

Abstract
A high hepatitis B virus (HBV) load and chronic hepatitis B infection are well-recognized risk factors for the development of hepatocellular carcinoma (HCC), highlighting the need for research into the mechanisms underlying the role of HBV infection in HCC. Because phosphatase and tensin homolog (PTEN) has been implicated in HCC development, we explored whether PTEN has a role in HBV-related hepatocarcinogenesis. We found that PTEN expression was correlated with advanced clinicopathological features and that HBV infection exacerbates PTEN defects in HCC. Using an integrated approach, we then investigated if miRNAs linked HBV infection to PTEN downregulation in HCC and found that PTEN was a target of miR-181a/382/362/19a. We also show that miR-181a/382/362/19a-mediated inhibition of PTEN led to an enhanced malignant phenotype and stimulation of AKT signaling in HCC cells. Collectively, our results indicate that HBV infection exacerbates PTEN defects in hepatocellular carcinoma through upregulation of miR-181a/362/382/19a. Our work implicates miR-181a/362/382/19a and PTEN as potential biomarkers and targets for novel prognostic, diagnostic, and therapeutic strategies targeting HBV-related HCC.
Keywords: HBV, phosphatase and tensin homolog deleted on chromosome 10, microRNA, hepatocellular carcinoma
Introduction
The etiology of hepatocellular carcinoma (HCC) involves multiple factors, including multiple viral infections, chronic inflammation, obesity, chronic alcohol abuse, and diabetes [1]. Of these, hepatitis B virus (HBV) infection is the most common cause, with 50%~55% of cases of primary liver cancer being attributable to persistent HBV infection [2-4]. HBV causes HCC by both direct and indirect pathways. It can integrate its DNA into the host cells and act as a mutagenic agent, thereby increasing genomic instability [5]. Nevertheless, the precise mechanisms underlying the role of HBV infection in the development and progression of HCC require further investigation.
Phosphatase and tensin homolog (PTEN, also known as MMAC1 or TEP1), is one of the most frequently mutated tumor suppressor genes in human cancers [6]. The PTEN protein functions primarily as a lipid phosphatase in the maintenance of homeostasis in the phosphatidylinositol 3 kinase (PI3K)/serine-threonine protein kinase (AKT) signaling cascade, thereby regulating crucial cell processes, such as growth, adhesion, genomic stability, stem cell self-renewal, migration and the tumor microenvironment [7]. PTEN activity can be modulated by mutations, epigenetic silencing, transcriptional repression, aberrant protein localization, and posttranslational modifications [8]. Studies have shown that reduced PTEN expression is a frequent event in HCC and is correlated with disease stage, tumor grade, tumor size, and increased expression of tumor markers [9-11]. However, the precise role that PTEN plays in the development and progression of HBV-related HCC remains unclear. To address this, we examined the expression of PTEN in HBV-infected HCC tissues and assessed whether miRNAs might link HBV infection to reduced PTEN levels in HCC. We present evidence that HBV infection exacerbates PTEN defects in HCC and that PTEN is a target of miR-181a/382/362/19a. Furthermore, we show that miR-181a/382/362/19a-mediated PTEN downregulation enhanced the malignant phenotype and stimulated AKT signaling in HCC cells. Our work implicates miR-181a/362/382/19a and PTEN as potential biomarkers and targets in novel prognostic, diagnostic, and therapeutic strategies targeting HBV-related HCC.
Materials and methods
Immunohistochemistry
HBV-infected and non-HBV infected liver tissue specimens were obtained from patients at the Department of Hepatobiliary Surgery at Tongji Hospital (affiliated with Huazhong University of Science and Technology, Wuhan, Hubei, China). Informed consent was obtained from all the subjects in accordance with the Declaration of Helsinki. Diagnosis was based on World Health Organization criteria. The samples were snap-frozen in liquid nitrogen and stored at -80°C for subsequent RNA extraction or formalinfixed and paraffin-embedded for immunohistochemistry (IHC). The IHC protocol was as previously described [12].
Cells and transfection
The hepatoma cell lines HepG2 and HepG2.215 (a derivative of the HepG2 line that has been stably transfected with a head-to-tail dimer of HBV DNA) were cultured in DMEM while the human normal liver cell line LO2 was cultured in RPMI1640 (Gibco-BRL, Invitrogen, Carlsbad, CA, USA). Transfections were performed using the jetPRIME kit (Polyplus-transfection, Strasbourg, France) according to the manufacturer’s instructions. Mimics or inhibitors of miR-181a/362/382/19a and their corresponding negative control RNAs (GenePharma, Shanghai, China) were synthesized and introduced into cells at a final concentration of 200 nM.
Dual-luciferase reporter assay
Wild-type (WT) or mutant (MUT) PTEN 3’ untranslated region (UTR) sequences were inserted into the XbaI and XbaI sites of the GV272 vector. HepG2 cells were cotransfected with miR-181a/362/382/19a mimics and the PTEN 3’UTR (800 ng) according to the manufacturer’s protocol (Promega, Madison, WI, USA). Firefly and Renilla luciferase activity were detected using Dual-Luciferase Reporter Assay Kits (Promega, Madison, WI, USA) on a Lumat LB 9507 Tube Luminometer machine (Berthold, Dettenheim, Germany).
HBV cccDNA quantification
Liver tissue DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, Dusseldorf, Germany) and HBV cccDNA was detected with a HBV cccDNA Probe Detection Kit (Fuxing, Shanghai, China) using a LightCycler® (Bio-Rad Laboratories, Hercules, CA, USA).
Quantitative real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First-strand complementary DNA was synthesized using a HiFiScript cDNA Synthesis Kit (Cwbiotech, Beijing, China). Stem-loop reverse transcription and real-time PCR for miR-181a/362/382/19a and U6 were performed with a HairpinitTM miRNAs RT-PCR Quantitation Kit (GenePharma, Shanghai, China) on a real-time PCR detection machine Mx3000P (Stratagene, La Jolla, CA, USA) using U6 RNA as a miRNA internal control. Real-time PCR was carried out using a standard SYBR-Green real-time PCR kit and protocol. GAPDH was used as an endogenous control to normalize the amount of total mRNA in each sample. The 2-ΔΔC(T) method was used to analyze the relative changes in gene expression.
Western blot analysis
Equimolar amounts of extracted protein were fractionated by 10% SDS-PAGE, blotted, and probed with antibodies specific for PTEN, phosphorylated AKT, total AKT or β-actin (Cell Signaling Technology, Danvers, MA, USA). After incubation with goat anti-rabbit or anti-mouse secondary antibody, the blots were visualized with enhanced chemiluminescence. Expression intensities were determined with the ChemiScope Analysis software (Clinx, Shanghai, China).
Cell Counting Kit 8 assay
The in vitro proliferation potential of HCC cells was measured using the Cell Counting Kit 8 (CCK-8) assay. A total of 1 × 104 HCC cells cotransfected with miR-181a/362/382/19a mimics or inhibitors plus PTEN, shPTEN or control vectors were treated by CCK-8 solution at 37°C for 1 h. The absorbance was then measured at 450 nm with a microplate reader (Tecan, Männedorf, Switzerland).
Transwell migration assay
HCC cells were synchronized by serum deprivation for 24 h. A total of 5 × 104 synchronized HCC cells were seeded into the upper chamber of a 24-well plate, while medium containing 10% fetal bovine serum (FBS) was added into the lower chamber. After incubation at 37°C for 24 h (for HepG2 cells) or 48 h (for HepG2.215 cells), the cells in the upper chamber were carefully removed. Then cells adhering to the underside of the membrane were fixed in 4% paraformaldehyde and stained with Hoechst 33342 (Abcam, Cambridge, UK). Cells were counted under a fluorescence microscope (Olympus, Tokyo, Japan).
Wound healing assay
A total of 1 × 106 synchronized HCC cells were seeded into a 6-well plate and cultured until almost 100% confluence. A scraped line was created with a 200-ul pipette tip. The speed of wound closure was imaged with a fluorescence microscope (Olympus, Tokyo, Japan) and the rate of closure was calculated.
Transwell invasion assay
A total of 5 × 104 synchronized HCC cells were added into the upper chamber on a Matrigel (BD Biosciences, Franklin Lakes, NJ, USA)-coated Transwell membrane, while medium containing 10% FBS was added into the lower chamber. After incubation at 37°C for 24 h (for HepG2 cells) or 48 h (for HepG2.215 cells), the cells in the upper chamber were carefully removed. Then cells adhering to the underside of the membrane were fixed in 4% paraformaldehyde and stained with Hoechst 33342 (Abcam, Cambridge, UK). Cells were counted under a fluorescence microscope (Olympus, Tokyo, Japan).
Statistical analysis
Data were expressed as the means ± standard deviation (SD) of at least three independent experiments. Differences between two groups were analyzed by the Student’s t-test while one-way analysis of variance (ANOVA) was used for comparisons between more than two groups. Differences in miRNA expression in tissue specimens from HCC patients were evaluated by the chi-squared test. A two-tailed P-value < 0.05 obtained with the SPSS 20.0 software package (SPSS, Chicago, IL, USA) was considered significant. *P < 0.05, **P < 0.01, and ***P < 0.001 were assumed.
Results
HBV infection exacerbated PTEN defects in hepatocellular carcinoma
To investigate the possible role of PTEN in hepatocarcinogenesis, PTEN expression was compared between cancerous and corresponding paracancerous tissues from HCC patients by IHC. The results indicated that PTEN expression was markedly reduced in cancerous tissues when compared with that of paracancerous tissues and liver hemangioma tissues (Figure 1A). Furthermore, PTEN levels gradually decreased with decreasing levels of HCC tissue differentiation (Figure 1B). As HBV infection is a major risk factor for HCC [13], we next investigated whether HBV infection exacerbated PTEN defects. For this, the above specimens were classified into HBV+ and HBV- subgroups and again evaluated for PTEN expression. We found that PTEN expression was markedly lower in HBV+ tissue than in HBV- tissue, irrespective of whether the tissue was cancerous or paracancerous (Figure 1C). Accordingly, PTEN expression was also decreased in HBsAg+ HCC tissue, as well as in tissue with a heavy HBV cccDNA load (Figure 1D and 1E). Consistent with the data from human tissue specimens, PTEN expression was also lower in HepG2.215 cells than in HepG2 cells (Figure 1F). Together, our data suggest that PTEN expression is downregulated in HCC and HBV infection exacerbates PTEN defects.
Figure 1.
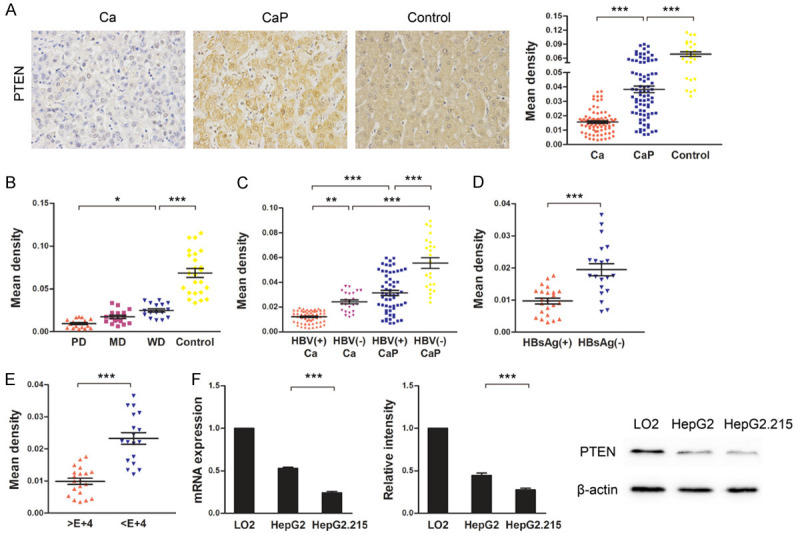
HBV infection decreased PTEN expression in HCC tissues and cell lines. A. PTEN expression in cancerous (Ca) and paracancerous (CaP) tissues from HCC patients was detected by immunohistochemistry (IHC). Hepatic hemangioma tissues were used as controls. Images were obtained using a microscope at × 400 magnification. Three random fields of view were used to calculate mean densities of PTEN-positive areas with Image-Pro Plus software. B. PTEN expression in poorly differentiated (PD), moderately differentiated (MD), and well differentiated (WD) cancerous tissues and hepatic hemangioma tissues (control) detected by IHC. C. PTEN expression in Ca and CaP tissues with HBV infection (HBV+) or without HBV infection (HBV-) detected by IHC. D. PTEN expression in HBsAg+ and HBsAg- cancerous tissues detected by IHC. E. PTEN expression in the cancerous tissues with a high HBV cccDNA load (> 1 × 104) or low HBV cccDNA load (< 1 × 104) detected by IHC. F. PTEN expression in LO2, HepG2, and HepG2.215 cells was detected by quantitative real-time PCR (left) and western blot (middle and right). The mRNA expression was normalized to that of GAPDH and protein expression was normalized to that of β-actin. PTEN expression values in LO2 cells were designated as 1 and data were expressed as fold changes. Data represent means ± standard deviation from triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Expression of miR-181a/362/382/19a was aberrant in HBV-infected HCC
Next, we elucidated the mechanisms by which HBV affects PTEN expression in HCC. As miRNA dysregulation is causal in many types of cancer and miRNAs can target tumor suppressors and act as oncogenes [14], we profiled miRNA expression in the HepG2 and HepG2.215 cell lines and referred to miRNA prediction databases. Among the predicted miRNAs, miR-181a/362/382/19a seemed to be of particular interest. To test whether these miRNAs could link HBV infection to PTEN downregulation, we analyzed the expression of these miRNAs by quantitative real-time PCR in HCC cell lines and tissues. The expression levels of miR-181a/362/382/19a were higher in HepG2 cells than in LO2 cells, and were also higher in HepG2.215 cells than in HepG2 cells (Figure 2A). Similar differences and trends were found between HBV+ and HBV- hepatocellular carcinoma tissues (high miR-181a subgroup: 8/13 (62%) vs 2/6 (33%), χ2 = 16.68, df = 1, P < 0.0001; high miR-362 subgroup: 7/13 (54%) vs 1/6 (17%), χ2 = 29.89, df = 1, P < 0.0001; high miR-382 subgroup: 9/13 (69%) vs 2/6 (33%), χ2 = 25.93, df = 1, P < 0.0001; high miR-19a subgroup: 8/13 (62%) vs 1/6 (17%), χ2 = 42.37, df = 1, P < 0.0001) (Figure 2B). Taken together, these data imply that the expression levels of miR-181a/362/382/19a are upregulated in HCC and HBV infection enhances this upregulation.
Figure 2.
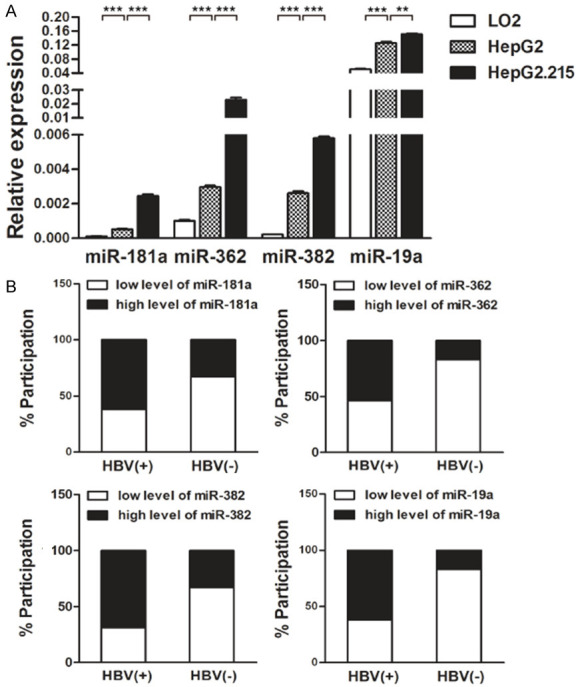
HBV infection increased miR-181a/362/382/19a levels in HCC tissues and cell lines. A. The expression levels of miR-181a/362/382/19a in LO2, HepG2, and HepG2.215 cells were determined by quantitative real-time PCR and normalized to that of U6. Data represent means ± standard deviation from triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. B. The expression levels of miR-181a/362/382/19a in cancerous tissues with HBV infection (HBV+) or without HBV infection (HBV-) were determined by quantitative real-time PCR and normalized to that of U6. The median of relative miR-181a/362/382/19a values was included for subgroup separation for both HBV+ and HBV- cancerous tissues. Differences in miR-181a/362/382/19a expression levels were evaluated by the chi-squared test. (miR-181a: χ2 = 16.68, df = 1, P < 0.0001; miR-362: χ2 = 29.89, df = 1, P < 0.0001; miR-382: χ2 = 25.93, df = 1, P < 0.0001; miR-19a: χ2 = 42.37, df = 1, P < 0.0001).
PTEN is a target of miR-181a/362/382/19a
To verify whether PTEN is a target of miR-181a/362/382/19a, we constructed wild-type or mutant PTEN 3’UTR dual-luciferase reporter vectors and cotransfected the vectors into HepG2 cells with miR-181a/362/382/19a mimics. The dual-luciferase reporter assay showed that cells cotransfected with wild-type PTEN 3’UTR and miR-181a/362/382/19a mimics displayed significantly lower luciferase activity than the controls, whereas cells cotransfected with mutant PTEN 3’UTR and miR-181a/362/382/19a mimics showed luciferase activity comparable with that of the controls (Figure 3A-D). These results were corroborated by quantitative real-time PCR and western blot, showing that miR-181a/362/382/19a inhibited both the mRNA and protein expression of PTEN in HepG2 cells (Figure 4A-D). Therefore, our data indicate that miR-181a/362/382/19a negatively regulate PTEN expression by directly targeting its 3’UTR.
Figure 3.

PTEN is a target of miR-181a/362/382/19a. Diagrams show the putative binding sites (red) for miR-181a (A), miR-362 (B), miR-382 (C) and miR-19a (D) in PTEN. The wild-type (WT) 3’UTR of PTEN is shown in blue and the corresponding mutant (MUT) sites in green. Bar graphs show the relative fluorescence intensity of HepG2 cells transfected with dual-luciferase reporter vectors containing either the WT or MUT 3’UTR of PTEN plus miR-181a/362/382/19a mimics (miR-181a/362/382/19a-m) or negative control mimics (NC-m). Data represent means ± standard deviation from triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 4.

MiR-181a/362/382/19a downregulated PTEN expression in HepG2 cells. The mRNA (left) and protein (middle and right) expression of PTEN in HepG2 cells transfected with miR-181a (A), miR-362 (B), miR-382 (C), miR-19a (D) mimics or negative control mimics (NC-m) were examined by quantitative real-time PCR and western blot. The mRNA expression levels were normalized to that of GAPDH and protein expression levels were normalized to that of β-actin. Values of PTEN expression were designated as 1 and data were expressed as fold changes. Data represent means ± standard deviation from triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
MiR-181a/382/362/19a-mediated inhibition of PTEN enhanced the malignant phenotype of HCC cells
As we found an association between miR-181a/362/382/19a and PTEN expression in HCC, we then functionally characterized the effects of miR-181a/362/382/19a on the behaviors of HCC cells. To examine whether miR-181a/362/382/19a exerted their effects on HCC through PTEN, we overexpressed PTEN in HepG2.215 cells and silenced its expression in HepG2 cells. CCK-8 assays showed that silencing of PTEN increased the proliferative ability of HepG2 cells when compared with that of untreated controls, and a similar result was observed with miR-181a/362/382/19a mimics. However, the increased proliferation was abrogated when PTEN was overexpressed in HepG2.215 cells, similar to that observed with miR-181a/362/382/19a inhibitors. Nevertheless, these effects were reversed by PTEN silencing (Figure 5A). The wound-healing and Transwell (with or without Matrigel) assays also confirmed that overexpression of PTEN blocked the effect of miR-181a/362/382/19a mimics, while PTEN silencing rescued the suppression of HCC cell migration and invasion induced by miR-181a/362/382/19a inhibitors (Figure 5B-D). In summary, our data demonstrated that miR-181a/382/362/19a-mediated inhibition of PTEN enhances the malignant phenotype of HCC cells.
Figure 5.

MiR-181a/362/382/19a enhanced the activities of HCC cells, and overexpression/silencing of PTEN antagonized/mimicked their effects. HepG2 cells were transfected with miR-181a/362/382/19a mimics (miR-181a/362/382/19a-m) or negative control mimics (NC-m) plus PTEN, shPTEN or control vectors. HepG2.215 cells were transfected with miR-181a/362/382/19a inhibitors (miR-181a/362/382/19a-i) or negative control inhibitors (NC-i) plus PTEN, shPTEN or control vectors. A. Proliferation of HepG2 (top) and HepG2.215 (bottom) cells was determined by CCK-8 assay. B. Migration of HepG2 (top) and HepG2.215 (bottom) cells was assessed by Transwell assay. Representative images and bar graphs are shown. C. Migration of HepG2 (left) and HepG2.215 (right) cells was examined by wound-healing assay. Representative images at the indicated time points following scratch initiation and bar graphs are shown. Values at the 0 h-timepoint were designated as 1 and the rate of closure was assessed. D. Invasion of HepG2 (top) and HepG2.215 (bottom) cells was determined by Transwell-Matrigel assay. Representative images and bar graphs are shown. Data represent means ± standard deviation from triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
MiR-181a/362/382/19a inhibited PTEN-mediated deactivation of AKT signaling
PTEN can reduce the phosphorylation status of AKT and inhibit signaling downstream of AKT, which plays an important role in regulating multiple biological processes [15]. To examine whether miR-181a/362/382/19a-mediated PTEN downregulation could affect AKT signaling, we used western blot to assess the influence of these miRNAs on AKT phosphorylation. In HepG2 cells, transfection of miR-181a/362/382/19a mimics suppressed PTEN expression while increasing AKT phosphorylation, and these effects were antagonized by overexpression of PTEN. In contrast, in HepG2.215 cells, transfection of miR-181a/362/382/19a inhibitors enhanced PTEN expression while decreasing AKT phosphorylation, and these effects were antagonized by PTEN silencing (Figure 6A-D). Collectively, these findings indicate that miR-181a/362/382/19a inhibited PTEN-mediated deactivation of AKT signaling.
Figure 6.
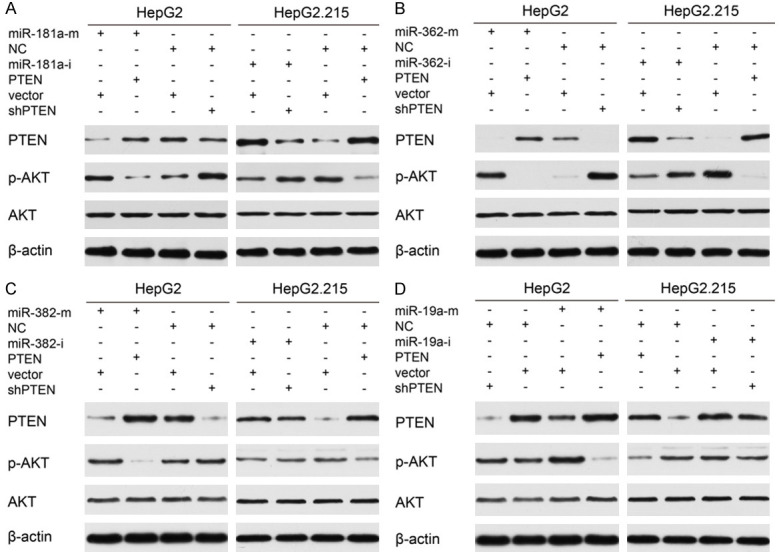
MiR-181a/362/382/19a upregulated AKT phosphorylation, and overexpression/silencing of PTEN antagonized/mimicked their effects. A-D. HepG2 cells were transfected with miR-181a/362/382/19a mimics (miR-181a/362/382/19a-m) or negative control mimics (NC-m) plus PTEN, shPTEN or control vectors. HepG2.215 cells were transfected with miR-181a/362/382/19a inhibitors (miR-181a/362/382/19a-i) or negative control inhibitors (NC-i) plus PTEN, shPTEN or control vectors. The expression levels of PTEN, phosphorylated AKT (p-AKT), and total AKT were determined by western blot.
Discussion
A high HBV load and chronic HBV infection are well-recognized risk factors for developing HCC, highlighting the need for research into the mechanisms underlying the role of HBV infection in HCC [16]. PTEN is a potent and widely expressed tumor suppressor, and PTEN dysregulation is a well-documented key event in the pathogenesis and progression of several human cancers. Consequently, in this study, we focused on evaluating the role of PTEN in HBV-related HCC.
In agreement with previous studies [17,18], we found that PTEN expression was markedly downregulated in cancerous tissues when compared with that in paracancerous tissues or hepatic hemangioma tissues, and was correlated with advanced clinicopathological characteristics. Furthermore, we also found that PTEN defects were exacerbated in HBV-infected HCC tissues and cell lines when compared with HBV-uninfected ones, suggesting that PTEN plays a role in HBV-related hepatocarcinogenesis.
HBV causes chronic hepatitis both by synthesizing some of its own proteins (HBx, HBs, HBc, and others) and by inducing genetic alterations and activating several cell signaling pathways (NF-κB, PI3K, and others), which eventually leads to HCC. Tumor suppressor genes/oncogenes are often inactivated/upregulated by genetic and epigenetic mechanisms in the initiation and progression of HCC. However, miRNA-mediated silencing is also considered to be an important mechanism influencing the loss/gain of tumor suppressor gene/oncogene function [19-21]. For instance, HBx-induced upregulation of miR-21 targets the tumor suppressor PDCD4 and promotes hepatocarcinogenesis [22,23], while decreased expression of let-7 in the presence of HBx results in the upregulation of its target oncogene STAT3 and a consequent enhancement of hepatocarcinogenesis [24].
Short noncoding microRNAs (miRNAs) play crucial roles in numerous diseases [25-28], regulating essential biological processes [29-31] and functioning both as oncogenes and tumor suppressor genes [32-35]. MiR-21 was identified as an endogenously expressed miRNA that specifically targets PTEN, and upregulated miR-21 expression has been linked to the promotion of tumorigenesis in liver cancer [36,37]. Other miRNAs that target PTEN have also been identified [38]. Here, we investigated how HBV affects PTEN expression from the perspective of miRNAs. We employed an integrated approach that involved profiling miRNA expression in HepG2 and HepG2.215 cell lines and referring to miRNA prediction databases. Among the predicted miRNAs, miR-181a/362/382/19a gained our interest. Our results indicated that miR-181a/362/382/19a expression was upregulated in HCC, and HBV infection further increased their expression. The inverse correlation observed between miR-181a/362/382/19a levels and PTEN expression suggested that HBV-mediated upregulation of these miRNAs may result in the suppression of PTEN expression.
MiRNAs are known to alter target gene expression at a posttranscriptional level [39]. Using bioinformatics analysis, we identified sequence complementarity between miR-181a/362/382/19a and the 3’UTR of PTEN. The data from the dual-luciferase reporter and western blot assays confirmed that miR-181a/362/382/19a negatively regulate PTEN expression by directly targeting its 3’UTR. Our data were in line with those of previous studies showing that miR-382 and miR-19a target PTEN [40-44] and revealed for the first time that PTEN is a target of miR-181a and miR-362.
As we found an association between miR-181a/362/382/19a and PTEN expression in HCC, we then functionally characterized the effects of miR-181a/362/382/19a on the behaviors of HCC cells. The combined loss- and gain-of-function approaches confirmed that overexpression of PTEN blocked the effects of miR-181a/362/382/19a, whereas PTEN silencing rescued the suppression of proliferation, migration, and invasion induced by miR-181a/362/382/19a inhibitors in HCC cells. The results indicated that PTEN is a primary functional target of miR-181a/362/382/19a in HCC. The PTEN protein is reported to exert its effects by regulating AKT signaling [45]. Our data suggest that miR-181a/362/382/19a enhances AKT phosphorylation through inhibition of PTEN expression, thereby activating AKT signaling in HCC. Combined, our data indicate that miR-181a/382/362/19a-mediated inhibition of PTEN leads to an enhanced malignant phenotype and stimulation of AKT signaling in HCC cells.
The development of HCC can be considered as an end-stage outcome of HBV infection. Early prognostic and diagnostic markers are needed to allow effective prevention and intervention. Our findings indicated that miR-181a/362/382/19a may serve as potential biomarkers and novel targets in HCC prognosis and diagnosis. MiRNAs play important roles in tumor development, and miRNA mimics (for downregulated miRNAs) or inhibitors (for upregulated miRNAs) have potential as a new class of molecular therapeutic targets [46]. Several studies have confirmed the effectiveness of mimics and inhibitors as a novel method of therapeutic intervention targeting disease-specific miRNAs [47-49]. In this study, we provided a rationale for the therapeutic targeting of miR-181a/362/382/19a using synthetic constructs targeting these miRNAs, alone or in conjunction with other treatments, to activate tumor suppressor genes such as PTEN and reverse the malignant phenotype of HCC cells.
In summary, we presented evidence that HBV infection exacerbates PTEN defects in HCC and found that PTEN is a target of miR-181a/382/362/19a. We further showed that miR-181a/382/362/19a-mediated PTEN inhibition led to an enhanced malignant phenotype and stimulated AKT signaling in HCC cells. Our work provides new insights into the regulatory mechanisms underlying the development and progression of HBV-related HCC and implicates miR-181a/362/382/19a and PTEN as potential biomarkers and targets for novel prognostic, diagnostic, and therapeutic strategies targeting HBV-related HCC.
Acknowledgements
This study was supported by grants from the Chinese National Natural Science Foundation Grant No. 81373152.
Disclosure of conflict of interest
None.
References
- 1.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273. e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15:14–22. doi: 10.1634/theoncologist.2010-S4-14. [DOI] [PubMed] [Google Scholar]
- 3.Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. Biomed Res Int. 2013;2013:810461. doi: 10.1155/2013/810461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertoletti A, Kennedy PTF, Durantel D. HBV infection and HCC: the ‘dangerous liaisons’. Gut. 2018;67:787–788. doi: 10.1136/gutjnl-2017-315528. [DOI] [PubMed] [Google Scholar]
- 5.Chen C, Wang G. Mechanisms of hepatocellular carcinoma and challenges and opportunities for molecular targeted therapy. World J Hepatol. 2015;7:1964–1970. doi: 10.4254/wjh.v7.i15.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 7.Hopkins BD, Parsons RE. Molecular pathways: intercellular PTEN and the potential of PTEN restoration therapy. Clin Cancer Res. 2014;20:5379–5383. doi: 10.1158/1078-0432.CCR-13-2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bermudez Brito M, Goulielmaki E, Papakonstanti EA. Focus on PTEN regulation. Front Oncol. 2015;5:166. doi: 10.3389/fonc.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sze KM, Wong KL, Chu GK, Lee JM, Yau TO, Ng IO. Loss of phosphatase and tensin homolog enhances cell invasion and migration through AKT/Sp-1 transcription factor/matrix metalloproteinase 2 activation in hepatocellular carcinoma and has clinicopathologic significance. Hepatology. 2011;53:1558–1569. doi: 10.1002/hep.24232. [DOI] [PubMed] [Google Scholar]
- 10.Hu TH, Huang CC, Lin PR, Chang HW, Ger LP, Lin YW, Changchien CS, Lee CM, Tai MH. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer. 2003;97:1929–1940. doi: 10.1002/cncr.11266. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe S, Horie Y, Kataoka E, Sato W, Dohmen T, Ohshima S, Goto T, Suzuki A. Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J Gastroenterol Hepatol. 2007;22:S96–S100. doi: 10.1111/j.1440-1746.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Zhang R, Yang H, Xiang Q, Jiang Q, He Q, Zhang T, Chen C, Zhu H, Wang Q, Ning Q, Li Y, Lei P, Shen G. Hepatitis B virus enhances cisplatin-induced hepatotoxicity via a mechanism involving suppression of glucose-regulated protein of 78 Kda. Chem Biol Interact. 2016;254:45–53. doi: 10.1016/j.cbi.2016.05.030. [DOI] [PubMed] [Google Scholar]
- 13.Niu D, Feng H, Chen WN. Proteomic analysis of HBV-associated HCC: insights on mechanisms of disease onset and biomarker discovery. J Proteomics. 2010;73:1283–1290. doi: 10.1016/j.jprot.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet. 2016;17:719–732. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 15.Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24. doi: 10.3389/fonc.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 17.Zhu X, Qin X, Fei M, Hou W, Greshock J, Bachman KE, Wooster R, Kang J, Qin CY. Combined phosphatase and tensin homolog (PTEN) loss and fatty acid synthase (FAS) overexpression worsens the prognosis of Chinese patients with hepatocellular carcinoma. Int J Mol Sci. 2012;13:9980–9991. doi: 10.3390/ijms13089980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su R, Nan H, Guo H, Ruan Z, Jiang L, Song Y, Nan K. Associations of components of PTEN/AKT/mTOR pathway with cancer stem cell markers and prognostic value of these biomarkers in hepatocellular carcinoma. Hepatol Res. 2016;46:1380–1391. doi: 10.1111/hepr.12687. [DOI] [PubMed] [Google Scholar]
- 19.Liu WH, Yeh SH, Chen PJ. Role of microRNAs in hepatitis B virus replication and pathogenesis. Biochim Biophys Acta. 2011;1809:678–685. doi: 10.1016/j.bbagrm.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar N, Chakravarty R. Hepatitis B virus infection, microRNAs and liver disease. Int J Mol Sci. 2015;16:17746–17762. doi: 10.3390/ijms160817746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie KL, Zhang YG, Liu J, Zeng Y, Wu H. MicroRNAs associated with HBV infection and HBV-related HCC. Theranostics. 2014;4:1176–1192. doi: 10.7150/thno.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Yu J, Yu S, Lavker RM, Cai L, Liu W, Yang K, He X, Chen S. MicroRNA-21 acts as an oncomir through multiple targets in human hepatocellular carcinoma. J Hepatol. 2010;53:98–107. doi: 10.1016/j.jhep.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 23.Qiu X, Dong S, Qiao F, Lu S, Song Y, Lao Y, Li Y, Zeng T, Hu J, Zhang L, Zhang L, Fan H. HBx-mediated miR-21 upregulation represses tumor-suppressor function of PDCD4 in hepatocellular carcinoma. Oncogene. 2013;32:3296–3305. doi: 10.1038/onc.2013.150. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, Chung AY, Jooi LL, Lee CG. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J Hepatol. 2010;53:57–66. doi: 10.1016/j.jhep.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 25.Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12:580–587. doi: 10.1016/j.molmed.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 27.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giordano S, Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57:840–847. doi: 10.1002/hep.26095. [DOI] [PubMed] [Google Scholar]
- 29.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 30.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 31.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 33.Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song JS, Tang LH, Levine AJ, Feng Z. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell. 2010;38:689–699. doi: 10.1016/j.molcel.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, Sacco A, Liu Y, Ngo H, Quang P, Azab AK, Azab F, Maiso P, Reagan M, Brown JR, Thai TH, Kauppinen S, Ghobrial IM. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012;120:1678–1686. doi: 10.1182/blood-2012-02-410647. [DOI] [PubMed] [Google Scholar]
- 36.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao L, Yan Y, Xu C, Ji W, Shen S, Xu G, Zeng Y, Sun B, Qian H, Chen L, Wu M, Su C, Chen J. MicroRNA-21 suppresses PTEN and hSulf-1 expression and promotes hepatocellular carcinoma progression through AKT/ERK pathways. Cancer Lett. 2013;337:226–236. doi: 10.1016/j.canlet.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Han ZB, Zhong L, Teng MJ, Fan JW, Tang HM, Wu JY, Chen HY, Wang ZW, Qiu GQ, Peng ZH. Identification of recurrence-related microRNAs in hepatocellular carcinoma following liver transplantation. Mol Oncol. 2012;6:445–457. doi: 10.1016/j.molonc.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Sun XH, Xu HY, Pan HS, Liu Y, He L. Circ_ORC2 enhances the regulatory effect of miR-19a on its target gene PTEN to affect osteosarcoma cell growth. Biochem Biophys Res Commun. 2019;514:1172–1178. doi: 10.1016/j.bbrc.2019.04.188. [DOI] [PubMed] [Google Scholar]
- 41.Hou C, Chen Y, Huang X, Huang Q, Li M, Tan X. miR-19 targets PTEN and mediates high mobility group protein B1(HMGB1)-induced proliferation and migration of human airway smooth muscle cells. PLoS One. 2019;14:e0219081. doi: 10.1371/journal.pone.0219081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang Z, Li Y, Huang K, Wagar N, Shim H. Regulation of miR-19 to breast cancer chemoresistance through targeting PTEN. Pharm Res. 2011;28:3091–3100. doi: 10.1007/s11095-011-0570-y. [DOI] [PubMed] [Google Scholar]
- 43.Seok JK, Lee SH, Kim MJ, Lee YM. MicroRNA-382 induced by HIF-1alpha is an angiogenic miR targeting the tumor suppressor phosphatase and tensin homolog. Nucleic Acids Res. 2014;42:8062–8072. doi: 10.1093/nar/gku515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu D, Zhong L, Yuan Z, Yao J, Zhong P, Liu J, Yao S, Zhao Y, Liu L, Chen M, Li L, Liu B. miR-382-5p modulates the ATRA-induced differentiation of acute promyelocytic leukemia by targeting tumor suppressor PTEN. Cell Signal. 2019;54:1–9. doi: 10.1016/j.cellsig.2018.11.012. [DOI] [PubMed] [Google Scholar]
- 45.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 46.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 47.Park JK, Kogure T, Nuovo GJ, Jiang J, He L, Kim JH, Phelps MA, Papenfuss TL, Croce CM, Patel T, Schmittgen TD. miR-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer Res. 2011;71:7608–7616. doi: 10.1158/0008-5472.CAN-11-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, Fornari F, Giacomelli L, D’Abundo L, Ferracin M, Bassi C, Zagatti B, Corra F, Miotto E, Lupini L, Bolondi L, Gramantieri L, Croce CM, Sabbioni S, Negrini M. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012;56:1025–1033. doi: 10.1002/hep.25747. [DOI] [PubMed] [Google Scholar]
- 49.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]