

Abstract
Cholangiocarcinoma (CCA) is an aggressive tumour with a poor prognosis due to its late clinical presentation and the lack of effective non-surgical therapies. Previous studies have reported that platelets are implicated in tumour invasion and metastasis, while their role and the underlying mechanism in CCA remain unclear. Here, we show that platelets are hyperactivated in patients with CCA and that platelet-derived growth factor (PDGF) promotes the migration of CCA tumour cells both in vitro and in vivo. Further investigations revealed that PDGF can upregulate the expression of MMP2/MMP9 and induce epithelial-mesenchymal transition (EMT) by activating the p38/MAPK signalling pathway in CCA cells. In addition, the expression of MMP2/MMP9 was associated with lymph node metastasis and poor prognosis in CCA patients after surgical resection. In conclusion, our findings demonstrate that platelets play an important role in facilitating the invasion and metastasis of CCA cells by secreting PDGF, which may provide a novel target for CCA treatment.
Keywords: Cholangiocarcinoma, metastasis, platelet-derived growth factor, p38/MAPK pathway
Introduction
Cholangiocarcinoma that originates from bile duct epithelial cells is famous for its high recurrence rate and high mortality rate [1,2]. It is the second most common liver malignancy after hepatocellular carcinoma (HCC) [3]. Because of the difficulty of early diagnosis and the higher possibility of lymph node metastasis, many patients miss the opportunity for surgical resection [4,5]. Even if patients can undergo surgical resection and liver transplantation in the early stage, the clinical prognosis is also poor, and the 5-year survival rate is still very low [4]. Therefore, there is an urgent need to find effective treatments and strategies.
Tumour cell metastasis and invasion involve multiple different processes, including loss of cell adhesion, extracellular matrix (ECM) degradation, and induction of cell movement [6,7]. Studies have reported that matrix metalloproteinases (MMPs) released from tumour cells play a key role in tumour invasion and migration during the process of ECM degradation [8,9]. For example, a study found that MMP9 promotes the invasion and metastasis of colon cancer [10,11]. On the other hand, the epithelial-mesenchymal-like transition (EMT) is a critical step in distant tumour metastasis [12,13]. In the process of EMT, tumour cells change from a polarized epithelial phenotype to a depolarized and migratory phenotype [14,15]. Meanwhile, the expression of E-cadherin is downregulated, and the expression of N-cadherin, vimentin, slug and snail is upregulated, which reduces tumour cell adhesion [16]. However, the exact mechanism of CCA invasion and metastasis has not been fully uncovered.
A large number of studies have shown that platelets are a stimulative factor for distant tumour metastasis in the early stage [17,18]. The interaction between tumour cells and platelets allows histocompatibility complexes on platelets to adhere to tumour cells, which prevents natural killer (NK) cells from recognizing and killing them and promotes EMT, thereby promoting tumour progression [19,20]. Studies have reported that multiple cytokines present in the tumour microenvironment mediate the metastasis and invasion of tumour cells [21,22]. It has been well established that platelets are the main source and carrier of vascular growth factors, such as transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) [23,24]. However, the role and underlying mechanism of platelets in CCA invasion and metastasis have not been explored.
In this study, we mainly investigated the effect of platelets on CCA. First, we found that platelets can secrete PDGF to promote the migration metastasis of CCA tumour cells. Furthermore, we confirmed that PDGF promotes the invasion and metastasis of CCA by upregulating MMP2/MMP9 expression and inducing EMT via the p38/MAPK signalling pathway. Collectively, these findings provide a new clue for the development of effective strategies for the treatment of CCA.
Materials and methods
CCA tissue specimens
Pathological specimens were obtained from 107 patients with hilar cholangiocarcinoma who underwent surgical treatment in the Department of Hepatobiliary Surgery at the Southwest Hospital Affiliated with the Third Military Medical University (Chongqing, China) from January 2011 to December 2015. In total, 56 pairs of cancer and adjacent tissues were included in the study. All patients were re-examined using upper abdominal colour Doppler ultrasound and computed tomography angiography (CTA) every three months after discharge. Overall survival was calculated from the date of surgery until the date of last contact. All patients provided written informed consent for the collection of samples and subsequent analyses. The present study was approved by the local ethics committee of the Third Military Medical University following the guidelines of the Declaration of Helsinki.
Animals
Male BALB/c-nu mice were purchased from the Institute of Laboratory Animal Sciences of the Chinese Academy of Sciences. A total of 15 male nude (BALB/c-nude) mice (age, 4-6 weeks; weight, 18-22 g) were randomly divided into 3 groups and housed under specific pathogen free conditions (temperature, 24-26°C; humidity, 40-60%; ventilation, 15 times/h; 12:12-h light/dark cycle; free access to food and water). The animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University.
Cell culture
The human CCA cell lines RBE and HCCC-9810 were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences. The cells were cultured in RPMI 1640 (Gibco; Thermo Fisher Scientific, Inc.) medium containing 10% foetal bovine serum (FBS; Zeta Life) and maintained in a humidified incubator containing 5% CO2 at 37°C.
Reagents and inhibitors
The P38MAPK inhibitor SB203580 (cat. no. HY-10256, MCE) and the PDGFR inhibitor CP-673451 (cat. no. HY12050, MCE) were purchased from MedChemExpress. The SB203580 and CP-673451 doses used in the present study were 20 μmol/l.
Immunoblotting
Total protein was extracted from cells and CCA tissue samples using RIPA lysis buffer (Sigma-Aldrich; Merck KGaA) containing protease inhibitors (Roche Diagnostics); the protein concentration was measured using a BCA reagent kit (Beyotime Institute of Biotechnology). Immunoblotting Specific analyses were performed as described previously [25]. Antibodies against MMP2 (ab92536; Abcam), MMP9 (ab38898; Abcam), phospho-p38MAPK (Cell Signaling Technology, Inc.; #4451), p38MAPK (Cell Signaling Technology, Inc.; #8690), E-cadherin (Cell Signaling Technology, Inc.; #3195), N-cadherin (Cell Signaling Technology, Inc.; #13116), ZEB1/TCF8 (Cell Signaling Technology, Inc.; #3396), Snail (Cell Signaling Technology, Inc.; #3879), Slug (Cell Signaling Technology, Inc.; #9585) and vimentin (Cell Signaling Technology, Inc.; #5741) were used at 1:1,000 dilution. Membranes were washed with TBST for 30 min and incubated with an HRP-conjugated anti-rabbit secondary antibody (1:4,000; cat. no. ab6721; Abcam) for 2 h at room temperature.
Platelet isolation from human blood
Blood samples were collected from healthy volunteers who had not taken any drugs known to affect platelet function for at least 14 days prior to the study. This study was conducted according to the principles of the Declaration of Helsinki. The study was approved by the Committee of the Third Military Medical University. All patients signed informed consent forms for the collection of blood and the analysis of clinical data. Blood from 67 cholangiocarcinoma patients and 60 healthy volunteers was collected in ACD buffer and centrifuged at 1500 g for 20 minutes. The obtained platelet-rich plasma was added to modified Tyrode-HEPES buffer (137 mM NaCl, 2.8 mM KCl, 12 mM NaHCO3, 5 mM glucose, 0.4 mM Na2HPO4, 10 mM HEPES, 0.1% BSA, pH 6.5). After centrifugation at 900 g for 10 minutes and removal of the supernatant, the resulting platelet pellet was resuspended in Tyrode-HEPES buffer (pH 7.4, supplemented with 1 mM CaCl2).
Tumour cell invasion and migration assay
Tumour cell invasion assays were performed using Transwell chambers (8 μm, 24-well format) (Corning Costar, NY) coated with 30 μl of Basement Membrane Matrigel (diluted 1:6 in 1640; Corning Life Science, USA) for 5 h in a 37 C incubator. Tumour cells (2×105) in 0.2 ml serum-free medium were seeded in the insert (upper chamber). The lower chamber was supplemented with 0.8 ml medium containing 10% FBS. Additionally, tumour cells (2×105) in 0.2 ml serum-free medium were seeded in the insert (upper chamber) and treated with 1.5×108 platelets per ml in the absence or presence of the inhibitor. The lower chamber was not changed. After 24 h (RBE) or 36 h (HCCC-9810), cells remaining in the upper part of the Transwell insert were removed with a cotton swab. Migrated cells were then stained with 0.5% crystal violet, and the number of cells was counted per high field (x200) with an Olympus microscope.
Tumour cell migration assay: Transwell migration assays were performed according to the protocol of the cell invasion assay, except that the 24-well chambers (Corning Costar, NY) were not coated with Matrigel. HCCC-9810 and RBE cells were incubated for an additional 12 h for migration studies. Migrated cells were then stained with 0.5% crystal violet, and the number of cells per field was determined using a light microscope (Olympus Corporation; magnification, ×200).
Immunohistochemistry (IHC)
Haematoxylin and eosin (H&E) staining of liver tumour tissue sections from mice was performed as previously described [26]. Each immunostained slide was scored by two pathologists in a double-blind manner using a light microscope.
ELISA
Total protein was extracted from CCA tissue samples using RIPA lysis buffer (Sigma-Aldrich; Merck KGaA) containing protease inhibitors (Roche Diagnostics); the protein concentration was measured using a BCA reagent kit (Beyotime Institute of Biotechnology). MMP2 and MMP9 levels in the CCA tissue samples were determined using an ELISA kit (cat. no. MMP200; R&D Systems, Inc.) and (cat. no. DMP900; R&D Systems, Inc.), according to the manufacturer’s instructions. Samples were divided into two groups (high and low expression) based on the mean expression levels of MMP2 and MMP9.
Immunohistochemical staining and platelet adhesion assay
In brief, RBE or HCCC-9810 cells preloaded with VybrantTM DIO cell-labelling solution (V22886, Invitrogen by Thermo Fisher Scientific) for 15 min at 37°C following the manufacturer’s instructions were seeded in a 24-well plate (black with clear bottom; Corning, NY) at a concentration of 5×104 per 100 µl and grown until confluent. The platelets (1.5×108 per ml) were preloaded with VybrantTM DIO cell-labelling solution (V22885, Invitrogen by Thermo Fisher Scientific) for 15 min at 37°C and then washed three times with PBS; then they were allowed to adhere to the cells for 45 min at 37°C. Next, the cells were washed three times with PBS and fixed with 4% formaldehyde. The adherent platelets around tumour cells were photographed with a Leica confocal microscope.
Platelet-derived microparticle (PMP) detection
Platelet-free plasma was prepared using serial centrifugation as previously reported [27]. Then, the plasma was diluted with annexin V binding buffer and incubated with antibodies against annexin V (eBioscience) and CD41. Equal amounts of 1 µm beads (Invitrogen) were added to the sample as a size standard and analysed by a FACSVerse flow cytometer. PMPs. particles less than 1 µm in size that stained positive for CD41 and annexin V, were detected. Data were converted to the number of PMPs per 2 µL of whole blood.
Tumour cell-platelet co-incubation experiments
Platelets (1.5×108 platelets/ml) were added to 1640 medium containing subconfluent RBE and HCCC-9810. Platelets were allowed to interact with RBE and HCCC-9810 cells for 24 h and 12 h in the presence or absence of inhibitors. Both platelets and tumour cells produce MMPs, but only tumour cells can synthesize new MMPs. We used the p38MAPK pathway inhibitor SB203580 (20 μM) to selectively inhibit the p38 pathway to detect changes in the expression of MMPs when tumour cells (RBE or HCCC-9810) activated platelets. Afterwards, the conditioned media were discarded, and the attached cells were rinsed (phosphate-buffered saline ×3), detached and homogenized. Finally, tumour cell proteins were extracted. In addition, using the same procedure, we used the PDGFR inhibitor CP-673451 (20 μM) to selectively inhibit PDGF to detect changes in the expression of MMPs when tumour cells (RBE or HCCC-9810) activated platelets.
In addition, morphology and EMT expression analyses were carried out in CCA cells, platelet-stimulated CCA cells, and platelet-stimulated CCA cells in the presence of CP-673451 and SB203580 inhibitors at 12 hours (HCCC-9810) and 24 hours (RBE).
In vivo experiments
After anaesthetizing with 1% sodium pentobarbital 75 mg/kg (Sigma-Aldrich; Merck KGaA) by intraperitoneal injection, a median abdominal incision of 1 cm was made under the xiphoid of nude mice. To assess experimental liver metastasis, RBE cells were trypsinized and resuspended in PBS, and 5×105 cells or the indicated number of cells (in 0.1 ml of PBS) were injected into the spleen of the mice. Mice were sacrificed at 45 days after injection (before, we performed the time course of tumorigenesis in mice), and livers were isolated and fixed in 10% neutral-buffered formalin for 24 h at room temperature and then dehydrated and embedded in paraffin. Metastatic foci on the surface of the livers were counted under a dissecting microscope, and the nodule size was measured.
To assess tumour volumes, the longest (length) and shortest (width) diameters of the metastatic tumour foci in the liver were measured, and the volumes were calculated using the following formula: Length × width2 ×0.52. The maximum volume of liver tumours was 87 mm3, and the maximum diameter was 6 mm.
In vivo assay of tumour cells pretreated with platelets
RBE cells were seeded in a 10-cm plate. After growth to 60-70% confluence, purified platelets (1.5×108 ml-1) were added to the plate, coincubated for 40 h in vitro, and then washed away. A suspension of tumour cells (5×106 ml-1) in PBS (0.1 ml) was injected into the spleen of mice. In addition, before injection, RBE cells were treated with or without the PDGFR inhibitor CP-673451 for 24 h. To further block PDGF, mice were injected with 1 mg kg-1 CP-673451 1 h before tumour cell infusion. Washed platelets (1.5×109 in 0.2 ml PBS) and 1 mg kg-1 CP-673451 were injected into mice via the lateral tail vein every 5 days [28].
Statistical analysis
Statistical analyses were conducted by SPSS 19.0 (SPSS Inc., USA) for Windows and Prism 7 (GraphPad, USA). Student’s t-test was used to determine the signifcance of differences in platelet hyperactivation of CCA, platelets promote the invasion and metastasis of CCA cells and MMP2/MMP9 expression levels. Pearson’s chi-square analysis or Fisher’s exact test was performed to analyze the correlation between MMP2/MMP9 expression level and the clinicopathological parameters. Survival was analyzed by Kaplan-Meier method, while factors associated with survival were identifed by the Cox proportional hazard regression model. Statistical comparisons were performed using a one-way ANOVA of qualitative data to compare differences between groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Platelet hyperactivation is detected in CCA
To detect the activation of platelet microparticles in patients with CCA, we extracted platelets from patients’ peripheral venous blood for flow cytometry. The circulating level of PMPs significantly increased in the blood of CCA, indicating a hyperactivation state of platelets in CCA (Figure 1A). These data indicate that platelet activity is enhanced significantly in CCA, and we speculate that platelet activation may play a role in the progression of cholangiocarcinoma. In addition, we found that CCA cell lines adhered well to platelets (Figure 1B).
Figure 1.
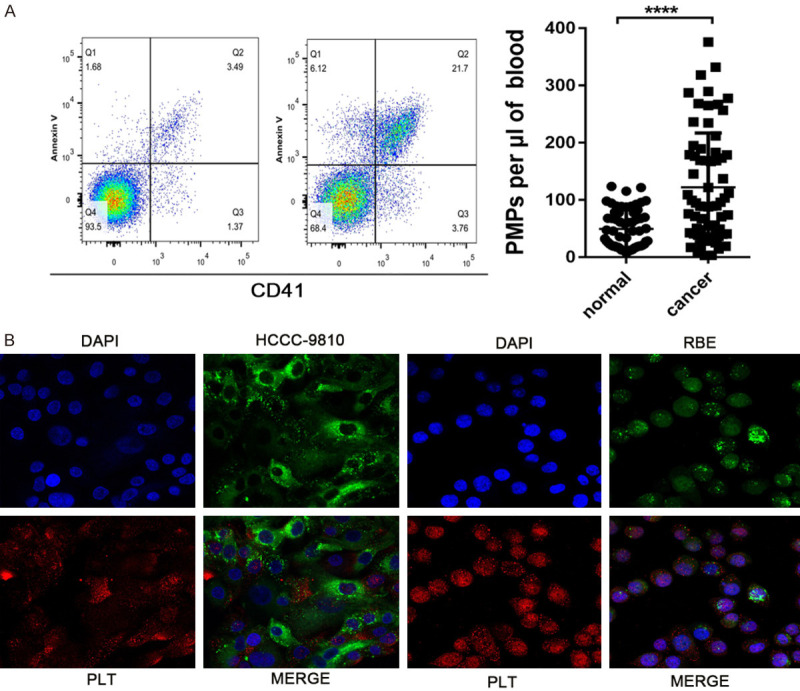
Platelet hyperactivation is detected in CCA and CCA cell-platelet aggregates. A. Flow cytometric analysis of the concentration of PMPs in whole blood. The plasma was diluted with annexin V binding buffer and incubated with antibodies against annexin V (eBioscience) and CD41. Equal amounts of 1 µm beads (Invitrogen) were added to the sample as a size standard and analysed by a FACSVerse flow cytometer. B. Representative immunofluorescence staining of HCCC-9810 and RBE cell-platelet aggregates as described in the Materials and Methods. ****P<0.0001.
Platelets promote the invasion and metastasis of CCA cells
Platelets are highly activated in patients with CCA, and we speculate that platelets may be an important factor in the invasion and metastasis of CCA. Previous studies have reported that platelets can promote tumour invasion and metastasis [28]. Therefore, we wanted to confirm this hypothesis in vitro. HCCC-9810 and RBE cells with or without treatment with platelets were allowed to migrate and invade in Transwell chambers for 12 h and 24 h, respectively, and then the numbers of migrated cells were determined by microscopy (magnification, ×200). In vitro Transwell assays revealed that HCCC-9810 and RBE cells treated with platelets displayed a significant increase in invasion and migration ability compared with that in control cells (Figure 2A-D).
Figure 2.

Platelets promote invasion and metastasis in CCA cells. A and B. Transwell migration and invasion assays of control HCCC-9810 and platelet-stimulated HCCC-9810 cells incubation for 12 h and 36 h. C and D. Transwell migration and invasion assays of control RBE and platelet-stimulated RBE cells incubation for 12 h and 24 h. *P<0.05 and **P<0.01.
PDGF upregulates MMP2/MMP9 in platelet-stimulated CCA cells
Several signalling molecules, including TGFβ, PDGF, VEGF and angiopoietin, are abundant in platelets and may therefore impact tumour cell invasion and metastasis. In addition, previous studies have shown that PDGF can promote the invasion and metastasis of malignant tumours [29,30]. We next investigated whether platelets promote invasion and metastasis in a PDGF-dependent manner. We added a PDGFR inhibitor (CP-673451) to the co-culture of tumour cells and platelets. We found that the PDGF inhibitor CP-673451 completely abrogated platelet-induced invasion and metastasis (Figure 3A and 3B).
Figure 3.

PDGF upregulates MMP2/MMP9 in platelet-stimulated CCA cells. (A) Transwell migration and invasion assays of control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without CP inhibitors (20 μmol/l) incubation for 12 h and 36 h. (B) Transwell migration and invasion assays of control RBE and platelet-stimulated RBE cells with or without CP inhibitors (20 μmol/l) incubation for 12 h and 24 h. (C) Protein expression levels of MMP2 and MMP9 were detected by western blotting in 56 tumour tissues and adjacent normal tissues from patients with CCA. Representative blots from 4 paired samples are shown. (D) Kaplan-Meier plot of the overall survival of 107 patients with CCA with low or high MMP2 or (E) MMP9 expression. Patients were divided into two groups based on the mean expression level. (F) MMP2 and MMP9 protein expression levels were determined by western blotting in control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without CP inhibitors (20 μmol/l) incubation for 12 h. (G) MMP2 and MMP9 protein expression levels were determined by western blotting in control RBE and platelet-stimulated RBE cells with or without CP inhibitors (20 μmol/l) incubation for 24 h. *P<0.05; **P<0.01; ***P<0.001 and ****P<0.0001.
MMP2/MMP9 is an important step in the invasion and metastasis of malignant tumours. The present study next evaluated MMP2/MMP9 expression levels in patient tissues. The results demonstrated that MMP2/MMP9 was more highly expressed in CCA tissues than in adjacent normal tissues from patients with CCA (Figure 3C). Univariate and multivariate analysis results showed that MMP2/MMP9 expression was also significantly associated with lymph node metastasis and poor prognosis in patients with CCA following surgical resection (Tables 1, 2, 3 and 4). In accordance with this finding, MMP2/MMP9 expression levels were also significantly associated with poor prognosis in patients with CCA following surgical resection, as evaluated by multivariate Cox proportional hazards regression analysis (Tables 2 and 4). In addition, Kaplan-Meier survival analysis revealed that high MMP2/MMP9 expression was associated with a significant decrease in the overall survival of patients with CCA (Figure 3D and 3E). Furthermore, multivariate Cox proportional hazards regression analyses revealed that combined high MMP2 and high MMP9 expression levels were significantly associated with poor prognosis in patients with CCA following surgical resection (Table 5).
Table 1.
Association between MMP2 expression levels and clinicopathological features in 107 patients with cholangiocarcinoma
| Features | MMP2 Expression | P-value | |
|---|---|---|---|
|
| |||
| Low 48 (%) | High 59 (%) | ||
| Gender | 0.172 | ||
| Male | 29 (60%) | 43 (73%) | |
| Female | 19 (40%) | 16 (27%) | |
| Age (years) | 0.732 | ||
| <55 | 22 (46%) | 29 (49%) | |
| ≥55 | 26 (54%) | 30 (51%) | |
| Differentiation | 0.004 | ||
| Poorly | 9 (19%) | 23 (39%) | |
| Moderately | 33 (69%) | 36 (61%) | |
| Highly | 6 (12%) | 0 (0%) | |
| Vascular invasion | 0.314 | ||
| Yes | 15 (31%) | 24 (41%) | |
| No | 33 (69%) | 35 (59%) | |
| Lymph node metastasis | 0.011 | ||
| Yes | 20 (42%) | 39 (66%) | |
| No | 28 (58%) | 20 (34%) | |
MMP2, Matrix metalloproteinase-2.
Table 2.
Multivariate Cox proportional hazards regression analysis for MMP2 expression levels and overall survival in 107 patients with cholangiocarcinoma
| Factors | Overall survival | P-value |
|---|---|---|
|
| ||
| Hazard ratio (95% confidence interval (CI)) | ||
| Expression of MMP2 (Low/High) | 1.743 (1.103-2.756) | 0.017 |
| Gender | 0.810 (0.511-1.283) | 0.369 |
| Age (years) | 1.197 (0.788-1.819) | 0.399 |
| Differentiation | 0.995 (0.651-1.521) | 0.982 |
| Vascular invasion | 0.425 (0.268-0.675) | <0.001 |
| Lymph node metastasis | 0.423 (0.264-0.677) | <0.001 |
MMP2, Matrix metalloproteinase-2; CI, confidence interval.
Table 3.
Association between MMP9 expression levels and clinicopathological features in 107 patients with cholangiocarcinoma
| Features | MMP9 Expression | P-value | |
|---|---|---|---|
|
| |||
| Low 51 (%) | High 56 (%) | ||
| Gender | 0.587 | ||
| Male | 33 (65%) | 39 (70%) | |
| Female | 18 (35%) | 17 (30%) | |
| Age (years) | 0.905 | ||
| <55 | 24 (47%) | 27 (48%) | |
| ≥55 | 27 (53%) | 29 (52%) | |
| Differentiation | 0.019 | ||
| Poorly | 12 (23%) | 20 (36%) | |
| Moderately | 33 (65%) | 36 (64%) | |
| Highly | 6 (12%) | 0 (0%) | |
| Vascular invasion | 0.523 | ||
| Yes | 17 (33%) | 22 (39%) | |
| No | 34 (67%) | 34 (61%) | |
| Lymph node metastasis | 0.017 | ||
| Yes | 22 (43%) | 37 (66%) | |
| No | 29 (57%) | 19 (34%) | |
MMP9, Matrix metalloproteinase-9.
Table 4.
Multivariate Cox proportional hazards regression analysis for MMP9 expression levels and overall survival in 107 patients with cholangiocarcinoma
| Factors | Overall survival | P-value |
|---|---|---|
|
| ||
| Hazard ratio (95% confidence interval (CI)) | ||
| Expression of MMP9 (Low/High) | 1.869 (1.196-2.922) | 0.006 |
| Gender | 0.879 (0.558-1.385) | 0.578 |
| Age (years) | 1.188 (0.782-1.806) | 0.42 |
| Differentiation | 1.009 (0.663-1.534) | 0.968 |
| Vascular invasion | 0.400 (0.249-0.642) | <0.001 |
| Lymph node metastasis | 0.438 (0.272-0.704) | 0.001 |
MMP9, Matrix metalloproteinase-9; CI, confdence interval.
Table 5.
Multivariate Cox proportional hazards regression analysis for MMP2*MMP9 expression levels and overall survival in 107 patients with cholangiocarcinoma
| Factors | Overall survival | P-value |
|---|---|---|
|
| ||
| Hazard ratio (95% confidence interval (CI)) | ||
| Expression of MMP2*MMP9 (Low/High) | 1.333 (1.111-1.601) | 0.002 |
| Gender | 0.828 (0.525-1.306) | 0.417 |
| Age (years) | 1.163 (0.766-1.765) | 0.478 |
| Differentiation | 1.091 (0.708-1.683) | 0.692 |
| Vascular invasion | 0.413 (0.259-0.658) | <0.001 |
| Lymph node metastasis | 0.443 (0.275-0.713) | 0.001 |
MMP9, Matrix metalloproteinase-9; MMP2, Matrix metalloproteinase-2; CI, confidence interval.
Moreover, in our CCA cell lines, we further studied whether the expression of MMP2/MMP9 was upregulated in platelet-stimulated RBE and HCCC-9810 cells. Our immunoblotting results showed that the expression of MMP2/MMP9 was significantly upregulated in platelet-stimulated RBE and HCCC-9810 cells compared to control cells (Figure 3F and 3G). In addition, to further confirm whether platelet PDGF plays a major role in this process, we used the PDGFR receptor inhibitor CP-673451 to pretreat the CCA cell lines before co-incubation with platelets and found that selective inhibition of platelet PDGF also significantly reduced the expression of MMP2/MMP9 (Figure 3F and 3G).
PDGF induces EMT in platelet-stimulated CCA cells
Platelets were previously reported to promote EMT in epithelial cells [17,24]. We speculate that PDGF plays an important role in the induction of EMT in CCA. To further confirm this feature of platelets in CCA cell lines, we used platelets with RBE and HCCC-9810 cells to incubate in serum medium 1640 for 24 hours and 12 hours, respectively. Platelet induction of EMT in RBE and HCCC-9810 cells was confirmed at the protein level by protein expression analysis of the mesenchymal markers N-cadherin, vimentin, Slug, Snail and ZEB1 after 24 and (Figure 4A and 4B). Immunoblotting analysis revealed that compared with that at 0 h, N-cadherin, vimentin, Slug, Snail and ZEB1 expression was significantly higher at 12 h after platelet coculture with HCCC-9810 cells (Figure 4A) and was significantly higher at 24 h after platelet coculture with RBE cells (Figure 4B). However, the epithelial marker E-cadherin expression was significantly lower at 12 h and 24 h after platelet coculture with HCCC-9810 and RBE, respectively (Figure 4A and 4B). Altogether, these results show that platelets induce EMT in both HCCC-9810 and RBE cells in vitro. In addition, we investigated whether platelet-derived PDGF could be activated in the co-culture of tumour cells and platelets. Interestingly, adding a PDGFR inhibitor (CP-673451) to block PDGF abolished platelet-induced EMT in RBE and HCCC-9810 cells (Figure 4A and 4B).
Figure 4.
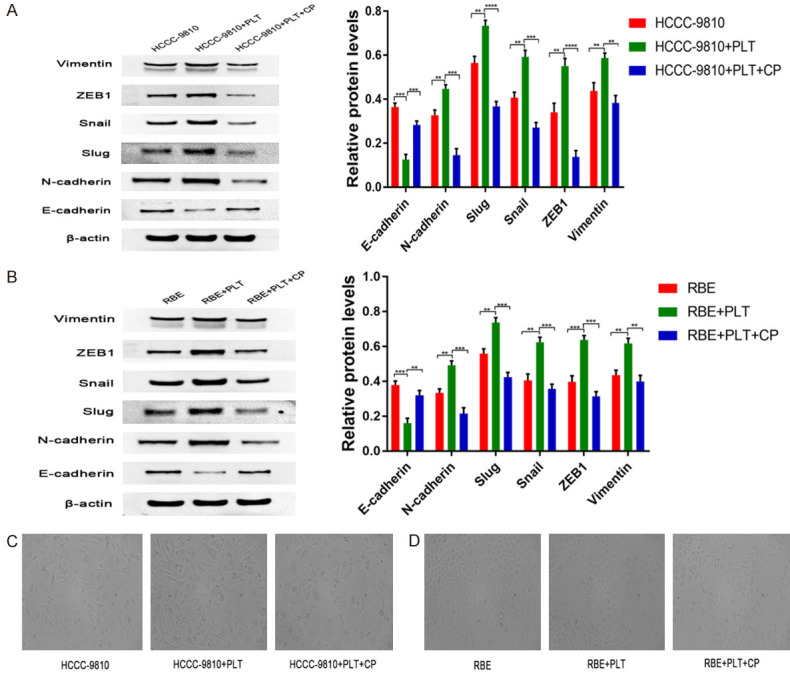
PDGF induces EMT in platelet-stimulated CCA cells. (A) E-cadherin, N-cadherin, Slug, Snail, ZEB1 and vimentin protein expression levels were determined by western blotting in control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without CP inhibitors (20 μmol/l) incubation for 12 h. (B) E-cadherin, N-cadherin, Slug, Snail, ZEB1 and vimentin protein expression levels were determined by western blotting in control RBE and platelet-stimulated RBE cells with or without CP inhibitors (20 μmol/l) incubation for 24 h. (C) Phase-contrast micrographs of the morphological changes of tumour cells in control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without CP inhibitors (20 μmol/l) were observed at 12 h. Scale bar =50 μm. (D) RBE as in (C) for 24 h. Scale bar =50 μm. **P<0.01; ***P<0.001 and ****P<0.0001.
Furthermore, platelets were previously reported to promote EMT epithelial cells [20,31]. To verify this finding in our cell lines and to address whether platelet-induced EMT is a feature of CCA cells. We cultured both CCA cell lines HCCC-9810 and RBE and observed morphological changes reminiscent of epithelial-mesenchymal transition (EMT) when treated with platelets for 12 and 24 h. Compared with the control group, the tumour cells treated with platelets had significantly increased branches and intercellular space instead of a spindle shape (Figure 4C and 4D). To detect morphological changes induced by PDGF, we added a PDGFR inhibitor (CP-673451) to the co-culture of tumour cells and platelets. We found that the PDGF inhibitor CP673451 completely abrogated the platelet-induced morphological changes (Figure 4C and 4D).
PDGF upregulates the expression of MMP2/MMP9 and induces EMT by activating the p38MAPK pathway
A large number of studies have shown that the p38MAPK signalling pathway is significantly related to the invasion and metastasis of malignant tumours [32,33]. We next investigated whether the promotion of invasion and metastasis is dependent on P38MAPK signalling. We added a P38MAPK inhibitor (SB203580) to the co-culture of CCA cells and platelets. We found that the P38MAPK inhibitor (SB203580) completely abrogated platelet-induced invasion and metastasis of RBE and HCCC-9810 cells (Figure 5A and 5B).
Figure 5.

PDGF upregulates the expression of MMP2/MMP9 and induces EMT by activating the p38MAPK pathway. A. Transwell migration and invasion assays of control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without SB inhibitors (20 μmol/l) incubation for 12 h and 36 h. B. Transwell migration and invasion assays of control RBE and platelet-stimulated RBE cells with or without SB inhibitors (20 μmol/l) incubation for 12 h and 24 h. C. P-P38MAPK, P38MAPK, MMP2 and MMP9 protein expression levels were determined by western blotting in control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without SB inhibitors (20 μmol/l) incubation for 12 h. D. P-P38MAPK, P38MAPK, MMP2 and MMP9 protein expression levels were determined by western blotting in control RBE and platelet-stimulated RBE cells with or without SB inhibitors (20 μmol/l) incubation for 24 h. E. P-P38MAPK, P38MAPK, E-cadherin, N-cadherin, Slug, Snail, ZEB1 and vimentin protein expression levels were determined by western blotting in control HCCC-9810 and platelet-stimulated HCCC-9810 cells with or without SB inhibitors (20 μmol/l) incubation for 12 h. F. P-P38MAPK, P38MAPK, E-cadherin, N-cadherin, Slug, Snail, ZEB1 and vimentin protein expression levels were determined by western blotting in control RBE and platelet-stimulated RBE cells with or without SB inhibitors (20 μmol/l) incubation for 24 h. *P<0.05; **P<0.01; ***P<0.001 and ****P<0.0001.
In addition, previous studies found that the p38MAPK pathway upregulates MMP9 and induces EMT [33,34]. Moreover, our immunoblotting results showed that the expression of MMP2/MMP9 was significantly upregulated in platelet-stimulated RBE and HCCC-9810 cells (Figure 5C and 5D). We speculated whether the MAPK pathway is involved in the upregulation of MMP2/MMP9 in CCA cells. To further test this hypothesis, we used the p38MAPK pathway inhibitor SB203580 to pretreat CCA cell lines in the presence of platelets. The results showed that selective inhibition of the p38MAPK pathway could significantly downregulate the expression of MMP2/MMP9 (Figure 5C and 5D). The p38MAPK pathway may control the production of MMP2/MMP9 in platelet-stimulated CCA cells. Our study results further confirmed that platelet activation indeed stimulated p38MAPK phosphorylation, and this effect was completely affected by SB203580 inhibitors in the presence of platelets (Figure 5C and 5D).
We next investigated whether platelet-induced tumour cell EMT is dependent on P38MAPK signalling. We added a P38MAPK inhibitor (SB203580) to the co-culture of tumour cells and platelets. We found that the P38MAPK inhibitor (SB203580) completely abrogated platelet-induced EMT (Figure 5E and 5F). Consistently, our study results also confirmed that platelet activation indeed stimulated p38MAPK phosphorylation, and this effect was completely affected by SB203580 inhibitors in the presence of platelets (Figure 5E and 5F).
PDGF can promote the invasion and metastasis of CCA in vivo
To investigate whether platelet PDGF increases CCA metastasis in vivo, a liver experimental metastasis model was established in mice. Control RBE and platelet-stimulated RBE cells in the absence and presence of the CP inhibitors were injected into the spleens of wild-type mice (specific experimental steps in Materials and Methods), and then, liver metastasis was monitored. Both RBE cells and treated RBE cells formed metastatic foci in the liver, but treatment of RBE cells co-incubated with platelets resulted in significantly increased numbers of metastatic foci (Figure 6A and 6B). By contrast, inhibition of platelet PDGF reduced the metastatic ability of RBE CCA cells in vivo (Figure 6A and 6B). To investigate whether platelet PDGF affects metastatic growth, the volumes of the liver metastatic foci were measured, and no significant difference was found between stimulated and un-stimulated cells (data not shown). These data suggested that PDGF may enhance the dissemination and metastasis ability of CCA cells in vivo.
Figure 6.
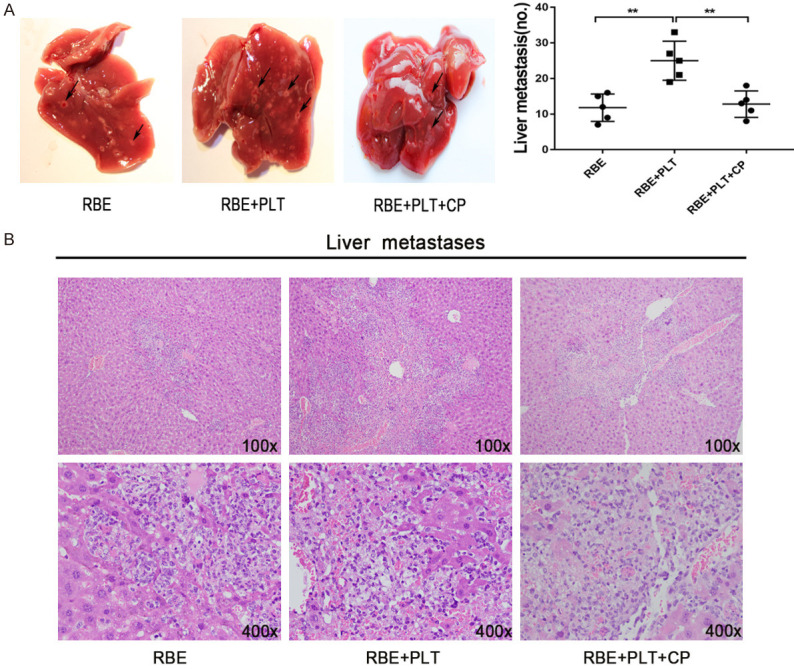
Platelet PDGF can promote the invasion and metastasis of CCA in vivo. A. Representative photographs of liver metastasis foci in mice 45 days after splenic injection of RBE cells and platelet-stimulated RBE cells in the absence and presence of CP inhibitors (n=5 per group). Quantification of the numbers of liver metastatic foci is shown on the right. B. Representative images of metastatic foci (magnification, ×100 and ×400) in haematoxylin and eosin-stained liver tissue sections from mice injected with control or platelet-stimulated RBE cells in the absence and presence of CP inhibitors. **P<0.01.
Discussion
CCA is characterized by a high recurrence rate, a high mortality rate and a low five-year survival rate, threatening the lives of human beings worldwide [1,35]. Although many advances have been made, the prognosis of this disease is still unsatisfactory [36,37]. Therefore, it is necessary to find more effective strategies to prevent CCA and to detect and treat CCA early. In this paper, we show for the first time that platelet-derived PDGF upregulates MMP2/MMP9 expression and induces EMT to promote the invasion and metastasis of cholangiocarcinoma via the p38/MAPK signalling pathway.
Increasing evidence has shown that the interaction between platelets and tumour cells promotes the metastasis of cancer cells to distant sites [19,38], but the specific mechanism of metastasis is not completely clear. In our study, we found that platelets are hyperactivated in patients with CCA. Consistently, in vitro experiments showed that RBE and HCCC-9810 cells can induce platelet activation and promote platelet aggregation. These data suggest that platelet hyperactivation may be involved in CCA progression. Further investigations revealed that platelets can secrete PDGF, which is involved in the invasion and metastasis of CCA both in vitro and in vivo. As previous studies reported, PDGF can promote the invasion and metastasis of malignant tumours [39,40], which is consistent with our results. It is well accepted that invasion and migration are the main causes of tumour deterioration. Hence, the inhibition of platelet hyperactivation or PDGF secretion may be a potential therapeutic measure for CCA.
It was found that the upregulation of MMP9 was an early event in colon cancer progression and that MMP9 activity was positively correlated with the size of colon polyps [34]. MMP2 and MMP9 are proteolytic enzymes involved in extracellular matrix degradation [41,42]. These studies highlight a critical role of MMPs in promoting tumour invasion and migration [42,43]. However, the role of MMPs in CCA is unknown. In the present study, we observed that platelet-derived PDGF significantly upregulated the expression of MMP2/MMP9 in CCA cell lines. Notably, MMP2/MMP9 expression was higher in CCA tissues than in adjacent normal tissues. At the same time, we also found that MMP2/MMP9 expression levels were significantly associated with lymph node metastasis and poor prognosis in patients with CCA following surgical resection. Of note, the survival rate of patients with high MMP2/MMP9 expression was significantly lower than that of patients with low MMP2/MMP9 expression. Therefore, these data suggest that the upregulation of MMP2/MMP9 is closely associated with CCA progression. On the other hand, we found that EMT-associated proteins, including N-cadherin, vimentin, Slug, Snail and ZEB1, were significantly increased in CCA cells after culture in platelet-containing medium. However, the expression of the epithelial marker E-cadherin was significantly lower. Interestingly, these effects were partly abrogated by the PDGF inhibitor, which suggests that platelets can induce EMT by secreting PDGF.
MAPK, a proline-directed serine/threonine kinase, is activated by a dual phosphorylation reaction to extracellular stimulation [44]. The MAPK family includes extracellular signal-regulated kinase (ERK), p38 and c-Jun NH(2)-terminal kinase (JNK) [45]. MAPKs are the main signal transduction molecules involved in regulating cell responses, including proliferation, differentiation, survival and apoptosis [46,47]. Notably, the p38MAPK pathway plays an important role in promoting the growth, survival and invasion of tumour cells [48]. In this study, we show that platelet-derived PDGF can significantly activate the p38MAPK pathway. More importantly, the inhibition of the p38MAPK pathway significantly abolished the upregulation of MMP2/MMP9 expression and EMT. These results indicate that platelet-derived PDGF contributes to CCA invasion and metastasis, at least in part, by activating the p38MAPK pathway, which is consistent with previous studies. However, whether other pathways also play a role still needs further research.
In summary, our results demonstrate that platelets play an important role in upregulating MMP2/MMP9 expression and inducing EMT through the secretion of PDGF, therefore promoting the invasion and metastasis of CCA. These data provide further evidence that platelet-tumour cell interaction supports the haematogenous dissemination of cancer cells to sites of distant metastasis. In addition, this finding provides new evidence for the subsequent exploration of the pathogenesis and treatment of CCA.
Acknowledgements
This work was supported by grants from the National Natural Science Fund of China (NO. 81725019, 81502755, 81500087) and the Scientific Research Project of PLA (AWS16J014).
Disclosure of conflict of interest
None.
References
- 1.Groot Koerkamp B, Wiggers JK, Allen PJ, Besselink MG, Blumgart LH, Busch OR, Coelen RJ, D’Angelica MI, DeMatteo RP, Gouma DJ, Kingham TP, Jarnagin WR, van Gulik TM. Recurrence rate and pattern of perihilar cholangiocarcinoma after curative intent resection. J Am Coll Surg. 2015;221:1041–1049. doi: 10.1016/j.jamcollsurg.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu YF, Liu ZL, Pan C, Yang XQ, Ning SL, Liu HD, Guo S, Yu JM, Zhang ZL. HMGB1 correlates with angiogenesis and poor prognosis of perihilar cholangiocarcinoma via elevating VEGFR2 of vessel endothelium. Oncogene. 2019;38:868–880. doi: 10.1038/s41388-018-0485-8. [DOI] [PubMed] [Google Scholar]
- 3.Tyson GL, El-Serag HB. Risk factors for cholangiocarcinoma. Hepatology. 2011;54:173–184. doi: 10.1002/hep.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology. 2003;37:961–969. doi: 10.1053/jhep.2003.50200. [DOI] [PubMed] [Google Scholar]
- 6.Lechuga S, Amin PH, Wolen AR, Ivanov AI. Adducins inhibit lung cancer cell migration through mechanisms involving regulation of cell-matrix adhesion and cadherin-11 expression. Biochim Biophys Acta Mol Cell Res. 2019;1866:395–408. doi: 10.1016/j.bbamcr.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haage A, Schneider IC. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014;28:3589–3599. doi: 10.1096/fj.13-245613. [DOI] [PubMed] [Google Scholar]
- 8.Marcus J, Bejerano-Sagie M, Patterson N, Bagchi S, Verkhusha VV, Connolly D, Goldberg GL, Golden A, Sharma VP, Condeelis J, Montagna C. Septin 9 isoforms promote tumorigenesis in mammary epithelial cells by increasing migration and ECM degradation through metalloproteinase secretion at focal adhesions. Oncogene. 2019;38:5839–5859. doi: 10.1038/s41388-019-0844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cepeda MA, Pelling JJ, Evered CL, Williams KC, Freedman Z, Stan I, Willson JA, Leong HS, Damjanovski S. Less is more: low expression of MT1-MMP is optimal to promote migration and tumourigenesis of breast cancer cells. Mol Cancer. 2016;15:65. doi: 10.1186/s12943-016-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin CY, Lee CH, Huang CC, Lee ST, Guo HR, Su SB. Impact of high glucose on metastasis of colon cancer cells. World J Gastroenterol. 2015;21:2047–2057. doi: 10.3748/wjg.v21.i7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao H, Peng C, Liang B, Shahbaz M, Liu S, Wang B, Sun Q, Niu Z, Niu W, Liu E, Wang J, Lin P, Wang J, Niu J. beta6 integrin induces the expression of metalloproteinase-3 and metalloproteinase-9 in colon cancer cells via ERK-ETS1 pathway. Cancer Lett. 2014;354:427–437. doi: 10.1016/j.canlet.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645–654. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Liu J, Xue H, Li C, Liu Q, Zhou Y, Wang T, Wang H, Qian H, Wen T. A TGF-beta-MTA1-SOX4-EZH2 signaling axis drives epithelial-mesenchymal transition in tumor metastasis. Oncogene. 2020;39:2125–2139. doi: 10.1038/s41388-019-1132-8. [DOI] [PubMed] [Google Scholar]
- 14.Zinn R, Otterbein H, Lehnert H, Ungefroren H. RAC1B: a guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells. 2019;8:1569. doi: 10.3390/cells8121569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barry S, Carlsen E, Marques P, Stiles CE, Gadaleta E, Berney DM, Roncaroli F, Chelala C, Solomou A, Herincs M, Caimari F, Grossman AB, Crnogorac-Jurcevic T, Haworth O, Gaston-Massuet C, Korbonits M. Tumor microenvironment defines the invasive phenotype of AIP-mutation-positive pituitary tumors. Oncogene. 2019;38:5381–5395. doi: 10.1038/s41388-019-0779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu J, Lv X, Lin H, Wu L, Wang R, Zhou Z, Zhang B, Wang YL, Tsang BK, Zhu C, Wang H. Ubiquitin ligase cullin 7 induces epithelial-mesenchymal transition in human choriocarcinoma cells. J Biol Chem. 2010;285:10870–10879. doi: 10.1074/jbc.M109.004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cedervall J, Zhang Y, Ringvall M, Thulin A, Moustakas A, Jahnen-Dechent W, Siegbahn A, Olsson AK. HRG regulates tumor progression, epithelial to mesenchymal transition and metastasis via platelet-induced signaling in the pre-tumorigenic microenvironment. Angiogenesis. 2013;16:889–902. doi: 10.1007/s10456-013-9363-8. [DOI] [PubMed] [Google Scholar]
- 18.Lee CH, Lin YJ, Lin CC, Yen CL, Shen CH, Chang CJ, Hsieh SY. Pretreatment platelet count early predicts extrahepatic metastasis of human hepatoma. Liver Int. 2015;35:2327–2336. doi: 10.1111/liv.12817. [DOI] [PubMed] [Google Scholar]
- 19.Zuo XX, Yang Y, Zhang Y, Zhang ZG, Wang XF, Shi YG. Platelets promote breast cancer cell MCF-7 metastasis by direct interaction: surface integrin alpha2beta1-contacting-mediated activation of Wnt-beta-catenin pathway. Cell Commun Signal. 2019;17:142. doi: 10.1186/s12964-019-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Cui W, Pei Y, Xu D. Platelets promote invasion and induce epithelial to mesenchymal transition in ovarian cancer cells by TGF-beta signaling pathway. Gynecol Oncol. 2019;153:639–650. doi: 10.1016/j.ygyno.2019.02.026. [DOI] [PubMed] [Google Scholar]
- 21.Smith BD, Kaufman MD, Leary CB, Turner BA, Wise SC, Ahn YM, Booth RJ, Caldwell TM, Ensinger CL, Hood MM, Lu WP, Patt TW, Patt WC, Rutkoski TJ, Samarakoon T, Telikepalli H, Vogeti L, Vogeti S, Yates KM, Chun L, Stewart LJ, Clare M, Flynn DL. Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2. Mol Cancer Ther. 2015;14:2023–2034. doi: 10.1158/1535-7163.MCT-14-1105. [DOI] [PubMed] [Google Scholar]
- 22.Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med. 2010;14:2037–2044. doi: 10.1111/j.1582-4934.2010.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha CW, Park YB, Jang JW, Kim M, Kim JA, Park YG. Variability of the composition of growth factors and cytokines in platelet-rich plasma from the knee with osteoarthritis. Arthroscopy. 2019;35:2878–2884. e2871. doi: 10.1016/j.arthro.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gan L, Pan S, Cui J, Bai J, Jiang P, He Y. Functional analysis of the correlation between ABCB11 gene mutation and primary intrahepatic stone. Mol Med Rep. 2019;19:195–204. doi: 10.3892/mmr.2018.9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Wang D, Ji TF, Shi L, Yu JL. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget. 2017;8:17347–17359. doi: 10.18632/oncotarget.14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robert S, Poncelet P, Lacroix R, Arnaud L, Giraudo L, Hauchard A, Sampol J, Dignat-George F. Standardization of platelet-derived microparticle counting using calibrated beads and a cytomics FC500 routine flow cytometer: a first step towards multicenter studies? J Thromb Haemost. 2009;7:190–197. doi: 10.1111/j.1538-7836.2008.03200.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu LX, Yan L, Yang W, Wu FQ, Ling Y, Chen SZ, Tang L, Tan YX, Cao D, Wu MC, Yan HX, Wang HY. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat Commun. 2014;5:5256. doi: 10.1038/ncomms6256. [DOI] [PubMed] [Google Scholar]
- 29.Wang JC, Li GY, Wang B, Han SX, Sun X, Jiang YN, Shen YW, Zhou C, Feng J, Lu SY, Liu JL, Wang MD, Liu PJ. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J Exp Clin Cancer Res. 2019;38:235. doi: 10.1186/s13046-019-1211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, Fingas C, Cristina Malerba M, Nardo G, Dall’Olmo L, Milani E, Mariotti V, Stecca T, Massani M, Spirli C, Fiorotto R, Indraccolo S, Strazzabosco M, Fabris L. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. 2019;70:700–709. doi: 10.1016/j.jhep.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Duan J, Liu X, Guo SW. Platelets drive smooth muscle metaplasia and fibrogenesis in endometriosis through epithelial-mesenchymal transition and fibroblast-to-myofibroblast transdifferentiation. Mol Cell Endocrinol. 2016;428:1–16. doi: 10.1016/j.mce.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Unnithan RVM, Hamidi A, Caja L, Saupe F, Moustakas A, Cedervall J, Olsson AK. TANK-binding kinase 1 is a mediator of platelet-induced EMT in mammary carcinoma cells. FASEB J. 2019;33:7822–7832. doi: 10.1096/fj.201801936RRR. [DOI] [PubMed] [Google Scholar]
- 33.Zhu J, Zheng Y, Zhang H, Liu Y, Sun H, Zhang P. Galectin-1 induces metastasis and epithelial-mesenchymal transition (EMT) in human ovarian cancer cells via activation of the MAPK JNK/p38 signalling pathway. Am J Transl Res. 2019;11:3862–3878. [PMC free article] [PubMed] [Google Scholar]
- 34.Radziwon-Balicka A, Santos-Martinez MJ, Corbalan JJ, O’Sullivan S, Treumann A, Gilmer JF, Radomski MW, Medina C. Mechanisms of platelet-stimulated colon cancer invasion: role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis. 2014;35:324–332. doi: 10.1093/carcin/bgt332. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe A, Araki K, Hirai K, Kubo N, Igarashi T, Tsukagoshi M, Ishii N, Hoshino K, Kuwano H, Shirabe K. A novel clinical factor, D-Dimer platelet multiplication, may predict postoperative recurrence and prognosis for patients with cholangiocarcinoma. Ann Surg Oncol. 2016;23:886–891. doi: 10.1245/s10434-016-5422-x. [DOI] [PubMed] [Google Scholar]
- 36.Blechacz B. Cholangiocarcinoma: current knowledge and new developments. Gut Liver. 2017;11:13–26. doi: 10.5009/gnl15568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15:95–111. doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plantureux L, Mege D, Crescence L, Carminita E, Robert S, Cointe S, Brouilly N, Ezzedine W, Dignat-George F, Dubois C, Panicot-Dubois L. The interaction of platelets with colorectal cancer cells inhibits tumor growth but promotes metastasis. Cancer Res. 2020;80:291–303. doi: 10.1158/0008-5472.CAN-19-1181. [DOI] [PubMed] [Google Scholar]
- 39.Hosaka K, Yang Y, Seki T, Nakamura M, Andersson P, Rouhi P, Yang X, Jensen L, Lim S, Feng N, Xue Y, Li X, Larsson O, Ohhashi T, Cao Y. Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat Commun. 2013;4:2129. doi: 10.1038/ncomms3129. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Hu C, Dong R, Huang X, Qiu H. Platelet-derived growth factor-D promotes ovarian cancer invasion by regulating matrix metalloproteinases 2 and 9. Asian Pac J Cancer Prev. 2011;12:3367–3370. [PubMed] [Google Scholar]
- 41.Jablonska-Trypuc A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. 2016;31:177–183. doi: 10.3109/14756366.2016.1161620. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Avila G, Sommer B, Mendoza-Posada DA, Ramos C, Garcia-Hernandez AA, Falfan-Valencia R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit Rev Oncol Hematol. 2019;137:57–83. doi: 10.1016/j.critrevonc.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Shay G, Lynch CC, Fingleton B. Moving targets: emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015;44-46:200–206. doi: 10.1016/j.matbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J, Kim J, Shim J, Lee S, Kim J, Lim SS, Lee KW, Lee HJ. Licorice-derived dehydroglyasperin C increases MKP-1 expression and suppresses inflammation-mediated neurodegeneration. Neurochem Int. 2013;63:732–740. doi: 10.1016/j.neuint.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. [Google Scholar]
- 46.Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. 2015;89:867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- 47.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 48.Shi D, Wu F, Mu S, Hu B, Zhong B, Gao F, Qing X, Liu J, Zhang Z, Shao Z. LncRNA AFAP1-AS1 promotes tumorigenesis and epithelial-mesenchymal transition of osteosarcoma through RhoC/ROCK1/p38MAPK/Twist1 signaling pathway. J Exp Clin Cancer Res. 2019;38:375. doi: 10.1186/s13046-019-1363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]