

Abstract
Hyperuricemia may occur when there is an excess of uric acid in the blood. Hyperuricemia may result from increased production or decreased excretion of uric acid. Elevated uric acid levels are a risk factor for gout, and various risk factors, including some medications, alcohol consumption, kidney disease, high blood pressure, hypothyroidism, and pesticide exposure, as well as obesity, are associated with an elevated risk of hyperuricemia. Although the mechanisms underlying the pathogenesis of hyperuricemia are complex, previously reported studies have revealed that hyperuricemia is involved in a variety of biological processes and signaling pathways. In this review, we summarize common comorbidities related to hyperuricemia and describe an update of epidemiology, pathogenesis, and therapeutic options of hyperuricemia. This systematic review highlights the epidemiology and risk factors of hyperuricemia. Moreover, we discuss genetic studies on hyperuricemia to uncover current status and advances in the pathogenesis of hyperuricemia. Additionally, we conclude with a reflection on the underlying mechanisms of hyperuricemia and present the alternative drug strategies for the treatment of hyperuricemia to offer more effective clinical interventions.
Keywords: Hyperuricemia, epidemiology, genetics, mechanisms, treatment
Introduction
Uric acid, a nitrogenous component of urine, is a poorly soluble final product of protein and purine metabolism in humans. The level of serum uric acid maintained the balance between its production and excretion [1-3]. Uric acid is a by-product of amino acid metabolism, and the breakdown of amino acids produces uric acid in the liver. Moreover, the breakdown of purines releases uric acid in small quantities. Uric acid in human urine and the blood may form sharp crystals and bring about an increased risk of gout [3-6]. Hyperuricemia mainly caused by metabolic disorders of purine and closely associated with the increases in the risk of cardiovascular disease, kidney disease, diabetes, obesity can be congenital or acquired [7-9]. Hyperuricemia is the main factor that leads to long-term systemic inflammation in patients with gout [10]. Inflammatory reactions in patients with asymptomatic hyperuricemia induced by urate can contribute to the development of obesity, chronic kidney disease, diabetes mellitus, and hypertension [11]. Hyperuricemia may occur when there are high levels of uric acid in the blood due to excess production, or more commonly, inefficient excretion of uric acid. Defects in the key enzymes of purine metabolism lead to impaired purine utilization or enhanced purine oxidase activity. The increased reabsorption or decreased secretion of uric acid in proximal renal tubules results in a decrease in the rate of uric acid excretion. The regulation of the enzymes is responsible for uric acid breakdown and production. Multiple transport proteins related to uric acid transport are crucial in the treatment of hyperuricemia [12]. An absolute lack of comprehension of the mechanism is a limiting factor in the management of hyperuricemia. However, the cellular and molecular processes and its implications are still not completely elucidated. New insights towards understanding the mechanisms may provide more precise treatment options for hyperuricemia.
Epidemiology
Worldwide prevalence of hyperuricemia
In recent years, the disease burden of hyperuricemia is increasing, especially in high-income countries and economically developing world with a Western lifestyle [13,14]. The prevalence and incidence of hyperuricemia substantially differ across geographical areas [15]. According to the data from the National Health and Nutrition Examination Survey (NHANES) 2007-2016, a nationally representative survey showed that the prevalence rates of hyperuricemia were 20.2% among men and 20.0% among women between 2015 to 2016 in the United States and the incidence of hyperuricemia remained stable in 2007-2016 [16].
The prevalence of hyperuricemia increases in both men (19.7% to 25.0%) and women (20.5% to 24.1%) from 2006 to 2014 in Ireland [17]. In the United States, an epidemiological survey has shown that the prevalence of hyperuricemia substantially increased from 19.1% (1988-1994 years) to 21.5% (2007-2008 years). The National Health and Nutrition Examination Survey (NHANES) 2007-2008 found a similar prevalence of hyperuricemia between women (21.6%) and men (21.2%) [15]. Most epidemiological studies show that the prevalence of hyperuricemia is generally higher in high-income countries than economically developing world [15,17-20]. The reduction of hormone estrogen production in postmenopausal women can decrease the removal of urate from the body result in an increase in urate levels and an elevated risk of developing hyperuricemia [21].
Prevalence of hyperuricemia in China
China, as the largest developing country, is characterized by marked regional disparities and diverse populations [14,18]. A national cross-sectional survey has shown that the prevalence of hyperuricemia was 8.4% (9.9% in men and 7.0% in women) among Chinese adults from 2009 to 2010 [14]. A systematic review and meta-analysis systematically have shown that the pooled prevalence of hyperuricemia was 13.3% in Mainland China from 2000 to 2014 [18]. According to a large-scale population-based survey, the overall crude prevalence of hyperuricemia of the elderly and middle-aged population in Tibet Autonomous Region is 1.83%, relatively lower than other places in China. The prevalence in men is 2.86%, whereas it is only 0.75% in women [22]. Additionally, the age-standardized prevalence rate of hyperuricemia in Henan rural population is 12.60% from July 2015 to September 2017, consistent with the primary meta-analysis (11.7%). The prevalence and risk of hyperuricemia increase significantly with age. Interestingly, the decreased prevalence of hyperuricemia is observed in older men, while the contradictory trend is found in women [23].
Risk factors
The levels of urate hinge on the dynamic balance between purine-rich foods intake, synthesis of urate within the body, the excretion of urate via urine or the gastrointestinal tract (Figure 1) [24]. Prospective epidemiologic studies have pointed to obesity, hypertension, metabolic syndrome, diuretic use, dietary factors, and chronic kidney disease as risk factors for gout and hyperuricemia [9,25-29]. Recently, it turns out that iron overload can enhance serum uric acid levels, indicating a causal connection between hyperferritinaemia and hyperuricemia [30]. It is well known that chronic noncommunicable diseases, such as cardiovascular and rheumatic diseases, are associated with the development of hyperuricemia [31-33]. An onset sequence study revealed that hyperuricemia is an earlier-onset metabolic disorder than hypertriglyceridemia, diabetes mellitus, and hypertension [34]. The study of epidemiological aspects of hyperuricemia in Poland shows that doctors often underestimate the problem of hyperuricemia in patients with a high risk of cardiovascular disease [32]. Due to increased comorbidity, moderate hyperuricemia has associations with increased cardiovascular mortality [35]. A 5-year Japanese cohort study indicated that hyperuricemia is an independent risk factor for developing hypertension, especially in children and adolescents [36]. Uric acid associated with pro-inflammatory immune effects can not only contribute to microvascular injury within the intracellular environment but also conduce to increased blood pressure [37,38]. Hyperuricemia or gout has relationships with a substantial comorbidity burden in patients with rheumatoid arthritis [35]. Previous scientific investigations make general reference to the relationship between excess serum uric acid and gout [39,40]. Gout, a common rheumatic disease, characterized by the deposition of sodium monourate crystals in the periarticular joints, can mainly result from hyperuricemia [41]. Notably, some medicines used to treat chronic diseases have the side effects of elevated uric acid levels in the blood [24,39,42].
Figure 1.
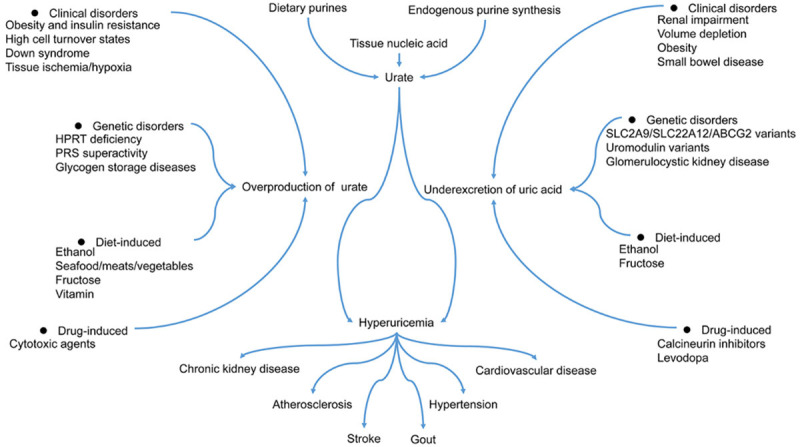
Mechanisms of hyperuricemia. HPRT, Hypoxanthine-guanine phosphoribosyltransferase; PRS, Phosphoribosylpyrophosphate synthetase.
Some high-purine foods, such as beer, seafood, sugar-sweetened beverages, dried beans, and meat, can help raise serum uric acid levels. When purines break down to produce uric acid, it can cause hyperuricemia in some individuals [43-45]. However, there is limited information on the exact content of purines contained in foods mainly because several factors, such as food processing procedures, can affect its content. Some research suggests that purine-rich foods intake is closely associated with the prevalence of hyperuricemia. There was a direct and significant correlation between seafood intake and the prevalence of hyperuricemia, and an antagonistic relationship between soy food consumption and hyperuricemia among middle-aged Chinese men. Moreover, protein intake from animal or plant sources has the opposite contribution to the prevalence of hyperuricemia. Additionally, reducing the consumption of high-protein animal foods can help lower the levels of plasma uric acid [46]. The association between hyperuricemia and the intake of vitamin B12, vitamin B6, and folate among American adults was assessed according to the data from the National Health and Nutrition Examination Survey (NHANES) 2001-2014. The result revealed that the intakes of vitamin B12 and folate, but not vitamin B6, were associated with a reduced risk of hyperuricemia in males. The only intakes of food folate, folate, and total folate had associations with a lower risk of hyperuricemia in females [47].
According to a nationally representative survey, body mass index, alcohol use, Dietary Approaches to Stop Hypertension (DASH) diet, and diuretic use are modifiable risk factors, and they can be applied to illustrate the prevalence of hyperuricemia [48,49]. Both endogenous and exogenous purine metabolism generates uric acid [42]. Previous studies capture much attention to fructose consumption because, during fructose metabolism, adenosine triphosphate (ATP) hydrolysis can generate adenosine diphosphate (ADP) and adenosine monophosphate (AMP), and the latter may stimulate the increase of serum uric acid concentrations and lead to hyperuricemia. Furthermore, fructose intake can contribute to the biosynthesis of uric acid from amino acid precursors [50,51]. Uric acid levels increase with the increase of body mass index (BMI), and obesity has an association with hyperuricemia. Additionally, hyperuricemia and obesity may ratchet up the likelihood of type 2 diabetes [52].
From the above, we can conclude that hyperuricemia is closely related to multiple risk factors. However, its pathophysiology has not yet been thoroughly investigated. We should pour enough attention to the adverse effects caused by hyperuricemia.
Genetics
Hyperuricemia, considered to result in a combination of environmental and genetic factors, is characterized by an excess of serum uric acid [53]. As shown in Figure 2, most uric acid disposal naturally occurs in the kidney. Insufficient excretion of urate in the kidney accounts for about 90% of individuals with hyperuricemia. Underexcretion associated with reduced glomerular filtration, impaired tubular secretion, and improved tubular reabsorption contributes to hyperuricemia. In the proximal tubular, URAT1 (uric acid transporter 1) manage uric acid reabsorption. Besides, multiple medications, extracellular fluid volume depletion, and organic acids facilitating transport lead to hyperuricemia. Furthermore, the kidney is held accountable for 60-65% of urate elimination associated with various transporters in the intestinal mucosa, salivary glands, and the proximal renal tubule. The filtered urate in the kidney via the glomerulus is mainly reabsorbed and secreted in the proximal tubule. Ultimately, only 3-10% of the filtered urate is eliminated in the urine [54].
Figure 2.
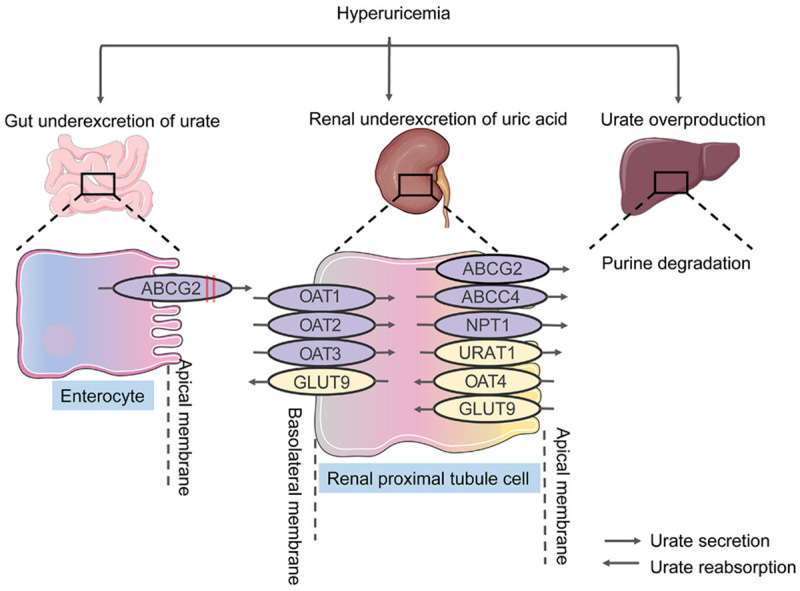
Urate transporters in humans. Overproduction and underexcretion of urate result in hyperuricemia. Underexcretion of urate occurs mainly in the proximal renal tubules. Urate overproduction in the liver and underexcretion of urate in the gut may lead to the occurrence of hyperuricemia. In the gut, Genetic variants in ABCG2 inhibit the excretion of urate and promote underexcretion. Numerous transmembrane transporters, such as ABCG2, URAT1, and GLUT9, have crucial roles in urate reuptake and secretion. OAT, organic anion transporter; NPT1, sodium-dependent phosphate cotransporter type 1; ABCG2, ABC transporter G family member 2; URAT1, urate anion exchanger 1; GLUT9, glucose transporter type 9; ABCC4, ATP-binding cassette sub-family C member 4.
Uric acid, the end oxidation product of purine breakdown, is primarily excreted in the urine. It is clear that uric acid, a weak organic acid, circulates in the ionized shape of urate under normal physiologic conditions. The metabolism of purine chiefly happens in the liver, but it can occur in the tissues with a wide distribution of xanthine oxidase [3]. The excretion of uric acid from the kidney accounts for about two-thirds, and the remaining proportion of uric acid is mainly excreted into the intestine. Most uric acid is filtered in a free manner, with roughly 90% of the filtrate reabsorbed [55]. Enhanced cellular breakdown, endogenous purine production, and high-purine diets promote the production of urate, responsible for a small portion of hyperuricemia. High Purine Foods chiefly include some meats, some fish, seafood and shellfish, and alcoholic beverages, strengthening the levels of uric acid by lowering renal excretion. The higher phosphoribosylpyrophosphate (PRPP) synthetase activity, as well as hypoxanthine phosphoribosyltransferase (HPRT) deficiency, can not only improve endogenous purine production but also lead to uric acid overproduction and accumulation. Cell turnover or breakdown, such as tumor lysis, rhabdomyolysis, and hemolysis, can improve urate production. Ultimately, both environmental and physiological changes can influence the production of urate [56].
Patients who suffered from gout or hyperuricemia have elevated urate reabsorption in the proximal tubule. According to renal urate excretion, hyperuricemia is usually divided into urate overproduction type and underexcretion type. The urate reabsorption levels are driven by genetic variation in urate transporters or upstream regulators. Previous studies have confirmed that various genes are associated with hyperuricemia [57-59]. The great mass of the genes is involved in transporting urate, which is a byproduct of natural biochemical processes. Various hyperuricemia-associated genes regulate excretion or reabsorption of uric acid according to the body’s needs. Some hyperuricemia-associated genes have relationships with the transport or breakdown of small molecules [57,59,60]. Researchers in mounting numbers tend to pay much attention to genetic studies, offering novel insights into the genetic effects on hyperuricemia. However, current research cannot fully account for the occurrence and progression of it. It is, therefore, crucial to conduct more extensive and intensive research on genetic studies.
Genome-wide association studies provide new perspectives on the genetic basis of hyperuricemia, which is dominated by loci containing urate transporters associated with urate excretion. Genome-wide association studies in hyperuricemia identify more genetic variants and delineate the pathogenesis of hyperuricemia, providing novel opportunities for underlying clinical translation [60]. A meta-analysis showed gout and asymptomatic hyperuricemia loci and reveal gout development by identifying the gout risk loci associated with crystal-induced inflammation [61,62].
Previous research has revealed that various transporter genes have associations with serum uric acid levels, such as urate transporter 1 (URAT1), glucose transporter 9 (GLUT9), organic anion transporter 4 (OAT4), ATP-binding cassette transporter, subfamily G, member 2 (BCRP), and sodium-dependent phosphate cotransporter type 1 (NPT1). Among them, GLUT9 (SLC2A9) and BCRP (ABCG2) appear to have the most significant impact on urate levels [63].
GLUT9
GLUT9 (SLC2A9), encoding a member of the SLC2A facilitative glucose transporter family to maintain glucose homeostasis, has an essential role in urate transporter and reabsorption. The protein encoded by GLUT9 helps excrete urate into the urine or reabsorb urate into the bloodstream. GLUT9 variants can reduce the excretion of urate into the urine and enhance the reabsorption of uric acid into the bloodstream, leading to hyperuricemia [64]. The rs7442295 single nucleotide polymorphism in the SLC2A9 gene robustly associated with hyperuricemia, increased plasma uric acid levels, gout, and urate excretion has been proposed as a proxy measurement for uric acid. It has been used to explore its causal associations with ischaemic heart disease and blood pressure [51,65]. Loss-of-function mutations in SLC2A9, interacting with the pore of the Glut9 urate transporter and the urate binding pocket, decreased the expression of Glut9 and reduced the activity of Glut9 transport [66].
BCRP
BCRP (ABCG2), a member of the superfamily of ATP-binding cassette (ABC) transporters, is involved in the transport of several molecules across extra- and intra-cellular membranes. BCRP, an ATP-driven efflux pump in the apical membrane of the proximal tubule epithelial cells, excretes endogenous and exogenous substrates. BCRP dysfunction reduces the excretion of extra-renal urate, which is a significant contributor to hyperuricemia. Besides, the protein encoded by BCRP helps excrete urate into the gut, and it, therefore, can be removed from the body. Various functional variants of BCRP were identified and enhanced the risk of hyperuricemia and gout [67]. BCRP variants can reduce the excretion of urate into the gut, leading to hyperuricemia. Moreover, BCRP contributes to the clearance of urate in the gut [68,69]. The BCRP 141K (rs2231142) variant contributes to hyperuricemia and has a role in the development of hyperuricemia to gout in Polynesian [70,71]. Mutations in ABCG2 mediating the intestinal excretion of uric acid lead to hyperuricemia. In hereditary hemochromatosis patients, iron/heme overload enhances the activity of xanthine oxidase and accelerates the degradation of p53, causing the reduction of ABCG2 expression. In consequence, intestinal excretion of uric acid through ABCG2 is reduced and the production of uric acid is enhanced, leading to the accumulation of uric acid in tissue and serum and promoting the progression of hereditary hemochromatosis-associated arthritis [72].
URAT1
URAT1 (SLC22A12) as urate transporter gene to adjust and control blood urate levels is a disease-causing gene for renal hypouricemia type 1 [73,74]. Glucocorticoids are crucial to maintaining uric acid homeostasis through the glucocorticoid receptor signaling pathway. Moreover, Glucocorticoids promote renal urate excretion via downregulating URAT1 in mouse kidney [75]. In a meta-analysis, the rs475688 polymorphism in SLC22A12 is associated with gout susceptibility and a significant association between hyperuricemia susceptibility and the rs3825016 polymorphism in SLC22A12 was observed [76]. The increased expression of ALPK1 reduced the expression of URAT1. The rs11726117 polymorphism in ALPK1 suppressed urate reuptake and reduced the risk of gout via SLC22A12 [77].
OAT4
OAT4 (SLC22A11), encoding an integral membrane protein, is associated with the sodium-independent transport and excretion of organic anions [51]. NPT1 (SLC17A1), a urate exporter, associated with the transport of phosphate into cells via Na (+) cotransport has crucial roles in the resorption of phosphate by the proximal tubule of the kidney [78].
OAT10
Organic anion transporter 10 (OAT10), also known as SLC22A13, encodes a urate reabsorption transporter protein on the apical side of the renal proximal tubular cells. OAT10 acts as a critical part of urate transport from urine to the blood and the dysfunctional variants of OAT10 reduce serum uric acid levels [79].
LDHD
Lactate Dehydrogenase D (LDHD) as a member of the D-isomer specific 2-hydroxyacid dehydrogenase family is involved in autosomal recessive gout with hyperuricemia and decreased excretion of uric acid. The mutations of LDHD result in the excessive production of blood D-lactate in exchange for uric acid reabsorption, eventually contributing to gout and hyperuricemia [80].
UMOD
Uromodulin encoded by UMOD is characterized as one of the most abundant proteins secreted in mammalian urine. Lower concentrations of urinary uromodulin associated with distal tubular cell damage are observed in individuals with UMOD-related diseases, such as familial juvenile hyperuricemic nephropathy (FJHN), renal disorders medullary cystic kidney disease-2 (MCKD2) and glomerulocystic kidney disease with hyperuricemia and isosthenuria (GCKDHI). Besides, uromodulin excretion in urine prevents urinary tract infections from uropathogenic bacteria. UMOD mutations impair uromodulin protein folding and accumulate misfolded proteins in the endoplasmic reticulum (ER) of renal tubular cells. The disruption of ER function contributes to hyperuricemia and tubulointerstitial nephritis [81,82].
HPRT1
Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) encoded by hypoxanthine phosphoribosyltransferase 1 (HPRT1) is a transferase, which catalyzes the conversion of hypoxanthine to inosine monophosphate and guanine to guanosine monophosphate. The catalytic reaction transfers the 5-phosphoribosyl group from 5-phosphoribosyl 1-pyrophosphate (PRPP) to the purine. HPRT1 exerts a vital part in purine nucleotide biosynthesis via the purine salvage pathway. HGPRT deficiency caused by HPRT1 mutation leads to elevated uric acid levels in the blood, which is associated with Kelley-Seegmiller syndrome, Lesch-Nyhan syndrome, and hyperuricemia [83,84].
SARS2
The mitochondrial seryl-tRNA synthetase precursor, a member of the class II tRNA synthetase family, encoded by Seryl-TRNA Synthetase 2, Mitochondrial (SARS2), which catalyzes the ligation of Serine to tRNA (Ser) and is involved in selenocysteinyl-tRNA (sec) biosynthesis in mitochondria. Mutations in SARS2 have been identified in Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis syndrome (HUPRAS), which is a multi-system involvement disease including pulmonary hypertension, hyperuricemia, renal failure in infancy and alkalosis [85].
G6PC
Glucose-6-Phosphatase Catalytic Subunit (G6PC), a multi-subunit integral membrane protein of the endoplasmic reticulum, catalyzes the hydrolysis of D-glucose 6-phosphate to D-glucose and orthophosphate in the endoplasmic reticulum and is one of the critical enzymes in blood glucose homeostasis, exerting a critical part in the regulation of gluconeogenesis and glycogenolysis. Mutations in G6PC have been identified in Glycogen storage disease type I (GSD1), a metabolic disorder, is marked by hypoglycemia, hyperlipidemia, hyperuricemia, and lactic acidemia [86].
XDH
Xanthine Dehydrogenase (XDH), a member of the family of oxidoreductases, participates in the oxidative metabolism of purines, catalyzing the oxidation of hypoxanthine to xanthine and the oxidation of xanthine to uric acid. Sulfhydryl oxidation or proteolytic modification can convert xanthine dehydrogenase to xanthine oxidase. Xanthine dehydrogenase deficiency results in xanthinuria, leading to adult respiratory stress syndrome [87]. Downregulated XDH expression can reduce the levels of xanthine oxidoreductase, and it may be an underlying treatment for hyperuricemia [88,89].
INS
Insulin (INS) encoded by INS is considered as an essential anabolic hormone in the metabolic process, participating in the control of carbohydrate and lipid metabolism. High levels of insulin in the blood hinder the production and secretion of glucose. As an anabolic hormone, it consolidates the conversion of small molecular substances in the blood into abundant molecular substances inside the cells. Low concentrations of insulin in the blood strengthen extensive catabolism. Insulin not only induces the change of cell permeability to fatty acids, amino acids, and monosaccharides, but also facilitates the synthesis of glycogen, the pentose phosphate cycle, and glycolysis in the liver. Insulin resistance may be a potential metabolic disturbance explaining the relationship among diverse elements of the metabolic syndrome associated with glucose intolerance, hyperlipidemia, hypertension, and obesity. Besides, hyperuricemia may be an underlying marker of insulin resistance [90,91].
REN
REN encoding renin causes autosomal dominant tubulointerstitial kidney disease, REN-related (ADTKD-REN) characterized by low plasma renin activity, bland urinary sediment, decreased fractional excretion of urinary uric acid, hyperuricemia, and hypoproliferative anemia. Hyperuricemia is depicted in 80% of patients with ADTKD-REN beginning in childhood due to reduced renal excretion of uric acid [92,93].
GPATCH8
G-Patch Domain Containing 8 (GPATCH8), a member of the G-patch domain family, is encoded by GPATCH8. A potentially pathogenic mutation in GPATCH8 is a conceivable candidate for hyperuricemia [94].
Drugs for hyperuricemia
According to the clinical scenarios, hyperuricemia can be divided into asymptomatic or symptomatic hyperuricemia. Decreasing unnecessary costs and avoiding underlying side effects take precedence over taking medication in careful consideration of the interests of the asymptomatic hyperuricemia patients. There seems no need to carry medical therapy for a multitude of asymptomatic hyperuricemia patients. They accompany with an elevated serum urate level but never experience signs or symptoms of monosodium urate crystal deposition disease, such as nephrolithiasis, gout, and uric acid renal disease. Urate-lowering drugs are likely to appeal to the asymptomatic hyperuricemia patients subjected to cytolytic therapy for a malignant tumor to restrict tumor lysis syndrome. It is sufficient to change in lifestyles, such as exercise, dietary changes, and alcohol consumption, for asymptomatic hyperuricemia patients to decrease the levels of uric acid [95]. The clinical symptoms of symptomatic hyperuricemia can be gout, nephrolithiasis, and uric acid renal disease [39].
To effectively manage hyperuricemia, inhibiting uric acid synthesis and reabsorption, as well as facilitating the excretion of uric acid, can be alternative strategies. As shown in Figure 3, urate-lowering medications can be roughly divided into three main categories: reducing the synthesis of uric acid (xanthine oxidase inhibitors), enhancing the excretion of uric acid (URAT1 inhibitors), and regulating the metabolic hydrolysis of uric acid (uricase inhibitors). Xanthine oxidase inhibitors classified as purine analogs (including allopurinol) and non-purine analog agents (including febuxostat and topiroxostat) can lower endogenous uric acid production and further decrease the levels of uric acid [96,97].
Figure 3.
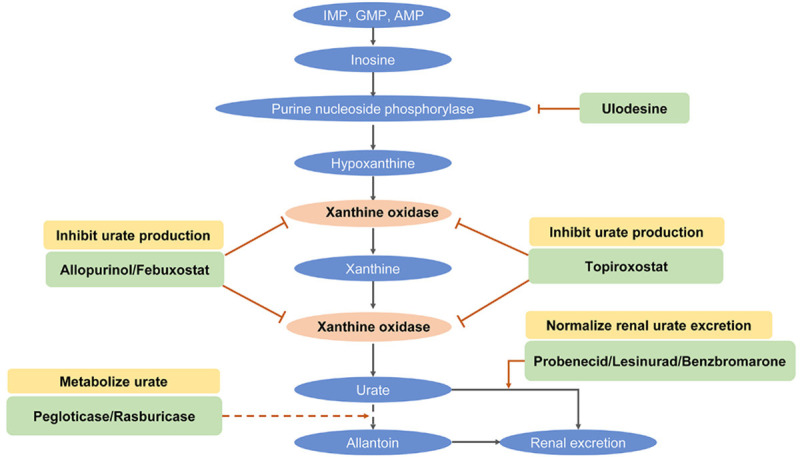
Mechanism of urate-lowering therapy for hyperuricemia. Numerous agents can be applied to inhibit urate production, promote renal urate excretion, and increase purine metabolism to allantoin.
Allopurinol
Allopurinol, a purine-based competitive xanthine oxidase inhibitor, can be metabolized to alloxanthine, an inhibitor of xanthine oxidase enzyme. Both allopurinol and alloxanthine can restrain xanthine oxidase, converting hypoxanthine to xanthine and xanthine to uric acid. Allopurinol promotes the secondary utilization of hypoxanthine and xanthine for the synthesis of nucleic acid and nucleotide by a metabolic reaction associated with hypoxanthine-guanine phosphoribosyltransferase (HGPRTase). This metabolic reaction accounts for an elevated level of nucleotide, which leads to feedback suppression of de novo synthesis of purine. Eventually, the reduced levels of urine and serum uric acid are responsible for a reduction in the incidence of hyperuricemia [98,99]. Based on the data from the Taiwan National Health Insurance Research Database, a retrospective nationwide population-based study showed that allopurinol could cause hypersensitivity reactions in patients with asymptomatic hyperuricemia accompanied by cardiovascular or renal diseases [100].
Febuxostat
Febuxostat, a non-purine xanthine oxidase inhibitor, can reduce the levels of serum uric acid, but not restrain different enzymes associated with the metabolism and synthesis of pyrimidine and purine. The metabolism of Febuxostat is dependent on uridine diphosphate glucuronosyltransferase (UGT) enzymes (including UGT1A1, UGT1A3, UGT1A9, and UGT2B7), cytochrome P450 (CYP) enzymes (CYP1A2, CYP2C8, and CYP2C9) and non-P450 enzymes [99,101]. Based on US Medicare claims data (2008-2013), a cohort study on the assessment of cardiovascular risk in 99744 older medicare patients with gout showed that there was almost no difference in the risk of all-cause mortality, new-onset heart failure, myocardial infarction, coronary revascularization, or stroke between patients who used febuxostat compared with allopurinol initiators. Nevertheless, the risk of heart failure exacerbation was slightly lower in patients initiating febuxostat compared with allopurinol [102]. A randomized and controlled trial showed that treatment with febuxostat did not delay carotid atherosclerosis progression in Japanese patients with asymptomatic hyperuricemia and it did not support the use of febuxostat to delay the progression of carotid atherosclerosis in patients with asymptomatic hyperuricemia [103].
Topiroxostat
Topiroxostat, a non-purine xanthine oxidase inhibitor, makes interaction with numerous amino acid residues in the solvent channel and bonds covalently to molybdenum (IV) ion, generating a hydroxylated 2-pyridine metabolite to inhibit xanthine oxidase. Additionally, Topiroxostat can restrain ATP-binding cassette transporter G2 (ABCG2), which is involved in the restoration of renal uric acid and uric acid secretion from the intestines [104].
Probenecid
Probenecid, a prototypical uricosuric agent, hinders the renal elimination of organic anions and impairs tubular urate reabsorption. Additionally, it can decrease the renal elimination of other drugs, and therefore it is a promising uric acid transporter 1 (URAT1) inhibitor for the treatment of renal impairment. Probenecid restrains the tubular urate reabsorption, facilitating urinary uric acid excretion, as well as reducing serum urate concentrations. Besides, Probenecid possibly decreases the binding of urate by plasma proteins and reduces uric acid secretion in the renal tubule [97,105].
Lesinurad
Lesinurad, a common URAT1 inhibitor, restrains the levels of serum uric acid via the suppression of URAT1 and OAT4. URAT1, uric acid transporter, is associated with uric acid reabsorption from the renal tubule. Organic anion transporter 4 (OAT4), uric acid transporter, is linked with the sodium-independent transport and excretion of organic anions, involved in diuretic-induced hyperuricemia [98]. The administration of Lesinurad in combination with allopurinol causes a considerable reduction in serum concentrations of inflammatory cytokines, glutathione peroxidase, catalase, urea nitrogen, and uric acid in a hyperuricemic mouse model, ameliorating renal function of the hyperuricemic mice [106]. In vitro, lesinurad suppressing the activity of OAT4 and URAT1 but not GLUT9, OAT1, OAT3, or ABCG2 can reduce the levels of serum uric acid via restraint of urate transporters in the human kidney [107].
Rasburicase
Rasburicase, a recombinant uricase, catalyzes the conversion of uric acid to allantoin, which is a metabolite existing in an inactive and soluble form [108]. Rasburicase seems to have an advantage in the speedy correction of hyperuricemia compared with allopurinol. However, its clinical benefit in cancer patients with tumor lysis syndrome (TLS) is still confusing, especially for patients with concurrent renal failure and hyperuricemia [109].
Conclusions
Hyperuricemia seems to be rising steadily in prevalence over the last decades. High uric acid concentrations are involved in the elevated risk of developing hyperuricemia. Hyperuricemia is a metabolic disease connected with Lesch-Nyhan syndrome and glycogen storage disease-ia. Substantial evidence suggests that hyperuricemia is an underlying risk factor for gout, and it can forecast the evolution of chronic kidney disease, obesity, diabetes, and hypertension. Although numerous studies have demonstrated close correlations between hyperuricemia and multiple comorbidities such as acute and chronic kidney disease, diabetes, metabolic syndrome, cardiovascular disease, hypertension, and dyslipidemia, it is currently unclear that there is a causal relationship between hyperuricemia and multiple comorbidities. Genetic characteristics provide novel perspectives on the physiology and pathophysiology of hyperuricemia. More importantly, genetic studies may provide more precision medicine for individuals. These are various approaches that help manage hyperuricemia. Nevertheless, patient and health-care provider education is the foundation of the successful treatment of hyperuricemia. As a general rule, it is not indispensable to treat most patients with asymptomatic hyperuricemia in the absence of kidney stones or gout. To effectively treat hyperuricemia, reducing the levels of uric acid is crucial, achieved by inhibiting uric acid synthesis and reabsorption, as well as facilitating the excretion of uric acid.
Acknowledgements
The study was supported by the National Natural Science Foundation of China [No. 81660755], and the Science and Technology Project of Shenzhen of China [No. JCYJ20170307160524377].
Disclosure of conflict of interest
None.
References
- 1.Borghi C, Piani F. Uric acid and estimate of renal function. Let’s stick together. Int J Cardiol. 2020;310:157–158. doi: 10.1016/j.ijcard.2020.01.046. [DOI] [PubMed] [Google Scholar]
- 2.Pan J, Shi M, Ma L, Fu P. Mechanistic insights of soluble uric acid-related kidney disease. Curr Med Chem. 2018 doi: 10.2174/0929867326666181211094421. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3.Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14. doi: 10.1016/j.ijcard.2015.08.109. [DOI] [PubMed] [Google Scholar]
- 4.Yu TY, Jin SM, Jee JH, Bae JC, Lee MK, Kim JH. The protective effects of increasing serum uric acid level on development of metabolic syndrome. Diabetes Metab J. 2019;43:504–520. doi: 10.4093/dmj.2018.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao T, Lv X, Cao L, Guo M, Zheng S, Xue Y, Zou H, Wan W, Zhu X. Renal excretion is a cause of decreased serum uric acid during acute gout. Int J Rheum Dis. 2018;21:1723–1727. doi: 10.1111/1756-185X.13348. [DOI] [PubMed] [Google Scholar]
- 6.Andrade Sierra J, Flores Fonseca MM. Renal handling of uric acid. Contrib Nephrol. 2018;192:1–7. doi: 10.1159/000484271. [DOI] [PubMed] [Google Scholar]
- 7.Lima WG, Martins-Santos ME, Chaves VE. Uric acid as a modulator of glucose and lipid metabolism. Biochimie. 2015;116:17–23. doi: 10.1016/j.biochi.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 8.Cai Z, Xu X, Wu X, Zhou C, Li D. Hyperuricemia and the metabolic syndrome in Hangzhou. Asia Pac J Clin Nutr. 2009;18:81–87. [PubMed] [Google Scholar]
- 9.Wang H, Zhang H, Sun L, Guo W. Roles of hyperuricemia in metabolic syndrome and cardiac-kidney-vascular system diseases. Am J Transl Res. 2018;10:2749–2763. [PMC free article] [PubMed] [Google Scholar]
- 10.Cabau G, Crisan TO, Kluck V, Popp RA, Joosten LAB. Urate-induced immune programming: consequences for gouty arthritis and hyperuricemia. Immunol Rev. 2020;294:92–105. doi: 10.1111/imr.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joosten LAB, Crisan TO, Bjornstad P, Johnson RJ. Asymptomatic hyperuricaemia: a silent activator of the innate immune system. Nat Rev Rheumatol. 2020;16:75–86. doi: 10.1038/s41584-019-0334-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Annu Rev Physiol. 2015;77:323–345. doi: 10.1146/annurev-physiol-021113-170343. [DOI] [PubMed] [Google Scholar]
- 13.Singh G, Lingala B, Mithal A. Gout and hyperuricaemia in the USA: prevalence and trends. Rheumatology (Oxford) 2019;58:2177–2180. doi: 10.1093/rheumatology/kez196. [DOI] [PubMed] [Google Scholar]
- 14.Liu H, Zhang XM, Wang YL, Liu BC. Prevalence of hyperuricemia among Chinese adults: a national cross-sectional survey using multistage, stratified sampling. J Nephrol. 2014;27:653–658. doi: 10.1007/s40620-014-0082-z. [DOI] [PubMed] [Google Scholar]
- 15.Roman YM. The Daniel K. Inouye college of pharmacy scripts: perspectives on the epidemiology of gout and hyperuricemia. Hawaii J Med Public Health. 2019;78:71–76. [PMC free article] [PubMed] [Google Scholar]
- 16.Chen-Xu M, Yokose C, Rai SK, Pillinger MH, Choi HK. Contemporary prevalence of gout and hyperuricemia in the united states and decadal trends: the national health and nutrition examination survey, 2007-2016. Arthritis Rheumatol. 2019;71:991–999. doi: 10.1002/art.40807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar AUA, Browne LD, Li X, Adeeb F, Perez-Ruiz F, Fraser AD, Stack AG. Temporal trends in hyperuricaemia in the Irish health system from 2006-2014: a cohort study. PLoS One. 2018;13:e0198197. doi: 10.1371/journal.pone.0198197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R, Han C, Wu D, Xia X, Gu J, Guan H, Shan Z, Teng W. Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: a systematic review and meta-analysis. Biomed Res Int. 2015;2015:762820. doi: 10.1155/2015/762820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson MD. Insights in public health: hyperuricemia and gout in Hawai’I. Hawaii J Med Public Health. 2018;77:121–124. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the national health and nutrition examination survey 2007-2008. Arthritis Rheum. 2011;63:3136–3141. doi: 10.1002/art.30520. [DOI] [PubMed] [Google Scholar]
- 21.Cho SK, Winkler CA, Lee SJ, Chang Y, Ryu S. The prevalence of hyperuricemia sharply increases from the late menopausal transition stage in middle-aged women. J Clin Med. 2019;8:296. doi: 10.3390/jcm8030296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Gong H, Lin C, Liu Q, Baima Y, Wang Y, Lin J. The prevalence of gout and hyperuricemia in middle-aged and elderly people in Tibet autonomous region, China: a preliminary study. Medicine (Baltimore) 2020;99:e18542. doi: 10.1097/MD.0000000000018542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Zhang H, Wang F, Liu X, Yang K, Tu R, Wei M, Wang L, Mao Z, Zhang G, Wang C. Epidemiology and prevalence of hyperuricemia among men and women in Chinese rural population: the henan rural cohort study. Mod Rheumatol. 2019;5:1–24. doi: 10.1080/14397595.2019.1660048. [DOI] [PubMed] [Google Scholar]
- 24.Robinson PC. Gout-an update of aetiology, genetics, co-morbidities and management. Maturitas. 2018;118:67–73. doi: 10.1016/j.maturitas.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40:155–175. doi: 10.1016/j.rdc.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni Q, Lu X, Chen C, Du H, Zhang R. Risk factors for the development of hyperuricemia: a STROBE-compliant cross-sectional and longitudinal study. Medicine (Baltimore) 2019;98:e17597. doi: 10.1097/MD.0000000000017597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhary NS, Bridges SL Jr, Saag KG, Rahn EJ, Curtis JR, Gaffo A, Limdi NA, Levitan EB, Singh JA, Colantonio LD, Howard G, Cushman M, Flaherty ML, Judd S, Irvin MR, Reynolds RJ. Severity of hypertension mediates the association of hyperuricemia with stroke in the REGARDS case cohort study. Hypertension. 2020;75:246–256. doi: 10.1161/HYPERTENSIONAHA.119.13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiger S, Ma Q, Anders HJ. The case for evidence-based medicine for the association between hyperuricaemia and CKD. Nat Rev Nephrol. 2020;16:422. doi: 10.1038/s41581-020-0288-3. [DOI] [PubMed] [Google Scholar]
- 29.Sato Y, Feig DI, Stack AG, Kang DH, Lanaspa MA, Ejaz AA, Sanchez-Lozada LG, Kuwabara M, Borghi C, Johnson RJ. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat Rev Nephrol. 2019;15:767–775. doi: 10.1038/s41581-019-0174-z. [DOI] [PubMed] [Google Scholar]
- 30.Richette P, Latourte A. Hyperferritinaemia and hyperuricaemia-a causal connection? Nat Rev Rheumatol. 2018;14:628–629. doi: 10.1038/s41584-018-0100-y. [DOI] [PubMed] [Google Scholar]
- 31.Poletto J, Harima HA, Ferreira SR, Gimeno SG. Hyperuricemia and associated factors: a cross-sectional study of Japanese-Brazilians. Cad Saude Publica. 2011;27:369–378. doi: 10.1590/s0102-311x2011000200018. [DOI] [PubMed] [Google Scholar]
- 32.Kostka-Jeziorny K, Widecka K, Tykarski A. Study of epidemiological aspects of hyperuricemia in Poland. Cardiol J. 2019;26:241–252. doi: 10.5603/CJ.a2019.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McHugh J. Hyperuricaemia or gout in patients with RA. Nat Rev Rheumatol. 2019;15:384. doi: 10.1038/s41584-019-0248-0. [DOI] [PubMed] [Google Scholar]
- 34.Chiang KM, Tsay YC, Vincent Ng TC, Yang HC, Huang YT, Chen CH, Pan WH. Is hyperuricemia, an early-onset metabolic disorder, causally associated with cardiovascular disease events in Han Chinese? J Clin Med. 2019;8:1202. doi: 10.3390/jcm8081202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiou A, England BR, Sayles H, Thiele GM, Duryee MJ, Baker JF, Singh N, Cannon GW, Kerr GS, Reimold A, Gaffo A, Mikuls TR. Coexistent hyperuricemia and gout in rheumatoid arthritis: associations with comorbidities, disease activity and mortality. Arthritis Care Res (Hoboken) 2019;72:950–958. doi: 10.1002/acr.23926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuwabara M, Hisatome I, Niwa K, Hara S, Roncal-Jimenez CA, Bjornstad P, Nakagawa T, Andres-Hernando A, Sato Y, Jensen T, Garcia G, Rodriguez-Iturbe B, Ohno M, Lanaspa MA, Johnson RJ. Uric acid is a strong risk marker for developing hypertension from prehypertension: a 5-year japanese cohort study. Hypertension. 2018;71:78–86. doi: 10.1161/HYPERTENSIONAHA.117.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tatsumi Y, Asayama K, Morimoto A, Satoh M, Sonoda N, Miyamatsu N, Ohno Y, Miyamoto Y, Izawa S, Ohkubo T. Hyperuricemia predicts the risk for developing hypertension independent of alcohol drinking status in men and women: the Saku study. Hypertens Res. 2020;43:442–449. doi: 10.1038/s41440-019-0361-0. [DOI] [PubMed] [Google Scholar]
- 38.Stewart DJ, Langlois V, Noone D. Hyperuricemia and hypertension: links and risks. Integr Blood Press Control. 2019;12:43–62. doi: 10.2147/IBPC.S184685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F, Stamp LK. Gout. Nat Rev Dis Primers. 2019;5:69. doi: 10.1038/s41572-019-0115-y. [DOI] [PubMed] [Google Scholar]
- 40.Chen X, Ding X. Reference level of serum urate for clinically evident incident gout. Ann Rheum Dis. 2019;78:e41. doi: 10.1136/annrheumdis-2018-213355. [DOI] [PubMed] [Google Scholar]
- 41.Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039–2052. doi: 10.1016/S0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- 42.Benn CL, Dua P, Gurrell R, Loudon P, Pike A, Storer RI, Vangjeli C. Physiology of hyperuricemia and urate-lowering treatments. Front Med (Lausanne) 2018;5:160. doi: 10.3389/fmed.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kan Y, Zhang Z, Yang K, Ti M, Ke Y, Wu L, Yang J, He Y. Influence of d-Amino acids in beer on formation of uric acid. Food Technol Biotechnol. 2019;57:418–425. doi: 10.17113/ftb.57.03.19.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakse B, Jakse B, Pajek M, Pajek J. Uric acid and plant-based nutrition. Nutrients. 2019;11:1736. doi: 10.3390/nu11081736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi HK, Liu S, Curhan G. Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the third national health and nutrition examination survey. Arthritis Rheum. 2005;52:283–289. doi: 10.1002/art.20761. [DOI] [PubMed] [Google Scholar]
- 46.Villegas R, Xiang YB, Elasy T, Xu WH, Cai H, Cai Q, Linton MF, Fazio S, Zheng W, Shu XO. Purine-rich foods, protein intake, and the prevalence of hyperuricemia: the shanghai men’s health study. Nutr Metab Cardiovasc Dis. 2012;22:409–416. doi: 10.1016/j.numecd.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Qiu H. Folate, vitamin b6 and vitamin b12 intake in relation to hyperuricemia. J Clin Med. 2018;7:210. doi: 10.3390/jcm7080210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi HK, McCormick N, Lu N, Rai SK, Yokose C, Zhang Y. Population impact attributable to modifiable risk factors for hyperuricemia. Arthritis Rheumatol. 2020;72:157–165. doi: 10.1002/art.41067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rai SK, Fung TT, Lu N, Keller SF, Curhan GC, Choi HK. The dietary approaches to stop hypertension (DASH) diet, western diet, and risk of gout in men: prospective cohort study. BMJ. 2017;357:1794. doi: 10.1136/bmj.j1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018;128:545–555. doi: 10.1172/JCI96702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Cui T, Ci X, Zhao F, Sun Y, Li Y, Liu R, Wu W, Yi X, Liu C. The effect of polymorphism of uric acid transporters on uric acid transport. J Nephrol. 2019;32:177–187. doi: 10.1007/s40620-018-0546-7. [DOI] [PubMed] [Google Scholar]
- 52.Han T, Meng X, Shan R, Zi T, Li Y, Ma H, Zhao Y, Shi D, Qu R, Guo X, Liu L, Na L, Li Y, Sun C. Temporal relationship between hyperuricemia and obesity, and its association with future risk of type 2 diabetes. Int J Obes (Lond) 2018;42:1336–1344. doi: 10.1038/s41366-018-0074-5. [DOI] [PubMed] [Google Scholar]
- 53.Rivera-Paredez B, Macias-Kauffer L, Fernandez-Lopez JC, Villalobos-Comparan M, Martinez-Aguilar MM, de la Cruz-Montoya A, Ramirez-Salazar EG, Villamil-Ramirez H, Quiterio M, Ramirez-Palacios P, Romero-Hidalgo S, Villarreal-Molina MT, Denova-Gutierrez E, Flores YN, Canizales-Quinteros S, Salmeron J, Velazquez-Cruz R. Influence of genetic and non-genetic risk factors for serum uric acid levels and hyperuricemia in Mexicans. Nutrients. 2019;11:1336. doi: 10.3390/nu11061336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taniguchi A, Kamatani N. Control of renal uric acid excretion and gout. Curr Opin Rheumatol. 2008;20:192–197. doi: 10.1097/BOR.0b013e3282f33f87. [DOI] [PubMed] [Google Scholar]
- 55.Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19:358–371. doi: 10.1053/j.ackd.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Oliveira EP, Burini RC. High plasma uric acid concentration: causes and consequences. Diabetol Metab Syndr. 2012;4:12. doi: 10.1186/1758-5996-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merriman TR. An update on the genetic architecture of hyperuricemia and gout. Arthritis Res Ther. 2015;17:98. doi: 10.1186/s13075-015-0609-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cameron JS, Simmonds HA. Hereditary hyperuricemia and renal disease. Semin Nephrol. 2005;25:9–18. doi: 10.1016/j.semnephrol.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 59.George RL, Keenan RT. Genetics of hyperuricemia and gout: implications for the present and future. Curr Rheumatol Rep. 2013;15:309. doi: 10.1007/s11926-012-0309-8. [DOI] [PubMed] [Google Scholar]
- 60.Major TJ, Dalbeth N, Stahl EA, Merriman TR. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14:341–353. doi: 10.1038/s41584-018-0004-x. [DOI] [PubMed] [Google Scholar]
- 61.Kawamura Y, Nakaoka H, Nakayama A, Okada Y, Yamamoto K, Higashino T, Sakiyama M, Shimizu T, Ooyama H, Ooyama K, Nagase M, Hidaka Y, Shirahama Y, Hosomichi K, Nishida Y, Shimoshikiryo I, Hishida A, Katsuura-Kamano S, Shimizu S, Kawaguchi M, Uemura H, Ibusuki R, Hara M, Naito M, Takao M, Nakajima M, Iwasawa S, Nakashima H, Ohnaka K, Nakamura T, Stiburkova B, Merriman TR, Nakatochi M, Ichihara S, Yokota M, Takada T, Saitoh T, Kamatani Y, Takahashi A, Arisawa K, Takezaki T, Tanaka K, Wakai K, Kubo M, Hosoya T, Ichida K, Inoue I, Shinomiya N, Matsuo H. Genome-wide association study revealed novel loci which aggravate asymptomatic hyperuricaemia into gout. Ann Rheum Dis. 2019;78:1430–1437. doi: 10.1136/annrheumdis-2019-215521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakayama A, Nakaoka H, Yamamoto K, Sakiyama M, Shaukat A, Toyoda Y, Okada Y, Kamatani Y, Nakamura T, Takada T, Inoue K, Yasujima T, Yuasa H, Shirahama Y, Nakashima H, Shimizu S, Higashino T, Kawamura Y, Ogata H, Kawaguchi M, Ohkawa Y, Danjoh I, Tokumasu A, Ooyama K, Ito T, Kondo T, Wakai K, Stiburkova B, Pavelka K, Stamp LK, Dalbeth N, Eurogout C, Sakurai Y, Suzuki H, Hosoyamada M, Fujimori S, Yokoo T, Hosoya T, Inoue I, Takahashi A, Kubo M, Ooyama H, Shimizu T, Ichida K, Shinomiya N, Merriman TR, Matsuo H, Eurogout C. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann Rheum Dis. 2017;76:869–877. doi: 10.1136/annrheumdis-2016-209632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu L, Shi Y, Zhuang S, Liu N. Recent advances on uric acid transporters. Oncotarget. 2017;8:100852–100862. doi: 10.18632/oncotarget.20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, Wilson JF, Marinaki A, Riches PL, Shu X, Janicijevic B, Smolej-Narancic N, Gorgoni B, Morgan J, Campbell S, Biloglav Z, Barac-Lauc L, Pericic M, Klaric IM, Zgaga L, Skaric-Juric T, Wild SH, Richardson WA, Hohenstein P, Kimber CH, Tenesa A, Donnelly LA, Fairbanks LD, Aringer M, McKeigue PM, Ralston SH, Morris AD, Rudan P, Hastie ND, Campbell H, Wright AF. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40:437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 65.Kobylecki CJ, Vedel-Krogh S, Afzal S, Nielsen SF, Nordestgaard BG. Plasma urate, lung function and chronic obstructive pulmonary disease: a Mendelian randomisation study in 114,979 individuals from the general population. Thorax. 2018;73:748–757. doi: 10.1136/thoraxjnl-2017-210273. [DOI] [PubMed] [Google Scholar]
- 66.Ruiz A, Gautschi I, Schild L, Bonny O. Human mutations in SLC2A9 (Glut9) affect transport capacity for urate. Front Physiol. 2018;9:476. doi: 10.3389/fphys.2018.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stiburkova B, Pavelcova K, Zavada J, Petru L, Simek P, Cepek P, Pavlikova M, Matsuo H, Merriman TR, Pavelka K. Functional non-synonymous variants of ABCG2 and gout risk. Rheumatology (Oxford) 2017;56:1982–1992. doi: 10.1093/rheumatology/kex295. [DOI] [PubMed] [Google Scholar]
- 68.Kannangara DR, Phipps-Green AJ, Dalbeth N, Stamp LK, Williams KM, Graham GG, Day RO, Merriman TR. Hyperuricaemia: contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann Rheum Dis. 2016;75:1363–1366. doi: 10.1136/annrheumdis-2015-208111. [DOI] [PubMed] [Google Scholar]
- 69.Ichida K, Matsuo H, Takada T, Nakayama A, Murakami K, Shimizu T, Yamanashi Y, Kasuga H, Nakashima H, Nakamura T, Takada Y, Kawamura Y, Inoue H, Okada C, Utsumi Y, Ikebuchi Y, Ito K, Nakamura M, Shinohara Y, Hosoyamada M, Sakurai Y, Shinomiya N, Hosoya T, Suzuki H. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764. doi: 10.1038/ncomms1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wrigley R, Phipps-Green AJ, Topless RK, Major TJ, Cadzow M, Riches P, Tausche AK, Janssen M, Joosten LAB, Jansen TL, So A, Harre Hindmarsh J, Stamp LK, Dalbeth N, Merriman TR. Pleiotropic effect of the ABCG2 gene in gout: involvement in serum urate levels and progression from hyperuricemia to gout. Arthritis Res Ther. 2020;22:45. doi: 10.1186/s13075-020-2136-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Horvathova V, Bohata J, Pavlikova M, Pavelcova K, Pavelka K, Senolt L, Stiburkova B. Interaction of the p.Q141K variant of the ABCG2 gene with clinical data and cytokine levels in primary hyperuricemia and gout. J Clin Med. 2019;8:1965. doi: 10.3390/jcm8111965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ristic B, Sivaprakasam S, Narayanan M, Ganapathy V. Hereditary hemochromatosis disrupts uric acid homeostasis and causes hyperuricemia via altered expression/activity of xanthine oxidase and ABCG2. Biochem J. 2020;477:1499–1513. doi: 10.1042/BCJ20190873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 74.Sakiyama M, Matsuo H, Shimizu S, Nakashima H, Nakamura T, Nakayama A, Higashino T, Naito M, Suma S, Hishida A, Satoh T, Sakurai Y, Takada T, Ichida K, Ooyama H, Shimizu T, Shinomiya N. The effects of URAT1/SLC22A12 nonfunctional variants, R90H and W258X, on serum uric acid levels and gout/hyperuricemia progression. Sci Rep. 2016;6:20148. doi: 10.1038/srep20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li G, Han L, Ma R, Saeed K, Xiong H, Klaassen CD, Lu Y, Zhang Y. Glucocorticoids increase renal excretion of urate in mice by downregulating urate transporter 1. Drug Metab Dispos. 2019;47:1343–1351. doi: 10.1124/dmd.119.087700. [DOI] [PubMed] [Google Scholar]
- 76.Zou Y, Du J, Zhu Y, Xie X, Chen J, Ling G. Associations between the SLC22A12 gene and gout susceptibility: a meta-analysis. Clin Exp Rheumatol. 2018;36:442–447. [PubMed] [Google Scholar]
- 77.Kuo TM, Huang CM, Tu HP, Min-Shan Ko A, Wang SJ, Lee CP, Ko YC. URAT1 inhibition by ALPK1 is associated with uric acid homeostasis. Rheumatology (Oxford) 2017;56:654–659. doi: 10.1093/rheumatology/kew463. [DOI] [PubMed] [Google Scholar]
- 78.Chiba T, Matsuo H, Kawamura Y, Nagamori S, Nishiyama T, Wei L, Nakayama A, Nakamura T, Sakiyama M, Takada T, Taketani Y, Suma S, Naito M, Oda T, Kumagai H, Moriyama Y, Ichida K, Shimizu T, Kanai Y, Shinomiya N. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal underexcretion gout. Arthritis Rheumatol. 2015;67:281–287. doi: 10.1002/art.38884. [DOI] [PubMed] [Google Scholar]
- 79.Higashino T, Morimoto K, Nakaoka H, Toyoda Y, Kawamura Y, Shimizu S, Nakamura T, Hosomichi K, Nakayama A, Ooyama K, Ooyama H, Shimizu T, Ueno M, Ito T, Tamura T, Naito M, Nakashima H, Kawaguchi M, Takao M, Kawai Y, Osada N, Ichida K, Yamamoto K, Suzuki H, Shinomiya N, Inoue I, Takada T, Matsuo H. Dysfunctional missense variant of OAT10/SLC22A13 decreases gout risk and serum uric acid levels. Ann Rheum Dis. 2020;79:164–166. doi: 10.1136/annrheumdis-2019-216044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Drabkin M, Yogev Y, Zeller L, Zarivach R, Zalk R, Halperin D, Wormser O, Gurevich E, Landau D, Kadir R, Perez Y, Birk OS. Hyperuricemia and gout caused by missense mutation in d-lactate dehydrogenase. J Clin Invest. 2019;129:5163–5168. doi: 10.1172/JCI129057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kottgen A, Hwang SJ, Larson MG, Van Eyk JE, Fu Q, Benjamin EJ, Dehghan A, Glazer NL, Kao WH, Harris TB, Gudnason V, Shlipak MG, Yang Q, Coresh J, Levy D, Fox CS. Uromodulin levels associate with a common UMOD variant and risk for incident CKD. J Am Soc Nephrol. 2010;21:337–344. doi: 10.1681/ASN.2009070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chavarriaga J, Ocampo M, Fakih N, Silva Herrera J. Kelley-seegmiller syndrome: urolithiasis, renal uric acid deposits, and gout: what is the role of the urologist? Urol Int. 2019;102:233–237. doi: 10.1159/000494360. [DOI] [PubMed] [Google Scholar]
- 84.Agrahari AK, Krishna Priya M, Praveen Kumar M, Tayubi IA, Siva R, Prabhu Christopher B, George Priya Doss C, Zayed H. Understanding the structure-function relationship of HPRT1 missense mutations in association with Lesch-Nyhan disease and HPRT1-related gout by in silico mutational analysis. Comput Biol Med. 2019;107:161–171. doi: 10.1016/j.compbiomed.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 85.Linnankivi T, Neupane N, Richter U, Isohanni P, Tyynismaa H. Splicing defect in mitochondrial Seryl-tRNA synthetase gene causes progressive spastic paresis instead of HUPRA syndrome. Hum Mutat. 2016;37:884–888. doi: 10.1002/humu.23021. [DOI] [PubMed] [Google Scholar]
- 86.Saeed A, Hoogerland JA, Wessel H, Heegsma J, Derks TGJ, van der Veer E, Mithieux G, Rajas F, Oosterveer MH, Faber KN. Glycogen storage disease type 1a is associated with disturbed vitamin A metabolism and elevated serum retinol levels. Hum Mol Genet. 2020;29:264–273. doi: 10.1093/hmg/ddz283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen C, Lu JM, Yao Q. Hyperuricemia-related diseases and xanthine oxidoreductase (XOR) inhibitors: an overview. Med Sci Monit. 2016;22:2501–2512. doi: 10.12659/MSM.899852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pang M, Fang Y, Chen S, Zhu X, Shan C, Su J, Yu J, Li B, Yang Y, Chen B, Liang K, Hu H, Lv G. Gypenosides inhibits xanthine oxidoreductase and ameliorates urate excretion in hyperuricemic rats induced by high cholesterol and high fat food (Lipid Emulsion) Med Sci Monit. 2017;23:1129–1140. doi: 10.12659/MSM.903217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schumacher HR Jr. Febuxostat: a non-purine, selective inhibitor of xanthine oxidase for the management of hyperuricaemia in patients with gout. Expert Opin Investig Drugs. 2005;14:893–903. doi: 10.1517/13543784.14.7.893. [DOI] [PubMed] [Google Scholar]
- 90.Vuorinen-Markkola H, Yki-Jarvinen H. Hyperuricemia and insulin resistance. J Clin Endocrinol Metab. 1994;78:25–29. doi: 10.1210/jcem.78.1.8288709. [DOI] [PubMed] [Google Scholar]
- 91.Han T, Lan L, Qu R, Xu Q, Jiang R, Na L, Sun C. Temporal relationship between hyperuricemia and insulin resistance and its impact on future risk of hypertension. Hypertension. 2017;70:703–711. doi: 10.1161/HYPERTENSIONAHA.117.09508. [DOI] [PubMed] [Google Scholar]
- 92.Zivna M, Hulkova H, Matignon M, Hodanova K, Vylet’al P, Kalbacova M, Baresova V, Sikora J, Blazkova H, Zivny J, Ivanek R, Stranecky V, Sovova J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gubler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet. 2009;85:204–213. doi: 10.1016/j.ajhg.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019;5:60. doi: 10.1038/s41572-019-0109-9. [DOI] [PubMed] [Google Scholar]
- 94.Kaneko H, Kitoh H, Matsuura T, Masuda A, Ito M, Mottes M, Rauch F, Ishiguro N, Ohno K. Hyperuricemia cosegregating with osteogenesis imperfecta is associated with a mutation in GPATCH8. Hum Genet. 2011;130:671–683. doi: 10.1007/s00439-011-1006-9. [DOI] [PubMed] [Google Scholar]
- 95.Brucato A, Cianci F, Carnovale C. Management of hyperuricemia in asymptomatic patients: a critical appraisal. Eur J Intern Med. 2020;74:8–17. doi: 10.1016/j.ejim.2020.01.001. [DOI] [PubMed] [Google Scholar]
- 96.Shekelle PG, Newberry SJ, FitzGerald JD, Motala A, O’Hanlon CE, Tariq A, Okunogbe A, Han D, Shanman R. Management of gout: a systematic review in support of an american college of physicians clinical practice guideline. Ann Intern Med. 2017;166:37–51. doi: 10.7326/M16-0461. [DOI] [PubMed] [Google Scholar]
- 97.Dong Y, Zhao T, Ai W, Zalloum WA, Kang D, Wu T, Liu X, Zhan P. Novel urate transporter 1 (URAT1) inhibitors: a review of recent patent literature (2016-2019) Expert Opin Ther Pat. 2019;29:871–879. doi: 10.1080/13543776.2019.1676727. [DOI] [PubMed] [Google Scholar]
- 98.Lesinurad/Allopurinol (Duzallo) for gout-associated hyperuricemia. JAMA. 2018;319:188–189. doi: 10.1001/jama.2017.20189. [DOI] [PubMed] [Google Scholar]
- 99.Battelli MG, Bortolotti M, Polito L, Bolognesi A. Metabolic syndrome and cancer risk: the role of xanthine oxidoreductase. Redox Biol. 2019;21:101070. doi: 10.1016/j.redox.2018.101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang CY, Chen CH, Deng ST, Huang CS, Lin YJ, Chen YJ, Wu CY, Hung SI, Chung WH. Allopurinol use and risk of fatal hypersensitivity reactions: a nationwide population-based study in Taiwan. JAMA Intern Med. 2015;175:1550–1557. doi: 10.1001/jamainternmed.2015.3536. [DOI] [PubMed] [Google Scholar]
- 101.Becker MA, Schumacher HR Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 102.Zhang M, Solomon DH, Desai RJ, Kang EH, Liu J, Neogi T, Kim SC. Assessment of cardiovascular risk in older patients with gout initiating febuxostat versus allopurinol: population-based cohort study. Circulation. 2018;138:1116–1126. doi: 10.1161/CIRCULATIONAHA.118.033992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanaka A, Taguchi I, Teragawa H, Ishizaka N, Kanzaki Y, Tomiyama H, Sata M, Sezai A, Eguchi K, Kato T, Toyoda S, Ishibashi R, Kario K, Ishizu T, Ueda S, Maemura K, Higashi Y, Yamada H, Ohishi M, Yokote K, Murohara T, Oyama JI, Node K, investigators Ps. Febuxostat does not delay progression of carotid atherosclerosis in patients with asymptomatic hyperuricemia: a randomized, controlled trial. PLoS Med. 2020;17:e1003095. doi: 10.1371/journal.pmed.1003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Strilchuk L, Fogacci F, Cicero AF. Safety and tolerability of available urate-lowering drugs: a critical review. Expert Opin Drug Saf. 2019;18:261–271. doi: 10.1080/14740338.2019.1594771. [DOI] [PubMed] [Google Scholar]
- 105.Pascart T, Richette P. Investigational drugs for hyperuricemia, an update on recent developments. Expert Opin Investig Drugs. 2018;27:437–444. doi: 10.1080/13543784.2018.1471133. [DOI] [PubMed] [Google Scholar]
- 106.Alghamdi YS, Soliman MM, Nassan MA. Impact of lesinurad and allopurinol on experimental hyperuricemia in mice: biochemical, molecular and immunohistochemical study. BMC Pharmacol Toxicol. 2020;21:10. doi: 10.1186/s40360-020-0386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miner JN, Tan PK, Hyndman D, Liu S, Iverson C, Nanavati P, Hagerty DT, Manhard K, Shen Z, Girardet JL, Yeh LT, Terkeltaub R, Quart B. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Res Ther. 2016;18:214. doi: 10.1186/s13075-016-1107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coiffier B, Mounier N, Bologna S, Fermé C, Tilly H, Sonet A, Christian B, Casasnovas O, Jourdan E, Belhadj K, Herbrecht R Groupe d’Etude des Lymphomes de l’Adulte Trial on Rasburicase Activity in Adult Lymphoma. Efficacy and safety of rasburicase (recombinant urate oxidase) for the prevention and treatment of hyperuricemia during induction chemotherapy of aggressive non-Hodgkin’s lymphoma: results of the GRAAL1 (Groupe d’Etude des Lymphomes de l’Adulte Trial on Rasburicase Activity in Adult Lymphoma) study. J. Clin. Oncol. 2003;21:4402–4406. doi: 10.1200/JCO.2003.04.115. [DOI] [PubMed] [Google Scholar]
- 109.Martens KL, Khalighi PR, Li S, White AA, Silgard E, Frieze D, Estey E, Garcia DA, Hingorani S, Li A. Comparative effectiveness of rasburicase versus allopurinol for cancer patients with renal dysfunction and hyperuricemia. Leuk Res. 2020;89:106298. doi: 10.1016/j.leukres.2020.106298. [DOI] [PubMed] [Google Scholar]