

Abstract
Melanoma-associated antigen A3 (MAGEA3), a member of the cancer-testis antigen (CTA) family, is aberrantly expressed in various cancer types. Accumulating evidence indicates that MAGEA3 plays a vital role in the pathogenesis and development of various cancers. However, the underlying mechanisms behind the tumor-promoting effect of MAGEA3 remain unclear, particularly in cervical cancer (CC). The present study investigated the effects of MAGEA3 on CC cell proliferation and apoptosis as well as the underlying molecular mechanism. Cell Counting Kit-8 (CCK-8), 5-ethynyl-2’-deoxyuridine (EdU), and flow cytometry assays were used to evaluate the effects of MAGE-A3 on proliferation, cell cycle, and apoptosis. Co-immunoprecipitation (Co-IP), dual-luciferase reporter, western blotting, and quantitative RT-PCR assays were performed to investigate the regulatory mechanisms of MAGEA3 in CC cells. Compared to the control, MAGE-A3 overexpression markedly promoted the proliferation of SiHa cells in vitro and in vivo, increased the proportion of cells in S phase, and suppressed apoptosis. However, MAGEA3 knockdown inhibited proliferation, blocked the cell cycle in G1 phase, and induced apoptosis in HeLa cells. Further mechanistic study revealed that MAGEA3 interacts with KAP1, thereby suppressing p53 transcriptional activity, thus suppressing p53-mediated regulation of the expression of genes involved in the cell cycle (p21, cyclin D1) and apoptosis (Bax, Bcl-2, and PUMA). Collectively, our results, both in vivo and in vitro, indicate that the expression of MAGEA3 contributes to CC cell proliferation and tumor growth and exerts tumor-promoting effects by regulating the KAP1/p53 signaling pathway.
Keywords: Melanoma-associated antigen A3 (MAGEA3), p53, KRAB domain-associated protein 1 (KAP1), cervical cancer, cell cycle, apoptosis
Introduction
As a common malignant tumor, cervical cancer ranks as the fourth leading cause of cancer-related death worldwide, and it remains one of the major causes of cancer-related death in women worldwide [1]. Although cervical cancer treatment options, including radiotherapy, chemotherapy, and surgery, have made great progress, the overall 5-year survival rate of patients with cervical cancer remains unfavorable because of recurrence and metastasis. Therefore, the development of new diagnosis and treatment strategies is required to reduce recurrence and improve the survival rate.
Melanoma-associated antigen A3 (MAGEA3) gene is a cancer-testis antigen (CTA) gene whose expression has been demonstrated in a wide array of malignancies, including melanoma, breast, colorectal, gastric, lung and pancreatic cancer [2-8]. Emerging data have reported that aberrant expression of MAGEA3 in many tumor types has been shown to correlate with poor clinical outcome [8-11]. As a cancer-testis antigen, MAGEA3 is only expressed in cancer and testes, making it an ideal candidate for cancer immunotherapy given its potential to target specific tumor cell types without affecting normal tissue [12,13].
To investigate whether MAGEA3 is involved in the tumorigenesis and progression of human cervical cancer, we previously detected the expression of MAGEA3 by quantitative RT-PCR and immunohistochemical methods in cervical lesion tissues compared with normal tissues. The results showed that the expression of MAGEA3 in cervical cancer (CC) tissues was significantly higher than that of the normal group. Moreover, the expression level of MAGEA3 was positively correlated with the clinical stage, pathological grade, and lymphatic metastasis of cervical cancer [14]. Based on this, we speculated that MAGEA3 plays a critical role during the development and progression of cervical cancer.
To verify our speculation, we performed the present study to determine whether MAGEA3 regulates the proliferation and apoptosis of CC cells. Recently, convincing evidence points to the capability of MAGE-A proteins to control the p53 tumor suppressor and regulate essential pathways associated with cell proliferation [15]. It has been shown that MAGEA3 is involved in the inhibition of apoptosis via p53-dependent suppression of Bax and preservation of surviving [16]. Another study indicated that Knockdown of MAGEA3 caused a reduction in proliferation in gastric cancer cells by regulating the cell cycle and apoptosis-related genes (p21, Bax) [17]. Thus, to investigate the underlying mechanism of MAGEA3 in carcinogenesis, we further explored the regulatory relationship between MAGEA3 and the P53 signaling pathway in cervical cancer.
Materials and methods
Cell culture
Human cervical cancer cell lines (HeLa, SiHa, C33A, and Caski) were purchased from the Cell Resource Center, Institute of Basic Medical Science (IBMS, Beijing, China). The cells were cultured in RPMI-1640 (GIBCO, NY, USA) medium supplemented with 10% fetal bovine serum (GIBCO), 100 U/ml penicillin and 100 µg/ml streptomycin (HyClone, UT, USA) and were incubated at 37°C with 5% CO2. The culture medium was replaced with fresh medium every 1-2 days. Cells passaging was performed when the cell cultures became 80-90% confluent.
Lentivirus transfection
The lentivirus for overexpressing MAGEA3 (Lv-MAGEA3) and the negative control lentivirus (Lv-NC) were obtained commercially from GenePharma (Shanghai, China). SiHa cells were seeded into a 6-well plate at 70-80% confluence. Cells were transfected with Lv-MAGEA3 or Lv-NC at an MOI (multiplicity of infection) of 5 and divided into three groups: blank control group (blank, untransfected cells), negative control group (Lv-NC, transfected with negative control lentivirus), and MAGEA3-overexpressing group (Lv-MAGEA3, transfected with lentivirus for overexpressing MAGEA3). Transfection was performed according to the manufacturer’s instructions. Cells were transfected 48-72 h prior to being harvested for use in subsequent analysis.
siRNA and plasmid transfection
MAGE-A3 knockdown was performed with siRNA in HeLa cells. Three siRNAs targeting different regions of the human MAGEA3 gene (siMAGEA3-1, 2, 3) and siRNA negative control (siRNA-NC) were designed and synthesized by RiboBio (Guangzhou, China). The siRNA sequences were as follows: siMAGEA3-1: sense 5’-3’ UGGCCAUAAUCGCAAGAGA dTdT; antisense 5’-3’ UCUCUUGCGAUUAUGGCCA dTdT. siMAGEA3-2: sense 5’-3’ CAGUGAUCCUGCAUGUUAU dTdT; antisense 5’-3’ AUAACAUGCAGGAUCACUG dTdT. siMAGEA3-3: sense 5’-3’ GAAGCUGCUCACCCAACAU dTdT; antisense 5’-3’ AUGUUGGGUGAGCAGCUUC dTdT; siRNA-NC: sense 5’-3’ UUCUCCGAACGUGUCACGU dTdT; antisense 5’-3’ ACGUGACACGUUCGGAGAA dTdT. HeLa cells were seeded into a 6-well plate at 70-80% confluence. The cells were divided into three groups: blank control group (blank, untransfected cells), negative control group (siRNA-NC, transfected with siRNA-NC), and MAGEA3 knockdown group (siMAGEA3, transfected with siMAGEA3-1, 2, or 3). Transfection was performed with Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s protocol. The medium was replaced with new culture medium 6 h after transfection. Transfected cells were then cultured for 24-48 h and harvested for gene expression and other assays. The most effective siRNA (siMAGEA3-1, siMAGEA3-2) identified by qRT-PCR was applied for further experiments.
KAP1 expression plasmids (pcDNA3.1-KAP1) and KAP1-specific siRNA (sense: GGAGAUGAUCCCUACUCAAtt; antisense: UUGAGUAGGGAUCAUCUCCtg) were synthesized by GenePharma (Shanghai, China). The process of plasmid transfection was similar to that of siRNA transfection.
CCK-8 assay
Cells were seeded into 96-well plates at a density of 5×103 cells/well. After transfection, CCK-8 (Beyotime, Shanghai, China) assays were performed at different time points (12 h, 24 h, and 48 h). Ten microliters of CCK-8 reagent was added to each well and incubated for an additional 2 h at 37°C. The optical density at 450 nm was read by a microplate reader (Biotex, VT, USA).
5-Ethynyl-2’-deoxyuridine (EdU) assay
Cells were seeded into 96-well plates (5×103 cells/well) and cultured before being transfected with siRNAs or overexpression lentivirus. Cell proliferation was measured using the kFluor488 EdU Kit (KeyGEN, Nanjing, China) according to the manufacturer’s instructions. At 48 h after transfection, EdU (30 μmol/L) was added, and the cells were cultured for an additional 2 h. The cells were then fixed in 4% paraformaldehyde at room temperature for 10 min, washed with glycine (2 mg/ml) for 5 min in a shaker, and treated with 0.2% Triton X-100 for 20 min. Cellular nuclei were stained with Hoechst 33342 (5 μg/ml) for 30 min at room temperature. The positive fluorescent signals were observed and taken under an inverted fluorescence microscope (LEICA DM5500 B, Germany). The proportion of EdU-positive cells was calculated with the following formula: (EdU-stained cells/Hoechst-stained cells) ×100%.
RNA extraction and quantitative RT-PCR
Total RNA from cells was extracted using TRIzol Reagent (Invitrogen, CA, USA), and the RNA concentration was detected with a NanoDrop 2000 spectrophotometer (Thermo Fisher, MA, USA). cDNA was synthesized using a FastQuant RT Kit (TIANGEN, Beijing, China). Quantitative RT-PCR was carried out in accordance with instructions provided by the manufacturer of the Talent qPCR PreMix kit (TIANGEN, Beijing, China) and the CFX96 PCR detection system (Bio-Rad, CA, USA). The primers used were as follows: MAGEA3 forward 5’-TACCCCTGTCTCAAACCGAG-3’, reverse 5’-GCTGACCTGAAGTCCCACTC-3’; and GAPDH forward 5’-GCACCGTCAAGGCTGAGAAC-3’, reverse 5’-TGGTGAAGACGCCAGTGGA-3’. All primers were synthesized by Invitrogen (Shanghai, China). The housekeeping gene GAPDH was used as the endogenous reference gene for RT-PCR. The data analysis was performed using the 2-ΔΔCt method.
Protein extraction and western blotting
Total cellular proteins were extracted by using RIPA lysis buffer (Beyotime, Shanghai, China). The protein concentration in the cell lysates was determined using a bicinchoninic acid (BCA) assay kit (Beyotime, Shanghai, China), and 20 μg of protein was loaded into each lane of a gel. After denaturation, proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, MA, USA). The membranes were blocked with 5% nonfat milk prepared with Tris-buffered saline Tween-20 (TBST) buffer for 2 h at room temperature and then incubated with the primary antibody at 4°C overnight and secondary antibody at room temperature for 2 h. The PVDF membranes were imaged using enhanced chemiluminescence (ECL) detection reagents (Applygen, Beijing, China) and visualized with a ChemiDoc XRS+ system (Bio-Rad, CA, USA). Relative band intensities were determined by ImageJ software. The antibodies used for western blotting were as follows: MAGEA3, KAP1, and p53 (Abcam, CA, USA); p21, Bax, Bcl-2, cyclin D1, cleaved caspase-3, PUMA, and GAPDH (Cell Signaling, MA, USA).
Co-immunoprecipitation (Co-IP) analysis
Co-IP experiments were performed using endogenous proteins in HeLa cells and overexpressed proteins in SiHa cells. The endogenous binding between MAGEA3 and KAP1 proteins was confirmed in HeLa cells by co-immunoprecipitation with anti-MAGEA3 or anti-KAP1 antibody followed by immunoblotting with anti-KAP1 or anti-MAGEA3 antibodies. SiHa cells were cotransfected with MAGEA3 overexpression lentivirus and pcDNA3.1-KAP1. At 24 h posttransfection, the co-IP assay results were conformed. Briefly, according to the instructions provided by the co-immunoprecipitation kit (Thermo Fisher, MA, USA), cells were lysed for 10 min on ice with lysis buffer. Following centrifugation at 14000 g for 10 min at 4°C, 1 ml of each cleared lysate was incubated with 5 µl of monoclonal anti-MAGEA3 antibody or 5 µl of preimmune rabbit IgG overnight with continuous rotation at 4°C. Protein A-sepharose beads (30 µl) were then added, and the samples were gently rocked at 4°C for 3 h. After five washes with lysis buffer, the beads were recovered and resuspended in 40 µl of 2× SDS sample buffer and then boiled for 5 min. The precipitated proteins were detected by western blotting.
Cell cycle analysis
The cell cycle was analyzed by a propidium iodide (PI) staining kit (BD Pharmingen, MA, USA) according to the manufacturer’s instructions. Cells were seeded in 6-well plates at a density of 3×105 cells/well and then transfected with Lv-MAGEA3 or siMAGEA3. At 48 h after transfection, cells were harvested and washed twice with PBS and then fixed in 70% ethanol overnight. Subsequently, the cell pellets were stained with 10 µg/ml PI and 10 µg/ml RNase in PBS buffer for 30 min at room temperature in the dark. The cell cycle distribution was evaluated by using a flow cytometer (BD Biosciences, MA, USA).
Apoptosis assay
Cell apoptosis was detected according to the instructions of an annexin V-FITC/PI apoptosis detection kit (BD, MA, USA). Cultured cells were harvested and washed twice with cold PBS. Cell pellets were resuspended in 500 µl of binding buffer containing 5 µl of annexin-V-FITC and 5 µl of PI in the dark for 30 min at room temperature. Finally, the rate of cell apoptosis was analyzed using a flow cytometer (BD Biosciences, MA, USA). The apoptosis rate was defined as follows: annexin V-positive/PI-negative (early apoptosis) and annexin V-positive/PI-positive (late apoptosis) cells.
Dual-luciferase reporter gene assay
Cells were cultured at a density of 5×104 cells/well in 12-well plates and cotransfected with the p53-responsive luciferase reporter plasmid (pp53-TA-luc, containing p53 response element, Beyotime, Shanghai, China) along with the Renilla luciferase reporter plasmid (pGMR-TK, Genomeditech, Shanghai, China) for 6 h. Some cells were further transfected with the indicated plasmids or lentivirus. At 48 h after transfection, the cells were lysed to measure luciferase activity using a dual-luciferase reporter assay kit (Beyotime, Shanghai, China). The relative fluorescence light unit (RLU) at 560 nm of the mixture consisting of 50 µl of total cell lysate and 100 µl of the firefly luciferase assay reagent was evaluated using a multimode microplate reader (ThermoFisher, MA, USA) for 10 sec. Then, 100 µl of Renilla luciferase assay reagent was added to the mixture above, and its fluorescence at 465 nm was measured. Renilla luciferase activity was standardized to firefly activity, and the ratio was used to be relative activity.
Tumor xenograft models
All animal experiments were authorized by the Animal Care Committee of Hebei University Medical College. In xenograft models, four-week-old BALB/c nude mice were used for the experiment and divided into two groups at random (Lv-MAGEA3 group and Lv-NC group, n=5 for each group). SiHa cells transfected with Lv-MAGEA3 or Lv-NC were suspended in 100 µl PBS and injected subcutaneously into the flanks of the mice (5×106 cells per mouse). The tumor volume was recorded every 3 days and calculated as follows: tumor volume (mm3) = (length × width2)×0.5. Thirty days after cell implantation, the mice were sacrificed, tumor specimens were removed, and related gene expression was examined.
Immunohistochemistry (IHC) analysis
The tumor tissue were paraffin-embedded and cut into 5 µm sections and placed onto glass slides. After antigen retrieval and blocking with goat serum albumin, the sections were incubated overnight with primary antibodies. After being washed with PBS three times, the sections were incubated with secondary antibodies at 37°C for 30 min and visualized with diaminobenzidine (DAB). Finally, the slides were counterstained with 10% hematoxylin and analyzed under a light microscope with a digital camera (LEICA DM2500, Germany).
Statistical analysis
Statistical analysis was performed using SPSS 18.0 software. All experiments were repeated more than three times independently. The date are displayed as the means ± standard deviation (SD). Differences in independent samples were compared by Student’s t-test, while one-way ANOVA was used to analyze the differences between groups. The significance data were expressed as p-values, and P<0.05 was deemed statistically significant.
Results
MAGE-A3 enhances CC cell proliferation in vitro
We previously detected the expression of MAGEA3 in cervical lesion tissues and found that the expression of MAGEA3 in CC tissues was significantly higher than that of the control group [14]. Here, we measured the expression levels of MAGEA3 in four CC cell lines, including Caski, HeLa, C33A, and SiHa. Western blotting and qRT-PCR analysis results showed that HeLa cells had the highest endogenous level of MAGEA3 expression, and SiHa cells had the lowest expression level (Figure 1A).
Figure 1.
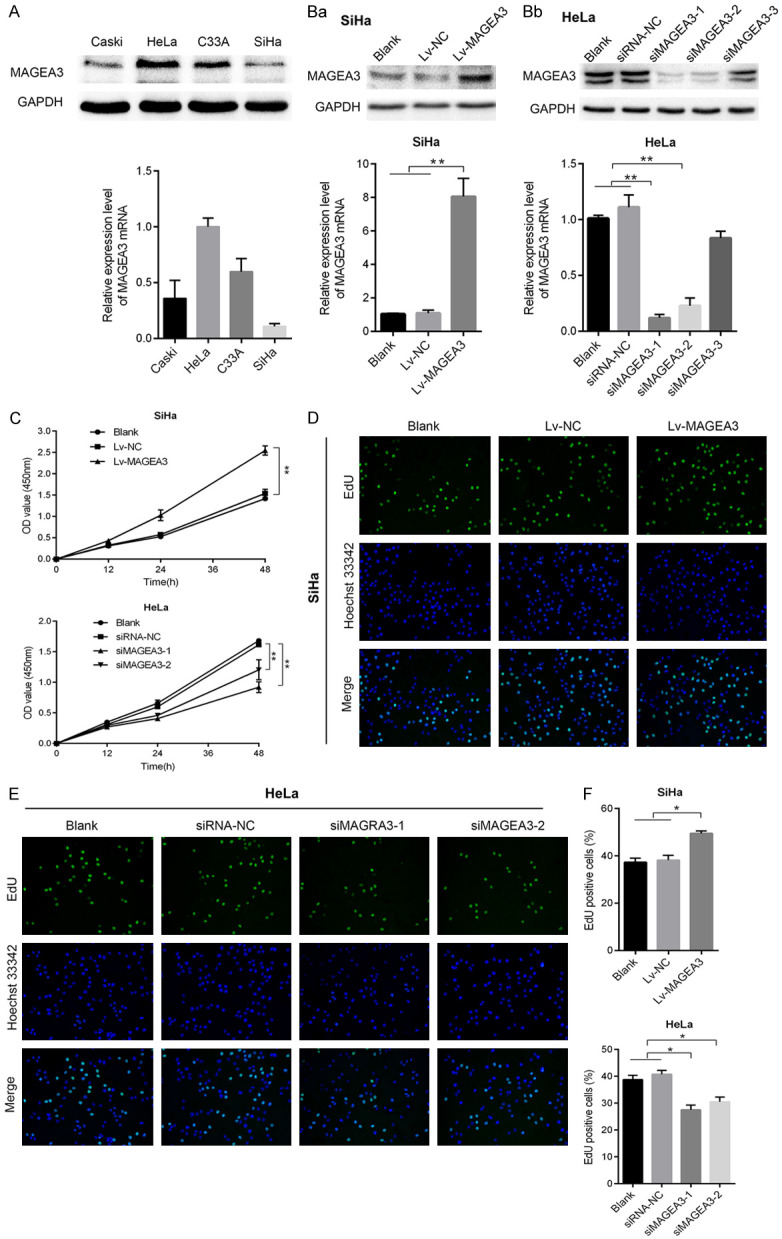
The effects of MAGEA3 on cervical cancer cell proliferation in vitro. (A) Western blotting and qRT-PCR analysis of MAGEA3 expression levels in four CC cell lines (Caski, HeLa, C33A, and SiHa). (B) Western blotting and qRT-PCR analysis of the expression of MAGEA3 in SiHa (Ba) cells transfected with MAGEA3 overexpression lentivirus and in HeLa (Bb) cells transfected with MAGEA3-specific siRNA. Blank: untransfected cells; Lv-NC or siRNA-NC: transfected with negative control lentivirus or siRNAs; Lv-MAGEA3: transfected with MAGEA3 overexpression lentivirus; siMAGEA3: transfected with MAGEA3-specific siRNA. (C) CCK-8 assay was performed to evaluate the cell viability in SiHa and Hela cells at different times. (D, E) EdU assay was utilized to measure cell proliferation in SiHa (D) and HeLa (E) cells. (F) Cell proliferation was assessed by EdU staining, and EdU-positive cell propotion was quantified by ImageJ and calculated as follows: EdU-positive cells (depicted as green fluorescence)/total number of cells (represented by blue nuclei stain, Hoechst 33342). *P<0.05, **P<0.01 vs. control groups.
To explore the biological functions of MAGEA3 in cervical cancer cells, we used a lentivirus-mediated expression system to stably overexpress MAGEA3 in SiHa cell lines, which harbor a low level of endogenous MAGEA3. MAGEA3 overexpression lentivirus (Lv-MAGEA3) or negative control lentivirus (Lv-NC) were transfected into SiHa cells, and compared with Lv-NC, Lv-MAGEA3 led to a marked increase in the expression of MAGEA3. In addition, the MAGEA3 gene was silenced by siRNA in HeLa cells, which harbor a high level of endogenous MAGEA3. The three siRNAs targeting MAGEA3 or siRNA-NC were transfected into HeLa cells. The gene knockdown efficiencies of siMAGEA3-1 and siMAGEA3-2 were the most pronounced in the qRT-PCR analysis, so they were applied for further experiments (Figure 1Ba, 1Bb).
CCK-8 (Figure 1C) and EdU (Figure 1D-F) assays were performed to detect the impacts of changes in MAGEA3 expression on the cell growth and proliferation of CC cell lines. As shown in Figure 1C and 1D-F, the overexpression of MAGEA3 significantly promoted cell viability and proliferation compared with that in the control group (blank, Lv-NC) (P<0.05). Conversely, knockdown of MAGEA3 markedly inhibited cell viability and proliferation. There was a significant difference between the MAGEA3 silencing group (siMAGEA3-1, siMAGEA3-2) and the control group (blank, siRNA-NC) (P<0.05).
MAGE-A3 increases tumor growth in xenograft models
To investigate the role of MAGEA3 in tumorigenesis, we performed an in vivo study by generating a SiHa xenograft model. SiHa cells infected with Lv-MAGEA3 or Lv-NC were subcutaneously injected into the flank of each nude mouse. Increased tumor growth and tumor weight were found in the Lv-MAGEA3 group compared with the control group (Figure 2A-C). In addition, the immunohistochemical analysis confirmed that the expression of MAGEA3 in the Lv-MAGEA3 group was significantly higher than that in the Lv-NC group (Figure 2D). These results indicate that MAGEA3 acts as a tumor promoter in the progression of cervical cancer.
Figure 2.
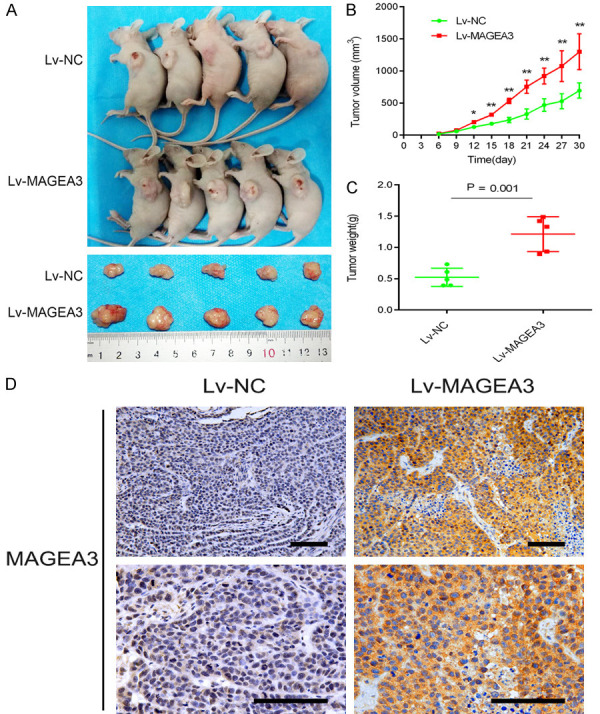
Overexpression of MAGEA3 promoted tumor growth in a xenograft mouse model. A. Representative images of xenografts derived from Lv-NC- and Lv-MAGEA3-transfected SiHa cells. B. Tumor growth was monitored by measuring tumor volumes every three days, and the tumor volumes were calculated by using the formula: tumor volume (mm3) = (length × width2)×0.5. C. Tumor weights measured after tumor dissection. *P<0.05, **P<0.01 vs. Lv-NC. D. MAGEA3 expression was detected by immunohistochemical staining of tumor sections in each group of nude mice. Bar length: 100 µm.
MAGEA3 promotes CC cell cycle progression and reduces cell apoptosis
To further demonstrate whether changes in MAGEA3 expression affected the cell cycle and apoptosis in human CC cells, we analyzed the cell cycle distribution and the percentage of apoptosis by flow cytometry in MAGEA3-overexpressing or MAGEA3-silenced cells. We found that the percentage of G1 phase was increased in HeLa cells with MAGEA3 knockdown, whereas the percentage of S phase was decreased in these cells compared with control cells (Figure 3B). Additionally, the overexpression of MAGEA3 in SiHa cells increased the percentage of cells in S phase (Figure 3A). These results indicated that MAGEA3 depletion induced cell cycle arrest at the G1 phase, and MAGEA3 overexpression promoted cell cycle progression and cell proliferation. The results of the cell apoptosis assay showed that the percentage of apoptotic cells was increased in HeLa cells with MAGEA3 knockdown (Figure 3B), while it was decreased in SiHa cells with MAGEA3 overexpression (Figure 3A). The results above indicated that MAGEA3 depletion induced cell apoptosis, while MAGEA3 overexpression suppressed cell apoptosis.
Figure 3.

MAGEA3 regulates cell cycle progression and inhibits apoptosis in CC cells. A. Flow cytometry analysis of the cell cycle and apoptosis in SiHa cells transfected with Lv-MAGEA3 or Lv-NC. B. Flow cytometry analysis of the cell cycle and apoptosis in HeLa cells transfected with siMAGEA3 or siRNA-NC. The apoptosis rate was defined as annexin V-positive/PI-negative (early apoptosis) and annexin V-positive/PI-positive (late apoptosis) cells. *P<0.05, **P<0.01 vs. control groups.
MAGE-A3 suppresses the transcriptional activity of p53 by interacting with KAP1
To investigate the underlying mechanism of MAGEA3 in promoting growth and suppressing apoptosis in human CC cells, the online software HitPredict was used to identify the interacting protein of MAGEA3. KAP1, a universal corepressor protein, directly interacts with MAGEA3 protein [18,19], and it can suppress p53 activity by forming a complex with MDM2 [20,21]. Therefore, we confirmed the protein-protein interactions between MAGEA3, KAP1, and p53 in CC cells by co-immunoprecipitation (co-IP) assay. Co-IP experiments were performed using endogenous proteins in HeLa cells and the overexpressed proteins in SiHa cells. As shown in Figure 4A, we pulled down KAP1 with MAGEA3 using the anti-MAGEA3 antibody and that we also pulled down both MAGEA3 and p53 with KAP1 using anti-KAP1 antibody. These results indicated the interactions of MAGEA3, KAP1, and p53.
Figure 4.
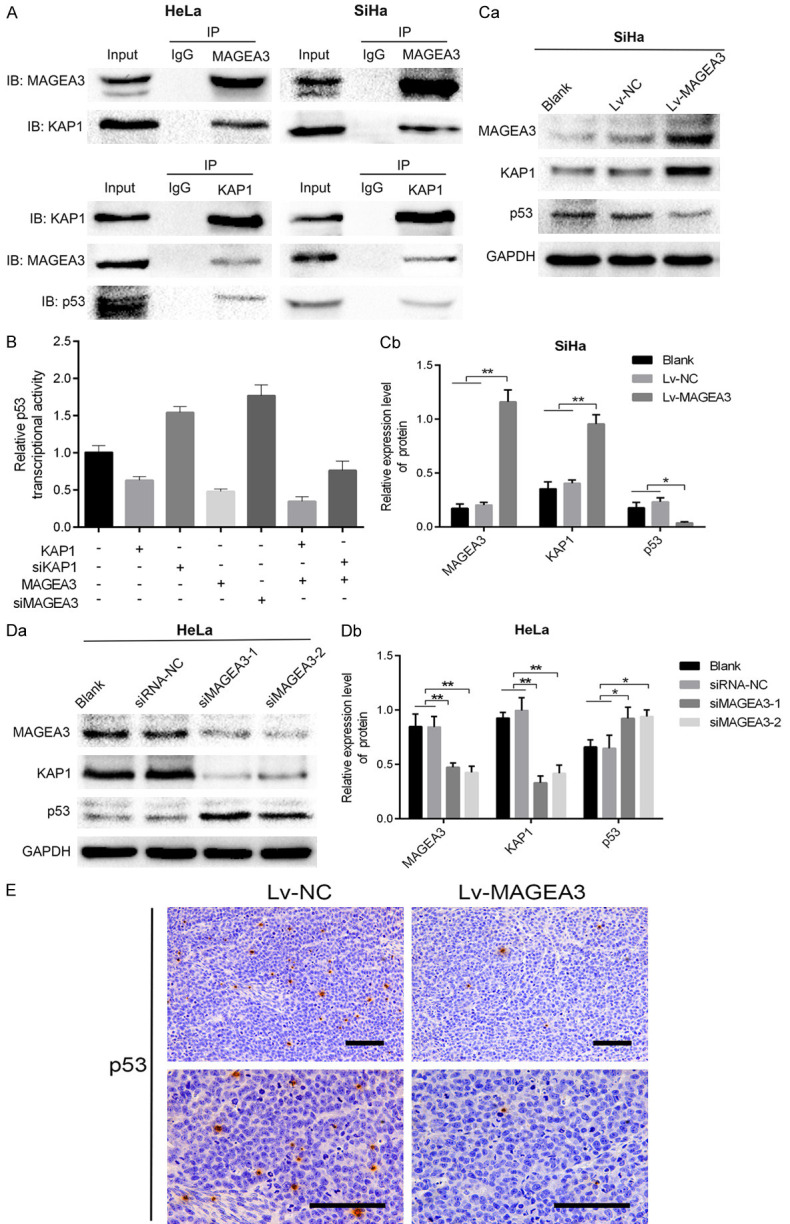
MAGEA3 interacts with KAP1 and inhibits p53 transcriptional activity. A. Co-IP assays were performed in HeLa cells and SiHa cells cotransfected with Lv-MAGEA3 and pcDNA3.1-KAP1. Cell lysates were immunoprecipitated (IP) with control IgG or the indicated antibody, and the precipitated protein was detected by immunoblotting (IB) analysis with the indicated antibody. Cell extracts were used as a positive control (Input). B. Effects of MAGEA3 or KAP1 on p53 activity, as indicated by the reporter plasmid pp53-TA-luc. HeLa cells (with endogenous wild-type p53) grown on a 12-well plate were cotransfected with the pp53-TA-luc and the indicated plasmids. Luciferase was measured at 48 h posttransfection and normalized according to Renilla luciferase activities. Data (means ± SD) are represented as fold differences relative to that observed in cells without transfecting the indicated plasmids. C, D. Western blotting analysis of the p53 and KAP1 proteins following overexpression or knockdown of MAGEA3 in SiHa (Ca, Cb) and HeLa (Da, Db) cells. *P<0.05, **P<0.01 vs. control groups. E. Immunohistochemical analysis of p53 expression in the Lv-MAGEA3 group and Lv-NC group of xenograft tumors. Bar length: 100 µm.
Then, we used the dual-luciferase assay to investigate the influence of MAGEA3 on p53 activity in HeLa cells (harboring wild-type p53). We found that the transfection of pcDNA3.1-KAP1 or Lv-MAGEA3 alone decreased p53 transcriptional activity, but the effect on p53 activity was more pronounced when both MAGEA3 and KAP1 were altered. Moreover, the cotransfection of Lv-MAGEA3 and siKAP1 into HeLa cells attenuated the suppressive effects of MAGEA3 on p53 activity (Figure 4B). These results suggested that MAGEA3 binding facilitates KAP1-mediated repression of p53 function.
We further examined whether MAGEA3 affected the protein expression levels of KAP1 and p53. Western blot analysis showed that MAGEA3 overexpression upregulated KAP1 expression and downregulated p53 expression in CC cells (Figure 4Ca, 4Cb), while MAGEA3 knockdown exerted the opposite effects (Figure 4Da, 4Db). In xenograft tumors, immunohistochemical analysis showed that the expression of p53 in the Lv-MAGEA3 group was lower than that in the Lv-NC group (Figure 4E). These results also indicated that the synergistic effect between MAGEA3 with KAP1 enhanced the inhibitory effect on p53 activity.
MAGEA3 regulates the protein expression of targets dpwnstream of p53
Next, we detected the effects of MAGEA3 on the protein levels of the transcriptional targets (p21, Bax, Bcl-2, and PUMA) of p53. As expected, overexpression of MAGEA3 significantly downregulated the protein levels of proapoptotic Bax and PUMA, cell cycle inhibitor p21 and apoptosis protein cleaved caspase-3, while upregulating the expression of antiapoptotic Bcl-2 and cell cycle regulator cyclin D1 in SiHa cells (Figure 5Aa, 5Ab), whereas MAGEA3 knockdown exerted the opposite effects in HeLa cells (Figure 5Ba, 5Bb). Consistent with this, immunohistochemical analysis of xenograft tumors showed that compared with the control group, the expression of p21 and Bax in the Lv-MAGEA3 group was decreased (Figure 5C). These results indicated that MAGEA3 promoted CC cell proliferation and inhibited apoptosis by inactivating the p53 signaling pathway.
Figure 5.
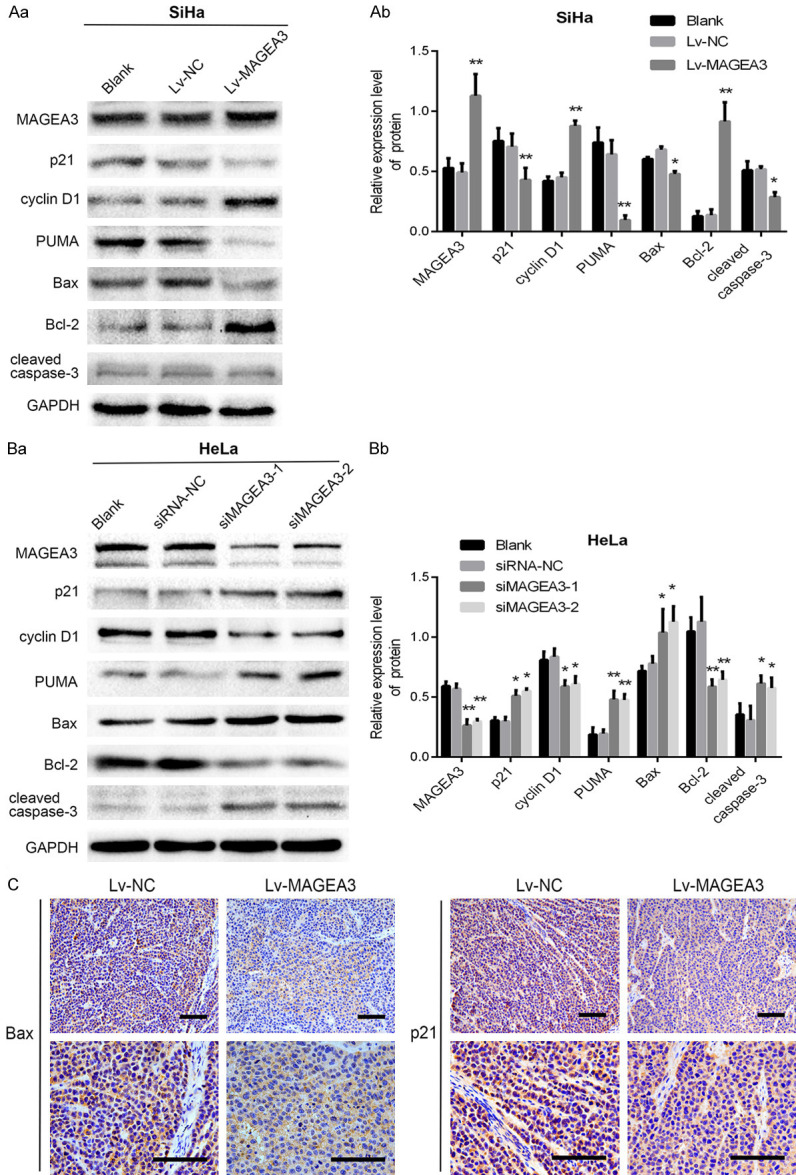
Effects of MAGEA3 expression on the protein levels of p53 downstream targets. A, B. Western blotting analysis of p53, p21, Bax, Bcl2, PUMA, cyclin D1, and cleaved caspase-3 proteins following overexpression or knockdown of MAGEA3 in SiHa (Aa, Ab) and HeLa (Ba, Bb) cells. *P<0.05, **P<0.01 vs. control groups. C. Immunohistochemical analysis of p21 and Bax expression in the Lv-MAGEA3 group and Lv-NC group of xenograft tumors. Bar length: 100 µm.
Nutlin3 reverses the effects of MAGEA3 overexpression
Nutlin3, as a p53 activator, could reactivate p53 by inhibiting the p53-MDM2 interaction [22]. To explore the effects of Nutlin3 (Sigma, MO, USA) on MAGEA3-overexpressing cells, flow cytometric analysis of the cell cycle and apoptosis was performed after treatment with Nutlin3 (10 μM) for 48 h. As shown in Figure 6A, Nutlin3 attenuated the effects of MAGEA3 overexpression on the cell cycle and apoptosis. Furthermore, western blotting analysis showed that the decreased expression of p53 and its target (p21, Bax) induced by MAGEA3 overexpression could also be reversed by Nutlin3 (Figure 6B). The above results indicated that MAGEA3 plays a tumor-promoting role by inhibiting the tumor-suppressive function of p53.
Figure 6.

Nutlin3 reverses the effects of MAGEA3 overexpression on the p53 signaling pathway, cell cycle, and apoptosis. A. Flow cytometric analysis of the cell cycle and apoptosis after treatment with Nutlin3 (10 μM) for 48 h. B. Western blotting analysis of the indicated proteins after treatment with Nutli3 (10 μM) for 48 h. *P<0.05, **P<0.01 vs. Lv-NC; ##P<0.01 vs. Lv-MAGEA3.
Discussion
MAGEA3 belongs to the cancer-testis antigen (CTA) group of tumor-associated genes and has been demonstrated to be expressed in a broad range of malignancies. The widespread expression of MAGEA3 in cancer suggests that MAGEA3 may have oncogenic activity and may be a class of therapeutic targets amenable to pharmacologic intervention [9,23,24]. Emerging evidence has shown that MAGEA3 promotes tumor cell survival and inhibits apoptosis. Atanackovic and colleagues reported that knockdown of MAGEA3 and MAGEC1/C7 by siRNA transfection led to the induction of apoptosis in malignant plasma cells [25]. Nardiello and colleagues proposed that silencing MAGEA3 in human myeloma cell lines did not impair cell cycle regulation, but it resulted in apoptosis through p53-dependent activation of Bax and Bak genes and by repression of survivin through p53-dependent and independent mechanisms [16]. More recently, Das and colleagues observed that depletion of MAGEA3 in pancreatic cancer cells reduced cell proliferation and increased apoptosis upon growth factor deprivation and in response to cytotoxic drugs [26]. Xie and coworker found that MAGEA3 was knocked down in gastric cancer cells, leading to a reduction in cell proliferation and colony forming capacities and causing an increased expression of cell cycle-related genes (p21, Bax) and apoptosis-related gene (PUMA, Noxa) [17]. In our present study, using the overexpression or knockdown approach, we demonstrated that overexpression of MAGEA3 in cervical cancer cells promoted cell proliferation and suppressed apoptosis in vivo as well as increased tumor growth in vitro. However, the depletion of MAGEA3 resulted in inhibited cell proliferation and induced apoptosis. These results are consistent with tumorigenic functions of MAGEA3.
P53 has been called the “guardian of the genome” because of its central role in sensing and reacting to DNA damage and oncogenic signaling. Under cellular stress stimuli such as DNA damage, mitogenic oncogenes, or hypoxia, p53 accumulates in the cell and transcriptionally regulates the expression of a variety of target genes [27,28]. p53 transcriptional activity is essential for preventing abnormal cell proliferation and carcinogenesis [29]. Multiple signaling pathways and mechanisms regulate p53 activity. However, it is suppressed mainly by its major regulator MDM2 protein [30,31]. MDM2 interacts with a variety of regulatory factors. Among others, KAP1 is known as a corepressor of p53 that acts by binding to MDM2, thereby suppressing p53 expression, p53 acetylation, and p53 function [20,32].
Moreover, KAP1 protein was demonstrated in complex with MAGE protein. MAGE protein, together with KAP1, actively promotes tumor survival by facilitating MDM2-mediated suppression of p53 activity [20,21]. Others have also suggested that MAGE-A binds to p53’s DNA-binding domain directly, which may prevent its transcriptional activity [33,34]. In the present study, our findings supported the mechanism by which MAGEA3 proteins act as corepressors of p53 by binding to KAP1 and enhancing its suppression of p53. As shown in the co-IP assay, MAGEA3 could bind to KAP1; similarly, KAP1 could bind to MAGEA3. Additionally, KAP1 was detected to bind to p53. However, we did not detect the direct binding between MAGEA3 and P53. In other words, our findings indicated that the MAGEA3 protein might interact with the KAP1/P53 complex. The dual-luciferase reporter gene assay showed that overexpression of either KAP1 or MAGEA3 decreased p53 transcriptional activity, whereas overexpression of both MAGEA3 and KAP1 resulted in a more obvious inhibitory effect. Intriguingly, the cotransfection of MAGEA3 overexpression lentivirus and siKAP1 into HeLa cells attenuated the suppressive effects of MAGEA3 on p53 activity. These findings suggested that MAGEA3 binding facilitates KAP1 repression of p53 expression and function. In addition, western blotting analysis showed that overexpression of MAGEA3 led to increased expression of KAP1 and reduced expression of p53, whereas depletion of MAGEA3 exerted the opposite effects. These results also indicated that MAGEA3 cooperated with KAP1 to enhance the inhibitory effect on p53 activity.
Activation of p53 causes a variety of responses, including cell cycle arrest or apoptosis, thereby providing a critical barrier against tumor development [35]. Cell cycle arrest driven by p53 requires the transcription of p21, which is a cyclin-dependent kinase (CDK) inhibitor. DNA damage or stress increases the levels of p53 protein, which in turn induces p21 transcription and leads to cell-cycle arrest at G1 [36,37]. P53 mediates cell apoptosis by activating the mitochondrial pathway, which is mainly regulated by p53 effector Bcl-2 proteins such as Bax [38]. In addition, PUMA (p53 upregulated modulator of apoptosis), a primary target gene of p53, plays a critical role in DNA damage-induced cell apoptosis [29,39]. In the present study, we found that the MAGEA3 knockdown led to cell cycle arrest at G1 as well as an increase in the apoptosis rate compared with the control group. Consistent with this, increased levels of Bax, PUMA, cleaved caspase-3, and p21 were observed in MAGEA3 knockdown cells compared to control cells. Conversely, in MAGEA3-overexpressing cells, we found that the number of cells in S phase was increased and that the apoptosis rate was decreased. Moreover, the p53 activator Nutlin3 reversed the effects of MAGEA3 overexpression on the p53 signaling pathway, cell cycle, and apoptosis. The results above suggested that MAGEA3 exerts pro-proliferative and antiapoptotic effects on CC cells that harbor wild-type p53 in a p53-dependent manner. However, further study is needed to determine whether overexpression or knockdown of the MAGEA3 gene has effects on p53-null or p53-mutant CC cells.
In conclusion, using overexpression or knockdown approaches, we found that in cervical cancer cells, MAGEA3 promoted cell proliferation and suppressed apoptosis in vitro and increased xenograft tumor growth in vivo. Mechanistically, we demonstrated that MAGEA3 inhibits p53 activity by interacting with KAP1 and suppresses p53-mediated regulation of the expression of the genes involved in the cell cycle (p21, cyclin D1) and apoptosis (Bax, Bcl2, PUMA). Our study highlights the pro-proliferative and antiapoptotic role of MAGEA3 in cervical cancer cells and provides a potential prognostic marker and therapeutic target for cervical malignancies.
Acknowledgements
This work was supported by the project of expression and clinical significance of MAGE-A3, MAGE-A9 in cervical lesions (NO. 361007).
Disclosure of conflict of interest
None.
References
- 1.GLOBOCAN. Cervical cancer estimated incidence, mortality and prevalence worldwide. 2019 [Google Scholar]
- 2.Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001;61:5544–5551. [PubMed] [Google Scholar]
- 3.Wascher RA, Bostick PJ, Huynh KT, Turner R, Qi K, Giuliano AE, Hoon DS. Detection of MAGE-A3 in breast cancer patients’ sentinel lymph nodes. Br J Cancer. 2001;85:1340–1346. doi: 10.1054/bjoc.2001.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shantha Kumara HMC, Grieco MJ, Caballero OL, Su T, Ahmed A, Ritter E, Gnjatic S, Cekic V, Old LJ, Simpson AJ, Cordon-Cardo C, Whelan RL. MAGE-A3 is highly expressed in a subset of colorectal cancer patients. Cancer Immun. 2012;12:16. [PMC free article] [PubMed] [Google Scholar]
- 5.Otte M, Zafrakas M, Riethdorf L, Pichlmeier U, Loning T, Janicke F, Pantel K. MAGE-A gene expression pattern in primary breast cancer. Cancer Res. 2001;61:6682–6687. [PubMed] [Google Scholar]
- 6.Kim J, Reber HA, Hines OJ, Kazanjian KK, Tran A, Ye X, Amersi FF, Martinez SR, Dry SM, Bilchik AJ, Hoon DS. The clinical significance of MAGEA3 expression in pancreatic cancer. Int J Cancer. 2006;118:2269–2275. doi: 10.1002/ijc.21656. [DOI] [PubMed] [Google Scholar]
- 7.Honda T, Tamura G, Waki T, Kawata S, Terashima M, Nishizuka S, Motoyama T. Demethylation of MAGE promoters during gastric cancer progression. Br J Cancer. 2004;90:838–843. doi: 10.1038/sj.bjc.6601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, Ritter G, Simpson AJ, Chen YT, Old LJ, Altorki NK. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005;11:8055–8062. doi: 10.1158/1078-0432.CCR-05-1203. [DOI] [PubMed] [Google Scholar]
- 9.Esfandiary A, Ghafouri-Fard S. MAGE-A3: an immunogenic target used in clinical practice. Immunotherapy. 2015;7:683–704. doi: 10.2217/imt.15.29. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Lu Y, Li J, Wu Y, Che G. The association of melanoma-associated antigen-A gene expression with clinicopathological characteristics and prognosis in resected non-small-cell lung cancer: a meta-analysis. Interact Cardiovasc Thorac Surg. 2019;29:855–860. doi: 10.1093/icvts/ivz196. [DOI] [PubMed] [Google Scholar]
- 11.Bujas T, Marusic Z, Peric BM, Mijic A, Kruslin B, Tomas D. MAGE-A3/4 and NY-ESO-1 antigens expression in metastatic esophageal squamous cell carcinoma. Eur J Histochem. 2011;55:e7. doi: 10.4081/ejh.2011.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, van der Bruggen P, Klebanoff CA, Goff SL, Sherry RM, Kammula US, Yang JC, Rosenberg SA. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J. Clin. Oncol. 2017;35:3322–3329. doi: 10.1200/JCO.2017.74.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, Lopez-Brea M, Vanakesa T, Jassem J, Kalofonos H, Perdeus J, Bonnet R, Basko J, Janilionis R, Passlick B, Treasure T, Gillet M, Lehmann FF, Brichard VG. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J. Clin. Oncol. 2013;31:2396–2403. doi: 10.1200/JCO.2012.43.7103. [DOI] [PubMed] [Google Scholar]
- 14.Haipeng H, Xinping G, Yijuan L, Hui Z, Jinzhi Z, Yachai L, Yan A. Expression of melanoma-associated antigen-A3 in cervical lesions and its clinical significance. Chinese Journal of Clinical Obstetrics and Gynecology. 2017;18:55–56. [Google Scholar]
- 15.Ladelfa MF, Peche LY, Toledo MF, Laiseca JE, Schneider C, Monte M. Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 2012;325:11–17. doi: 10.1016/j.canlet.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 16.Nardiello T, Jungbluth AA, Mei A, Diliberto M, Huang X, Dabrowski A, Andrade VC, Wasserstrum R, Ely S, Niesvizky R, Pearse R, Coleman M, Jayabalan DS, Bhardwaj N, Old LJ, Chen-Kiang S, Cho HJ. MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin Cancer Res. 2011;17:4309–4319. doi: 10.1158/1078-0432.CCR-10-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie C, Subhash VV, Datta A, Liem N, Tan SH, Yeo MS, Tan WL, Koh V, Yan FL, Wong FY, Wong WK, So J, Tan IB, Padmanabhan N, Yap CT, Tan P, Goh LK, Yong WP. Melanoma associated antigen (MAGE)-A3 promotes cell proliferation and chemotherapeutic drug resistance in gastric cancer. Cell Oncol (Dordr) 2016;39:175–186. doi: 10.1007/s13402-015-0261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, Jiang J, Wu H, Huang H. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis. 2017;6:e312. doi: 10.1038/oncsis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010;39:963–974. doi: 10.1016/j.molcel.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher FJ 3rd, Chen J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279–3290. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang B, O’Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, Gravekamp C, Setaluri V, Peters N, Hoffmann FM, Peng H, Ivanov AV, Simpson AJ, Longley BJ. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007;67:9954–9962. doi: 10.1158/0008-5472.CAN-07-1478. [DOI] [PubMed] [Google Scholar]
- 22.Van Maerken T, Rihani A, Van Goethem A, De Paepe A, Speleman F, Vandesompele J. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2014;344:157–165. doi: 10.1016/j.canlet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 24.Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol. 2015;37:1–8. doi: 10.1016/j.ceb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atanackovic D, Hildebrandt Y, Jadczak A, Cao Y, Luetkens T, Meyer S, Kobold S, Bartels K, Pabst C, Lajmi N, Gordic M, Stahl T, Zander AR, Bokemeyer C, Kroger N. Cancer-testis antigens MAGE-C1/CT7 and MAGE-A3 promote the survival of multiple myeloma cells. Haematologica. 2010;95:785–793. doi: 10.3324/haematol.2009.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das B, Senapati S. Functional and mechanistic studies reveal MAGEA3 as a pro-survival factor in pancreatic cancer cells. J Exp Clin Cancer Res. 2019;38:294. doi: 10.1186/s13046-019-1272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 28.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 29.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 31.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto K, Kitabayashi I, Taya Y. KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem Biophys Res Commun. 2006;351:216–222. doi: 10.1016/j.bbrc.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 33.Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, Rodolfo M, Schneider C. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A. 2006;103:11160–11165. doi: 10.1073/pnas.0510834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcar L, Maclaine NJ, Hupp TR, Meek DW. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 2010;70:10362–10370. doi: 10.1158/0008-5472.CAN-10-1341. [DOI] [PubMed] [Google Scholar]
- 35.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 36.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 37.El-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- 38.Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C, Nieruchalski M, May E. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene. 2000;19:649–660. doi: 10.1038/sj.onc.1203366. [DOI] [PubMed] [Google Scholar]
- 39.Jeon BN, Yoon JH, Han D, Kim MK, Kim Y, Choi SH, Song J, Kim KS, Kim K, Hur MW. ZNF509S1 downregulates PUMA by inhibiting p53K382 acetylation and p53-DNA binding. Biochim Biophys Acta Gene Regul Mech. 2017;1860:962–972. doi: 10.1016/j.bbagrm.2017.07.008. [DOI] [PubMed] [Google Scholar]