

Abstract
Resistance to doxorubicin (DOX) is a major clinical challenge in triple-negative breast cancer (TNBC), which is highly diverse in different patients with variable outcomes. Apatinib is a new antiangiogenic agent, which has been reported to induce apoptosis. Nevertheless, the potential role and underlying mechanisms of apatinib in reversing DOX resistance of TNBC remain unknown. This work aims to evaluate the effects of apatinib on improving the sensitivity of TNBC cells to DOX and its underlying molecular basis. Our data indicate that apatinib treatment sensitizes DOX-resistant breast cancer cells to DOX, which is accompanied by significantly increased apoptosis. Additionally, this increased induction of apoptosis is associated with an enhancement of reactive oxygen species (ROS) accumulation. Importantly, it was found that followed by DOX treatment, apatinib could inhibit NF-κB signaling pathways, which have been validated to increase ROS production and reverse DOX resistance. Moreover, our in vivo results indicate the combination of DOX and apatinib exerted increased antitumor effects on TNBC cell xenograft models. Taken together, our study suggests that apatinib sensitizes TNBC cells to DOX in vitro and in vivo through inactivation of NF-κB signaling pathways, providing a rationale for the combined use of apatinib and DOX in TNBC chemotherapy.
Keywords: Apatinib, doxorubicin, chemoresistance, apoptosis, NF-κB, ROS
Introduction
Triple-negative breast cancer (TNBC) is classified as extremely aggressive breast cancer which is characterized by the lack of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) [1]. Although TNBC accounts for only 12-17% of all breast cancers [2], the associated malignancy and death rates of TNBC are relatively high. Very limited therapeutic options are available for TNBC patients, and conventional cytotoxic chemotherapy is still considered the most important systemic therapy [3]. Doxorubicin is a well-known anthracycline, which was used primarily in combination chemotherapy for numerous malignancies, notably breast cancer, particularly TNBC [4]. DOX kills tumor cells by inhibiting the synthesis of nucleic acids, raising the cell levels of ROS and nitrogen species, such as nitric oxide (NO), damaging the metabolism of mitochondria, producing endoplasmic reticulum (ER) stress and immunogenic cell death (ICD), and inhibiting topoisomerase II, leading to cell apoptosis [5]. The incidence of drug resistance is as high as 30-60% [6], despite its usage as a broad-spectrum antitumor drug, leading to frequent relapse and poor prognosis for TNBC. Therefore, it is of impelling importance to systematic analyzing and advancing the approaches to enhance DOX-based therapy.
Apatinib is a new antiangiogenic small molecule agent that highly and selectively inhibits the activity of vascular endothelial growth factor receptor-2 (VEGFR-2) tyrosine kinase, thus inhibiting tumor angiogenesis [7]. Recent studies have demonstrated the antitumor activity of apatinib in several solid tumors [8-12]. Apatinib was demonstrated curable effects and significant survival benefits in a variety of tumors that have undergone standard chemotherapy failure [3,13,14]. Thus, it is possible to suspect that apatinib could also sensitize breast cancer cells to DOX in TNBC by promoting cell apoptosis. However, the role of apatinib in DOX resistance in TNBC remains poorly defined.
The transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) can bind to DNA in the nucleus and activate a series of antiapoptotic genes, together with other mediators, leading to cancer progression as well as chemotherapy resistance [15,16]. NF-κB signaling is highly active in TNBC [17]. The activation of NF-κB inhibits apoptosis of breast cancer cells, while inhibition of NF-κB signaling causes breast cancer regression due to the stimulation of tumor cell apoptosis [18]. Previous studies showed there exists crosstalk between the production of reactive oxygen species (ROS) and inhibition of NF-κB signaling pathways [19,20]. Thus, it is worth studying if apatinib can induce apoptosis by targeting NF-κB/ROS signaling and overcome DOX resistance in TNBC.
This work involved evaluating the antitumor effect of apatinib in improving the DOX sensitivity of TNBC. We investigated both in vitro and in vivo combinatorial efficacy of apatinib and DOX in the TNBC model. Besides, we also analyzed the underlying mechanism of apatinib-mediated chemosensitization.
Materials and methods
Reagents
Apatinib was purchased in Hengrui (Jiangsu, Chian); doxorubicin (DOX) was purchased from Sigma-Aldrich (St Louis, MO, USA). Apatinib and DOX were both dissolved in DMSO, prepared in stock concentration at 20 mM and stored at -20°C.
Cell culture
Human breast cancer line MDA-MB-231 (ATCC) was cultured in DMEM (Corning) supplemented with 10% (v/v) FBS (Gibco) and 100 U/mL penicillin and 100 µg/mL streptomycin (Life Technologies). The DOX-resistant MDA-MB-231/ADR cell line was selected by exposing to gradually increasing concentrations, as described in earlier work [21]. The DOX-resistant MDA-MB-231/ADR cell line was maintained by exposure to 1M DOX every three passages. Cells were maintained at 37°C in a humidified 5% CO2 air incubator. All cell lines were routinely tested for mycoplasma contamination.
CCK-8 assay
The Cell-Counting Kit 8 (Dojindo Molecular Technologies, Japan) was utilized to evaluate the viability of cells according to the manufacturer’s protocols. Briefly, cells were seeded in 48-well plate and cultured for 24 hours, then siRNA or plasmids were transfected to the cells, and cultured for another up to six days. Afterward, the cell medium was replaced by 100 µL complete medium mixed with 10 µL CCK-8 solution and incubated at 37°C for 60 min. The absorption was measured by Multiskan Spectrum (Thermo Fisher, Rockford, IL, USA) at 450 nm and 630 nm.
Flow cytometry for the analysis of the cell cycle and apoptosis
Cells were harvested after transfected for 48 hours, rinsed with cold phosphate-buffered saline (PBS) and then fixed with pre-cooled 75% ethanol overnight at 4°C. Cellular RNA was removed by RNaseA (Sigma-Aldrich), incubation at 37°C for 30 min, followed by stained with Propidium iodide (PI) solution (Sigma-Aldrich), incubation at room temperature for another 30 min. The cell cycle was detected using FACS Aria I flow cytometer (BD Biosciences). Cell apoptosis was assessed after transfected for 72 hours using FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, USA) following the manufacturer’s instructions and analyzed using C6 Flow Cytometer (BD Biosciences).
Assessment of intracellular ROS accumulation
Levels of intracellular hydrogen peroxide were detected by the stained of DCFH-DA (6-carboxy-20,70-dichlorodihydrofluorescein diacetate; Molecular Probes, OR, USA) followed by analyzing using flow cytometry according to the previous report [22]. The levels of cellular ROS were proportionate to the uptake of the staining.
Luciferase reporter assay
For the Luciferase Assay 8×104 cells were plated on a 24-well plate. After 24 h, cells were co-transfected using Lipofectamine 2000 (Invitrogen) with 150 ng of pRL-TK Vector (Promega) containing the Renilla luciferase construct, used as a normalizer and internal control, and with 650 ng of reporter vector (p1242-3x-KB-L or 3xAP1pGL3), or with empty vector pGL2 or pGL3, respectively (Promega). After 24 h transfection cells were treated with TNF-α 10 nM and after 24 h Dual-Luciferase Reporter Assay were performed by Glomax instrument (Promega). Results are calculated as fold changes and shown as means of Firefly Luciferase activity normalized on Renilla luciferase activity.
In vivo tumor growth model
Eight-week-old male severe combined B- and T-cell immunodeficiency (SCID) mice were subcutaneously injected of 5×106/100 μl MDA-MB-231/ADR cells per mouse. Once the average tumor volume reached around 100 mm3, the mice were randomly and blinded assigned into four groups, each group containing at least five mice. Mice embedding with tumors were treated respectively with: saline (200 μl, i.p.), apatinib (100 mg/kg, oral administration once per day for 21 days), DOX (5 mg/kg, i.p., once a week for 3 consecutive weeks), and sequential combination of apatinib and doxorubicin together. The tumor size of each mouse was measured manually once every 2 days using a caliper. At the end of 28 days, all mice were sacrificed and tumors were isolated, the tumor volume and weight were measured and compared among each group. All mice were operated in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health recommendations. All animal studies were performed in strict accordance with the guidelines of the Committee on the Ethics of Animal Experiments of the Harbin Medical University Cancer Hospital.
Western blotting
The cells were washed in cold PBS and lysed in RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 5 mM EDTA, and proteasome inhibitor cocktail). The tumour tissue protein was purified according to the reported method [23]. After centrifugation, the cell supernatants were denatured in SDS-PAGE loading buffer and subjected to SDS-PAGE, and then transferred to PVDF membrane (Millipore). Afterward, the membranes were immunoblotted with specific antibodies: NF-κB p65 (8242, Cell Signaling Technology), NF-κB p50 (4764, Cell Signaling Technology), histone H3 (4499, Cell Signaling Technology), PARP (5625, Cell Signaling Technology), Caspase-3 (9664, Cell Signaling Technology), GAPDH (BM1623, Boster Biological Technology) and Tubulin (2148, Cell Signaling Technology).
Statistical analysis
All statistical analyses were performed using GraphPad Prism® version 6.0 (GraphPad Software). Data sets were analyzed for significance using Student’s two-tailed t-test for two groups, one-way ANOVA with subsequent Bonferroni post hoc test for multiple group comparisons, respectively. All data were expressed as means ± SEM. All data were collected from at least three independent replicate experiments. In all experiments, an only P-value of < 0.05 was considered to be statistically significant.
Results
Apatinib increases the DOX sensitivity of MDA-MB-231/ADR cells
Firstly, in order to confirm the DOX resistance of the induced MDA-MB-231/ADR cells, parental cell lines were treated with increasing concentration of DOX, and the cell viability was examined with CCK-8 assay. Results showed that the half-maximal inhibitory concentrations (IC50) of DOX against MDA-MB-231 and MDA-MB-231/ADR cells were 10 μM and 80 μM, respectively (Figure 1A), demonstrated the MDA-MB-231/ADR cell line showed significant drug resistance. Next, MDA-MB-231/ADR cell was treated with different concentrations of apatinib to explore the potential inhibition effects. The results revealed that apatinib showed a certain dose-independent inhibitory effect (Figure 1B). To further investigate whether apatinib and DOX can exert synergistically effects on DOX-resistance cell line, MDA-MB-231/ADR cell was cultured with a nonlethal dose of apatinib and gradient concentrations of DOX for 48 hours. Notably, apatinib and DOX exhibited significantly synergistic inhibition effect on MDA-MB-231/ADR cell viability with the increase of DOX dose. These results indicated that 10 μM apatinib in combination with 20 μM DOX displayed a high synergistic inhibition rate (Figure 1C). These results suggest that apatinib may synergise the growth inhibitory activities of DOX on TNBC cells.
Figure 1.
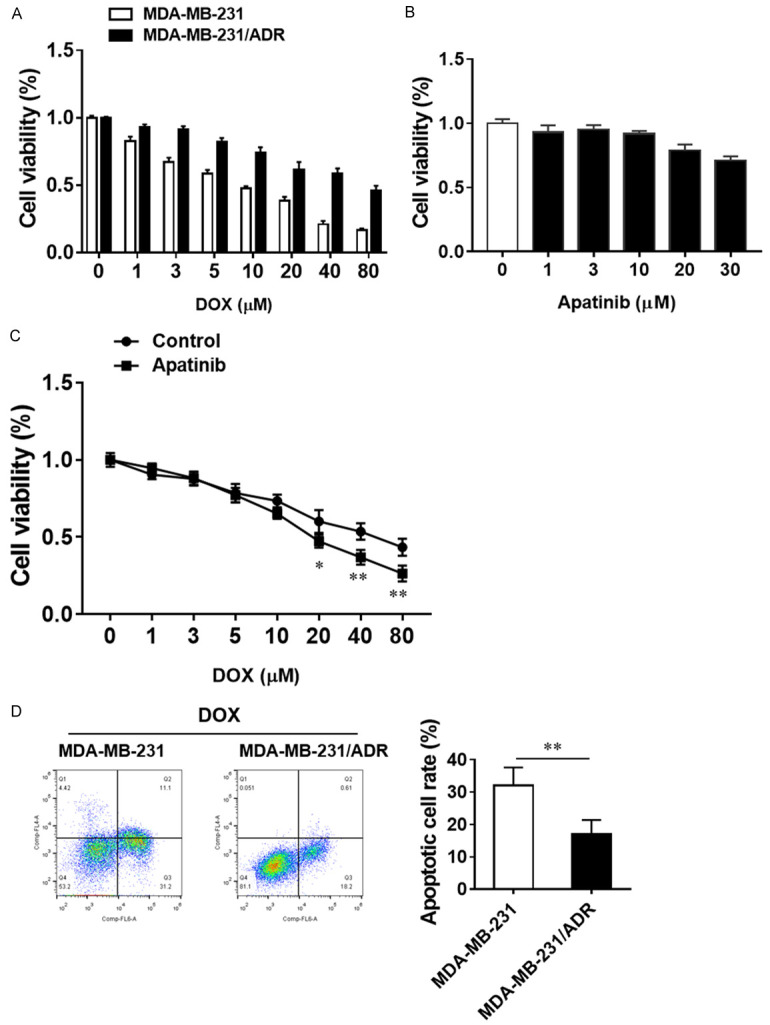
Apatinib sensitizes DOX-resistant TNBC cells to the cytotoxic effect of doxorubicin. A. MDA-MB-231 cells and MDA-MB-231/ADR cells were treated with the indicated concentrations of DOX for 48 h. B. MDA-MB-231/ADR cells were treated with the indicated concentrations of apatinib for 48 h. C. MDA-MB-231/ADR cells were treated with 10 μM apatinib combined with the indicated concentrations of DOX for 48 h. Cell viability was tested by CCK8 assay. The data are expressed as the mean ± SD obtained from 3 independent experiments. D. Apoptotic levels in MDA-MB-231/ADR cells and their parental cells treated with DOX were analyzed with Annexin V/PI staining. *P<0.05, **P<0.01 vs the indicated group.
DOX and apatinib increase apoptosis in MDA-MB-231/DOX cells
Previous studies have claimed that drug-resistance cells were insensitive to cell apoptosis, so we compared the levels of apoptosis in MDA-MB-231/ADR cells and parental cells in the presence of DOX, and the results showed dramatically lower apoptotic rate in MDA-MB-231/ADR cells than in the parental cell (Figure 1D). Since the apatinib can facilitate the DOX effects on the MDA-MB-231/ADR cell, we want to check the levels of apoptosis in both conditions. Our results demonstrated that apatinib alone did not significantly promote the cell apoptosis compared with the control and DOX alone group. However, the DOX and apatinib together could significantly enhance the cell apoptosis among all groups (Figure 2A). Besides, analysis of cell cycle under the treatment of each condition, we found that exposure to DOX increased the percentage of cells accumulation in the sub-G1 phase, while exposure to DOX and apatinib further caused the percentage of cells retained in sub-G1 phase up to 31.8% (Figure 2B). Furthermore, western blot analysis confirmed that apatinib combined with DOX could promote the apoptosis of MDA-MB-231/ADR cells by up-regulating the expression levels of cleaved caspase-3 and cleaved PARP (Figure 2C). Taken together, these results suggest that apatinib enhances DOX-induced MDA-MB-231/ADR cell apoptosis.
Figure 2.

The combination of DOX and apatinib enhances apoptosis in MDA-MB-231/DOX cells. A. MDA-MB-231/ADR cells treated with 10 μM apatinib, 20 μM DOX or the combination of DOX and apatinib for 24 h were analyzed by Annexin V/PI staining. B. Cells were stained with PI and then analyzed by flow cytometry. C. Activation levels of caspases and PARP (cleavage bands) were determined with western blot. The data shown are representative of 3 independent experiments. Data are presented as means ± SD (n = 3). **P<0.01 vs the indicated group.
Apatinib sensitizes MDA-MB-231/ADR cells to DOX-induced apoptosis through ROS generation
There is increasing evidence showing that ROS is critical for the induction of apoptosis in cancer cells. To determine the ROS production under the conditions of each treatment for MDA-MB-231/ADR cell, we used flow cytometry assay to measure the intracellular levels of ROS by examining the DCF fluorescence following the treatment of control, apatinib alone, DOX alone, apatinib and DOX for 24 hours. Cellular ROS production was elevated upon the treatment of DOX and apatinib, higher than treated with DOX or apatinib alone (Figure 3A). To confirm the intracellular ROS production stimulated by DOX and apatinib exposure, NAC, a ROS inhibitor, was introduced in cell culture. When cells were treated with NAC for 4 hours before performing the FACS assay, the results showed that intracellular ROS levels were reduced by the NAC pretreatment (Figure 3A). At the same time, the cell apoptotic rate was also inhibited (Figure 3B) and the cell survival rate was significantly upregulated with NAC treatment (Figure 3C). Overall, those results suggest that ROS mediates the ability of apatinib to enhance DOX-induced apoptosis.
Figure 3.

Apatinib sensitizes MDA-MB-231/ADR cells to DOX-induced apoptosis through ROS generation. A. MDA-MB-231/ADR cells were treated with apatinib, DOX and the combination with or without the ROS scavenger NAC for 24 h, and DCF fluorescence was recorded as a measure of intracellular ROS levels. B. Apoptosis was analyzed by Annexin V/PI staining. C. Cell viability was determined by CCK-8 assay. *P<0.05, **P<0.01 vs the indicated group.
Apatinib suppresses DOX-induced NF-κB activation
Upon the intracellular ROS production, the NF-κB signaling pathway could be stimulated to increase certain cellular antioxidant protein expression. Thus, we examined the involvement of the NF-κB signaling pathway upon the treatment of apatinib and DOX. After the treatment of apatinib and/or DOX, proteins in the cytosol and nucleus were subjected to immunoblot assay. The results showed that exposure of MDA-MB-231/ADR cells to DOX caused an increase in p65 and p50 in the nucleus, which could be reversed by the co-administration of apatinib (Figure 4A). Next, the effect of apatinib and DOX on induced NF-κB activity was assessed in cells stably transfected with the NF-κB-dependent luciferase reporter construct. Our data indicated that NF-κB activity was increased after DOX treatment, while inhibited with DOX and apatinib together (Figure 4B). Meanwhile, NF-κB downstream genes, survivin and IL-8 expression levels were also quantified. Real-time PCR results revealed that the levels of both targets were elevated after exposure to DOX alone, while reduced to the basal levels when treated with DOX and apatinib together (Figure 4C). Overall, it implied that DOX-induced NF-κB pathway activation was inhibited by apatinib in the MDA-MB-231/ADR cell line.
Figure 4.

Apatinib suppresses DOX-induced NF-κB activation. A. The cell extracts were analyzed for p65 and p50 levels by western blot analysis. B. Effect of DOX and apatinib on an NF-κB activity with NF-κB-dependant luciferase reporter assay. C. The mRNA levels were analyzed for IL-8 and survivin by quantitative PCR using specific primers. **P<0.01 vs the indicated group.
ROS release and apoptosis by DOX-Apatinib treatment involves the inhibition of the NF-κB pathway
Next, specific NF-κB pathway inhibitor PDTC was employed to further investigate thebefore subjected to the exposure to DOX or DOX/apatinib. Interestingly, the results showed that DOX combined with apatinib caused a distinct ROS increase in MDA-MB-231/ADR cells, while PDTC pretreatment showed no further enhancement (Figure 5A). Our data indicated that ROS release in the cell line studied involves the NF-κB pathway when exposed to the sequential combination of DOX and apatinib. Similar trends were also observed in the apoptosis and cell viability (Figure 5B-D). Taken together, our results confirm that inhibition NF-κB pathways contributes to the combination of DOX- and apatinib-induced ROS release and apoptosis in TNBC cells.
Figure 5.

Inhibition of the NF-κB pathway contributes to the sequential combination of DOX- and apatinib-induced ROS release and apoptosis. Specific NF-κB pathway inhibitors were applied, and ROS release, apoptotic rate, and cell death were analyzed. A. ROS release was assessed by flow cytometry analysis. B. Apoptosis was assessed by Annexin V/PI staining. C. Activation levels of caspases and PARP (cleavage bands) were determined with western blot. D. Cell viability was determined by CCK-8 assay vs indicates non-significance.
Apatinib and DOX combination treatment inhibits tumor growth in vivo
To further investigate whether apatinib synergizes DOX against tumour growth in vivo, TNBC cells were implanted in SCID mice. Our data showed that tumors were inhibited by DOX and apatinib treatment either in combination or individually (Figure 6A, 6B). Nevertheless, tumor volume was significantly smaller in combination group than that of apatinib group or DOX group (Figure 6A-C). These results demonstrate that the antitumour effect of apatinib combined with DOX is superior to that of the drugs used individually. To confirm the underlying mechanisms of the synergistic effect in vivo, we next assessed the effect of monotreatment or combined treatment of apatinib and DOX on the apoptosis and the expression levels of NF-κB-related proteins in tumor tissues from drug-administered mice. Our data indicate that the nuclear protein expression of p65 and p50 was remarkably decreased with increasing p65 and p50 expression in the cytoplasm in tumor tissues of combination treated mice (Figure 6D), consistent with the results in vitro. Collectively, our results demonstrated that the antitumor effect of DOX was enhanced by apatinib in vivo.
Figure 6.
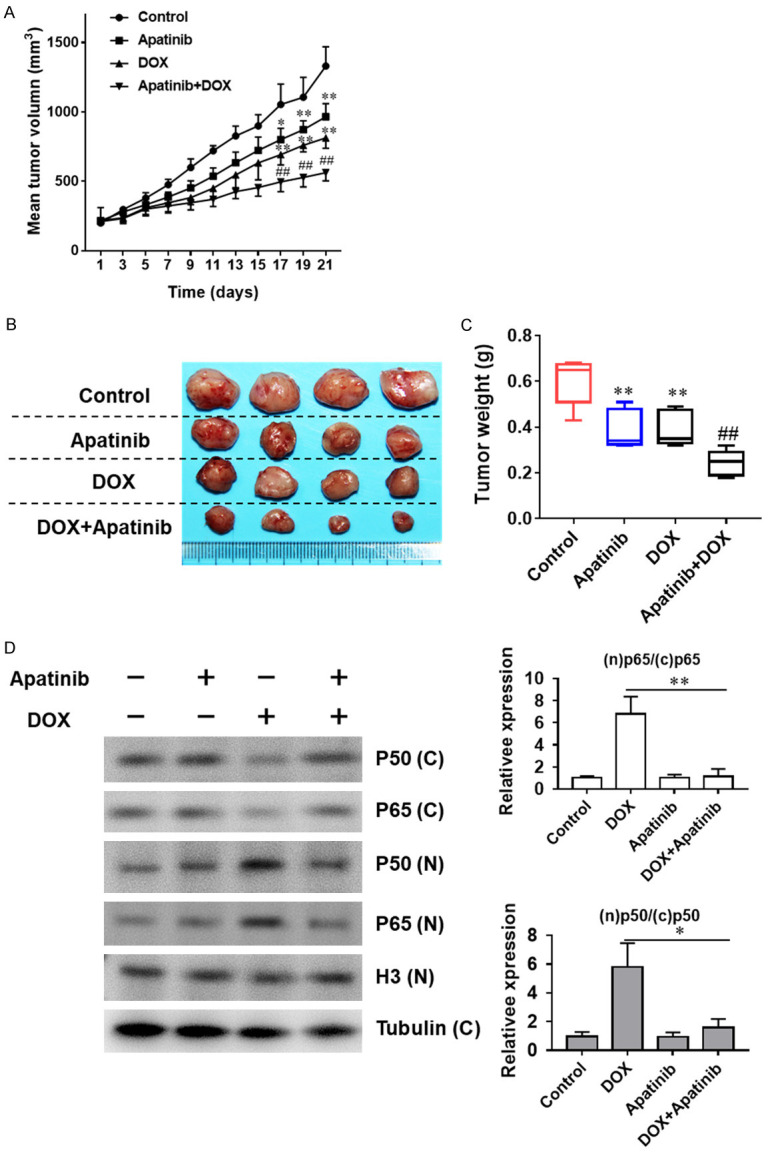
The combination of apatinib and DOX inhibits tumor growth in vivo. The antitumor effect of apatinib, DOX and the combination of both in an MDA-MB-231/ADR human xenograft model. A. Growth curves of xenograft tumors after the subcutaneous injection of MDA-MB-231/ADR cells. The tumor volumes were measured every 2 days after inoculation. B, C. Representative images and weights of tumors excised from mice after inoculation. D. Expression of p50 and p65 extracted from the tumor tissues of drug-administered mice of four groups were detected by western blot analysis. *P<0.05, **P<0.01, vs the control group. ##P<0.01 vs DOX single administration.
Discussion
TNBC is one of the most lethal subtype of breast cancer, and most of the time, TNBC could produce resistance to chemical treatment. In the current study, we elucidated that apatinib can sensitize TNBC cells to DOX by inducing apoptosis both in vitro and in vivo. Importantly, apatinib could inhibit the NF-κB signaling pathway accompanied by the accumulation of ROS, which induce apoptosis in TNBC cells. Furthermore, apatinib and DOX synergistically reduced the tumor burden in the MDA-MB-231 xenograft model. These findings suggest that the sensitization of the DOX-resistant cells by apatinib is due to the specific activation of apoptosis through the suppression of the NF-κB signaling pathway.
Resistance to apoptosis is the leading cause of chemotherapy resistance [24,25]. A recent study showed that DOX can cause apoptosis by mediating the production of reactive oxygen species and oxidative damage [26]. Thus, it is possible that DOX resistance could be induced by the inhibition of apoptosis. To assess this hypothesis, we compared the apoptosis level in DOX-resistant cells and DOX-sensitive cells and found that apoptosis was inhibited in DOX-resistant cells. Then, it was critical to find an approach to promote apoptosis to overcome DOX resistance. Apatinib has been reported to induce apoptosis in osteosarcoma, acute myeloid leukemia, and liver cancer, and showed satisfactory efficacy in TNBC [27-29]. Hence, it is reasonable to hypothesize that concomitant treatment with apatinib and DOX might enhance apoptosis in TNBC cells. In our study, apatinib was utilized in a combination approach as an adjuvant against a DOX resistant breast cancer line. This approach was found to lower the viability of the DOX resistant breast tumor cell line. Moreover, our data indicated that this DOX sensitization by apatinib occurred in parallel with the augmentation of apoptosis. Our data were consistent with previous studies in-dicating that apatinib promotes apoptosis. Next, we sought to further explore the reversal effect of apatinib on the resistance of TNBC to DOX and its underlying mechanisms.
Apoptosis induced by anthracyclines in breast tumors involves the generation of ROS [30,31] via the mutual effect and involvement of the NF-κB signaling pathway [19,20]. An increase in ROS and NF-κB inhibition can lead to caspase-dependent apoptosis [32-34]. We therefore further investigated whether apatinib regulates DOX chemoresistance in TNBC cells via NF-κB/ROS-apoptotic signaling. Our data indicate that apatinib promotes the accumulation of ROS in the resistant cell line studied. Additionally, apatinib and DOX further promote ROS release, thereby promoting apoptosis and reducing the viability of the cell line studied.
NF-κB plays a crucial role in chemotherapeutic resistance in TNBC cells via regulating antiapoptotic pathways [35]. In our study, DOX treatment alone activated NF-κB, but the combined DOX and apatinib treatment inhibited NF-κB activity. Furthermore, we detected the mRNA levels of the target genes of NF-κB pathway, IL-8, and survivin, and found them to be increased by exposure to only DOX; the combined treatment approach reduced the mRNA levels of these genes. Overall, we demonstrated that NF-κB pathway mediates the ability of apatinib to enhance DOX-induced apoptosis. Treatment with an NF-κB inhibitor, as well as apatinib, distinctly boosted the ROS levels in cells to significantly increase the percentage of apoptosis. Taken together, these results demonstrate that ROS mediates the ability of GA to enhance CDDP-induced apoptosis. Moreover, ROS accumulation and apoptosis in response to sequential and combinatorial treatment involved NF-κB pathway inhibition. In addition, the influence of the DOX and apatinib combination was further confirmed in a murine tumor xenograft model. The growth inhibition was consistent in cellular and animal models, demonstrating the antitumor efficacy of DOX plus apatinib. Therefore, our data suggest that apatinib sensitises DOX to TNBC cells through inhibiting DOX-induced NF-κB avtivation and then afterwards promote the generation of ROS.
Conclusions
Our study verified that apatinib sensitized TNBC cells to DOX in vitro and in vivo. Moreover, our rssults indicated that apatinib could inactivate NF-κB signaling pathway induced by DOX followed by enhanced ROS generation, thus potentiating the execution of apoptosis triggered by DOX. Overall, the results suggest that the combination of apatinib with DOX is an innovative approach for treatment of TNBC.
Acknowledgements
This study was supported by Heilongjiang Postdoctoral Financial Assistance, No. LBH-Z18169, by the China Postdoctoral Science Foundation, No. 2019M651306, and by National Natural Science Foundation of China, No. 81903063.
Disclosure of conflict of interest
None.
References
- 1.Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer--current status and future directions. Ann Oncol. 2009;20:1913–1927. doi: 10.1093/annonc/mdp492. [DOI] [PubMed] [Google Scholar]
- 2.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 3.Shen H, Yan W, Yuan J, Wang Z, Wang C. Nek2B activates the wnt pathway and promotes triple-negative breast cancer chemothezrapy-resistance by stabilizing beta-catenin. J Exp Clin Cancer Res. 2019;38:243. doi: 10.1186/s13046-019-1231-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krausz AE, Adler BL, Makdisi J, Schairer D, Rosen J, Landriscina A, Navati M, Alfieri A, Friedman JM, Nosanchuk JD, Rodriguez-Gabin A, Ye KQ, McDaid HM, Friedman AJ. Nanoparticle-encapsulated doxorubicin demonstrates superior tumor cell kill in triple negative breast cancer subtypes intrinsically resistant to doxorubicin. Precis Nanomed. 2018;1:173–182. doi: 10.33218/prnano1(3).181029.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 6.Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89:491–502. doi: 10.1038/clpt.2011.14. [DOI] [PubMed] [Google Scholar]
- 7.Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y, Li J, Lou L. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102:1374–1380. doi: 10.1111/j.1349-7006.2011.01939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu X, Cao J, Hu W, Wu C, Pan Y, Cai L, Tong Z, Wang S, Li J, Wang Z, Wang B, Chen X, Yu H. Multicenter phase II study of apatinib in non-triple-negative metastatic breast cancer. BMC Cancer. 2014;14:820. doi: 10.1186/1471-2407-14-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roviello G, Ravelli A, Fiaschi AI, Cappelletti MR, Gobbi A, Senti C, Zanotti L, Polom K, Reynolds AR, Fox SB, Generali D. Apatinib for the treatment of gastric cancer. Expert Rev Gastroenterol Hepatol. 2016;10:887–892. doi: 10.1080/17474124.2016.1209407. [DOI] [PubMed] [Google Scholar]
- 10.Jin Z, Cheng X, Feng H, Kuang J, Yang W, Peng C, Shen B, Qiu W. Apatinib inhibits angiogenesis via suppressing Akt/GSK3beta/ANG signaling pathway in anaplastic thyroid cancer. Cell Physiol Biochem. 2017;44:1471–1484. doi: 10.1159/000485583. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Feng B, Mei L, Sun R, Guo C, Zhu J. Clinical efficacy of TACE combined with Apatinib in the treatment of advanced hepatocellular carcinoma. J BUON. 2019;24:608–614. [PubMed] [Google Scholar]
- 12.Lin Y, Wu Z, Zhang J, Hu X, Wang Z, Wang B, Cao J, Wang L. Apatinib for metastatic breast cancer in non-clinical trial setting: satisfying efficacy regardless of previous anti-angiogenic treatment. Tumour Biol. 2017;39:1010428317711033. doi: 10.1177/1010428317711033. [DOI] [PubMed] [Google Scholar]
- 13.Peng H, Zhang Q, Li J, Zhang N, Hua Y, Xu L, Deng Y, Lai J, Peng Z, Peng B, Chen M, Peng S, Kuang M. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic cholangiocarcinoma. Oncotarget. 2016;7:17220–17229. doi: 10.18632/oncotarget.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Cao Y, Chen Y, Li G, Yu H. Apatinib promotes apoptosis of the SMMC-7721 hepatocellular carcinoma cell line via the PI3K/Akt pathway. Oncol Lett. 2018;15:5739–5743. doi: 10.3892/ol.2018.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celec P. Nuclear factor kappa B--molecular biomedicine: the next generation. Biomed Pharmacother. 2004;58:365–371. doi: 10.1016/j.biopha.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 16.Chen PM, Wu TC, Wang YC, Cheng YW, Sheu GT, Chen CY, Lee H. Activation of NF-κB by SOD2 promotes the aggressiveness of lung adenocarcinoma by modulating NKX2-1-mediated IKKβ expression. Carcinogenesis. 2013;34:2655–2663. doi: 10.1093/carcin/bgt220. [DOI] [PubMed] [Google Scholar]
- 17.Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:8542–8547. doi: 10.1073/pnas.97.15.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas DK, Martin KJ, McAlister C, Cruz AP, Graner E, Dai SC, Pardee AB. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res. 2003;63:290–295. [PubMed] [Google Scholar]
- 19.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, Prachand S, Miller R, Gordon LI, Evens AM. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res. 2009;15:3354–3365. doi: 10.1158/1078-0432.CCR-08-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol. 2012;180:2490–2503. doi: 10.1016/j.ajpath.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavithra PS, Mehta A, Verma RS. Aromadendrene oxide 2, induces apoptosis in skin epidermoid cancer cells through ROS mediated mitochondrial pathway. Life Sci. 2018;197:19–29. doi: 10.1016/j.lfs.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Zheng S, Lv P, Su J, Miao K, Xu H, Li M. Silencing of the long non-coding RNA RHPN1-AS1 suppresses the epithelial-to-mesenchymal transition and inhibits breast cancer progression. Am J Transl Res. 2019;11:3505–3517. [PMC free article] [PubMed] [Google Scholar]
- 24.Nyongesa CO, Park S. Chemotherapeutic resistance: a nano-mechanical point of view. Biol Chem. 2018;399:1433–1446. doi: 10.1515/hsz-2018-0274. [DOI] [PubMed] [Google Scholar]
- 25.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilco-Ferreto N, Calaf GM. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49:753–762. doi: 10.3892/ijo.2016.3558. [DOI] [PubMed] [Google Scholar]
- 27.Wen S, Shao G, Zheng J, Zeng H, Luo J, Gu D. Apatinib regulates the cell proliferation and apoptosis of liver cancer by regulation of VEGFR2/STAT3 signaling. Pathol Res Pract. 2019;215:816–821. doi: 10.1016/j.prp.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Pan Q, Wang J, Jiang X, Yang E, Dong L, Gu K. Apatinib enhances chemosensitivity of acute myeloid leukemia hl60 cells to cytarabine by inducing apoptosis. J BUON. 2019;24:374–381. [PubMed] [Google Scholar]
- 29.Hu X, Zhang J, Xu B, Jiang Z, Ragaz J, Tong Z, Zhang Q, Wang X, Feng J, Pang D, Fan M, Li J, Wang B, Wang Z, Zhang Q, Sun S, Liao C. Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer. Int J Cancer. 2014;135:1961–1969. doi: 10.1002/ijc.28829. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Gao H, Wu C, Xu QM, Lu JJ, Chen X. Diethyl blechnic, a novel natural product isolated from salvia miltiorrhiza bunge, inhibits doxorubicin-induced apoptosis by inhibiting ROS and activating JNK1/2. Int J Mol Sci. 2018;19:1809. doi: 10.3390/ijms19061809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei X, Liu L, Guo X, Wang Y, Zhao J, Zhou S. Light-activated ROS-responsive nanoplatform codelivering apatinib and doxorubicin for enhanced chemo-photodynamic therapy of multidrug-resistant tumors. ACS Appl Mater Interfaces. 2018;10:17672–17684. doi: 10.1021/acsami.8b04163. [DOI] [PubMed] [Google Scholar]
- 32.Lee SL, Son AR, Ahn J, Song JY. Niclosamide enhances ROS-mediated cell death through c-Jun activation. Biomed Pharmacother. 2014;68:619–624. doi: 10.1016/j.biopha.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Qin Y, Yang C, Zhang H, Li Y, Wu B, Huang J, Zhou X, Huang B, Yang K, Wu G. Cardamonin induces ROS-mediated G2/M phase arrest and apoptosis through inhibition of NF-kappaB pathway in nasopharyngeal carcinoma. Cell Death Dis. 2017;8:e3024. doi: 10.1038/cddis.2017.407. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Zhou DZ, Sun HY, Yue JQ, Peng Y, Chen YM, Zhong ZJ. Dihydromyricetin induces apoptosis and cytoprotective autophagy through ROS-NF-kappaB signalling in human melanoma cells. Free Radic Res. 2017;51:517–528. doi: 10.1080/10715762.2017.1328552. [DOI] [PubMed] [Google Scholar]
- 35.Godwin P, Baird AM, Heavey S, Barr MP, O’Byrne KJ, Gately K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol. 2013;3:120. doi: 10.3389/fonc.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]