tail vein. All mice were sacrificed 9 weeks after injection, and the lungs were isolated. Representative images of the lungs are shown. B. H&E staining was performed. Scale bar: 200 μm. C. The expression of LMP1, NTRK2 and cleaved PARP in the lungs was analyzed by immunohistochemistry. Scale bar: 50 μm. D. The model of the mechanism by which LMP1-induced NTRK2 promotes anoikis resistance and metastasis in NPC is shown.
Discussion
Metastasis of NPC is an important factor limiting the survival of patients [34,35]. Recently, anoikis has been found to be closely related to tumor metastasis and become a hot spot in the field of tumor metastasis [36]. A series of studies have shown that LMP1 can promote NPC metastasis [10,12,13], but the relationship between LMP1 and anoikis is not clear. Here, we found that EBV-LMP1 promoted resistance to anoikis and NPC metastasis. Notably, the present study found that when the culture density of NPC cells was 0.5×105 cells per well (6-well plate), the survival rate of LMP1-positive cells after suspension culture was significantly higher than that of LMP1-negative cells. When the culture density was 5×105 cells per well, the survival rate of LMP1-positive cells after suspension culture was still higher than that of LMP1-negative cells. However, when the culture density reached 1×106 cells per well, the survival rates of LMP1-positive cells and LMP1-negative cells after suspension culture were not significantly different, which is consistent with Wasil LR’s study [37]. These data imply that anoikis is associated with cell density. The reason may be that the signal transmission between cells can promote cell survival, when the cell culture density is low, the intercellular communication is reduced, so the possibility of inducing apoptosis is relatively obvious [27].
NTRK2 is a membrane-bound receptor that belongs to the tyrosine receptor kinase family. NTRK2 can promote anoikis resistance in some tumor cells [38-41]. Here, we showed that LMP1-induced NTRK2 could promote NPC cell resistance to anoikis and metastasis. Further research indicated that LMP1 upregulated NTRK2 expression through NF-κB p65. Tumor cells resist anoikis through a variety of mechanisms, including regulating EMT [19], autophagy [22], and oxidative stress [21]. Studies have shown that NTRK2 regulates EMT through twist and snail [12,13,19]. In the present study, we demonstrated that NTRK2 participated in EMT only through twist in NPC cells, which may be related to cell-type specificity. Meanwhile, our study also indicated that the expression of autophagy-related LC3 was not related to LMP1-induced NTRK2 in NPC cells. Whether NTRK2 participates in anoikis resistance through oxidative stress or other mechanisms in NPC cells needs further investigation.
NTRK2 involves in many signaling pathways, including the MAPK [31], AKT [32], JAK/STAT3 pathways [33]. The current study indicated that the expression of p-AKT and p-ERK was significantly downregulated after NTRK2 knocking down, whereas the total protein levels of AKT and ERK did not change, and the total protein and phosphorylation levels of p38 and JNK did not change as well. It is suggested that NTRK2 regulates twist by activating ERK and AKT signaling in NPC. Moreover, after treating cells with inhibitors of AKT and ERK, we found that twist and vimentin expression increased and E-cadherin expression decreased obviously. These results imply that NTRK2 promotes EMT by increasing the twist level through AKT/ERK signaling.
In conclusion, the present study demonstrate that the NPC tumorigenic protein LMP1 can promote anoikis resistance in NPC cells in vitro and in vivo, and identify that EBV-LMP1 in NPC cells mediates AKT and ERK signaling through NTRK2 and then upregulates twist expression to promote EMT, thereby inducing anoikis resistance and enhancing NPC metastasis (Figure 7D). These data will provide new molecular markers and potential targets for clinical intervention of NPC.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (No. 81372182 and 81672761 (L.Y.), 81672759 (D.F.)), Fundamental Research Funds for the Central Universities of Central South University (no. 1053320184236) and the Natural Science Foundation of Hunan Province, China (2018JJ2545).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, Xiang B, Zhou M, Li X, Wu X, Li Y, Li X, Li G, Xiong W, Zeng Z. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J Cancer. 2018;9:2852–2864. doi: 10.7150/jca.25460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsao SW, Tsang CM, Lo KW. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160270. doi: 10.1098/rstb.2016.0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yosra M, Sameh S, Randa G, Yassmine R, Sallouha G, Nadia B, Noureddine B, Abdelfattah Z, Elham H. Functional polymorphisms and gene expression of tlr9 gene as protective factors for nasopharyngeal carcinoma severity and progression. J Immunol Res. 2019;2019:2826563. doi: 10.1155/2019/2826563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kieser A, Sterz KR. The latent membrane protein 1 (LMP1) Curr Top Microbiol Immunol. 2015;391:119–149. doi: 10.1007/978-3-319-22834-1_4. [DOI] [PubMed] [Google Scholar]
- 5.Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang M, Chen L, Yang L, Tao Y, Jiang Y, Li J, Yi B, Li B, Fan S, You S, Deng X, Hu F, Feng L, Bode AM, Dong Z, Sun LQ, Cao Y. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33:4568–4578. doi: 10.1038/onc.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo X, Hong L, Cheng C, Li N, Zhao X, Shi F, Liu J, Fan J, Zhou J, Bode AM, Cao Y. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018;9:619. doi: 10.1038/s41419-018-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, Liu L, Xu Z, Liao W, Feng D, Dong X, Xu S, Xiao L, Lu J, Luo X, Tang M, Bode AM, Dong Z, Sun L, Cao Y. EBV-LMP1 targeted DNAzyme enhances radiosensitivity by inhibiting tumor angiogenesis via the JNKs/HIF-1 pathway in nasopharyngeal carcinoma. Oncotarget. 2015;6:5804–5817. doi: 10.18632/oncotarget.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, Tang M, Li H, Xu Z, Weng X, Li J, Yu X, Zhao L, Liu H, Hu Y, Tan Z, Yang L, Zhong M, Zhou J, Fan J, Bode AM, Yi W, Gao J, Sun L, Cao Y. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016;380:191–200. doi: 10.1016/j.canlet.2016.05.032. [DOI] [PubMed] [Google Scholar]
- 9.Terrin L, Dal Col J, Rampazzo E, Zancai P, Pedrotti M, Ammirabile G, Bergamin S, Rizzo S, Dolcetti R, De Rossi A. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J Virol. 2008;82:10175–10187. doi: 10.1128/JVI.00321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang LW, Jiang S, Gewurz BE. Epstein-barr virus LMP1-mediated oncogenicity. J Virol. 2017;91:e01718–16. doi: 10.1128/JVI.01718-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou YC, Chen CL, Yeh TH, Lin SJ, Chen MR, Doong SL, Lu J, Tsai CH. Involvement of recepteur d’origine nantais receptor tyrosine kinase in Epstein-Barr virus-associated nasopharyngeal carcinoma and its metastasis. Am J Pathol. 2012;181:1773–1781. doi: 10.1016/j.ajpath.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Horikawa T, Yoshizaki T, Kondo S, Furukawa M, Kaizaki Y, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br J Cancer. 2011;104:1160–1167. doi: 10.1038/bjc.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horikawa T, Yang J, Kondo S, Yoshizaki T, Joab I, Furukawa M, Pagano JS. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67:1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 14.Sun T, Zhong X, Song H, Liu J, Li J, Leung F, Lu WW, Liu ZL. Anoikis resistant mediated by FASN promoted growth and metastasis of osteosarcoma. Cell Death Dis. 2019;10:298. doi: 10.1038/s41419-019-1532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, Zhao Q, Wang ZX, Li T, Lu YX, Wu QN, Yu K, Wang F, Pu HY, Li B, Jia WH, Shi M, Xie D, Kang TB, Huang P, Ju HQ, Xu RH. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene. 2018;37:6025–6040. doi: 10.1038/s41388-018-0384-z. [DOI] [PubMed] [Google Scholar]
- 16.Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, Lyons YM, Nagaraja AS, Dood RL, Wen Y, Mangala LS, Hansen JM, Rupaimoole R, Gharpure KM, Rodriguez-Aguayo C, Yim SY, Lee JS, Ivan C, Hu W, Lopez-Berestein G, Wong ST, Karlan BY, Levine DA, Liu J, Afshar-Kharghan V, Sood AK. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8:310. doi: 10.1038/s41467-017-00411-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beausejour M, Thibodeau S, Demers MJ, Bouchard V, Gauthier R, Beaulieu JF, Vachon PH. Suppression of anoikis in human intestinal epithelial cells: differentiation state-selective roles of alpha2beta1, alpha3beta1, alpha5beta1, and alpha6beta4 integrins. BMC Cell Biol. 2013;14:53. doi: 10.1186/1471-2121-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luey BC, May FE. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: importance of the type I IGF receptor and PI3-kinase/Akt pathway. Mol Cancer. 2016;15:8. doi: 10.1186/s12943-015-0482-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smit MA, Peeper DS. Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene. 2011;30:3735–3744. doi: 10.1038/onc.2011.96. [DOI] [PubMed] [Google Scholar]
- 20.Zhao MY, Wang LM, Liu J, Huang X, Liu J, Zhang YF. MiR-21 suppresses anoikis through targeting PDCD4 and PTEN in human esophageal adenocarcinoma. Curr Med Sci. 2018;38:245–251. doi: 10.1007/s11596-018-1872-7. [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Kim JH. Activation of the leukotriene B4 receptor 2-reactive oxygen species (BLT2-ROS) cascade following detachment confers anoikis resistance in prostate cancer cells. J Biol Chem. 2013;288:30054–30063. doi: 10.1074/jbc.M113.481283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talukdar S, Pradhan AK, Bhoopathi P, Shen XN, August LA, Windle JJ, Sarkar D, Furnari FB, Cavenee WK, Das SK, Emdad L, Fisher PB. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc Natl Acad Sci U S A. 2018;115:5768–5773. doi: 10.1073/pnas.1721650115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoppe T, Kraus D, Probstmeier R, Jepsen S, Winter J. Stimulation with porphyromonas gingivalis enhances malignancy and initiates anoikis resistance in immortalized oral keratinocytes. J Cell Physiol. 2019;234:21903–21914. doi: 10.1002/jcp.28754. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y, Ramos da Silva S, He M, Liang Q, Lu C, Feng P, Jung JU, Gao SJ. An oncogenic virus promotes cell survival and cellular transformation by suppressing glycolysis. PLoS Pathog. 2016;12:e1005648. doi: 10.1371/journal.ppat.1005648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Luo H, Huo Z, Chen M, Han Z, Hung M, Su B, Li Y, Wang X, Guo X, Xiao H, Lee D, Zhao R, Yang H. Irradiation-induced dynamic changes of gene signatures reveal gain of metastatic ability in nasopharyngeal carcinoma. Am J Cancer Res. 2019;9:479–495. [PMC free article] [PubMed] [Google Scholar]
- 26.Fu XT, Shi YH, Zhou J, Peng YF, Liu WR, Shi GM, Gao Q, Wang XY, Song K, Fan J, Ding ZB. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018;412:108–117. doi: 10.1016/j.canlet.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Zhong X, Rescorla FJ. Cell surface adhesion molecules and adhesion-initiated signaling: understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal. 2012;24:393–401. doi: 10.1016/j.cellsig.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Saharat K, Lirdprapamongkol K, Chokchaichamnankit D, Srisomsap C, Svasti J, Paricharttanakul NM. Tumor susceptibility gene 101 mediates anoikis resistance of metastatic thyroid cancer cells. Cancer Genomics Proteomics. 2018;15:473–483. doi: 10.21873/cgp.20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ersing I, Bernhardt K, Gewurz BE. NF-kappaB and IRF7 pathway activation by Epstein-Barr virus latent membrane protein 1. Viruses. 2013;5:1587–1606. doi: 10.3390/v5061587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 31.MacFarland SP, Naraparaju K, Iyer R, Guan P, Kolla V, Hu Y, Tan K, Brodeur GM. Mechanisms of entrectinib resistance in a neuroblastoma xenograft model. Mol Cancer Ther. 2020;19:920–926. doi: 10.1158/1535-7163.MCT-18-1044. [DOI] [PubMed] [Google Scholar]
- 32.Jiang L, Wang Z, Liu C, Gong Z, Yang Y, Kang H, Li Y, Hu G. TrkB promotes laryngeal cancer metastasis via activation PI3K/AKT pathway. Oncotarget. 2017;8:108726–108737. doi: 10.18632/oncotarget.21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim MS, Lee WS, Jeong J, Kim SJ, Jin W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget. 2015;6:40158–40171. doi: 10.18632/oncotarget.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou Y, Yang R, Huang ML, Kong YG, Sheng JF, Tao ZZ, Gao L, Chen SM. NOTCH2 negatively regulates metastasis and epithelial-Mesenchymal transition via TRAF6/AKT in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2019;38:456. doi: 10.1186/s13046-019-1463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang MH, Sun R, Zhou XM, Zhang MY, Lu JB, Yang Y, Zeng LS, Yang XZ, Shi L, Xiao RW, Wang HY, Mai SJ. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018;9:2. doi: 10.1038/s41419-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481–3498. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 37.Wasil LR, Shair KHY. Modified anoikis assay that functionally segregates epstein-barr virus LMP1 strains into two groups. J Virol. 2018;92:e00557–18. doi: 10.1128/JVI.00557-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao W, Qiu H, Yang T, Luo X, Zhang H, Wan X. Upregulation of TrkB promotes epithelial-mesenchymal transition and anoikis resistance in endometrial carcinoma. PLoS One. 2013;8:e70616. doi: 10.1371/journal.pone.0070616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng YK, Wong EY, Lau CP, Chan JP, Wong SC, Chan AS, Kwan MP, Tsao SW, Tsang CM, Lai PB, Chan AT, Lui VW. K252a induces anoikis-sensitization with suppression of cellular migration in Epstein-Barr virus (EBV)--associated nasopharyngeal carcinoma cells. Invest New Drugs. 2012;30:48–58. doi: 10.1007/s10637-010-9513-4. [DOI] [PubMed] [Google Scholar]
- 40.Guo D, Hou X, Zhang H, Sun W, Zhu L, Liang J, Jiang X. More expressions of BDNF and TrkB in multiple hepatocellular carcinoma and anti-BDNF or K252a induced apoptosis, supressed invasion of HepG2 and HCCLM3 cells. J Exp Clin Cancer Res. 2011;30:97. doi: 10.1186/1756-9966-30-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731–747. doi: 10.1038/s41571-018-0113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.