

Abstract

Nucleophilic ring opening reactions of epoxides with aromatic amines are in the forefront of the synthetic organic chemistry research to build new bioactive scaffolds. Here, convenient, green, and highly efficient regioselective ring opening reactions of sterically hindered (2R,3S)-3-(N-Boc-amino)-1-oxirane-4-phenylbutane with various poorly reactive aromatic amines are accomplished under microwave irradiation in nitromethane. All the reactions effectively implemented for various aromatic amines involve the reuse of nitromethane that supports its dual role as a solvent and catalyst. The corresponding new β-alcohol analogs of hydroxyethylamine (HEA) are isolated in 41–98% yields. The reactions proceed under mild conditions for a broad range of less reactive and sterically hindered aromatic amines. Proton NMR experiments suggest that the nucleophilicity of amines is influenced by nitromethane, which is substantiated by the extensive computational studies. Overall, this methodology elucidates the first-time use of nitromethane as a solvent for the ring opening reactions under microwave conditions involving an equimolar ratio of epoxide and aromatic amine without any catalyst, facile ring opening of complex epoxide by less reactive aromatic amines, low reaction time, less energy consumption, recycling of the solvent, and simple workup procedures.
1. Introduction
Epoxides are invaluable building blocks, both in synthetic organic chemistry and medicinal chemistry as they facilitate the introduction of multiple functional groups.1 Nucleophilic ring opening of epoxides in the presence of amines is one of the important pathways to develop new chemical scaffolds with versatile functional groups, including β-amino alcohols.2 The β-amino alcohols are vital intermediates in medicinal chemistry and have been widely implemented for synthesis of numerous biologically active compounds.3,4 One of the most important scaffolds of β-amino alcohol is hydroxyethylamine (HEA)5 that has been extensively explored as a synthon for the discovery of antimalarials,6,7 antifungals,8 HIV protease inhibitors,9 and anti-Alzheimer agents,10,11 etc. In the literature, quite a few routes are available for synthesis of HEA that involves the nucleophilic ring opening of epoxide with amines under conventional heating or microwave irradiation. However, these procedures suffer from several drawbacks, viz., poor yields, a high molar ratio of epoxide and aniline, failure of reaction in case of sterically hindered epoxide and less reactive aromatic amines, prolonged reaction time, and tedious workup.12 The epoxide ring opening with less reactive aromatic amines is reported in the presence of catalysts such as zinc tetrafluoroborate hydrate in solvent-free conditions,13 Sc(OSO3C12H25)3 with a chiral bipyridine ligand at room temperature in water,14 zinc(II) perchlorate hexahydrate in solvent-free conditions,15 aluminum triflate,16 chiral zinc(II) and copper(II),17 lanthanide iodo binaphtholates,18 bismuth trichloride,19 tetrathiomolybdate,20 and antimony(III) chloride in dichloromethane at room temperature.21 The obstacles associated with the ring opening of complex epoxides have been tackled with the use of heterogeneous catalysts and metal triflates under microwave irradiation.2,22,23 However, the use of moisture and air-sensitive catalysts, recovery of catalysts, and requirement of a stoichiometric amount of catalysts collectively limit the efficiency of these procedures. Of late, Du et al.24 reported a microwave-assisted ring opening reaction of a simple epoxide with aniline (3:1 equivalents) in the absence of catalysts. To date, the available methods do not include the ring opening reaction of complex epoxides with less reactive and sterically hindered aromatic amines in equimolar ratios, particularly without the use of any catalysts. Therefore, new highly efficient, catalyst-free, and simpler procedures are needed to be explored for nucleophilic ring opening reaction in organic synthesis. In this paper, we report a facile procedure for the nucleophilic ring opening of epoxide, (2R,3S)-3-(N-Boc-amino)-1-oxirane-4-phenylbutane with less nucleophilic aromatic amines in nitromethane under microwave irradiation. Steric and electronic factors affecting ring opening of epoxide with aromatic amines in various solvents have been investigated, and the results are corroborated with the considerable computational studies.
2. Results and Discussion
2.1. Synthesis and Characterization
(2R,3S)-3-(N-Boc-amino)-1-oxirane-4-phenylbutane (1) is one the popular epoxides employed to prepare the high-valued compounds, viz., HEAs. The standard procedures for ring opening of the epoxide, 1, have been optimized that led to regioselective HEA analogs identified as scaffolds potent against malaria parasites,25−27 plasmepsin inhibitors,28,29 HIV inhibitors,30,31 etc. As a part of our ongoing research interest toward the discovery of new HEA scaffolds, synthesis of these analogs based on epoxide 1 was attempted following the standard conventional synthetic routes. Initially, ring opening reaction of epoxide 1 (1.0 mmol) with p-toluidine 2a (1.0 mmol) in isopropanol (50 mL) was carried out for 12 h at 80 °C as reported in the literature;32 however, thin-layer chromatography (TLC) did not indicate any product formation.
Next, we attempted the reaction of epoxide 1 (1.0 mmol) with p-toluidine 2a (1.0 mmol) under microwave irradiation following the reported procedures (Scheme 1).24 Various solvents were employed for this reaction such as ethanol, water, and a mixture of ethanol and water with different molar ratios (Table 1, entries 13–16) in the search for a suitable green solvent. The same reaction performed in water indicated no product formation probably due the insolubility of the reactants (Table 1, entry 16). The maximum yield, 70% of the product 3a, was isolated when ethanol was used as a solvent (Table 1, entry 13). Selection of the appropriate solvent was made on the basis of the optimization of the reaction in a broad range of polar solvents as depicted in Table 1. The unsuccessful reaction in aqueous media and less fruitful reaction in ethanol encouraged us to explore the polar organic solvents.33
Scheme 1. Reaction of Substrate 1 with Substituted Anilines (2a-m) to Give Products (3a-m).

Table 1. Optimization of Reaction Conditions in a Microwave at 80 °C.
| entry no. | molar ratio of compound 1 and 2a | solvent | power (W) | time (min) | yield (%)a |
|---|---|---|---|---|---|
| 1 | 1:1 | nitromethane | 80 | 20 | 56 |
| 2 | 1:1 | nitromethane | 100 | 20 | 64 |
| 3 | 1:1 | nitromethane | 150 | 20 | 71 |
| 4 | 1:1 | nitromethane | 200 | 20 | 77 |
| 5 | 1:1 | nitromethane | 250 | 20 | 80 |
| 6 | 1:1 | nitromethane | 300 | 5 | 53 |
| 7 | 1:1 | nitromethane | 300 | 10 | 62 |
| 8 | 1:1 | nitromethane | 300 | 15 | 72 |
| 9 | 1:1 | nitromethane | 300 | 20 | 90 |
| 10 | 1:1 | nitromethane | 300 | 25 | 90 |
| 11 | 1:1 | nitromethane | 300 | 30 | 90 |
| 12 | 1:1 | dimethyl sulfoxide | 300 | 20 | |
| 13 | 1:1 | ethanol | 300 | 20 | 70 |
| 14 | 1:1 | ethanol/water (1:1) | 300 | 20 | 62 |
| 15 | 1:1 | ethanol/water (30:70) | 300 | 20 | 41 |
| 16 | 1:1 | water | 300 | 20 | |
| 17 | 1:1 | DMF | 300 | 20 | |
| 18 | 1:1 | isopropanolb | |||
| 19 | 1:3 | nitromethanec | 43 | ||
| 20 | 2:1 | nitromethane | 300 | 30 | 98 |
| 21 | 1:1 | nitromethanec | 21 | ||
| 22 | 1:1 | nitromethaned | 300 | 20 | 85 |
| 23 | 1:1 | nitromethanee | 300 | 20 | 81 |
| 24 | 1:1 | nitromethanef | 300 | 20 | 76 |
Isolated yield after recrystallization of the product using ethyl acetate and hexane.
Reaction performed under reflux conditions for 12 h.
Reaction performed at room temperature for 36 h.
Reaction performed in nitromethane (II cycle).
Reaction performed in nitromethane (III cycle).
Reaction performed in nitromethane (IV cycle).
Two factors, the use of catalysts14,17,18 and a high molar ratio of the epoxide24 or amine,34 are broadly responsible for the efficiency of the ring opening reactions. Considering the complexities of these reactions, we attempted the ring opening reactions in the presence of polar solvents (i.e., dimethylformamide, dimethyl sulfoxide, and nitromethane) without any catalysts under microwave irradiation. We noted that the reaction progressed competently in nitromethane; however, no product formation was observed in dimethyl sulfoxide and dimethylformamide (Table 1, entries 12 and 17). Reports are available to support nitromethane as a good choice of solvent for the ring opening reactions with the limitation, i.e., a high molar ratio of epoxide and nucleophile (i.e., aniline), which is one of the major drawbacks of these reported reactions.34
Therefore, nitromethane was selected for the ring opening of tert-butyl(1-(oxiran-2-yl)-2-phenylethyl)carbamate (1) with p-toluidine (2a) (1.0 mmol) under microwave conditions, and the yield of the product (3a) was significantly improved. The optimization of reaction conditions (i.e., power and time) for nitromethane is represented in Table 1. The yield of the products was dependent on the reaction time. As the reaction time increases from 5 to 20 min, the yield of the product increases from 53 to 90% (Table 1, entries 6–9). The yield of 3a was also increased with the increase in power of the microwave irradiation (Table 1, entries 1–5, 9). A maximum yield, 90%, was observed at 300 W in 20 min (Table 1, entry 9); however, no further increment in yield was noted even after a 25 or 30 min reaction period (Table 1, entries 10 and 11). Apart from reaction conditions, different molar ratios of the reactants were investigated. While increasing the molar ratio of epoxide from one to two equivalents, the yield of the product 3a was significantly increased from 90 to 98% (Table 1, entries 11 and 20). The same reaction was performed at room temperature (Table 1, entries 19 and 21) that led to the poor yield of the product, which further supported the efficiency of microwave-assisted ring opening reaction. To explore the recyclability of the solvent, reactions were performed in recovered nitromethane for three consecutive cycles that afforded 85% (II recycle), 81% (III recycle), and 76% (IV recycle) yield of 3a (Table 1, entries 22–24), indicating the reuse and recyclability of the solvent.
Next, the yields of the products (3a-m) were compared in nitromethane and ethanol as depicted in Table 2. The solvent effect showed that the yield of all the listed new analogs was much better in nitromethane (a polar aprotic solvent) over ethanol (a protic solvent) possibly due to the improved nucleophilicity35−37 of aromatic amines in nitromethane as supported by the computational studies described in the next section. Although nitromethane is not a green solvent in comparison to ethanol, it was selected as a suitable solvent considering the high yields. Notably, a high yield of the products was obtained in nitromethane while using less nucleophilic anilines; however, similar reactions performed in ethanol led to reduced yields.
Table 2. Comparison of Yields for Products 3a-m in Ethanol and Nitromethane and the Charge on the Amino Group of Aromatic Rings in Nitromethane (QNH2)b.

Recrystallized yield (1:9, ethyl acetate/hexane).
Reaction conditions: reaction performed using a 1:1 molar ratio of 1 and 2a-m in a microwave for 20 min at 300 W and 80 °C to give 3a-m.
As an important part of the study, the effects of an electron donating group (EDG) and electron withdrawing group (EWG) on aromatic amines were investigated in the presence of both ethanol and nitromethane as listed in Table 2. In nitromethane, the effect of an EDG or EWG on the rate of the reaction was clearly noted in the case of the reactants 2a and 2c. Reactant 2a possessing a methyl group at the para position of aniline increased the electron density on −NH2 and enhanced the yield of the product 3a, i.e., 90% in comparison to the reactant 2c with a −CF3 group at the para position giving the product 3c in 54% yield (Table 2, entries 1 and 3). Further, the effect of one or two fluoro groups present at different positions of aromatic amines influencing the rate of the reaction was also studied. The observed trend for the yield of the product 3b > 3h > 3f > 3j (p > m > o > op) may be attributed to an −F group exerting −I and +M effects, the anomalous behavior shown by 3f may be due to the steric factor or involvement of H bonding between the −NH2 and −F group present at the ortho position. These results were further supported by the total charge on the amino group, i.e., QNH2 values calculated by the computational studies. It was observed that the greater the positive charge on −NH2, the lower is the yield of the product (Table 2, entries 2, 7, and 9). The chemical composition of all the listed new HEA analogs (3a-m) was confirmed by standard spectroscopic methods (Figures S1–S43, Supporting Information). An extensive NMR study (i.e., NOESY and DEPT) was also performed in order to confirm the regioselectivity. In 1H NMR of 3a (CDCl3), a multiplet appeared at 7.25 ppm due to the proton of the aromatic ring, also two doublets for two protons, each one at 6.96 ppm and the other at 6.52 ppm for p-toluidine ring protons. A doublet corresponding to the hydroxyl proton appeared at 4.89 ppm. The two-methylene moieties appeared at 3.90–3.79 (m) and 3.13–2.89 (m) ppm, respectively. The protons corresponding to chiral carbons appeared as a multiplet at 2.98–2.84 ppm, and the proton of Boc-protected nitrogen appeared as a broad peak at 2.76 ppm. The methyl protons of p-toluidine were observed at 2.22 (s), which are slightly deshielded due to the ring current effect in comparison to other methyl protons of 3a, which appeared at 1.40 (s) ppm. In addition, an extensive study of 19F-NMR was performed for the fluorine-containing analogs (Figure 1).
Figure 1.

Comparison of 19F NMR spectra of 3c, 3g, and 3i possessing an electron withdrawing group (CF3) at the para, ortho, and meta positions of aniline.
The effect of the −CF3 group present in aromatic amines at the para (3c), ortho (3g), and meta (3i) positions on the rate of reaction was also investigated. In 19F NMR, the peaks for 3c, 3g, and 3i were observed at δ −61.0, −62.4, and −62.8 ppm, respectively, as shown in Figure 1.38 The most shielded peak appeared at −62.8 ppm for the −CF3 group (meta position, 3i), causing the enhanced electron density at the −NH2 group, and hence resulted in a higher yield (66%) over the ortho (3g, 41%) and para (3c, 54%) substituents.
Density functional theory (DFT) calculations did not show significant dependence of the yields on the energy, orbital, or charge characteristics of both the reactants (1 and 2) and the reaction products (3). The best dependence was observed on the sum of the partial charges of the atoms of the −NH2 of reactants 2a-m at the DFT B3LYP 6-311G(d,p) level of theory; however, the correlation coefficient (R) was only 0.704 with the exclusion of the 2e molecule.
Next, a computational analysis of the yields within the MERA model39−41 showed that the yields in both ethanol and nitromethane solutions correlated well with the total charge of the −NH2 of reactants 2a-m. Dependencies are shown in Figure 2. The values of R were calculated 0.827 for ethanol and 0.815 for the nitromethane solvent system. Compound 3m was an outlier for both dependencies, possibly owing to the steric factors of methyl groups in the ortho positions. Without the compound 3m, R equals 0.906 and 0.902, correspondingly. Compound 3m is represented by filled markers as shown in Figure 2. The calculated charges for −NH2 and the product yields are depicted in Table 2.
Figure 2.

Dependencies of the yields on the charge of the amino group (QNH2) of reactants 2a-m: (a) in ethanol (• is the 2m compound); (b) in nitromethane (• is the 2m compound).
It should be noted that the yields in ethanol and nitromethane were correlated very well (correlation coefficient, 0.974), indicating the same mechanism of the process in different solvents, and the difference in yields could be related to the solvation effects.
The yields in both solvents can be described well by eq 1 obtained as a multiple regression model
| 1 |
Δ = 25.1%, in the case of nitromethane; Δ = 0%, in the case of ethanol.
R = 0.877; standard deviation S = 11%.
Therefore, the yields in nitromethane were greater than in ethanol by 25.1 ± 4.4%. The experimental (Yield (exp.)) and calculated yields by the equation (Yield (calc.)) are shown in Figure 3a (the outlier, compound 3m, is represented by filled markers). Without compound 3m, R = 0.927; S = 9.0%.
Figure 3.

Experimental and calculated. (a) Yields; (b) free energies (• is the 3m compound); (c) yields (excluding 3m); (d) Gibbs free energies (excluding 3m).
To clarify the energy characteristics of the reaction, it was necessary to calculate the equilibrium constant Ke for each of the reaction using experimental yields by the formula represented in eq 2 given below
| 2 |
where C is the concentration of the reactants (1.9 mM).
Then, it is possible to calculate the Gibbs free energy (ΔG) for each process using van’t Hoff eq 3, i.e.
| 3 |
The values of Ke and ΔG are presented in Table 3.
Table 3. Equilibrium Constants, Gibbs Free Energies (ET – in Ethanol, NM – in Nitromethane), Reactant-Accessible Area (RAA), and Amino Group Charges in Ethanol.
| compound | Ke (NM) | Ke (ET) | ΔG, kJ/mol (NM) | ΔG, kJ/mol (ET) | QNH2, a.u. (ET) | RAA, Å2 (NM) | RAA, Å2 (ET) |
|---|---|---|---|---|---|---|---|
| 3a | 4.073 × 104 | 4.080 × 103 | –26.30 | –20.60 | 0.0990 | 32.3345 | 30.8618 |
| 3b | 2.029 × 104 | 4.077 × 103 | –24.57 | –20.60 | 0.1597 | 32.4125 | 30.9070 |
| 3c | 1.346 × 103 | 1.990 × 102 | –17.85 | –13.11 | 0.216 | 32.3588 | 30.8691 |
| 3d | 2.071 × 103 | 2.426 × 102 | –18.92 | –13.61 | 0.230 | 32.3354 | 30.8564 |
| 3e | 1.234 × 103 | 1.690 × 102 | –17.64 | –12.71 | 0.2347 | 32.3090 | 31.0428 |
| 3f | 1.532 × 103 | 4.875 × 102 | –18.17 | –15.33 | 0.2220 | 32.4282 | 30.9562 |
| 3g | 6.257 × 102 | 2.916 × 101 | –15.95 | –8.36 | 0.3207 | 30.8295 | 29.5642 |
| 3h | 2.530 × 103 | 8.734 × 102 | –19.41 | –16.78 | 0.1636 | 32.4309 | 30.9200 |
| 3i | 3.009 × 103 | 5.657 × 102 | –19.84 | –15.70 | 0.2248 | 32.6209 | 30.8165 |
| 3j | 1.157 × 103 | 2.032 × 102 | –17.48 | –13.17 | 0.2671 | 32.4326 | 30.9541 |
| 3k | 1.096 × 104 | 1.787 × 103 | –23.05 | –18.55 | 0.0761 | 32.1514 | 30.7294 |
| 3l | 2.349 × 104 | 4.022 × 103 | –24.94 | –20.56 | 0.120 | 32.3260 | 30.8547 |
| 3m | 2.661 × 103 | 4.991 × 102 | –19.54 | –15.39 | 0.0905 | 30.5904 | 29.4223 |
A comparison of the Gibbs free energies (ΔG) showed that they were lowered by 4.6 ± 1.4 kJ/mol in ethanol when compared with nitromethane, leading to difference in yields. ΔG was also dependent on the −NH2 charge as per the following eq 4.
| 4 |
Δ1 = −4.55 kJ/mol, in the case of nitromethane; Δ1 = 0 kJ/mol in the case of ethanol.
R = 0.870; S = 2.1 kJ/mol.
The formation energy of products (3a-m) was also calculated and noted in the range of −30 ± 11 kJ/mol. Only compounds 3g and 3m were out of this range probably due to the steric hindrance of ortho substituents. The formation energy of these complexes was noted as −2.2 and −0.8 kJ/mol, correspondingly, that explained their low yield.
Next, studies were carried out to investigate the possible effects of steric obstacles for 3m and 3g, and the reactant-accessible area (RAA) of the amino group was calculated within the MERA approach. These values in ethanol and in nitromethane are presented in Table 3. It should be noted that the RAA of −NH2 in nitromethane is greater by 1.45 ± 0.15 Å2 than in ethanol. Hence, ethanol increased the charge on NH2 and decreased its RAA in comparison to nitromethane. The smallest RAA was observed for reagents, 2g and 2m, containing substituents at the ortho position. The abnormal low yield of 3g may be explained by both a higher charge of −NH2 and its low RAA provided by the electronegative −CF3 substituent at the ortho position.
ΔG was also well related to the RAA and charges of −NH2 in the corresponding solvents for these reactions given below in eq 5
| 5 |
R = 0.905; S = 1.8 kJ/mol. The experimental and calculated yields and ΔG are shown in Figure 3.
As mentioned above, the yields were higher in nitromethane in comparison to ethanol, and the ring opening of 1 was believed to proceed through the nucleophilic attack of aromatic amines (2) on the less hindered site (C atom) followed by proton transfer to yield regioselective products as shown in Figure 4a. Nitromethane is enhancing the rate of reaction possibly due to weak van der Waals interactions with 2. Further, to explore the role of nitromethane in the reaction mechanism (Figure 4a), 1H NMR and computational studies were carried out independently.
Figure 4.

(a) Possible mechanism for the ring opening of the epoxide (1) with aromatic amines (2); complexes of aromatic amine 2a with (b) nitromethane and (c) ethanol.
In 1H NMR spectroscopic studies, the weak van der Waals interactions42 between nitromethane and aromatic amine were supported by the shifting of peaks of 2a to the shielded region on addition of nitromethane. As shown in Figure 5, the aromatic protons and methyl protons shifted from δ 7.00 to 6.93 ppm, 6.64 to 6.57 ppm, and 2.28 to 2.21 ppm, respectively. However, there was no remarkable shifting for −NH2 protons due to the broadening of the peak. Likewise, 1H NMR experiments were carried out for aniline on addition of ethanol, and the shifting of the peaks suggested their interactions as shown in Figure S44 (Supporting Information). Further, to support the role of nitromethane in enhanced product yield, more computational studies were carried out. Complexes of reactants 2a-m with ethanol and nitromethane were simulated using the MOPS algorithm with continual account of the solvent influence.43−45 In the case of the nitromethane complex (Figure 4b), both the oxygen atoms of nitromethane exhibited interactions with both hydrogens of the −NH2. However, the O···H distances were significantly greater and in the range 2.62–2.63 Å, which approximately corresponds to the sum of the van der Waals radii confirming the weak intermolecular interactions. Meanwhile, in the case of the complex with ethanol (Figure 4c), a typical hydrogen bond with a length of 2.11–2.12 Å was observed as the distance was considerably low in comparison to the sum of the van der Waals radii of hydrogen (ethanol) and nitrogen (−NH2). The formation of hydrogen bonds led to an increase in the positive charge on −NH2 (Table 3) resulting in lesser yields. Also, the influence of hydrogen bond formation on the charges was in good agreement as per the reported literature.46,47 These observed studies further suggested that there could be an increase in the nucleophilicity of aromatic amines in nitromethane, as amines have been reported to possess the variable nucleophilic character with respect to the solvents.35,37 To confirm this hypothesis, more computational studies were carried out, i.e., the rate constant of these reactions was calculated along with the nucleophilicity of reactants 2a-m in both nitromethane and ethanol.
Figure 5.

1H NMR spectrum. (a) 2a in CDCl3; (b) 2a with nitromethane in CDCl3.
Assuming that the process yields were obtained under kinetic conditions, we calculated the second-order rate constants of the processes using the following eq 6.
| 6 |
where 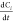 is the reaction rate, i.e., decreasing
of the initial compound concentration C1 in time t; k is the second-order
rate constant; C1 and C2 are the current concentrations of 1 and 2, respectively.
is the reaction rate, i.e., decreasing
of the initial compound concentration C1 in time t; k is the second-order
rate constant; C1 and C2 are the current concentrations of 1 and 2, respectively.
Since the concentrations of the components are equal (we denote them C), the abovementioned eq 6 is simplified to the following eq 7
| 7 |
Integration of this eq 7 leads to an eq 8 by which it is possible to calculate the second-order rate constants
| 8 |
where C0 is the initial concentration.
Since C = C0 – Cp, then
| 9 |
Dividing the numerator and denominator by C0 of eq 9, we obtain
Cp/C0 is called the extent of reaction ξ and equals Yield/100; then, finally, the abovementioned eq 9 has the form
| 10 |
The second-order rate constants calculated by eq 10 were significantly higher in nitromethane than in ethanol (Table 4). The logarithms of the rate constants are also related to the charge of the amino group; however, the reactants 2k and 2m strongly deviated from the dependence in both the solvents. As a result, the correlation coefficient of the logarithm of the rate constant with the charge of the −NH2 was only 0.699. The reasons for the deviation of the 2m have been discussed above. The reasons for the deviations of 2k in this case were difficult to explain. The best two-factor model included two characteristics: the charge of the −NH2 and the eigenvalue of the probability matrix of the association λVDW48,49 of complexes of reactants with a solvent is the following
| 11 |
Table 4. Second-Order Rate Constants, Eigenvalues of the Association Probability Matrix, and Relative Nucleophilicity of Reactants (ET – in Ethanol, NM – in Nitromethane).
| reactant | k, L·mol–1·s–1 NM | k, L·mol–1·s–1 ET | λVDW NM | λVDW ET | Nrel NM | Nrel ET |
|---|---|---|---|---|---|---|
| 2a | 3.6452 | 1.0214 | 76.24 | 124.87 | 0.3889 | –0.1165 |
| 2b | 2.5129 | 1.0210 | 66.94 | 100.29 | 0.2417 | –0.2980 |
| 2c | 0.5157 | 0.1283 | 69.40 | 99.78 | –0.0292 | –0.6281 |
| 2d | 0.6780 | 0.1505 | 81.08 | 118.67 | –0.1550 | –0.8455 |
| 2e | 0.4873 | 0.1121 | 86.05 | 132.63 | –0.3136 | –0.9671 |
| 2f | 0.5605 | 0.2564 | 76.20 | 107.34 | –0.1769 | –0.7013 |
| 2g | 0.3068 | 0.0231 | 87.11 | 119.34 | –0.7958 | –1.3765 |
| 2h | 0.7670 | 0.3868 | 68.52 | 100.81 | 0.2019 | –0.3246 |
| 2i | 0.8522 | 0.2855 | 72.22 | 103.02 | –0.1086 | –0.6986 |
| 2j | 0.4670 | 0.1305 | 81.90 | 104.11 | –0.3933 | –0.9543 |
| 2k | 1.7946 | 0.6180 | 105.32 | 168.72 | 0.2470 | –0.2940 |
| 2l | 2.7190 | 1.0127 | 91.89 | 129.95ss | 0.1919 | –0.2774 |
| 2m | 0.7910 | 0.2608 | 100.34 | 150.04 | 0.2174 | –0.2456 |
R = 0.915; S = 0.21.
The calculated and experimental values are presented in Figure 6a. The eigenvalues of the association probability matrix are presented in Table 4. It should be noted that the λVDW values in ethanol were significantly higher than in nitromethane, leading to the stabilization of the reactants in ethanol and in turn led to a decrease in their reactivity. In addition, just the reactants 2k and 2m had the maximum values of λVDW, which explains their deviations from the −NH2 charge regularity.
Figure 6.

(a) Calculated log k (calc) and experimental log k (exp) values of the second-order rate constants logarithms; (b) relationship of the relative nucleophilicities of the reagents with the yields of products.
According to Mayr and Patz,35−37 the nucleophilicity of the reactants is linearly related to the logarithm of the rate constant in accordance with eq 12
| 12 |
where N is the nucleophilicity of the nucleophilic reagent; E is the electrophilicity of an electrophilic reagent; s is a nucleophile-dependent slope parameter.
Then, in accordance with eq 11, the nucleophilicity of these reactions should also be exactly related to the charge of the −NH2 and λVDW since the values of s and E, in this case, are constant. However, exact nucleophilicity values cannot be determined since the electrophilicity of the epoxide is unknown (Mayr and Patz often took s = 1). However, it is possible to calculate the relative nucleophilicity (Nrel), in accordance with eq 11, as
| 13 |
The obtained relative nucleophilicities are different from the actual ones by the constant term E and are presented in Table 4. It should be noted that the relative nucleophilicities of the reactants in nitromethane are much higher than in ethanol, in which they all have negative values. The relationship of the relative nucleophilicities of the reactants with the product yields for all solvents is shown in Figure 6b. The correlation coefficient is 0.914.
Thus, the reaction yields are supported by the charges of the −NH2 of reactants 2a-m in the solvent. In addition, the hydroxyl-containing solvent stabilizes the reactants of these reactions and subsequently decreases the yields. Together, experimental and computational results supported the higher yield of products (3a-m) in nitromethane. These facts also supported the dual role of nitromethane, acting as a solvent and a catalyst.
3. Conclusions
In summary, we have demonstrated the synthesis of β-alcohols (i.e., HEA analogs) using highly deactivated anilines as a nucleophile for the ring opening reactions of sterically hindered epoxide. A mild and highly efficient procedure was optimized in nitromethane. Notably, the yield of new analogs was observed to be much higher in nitromethane as compared to ethanol. The low yields observed for ortho substituents may be due to the steric obstacles or H-bonding as supported by the reactant-accessible area (RAA) of the −NH2 group calculated by computational studies. Proton NMR experiments and complexes stimulated using the MOPS algorithm supported the role of nitromethane in the reaction mechanism for the epoxide ring opening. The rate constant and nucleophilicity of the reactants were much higher in nitromethane over ethanol, owing to weak van der Waals interactions. To the best of our knowledge, nitromethane was implemented for the first time as a suitable solvent as well as a catalyst for the ring opening of epoxide in microwave irradiation. Largely, this method offers various advantages such as regioselectivity, use of a 1:1 stoichiometric ratio of amine and epoxide, high yield of HEA analogs even for less nucleophilic aromatic amines and complex epoxides, low reaction time, less energy consumption, recycling of solvent, and simple workup procedures.
4. Experimental Section
4.1. General Method
The reagents and solvents were procured and used without any further purification. All reactions were performed in oven-dried glassware. Epoxide (2R,3S)-3-(N-Boc-amino)-1-oxirane-4-phenylbutane (CAS no. 98760-08-8) was purchased from GLR Innovation (New Delhi, India), and aromatic amines were purchased from AVRA Synthesis Pvt. Ltd. (Hyderabad, India). Nitromethane (AR grade) was purchased from Spectrochem (Mumbai, India), and ethanol (absolute) was purchased from Changshu Hongsheng Fine Chemical Co., Ltd. (Jiangsu, China). The reactions were carried out in a “Start Synth Microwave Synthesis Labstation” microwave for organic synthesis. The melting point of the isolated compounds was measured in a “BUCHI Labortechnik AG CH-9230”. The progress of reactions was examined by using thin-layer chromatography (TLC). Nuclear magnetic resonance (NMR) spectra were obtained using a JEOL ECX-400P NMR spectrometer. Chemical shifts were given in parts per million (ppm) downfield from an internal standard, tetramethylsilane (TMS). The molecular weight of all newly synthesized compounds was recorded at a high-resolution Biosystems Q-Star Elite time-of-flight electrospray mass spectrometer.
4.2. General Procedure
In a 50 mL round-bottom flask, aniline (2a-m) (1.9 mmol), tert-butyl(1-(oxiran-2-yl)-2-phenylethyl)carbamate (1) (1.9 mmol), and 5 mL of solvent were taken, and the contents were microwave-irradiated by controlled temperature programming by heating to 80 °C with a 2 min ramp and holding for 20 min at 80 °C and power not more than 300 W. The reaction mixture was allowed to attain room temperature, and the solvent was concentrated under reduced pressure. The crude product obtained was recrystallized from ethyl acetate and hexane in a 1:9 ratio to isolate the pure products (3a-3m). Synthesized compounds were characterized by 1H NMR, 13C NMR, 19F NMR, and HR-MS techniques. The regioselectivity of the product was confirmed by 1H NMR, 13C NMR, NOESY, DEPT-45, and DEPT-135 NMR.
4.3. Spectroscopic Data
4.3.1. tert-Butyl(3-hydroxy-1-phenyl-4-(p-tolylamino)butan-2-yl)carbamate (3a)
Rf value, 0.38 (3:1 hexane/ethyl acetate); yield, 90%; mp, 130–132 °C. 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 5H), 6.96 (d, J = 8.2 Hz, 2H), 6.52 (d, J = 8.4 Hz, 2H), 4.89, (d, J = 9.1 Hz, 1H), 3.90–3.79 (m, 2H), 3.13–2.89 (m, 2H), 2.98–2.84 (m, 2H), 2.76 (br, 1H), 2.22 (s, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.37 (s), 145.79 (s), 138.17 (s), 129.87 (s), 129.34 (s), 128.62 (s), 127.44 (s), 126.55 (s), 116.24 (s), 113.69 (s), 79.79 (s), 69.85 (s), 53.81 (s), 48.19 (s), 38.66 (s), 28.43 (s), 20.47 (s).
4.3.2. tert-Butyl(4-((4-fluorophenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3b)
Rf value, 0.33 (3:1 hexane/ethyl acetate); yield, 85%; mp, 129–131 °C. 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 5H), 6.84 (t, J = 8.7 Hz, 2H), 6.52 (dd, J = 9.0, 4.4 Hz, 2H), 4.85 (d, J = 9.1 Hz, 1H), 3.86 (dd, J = 15.9, 8.2 Hz, 1H), 3.79–3.74 (m, 1H), 3.11 (ddd, J = 18.1, 13.1, 5.0 Hz, 2H), 2.96–2.84 (m, 2H), 2.70 (br, 1H), 1.40 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −127.22 (s). 13C NMR (101 MHz, CDCl3) δ 156.42 (s), 144.38 (s), 138.02 (s), 129.27 (s), 128.67 (s), 126.63 (s), 115.89 (s), 115.67 (d, J = 22.3 Hz), 114.43 (d, J = 7.4 Hz), 79.93 (s), 69.82 (s), 53.82 (s), 48.45 (s), 38.55 (s), 28.41 (s).
4.3.3. tert-Butyl(3-hydroxy-1-phenyl-4-((4-(trifluoromethyl)phenyl)amino)butan-2-yl)carbamate (3c)
Rf value, 0.40 (3:1 hexane/ethyl acetate); yield, 54%; mp, 154–156 °C. 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.2 Hz, 2H), 7.30–7.17 (m, 5H), 6.62 (d, J = 8.4 Hz, 2H), 4.81 (d, J = 8.6 Hz, 1H), 3.86 (dt, J = 11.6, 6.8 Hz, 2H), 3.22 (ddd, J = 18.5, 13.4, 6.5 Hz, 2H), 2.92 (m, J = 20.8, 13.6, 8.1 Hz, 3H), 1.41 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −61.00 (s). 13C NMR (101 MHz, CDCl3) δ 129.17 (s), 128.72 (s), 126.73 (s), 112.83 (s), 69.76 (s), 54.15 (s), 39.44 (s), 28.37 (s).
4.3.4. tert-Butyl(3-hydroxy-1-phenyl-4-((4-(trifluoromethoxy)phenyl)amino)butan-2-yl)carbamate (3d)
Rf value, 0.40 (3:1 hexane/ethyl acetate); yield, 61%; mp, 148–150 °C. 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 6H), 6.98 (d, J = 8.6 Hz, 2H), 6.54 (d, J = 8.9 Hz, 2H), 4.82 (d, J = 9.1 Hz, 1H), 3.92–3.72 (m, 2H), 3.15 (dt, J = 13.2, 10.6 Hz, 2H), 2.99–2.83 (m, 2H), 2.58 (br, 1H), 1.40 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −58.47 (s). 13C NMR (101 MHz, CDCl3) δ 147.03 (s), 129.21 (s), 128.69 (s), 126.68 (s), 122.52 (s), 113.57 (s), 70.03 (s), 53.74 (s), 47.75 (s), 28.39 (s). ESI (HR-MS) m/z: C22H27F3N2O4 calcd, 441.1956; found, 441.2028.
4.3.5. tert-Butyl(4-((4-acetylphenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3e)
Rf value, 0.23 (3:1 hexane/ethyl acetate); yield, 53%; mp, 125–127 °C. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.5 Hz, 2H), 7.28–7.14 (m, 5H), 6.50 (d, J = 8.6 Hz, 2H), 4.97 (d, J = 9.0 Hz, 1H), 3.85 (dd, J = 40.9, 6.8 Hz, 2H), 3.31–3.26 (m, 2H), 3.20–3.15 (m, 1H), 2.96–2.83 (m, 2H), 2.46 (s, 3H), 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 196.91 (s), 156.66 (s), 152.33 (s), 137.95 (s), 130.98 (s), 129.56 (s), 129.26 (d, J = 14.9 Hz), 128.59 (d, J = 15.3 Hz), 126.65 (s), 113.80 (s), 111.67 (s), 80.05 (s), 69.77 (s), 53.74 (s), 46.58 (s), 38.35 (s), 28.40 (s), 26.15 (d, J = 8.4 Hz). ESI (HR-MS) m/z: C23H30N2O4 calcd, 399.2239; found, 399.2333.
4.3.6. tert-Butyl(4-((2-fluorophenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3f)
Rf value, 0.56 (3:1 hexane/ethyl acetate); yield, 56%; mp, 124–126 °C. 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 5H), 6.97–6.91 (m, 2H), 6.67–6.59 (m, 2H), 4.89 (d, J = 9.0 Hz, 1H), 3.86 (dd, J = 21.7, 13.7 Hz, 2H), 3.26–3.14 (m, 2H), 3.00–2.86 (m, 2H), 2.71 (br, 1H), 1.41 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −135.61 (s). 13C NMR (101 MHz, CDCl3) δ 156.22 (s), 153.03 (s), 150.69 (s), 138.06 (s), 136.44 (s), 129.33 (s), 128.68 (s), 126.63 (s), 124.66 (s), 117.46 (s), 114.68 (d, J = 18.5 Hz), 112.59 (s), 79.89 (s), 69.89 (s), 54.00 (s), 47.52 (s), 38.60 (s), 28.41 (s). ESI (HR-MS) m/z: C21H27FN2O3 calcd, 375.2039; found, 375.2089.
4.3.7. tert-Butyl(3-hydroxy-1-phenyl-4-((2-(trifluoromethyl)phenyl)amino)butan-2-yl)carbamate (3g)
Rf value, 0.56 (3:1 hexane/ethyl acetate); yield, 41%; mp, 100–102 °C. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.7 Hz, 1H), 7.33–7.19 (m, 6H), 6.75–6.65 (m, 2H), 4.90 (d, J = 8.3 Hz, 1H), 4.71 (s, 1H), 3.89–3.80 (m, 2H), 3.24 (s, 2H), 2.93 (td, J = 20.8, 7.4 Hz, 2H), 2.80 (br, 1H), 1.41 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −62.41 (s). 13C NMR (101 MHz, CDCl3) δ 156.37 (s), 145.55 (s), 137.97 (s), 133.18 (s), 129.28 (s), 128.71 (s), 126.66 (s), 116.58 (s), 112.25 (s), 79.96 (s), 69.72 (s), 54.18 (s), 47.24 (s), 38.49 (s), 28.39 (s). ESI (HR-MS) m/z: C22H27F3N2O3 calcd, 425.2007; found, 425.2050.
4.3.8. tert-Butyl(4-((3-fluorophenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3h)
Rf value, 0.56 (3:1 hexane/ethyl acetate); yield, 64%; mp, 131–133 °C. 1H NMR (400 MHz, CDCl3) δ 7.30–7.17 (m, 5H), 7.05 (dd, J = 15.0, 8.0 Hz, 1H), 6.40–6.32 (m, 2H), 6.27 (dt, J = 11.5, 2.2 Hz, 1H), 4.81 (d, J = 9.1 Hz, 1H), 3.91–3.75 (m, 2H), 3.15 (ddd, J = 18.7, 13.3, 6.6 Hz, 2H), 2.98–2.84 (m, 2H), 2.58 (br, 1H), 1.40 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −112.68 (s). 13C NMR (101 MHz, CDCl3) δ 165.40 (s), 156.51 (s), 149.91 (s), 137.95 (s), 130.36 (s), 129.22 (s), 128.70 (s), 126.67 (s), 109.13 (s), 104.40 (s), 100.03 (s), 80.03 (s), 69.95 (s), 53.71 (s), 47.46 (s), 38.42 (s), 28.39 (s).
4.3.9. tert-Butyl(3-hydroxy-1-phenyl-4-((3-(trifluoromethyl)phenyl)amino)butan-2-yl)carbamate (3i)
Rf value, 0.51 (3:1 hexane/ethyl acetate), yield, 66%; mp, 134–136 °C. 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 6H), 6.91 (d, J = 7.6 Hz, 1H), 6.76 (s, 1H), 6.72 (d, J = 8.2 Hz, 1H), 4.84 (d, J = 9.0 Hz, 1H), 3.93–3.76 (m, 2H), 3.18 (dt, J = 32.1, 9.4 Hz, 2H), 2.90 (dd, J = 28.8, 7.6 Hz, 2H), 2.68 (br, 1H), 1.41 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −62.79 (s). 13C NMR (101 MHz, CDCl3) δ 156.59 (s), 148.29 (s), 137.88 (s), 129.73 (s), 129.18 (s), 128.71 (s), 126.70 (s), 116.31 (s), 114.22 (s), 109.24 (s), 80.12 (s), 69.85 (s), 53.81 (s), 47.15 (s), 38.35 (s), 28.37 (s). ESI (HR-MS) m/z: C22H27F3N2O3 calcd, 424.1974; found, 424.1977.
4.3.10. tert-Butyl(4-((2,4-difluorophenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3j)
Rf value, 0.40 (3:1 hexane/ethyl acetate); yield, 52%; mp, 110–112 °C. 1H NMR (400 MHz, CDCl3) δ 7.44–7.08 (m, 5H), 6.74 (ddd, J = 20.5, 11.5, 4.8 Hz, 2H), 6.57 (td, J = 9.2, 5.5 Hz, 1H), 4.89 (d, J = 8.9 Hz, 1H), 3.93–3.74 (m, 2H), 3.24–3.09 (m, 2H), 3.00–2.85 (m, 2H), 2.76 (br, 1H), 1.42 (s, 9H). 19F NMR (376 MHz, CDCl3) δ −125.04 (s), −131.07 (s). 13C NMR (101 MHz, CDCl3) δ 129.30 (s), 128.69 (s), 126.66 (s), 69.68 (s), 54.14 (s), 48.19 (s), 38.10 (s), 28.02 (s). ESI (HR-MS) m/z: C21H26F2N2O3 calcd, 392.1911; found, 392.1956.
4.3.11. tert-Butyl(4-((4-(tert-butyl)phenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3k)
Rf value, 0.46 (3:1 hexane/ethyl acetate); yield, 80%; mp, 106–108 °C. 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 7H), 6.56 (d, J = 8.7 Hz, 2H), 4.91 (d, J = 9.2 Hz, 1H), 3.91–3.75 (m, 2H), 3.22–3.09 (m, 2H), 2.91 (3, J = 13.6, 10.7 Hz, 3H), 1.41 (s, 9H), 1.26 (s, 9H). 13C NMR (101 MHz, CDCl3δ 156.37 (s), 145.71 (s), 141.00 (s), 138.19 (s), 129.36 (s), 128.62 (s), 126.55 (s), 126.16 (s), 113.26 (s), 79.78 (s), 69.85 (s), 53.81 (s), 48.06 (s), 38.69 (s), 33.97 (s), 31.61 (s), 28.45 (s). ESI (HR-MS) m/z: C25H36N2O3 calcd, 412.2726; found, 412.2799.
4.3.12. tert-Butyl(3-hydroxy-4-((4-methoxyphenyl)amino)-1-phenylbutan-2-yl)carbamate (3l)
Rf value, 0.25 (3:1 hexane/ethyl acetate), yield, 86%; mp, 110–112 °C. 1H NMR (400 MHz, CDCl3) δ 7.30–7.14 (m, 5H), 6.77–6.56 (m, 4H), 4.89 (d, J = 9.3 Hz, 1H), 3.87–3.64 (m, 5H), 3.17–3.04 (m, 2H), 2.96–2.81 (m, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.34 (s), 152.69 (s), 143.06 (s), 142.13 (s), 138.16 (s), 129.34 (s), 128.56 (d, J = 11.9 Hz), 126.48 (d, J = 14.0 Hz), 120.51 (s), 114.99 (d, J = 11.8 Hz), 114.63 (s), 79.78 (s), 69.80 (s), 58.50 (s), 55.96 (s), 53.59 (s), 38.55 (s), 28.43 (s). ESI (HR-MS) m/z: C22H30N2O3 calcd, 386.2206; found, 386.2262.
4.3.13. tert-Butyl(4-((2,6-dimethylphenyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3m)
Rf value, 0.47 (3:1 hexane/ethyl acetate), yield, 64% mp, 99–101 °C. 1H NMR (400 MHz, CDCl3) δ 7.33–7.17 (m, 5H), 6.97 (d, J = 7.5 Hz, 2H), 6.83 (t, J = 7.4 Hz, 1H), 4.87 (d, J = 9.1 Hz, 1H), 3.87–3.67 (m, 2H), 3.20 (s, 1H), 3.04–3.00 (dd, 2H), 2.94–2.88 (m, 3H), 2.25 (s, 6H), 1.37 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.18 (s), 145.46 (s), 138.25 (s), 130.39 (s), 129.46 (s), 129.00 (s), 128.62 (s), 126.54 (s), 122.77 (s), 79.50 (s), 70.55 (s), 54.14 (s), 51.68 (s), 38.86 (s), 28.41 (s), 18.51 (s). ESI (HR-MS) m/z: C23H32N2O3 calcd, 384.2413; found, 384.2496.
4.4. Computation
Geometry optimization of individual molecules was carried out at the unrestricted DFT B3LYP 6-311G(d,p) level of theory. In order to simulate the solvate complexes, preliminarily, the MOPS43−45 algorithm was used, which finds the optimal geometry of a complex along all modes of translational, vibrational, and rotational movement in the combined force field MM3/MERA with a continual account of the solvent influence according to the MERA model. Energy, geometrical, surface, and charge characteristics were calculated within the MERA model.39−41
Acknowledgments
This work was supported by the Department of Science and Technology (DST/TDT/DDP-14/2018) and by Act 211 Government of the Russian Federation, contract 02.A03.21.0011 and by the Ministry of Science and Higher Education of Russia (grant FENU-2020-0019). We are grateful to the Department of Chemistry and University Science Instrumentation Center, University of Delhi for its instrumentation facility. S.K. and C.U. are grateful to CSIR, New Delhi for providing Junior Research Fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01760.
(Figures S1–S38) NMR spectra of 3a–m; (Figure S39–43) ESI (HR-MS) spectra of 3c, 3e, 3f, 3h, and 3i; (Figure S44) 1H NMR spectra of aniline in CDCl3 and aniline with ethanol in CDCl3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lin S.; Horsman G. P.; Shen B. Characterization of the epoxide hydrolase NcsF2 from the neocarzinostatin biosynthetic gene cluster. Org. Lett. 2010, 12, 3816–3819. 10.1021/ol101473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Asefa T. Epoxide ring-opening reactions with mesoporous silica-supported Fe (III) catalysts. ACS Catal. 2011, 1, 502–510. 10.1021/cs1001256. [DOI] [Google Scholar]
- Nakhate A. V.; Doke S. M.; Yadav G. D. Template Assisted Synthesis of Nanocrystalline Sulfated Titania: Active and Robust Catalyst for Regioselective Ring Opening of Epoxide with Aniline and Kinetic Modeling. Ind. Eng. Chem. Res. 2016, 55, 10829–10838. 10.1021/acs.iecr.6b02619. [DOI] [Google Scholar]
- Corey E. J.; Zhang F.-Y. re- and si-Face-Selective Nitroaldol Reactions Catalyzed by a Rigid Chiral Quaternary Ammonium Salt: A Highly Stereoselective Synthesis of the HIV Protease Inhibitor Amprenavir (Vertex 478). Angew. Chem., Int. Ed. 1999, 38, 1931–1934. . [DOI] [PubMed] [Google Scholar]
- Singh S.; Rajendran V.; He J.; Singh A. K.; Achieng A. O.; Vandana; Pant A.; Nasamu A. S.; Pandit M.; Singh J.; Quadiri A.; Gupta N.; Poonam; Ghosh P. C.; Singh B. K.; Narayanan L.; Kempaiah P.; Chandra R.; Dunn B. M.; Pandey K. C.; Goldberg D. E.; Singh A. P.; Rathi B. Fast-Acting Small Molecules Targeting Malarial Aspartyl Proteases, Plasmepsins, Inhibit Malaria Infection at Multiple Life Stages. ACS Infect. Dis. 2019, 5, 184–198. 10.1021/acsinfecdis.8b00197. [DOI] [PubMed] [Google Scholar]
- Zogota R.; Kinena L.; Withers-Martinez C.; Blackman M. J.; Bobrovs R.; Pantelejevs T.; Kanepe-Lapsa I.; Ozola V.; Jaudzems K.; Suna E.; Jirgensons A. Peptidomimetic plasmepsin inhibitors with potent anti-malarial activity and selectivity against cathepsin D. Eur. J. Org. Chem. 2019, 163, 344–352. 10.1016/j.ejmech.2018.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Sharma N.; Upadhyay C.; Kumar S.; Rathi B.; Poonam Small Molecules Effective Against Liver and Blood Stage Malarial Infection. Curr. Top. Med. Chem. 2018, 18, 2008–2021. 10.2174/1568026619666181129143623. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Obrai S.; Kaur A.; Hundal M. S.; Meehnian H.; Jana A. K. Synthesis, crystal structure investigation, DFT analyses and antimicrobial studies of silver(i) complexes with N,N,N′,N″-tetrakis(2-hydroxyethyl/propyl) ethylenediamine and tris(2-hydroxyethyl)amine. New J. Chem. 2014, 38, 1186–1198. 10.1039/c3nj00729d. [DOI] [Google Scholar]
- Dohnálek J.; Hašek J.; Dušková J.; Petroková H.; Hradilek M.; Souček M.; Konvalinka J.; Brynda J.; Sedláček J.; Fábry M. Hydroxyethylamine Isostere of an HIV-1 Protease Inhibitor Prefers Its Amine to the Hydroxy Group in Binding to Catalytic Aspartates. A Synchrotron Study of HIV-1 Protease in Complex with a Peptidomimetic Inhibitor. J. Med. Chem. 2002, 45, 1432–1438. 10.1021/jm010979e. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Braun H. A.; Narlawar R. Drug Development and PET-Diagnostics for Alzheimer’s Disease. Curr. Med. Chem. 2005, 12, 1677–1695. 10.2174/0929867054367130. [DOI] [PubMed] [Google Scholar]
- Orhan I. E.; Senol F. S. Designing Multi-Targeted Therapeutics for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 1889–1896. 10.2174/1568026616666160204121832. [DOI] [PubMed] [Google Scholar]
- Babič A.; Pečar S. Synthesis of Protected Hydroxyethylamine Transition-State Analogs of N-Ac-Muramyl-L-alanine. Synth. Commun. 2008, 38, 3052–3061. 10.1080/00397910802044280. [DOI] [Google Scholar]
- Pujala B.; Rana S.; Chakraborti A. K. Zinc Tetrafluoroborate Hydrate as a Mild Catalyst for Epoxide Ring Opening with Amines: Scope and Limitations of Metal Tetrafluoroborates and Applications in the Synthesis of Antihypertensive Drugs (RS)/(R)/(S)-Metoprolols. J. Org. Chem. 2011, 76, 8768–8780. 10.1021/jo201473f. [DOI] [PubMed] [Google Scholar]
- Azoulay S.; Manabe K.; Kobayashi S. Catalytic Asymmetric Ring Opening of meso-Epoxides with Aromatic Amines in Water. Org. Lett. 2005, 7, 4593–4595. 10.1021/ol051546z. [DOI] [PubMed] [Google Scholar]
- Shivani; Pujala B.; Chakraborti A. K. Zinc(II) Perchlorate Hexahydrate Catalyzed Opening of Epoxide Ring by Amines: Applications to Synthesis of (RS)/(R)-Propranolols and (RS)/(R)/(S)-Naftopidils. J. Org. Chem. 2007, 72, 3713–3722. 10.1021/jo062674j. [DOI] [PubMed] [Google Scholar]
- Williams D. B. G.; Cullen A. Al(OTf)3-Mediated Epoxide Ring-Opening Reactions: Toward Piperazine-Derived Physiologically Active Products. J. Org. Chem. 2009, 74, 9509–9512. 10.1021/jo9020437. [DOI] [PubMed] [Google Scholar]
- Kokubo M.; Naito T.; Kobayashi S. Chiral zinc(II) and copper(II)-catalyzed asymmetric ring-opening reactions of meso-epoxides with aniline and indole derivatives. Tetrahedron 2010, 66, 1111–1118. 10.1016/j.tet.2009.11.018. [DOI] [Google Scholar]
- Carrée F.; Gil R.; Collin J. Enantioselective Ring Opening of meso-Epoxides by Aromatic Amines Catalyzed by Lanthanide Iodo Binaphtholates. Org. Lett. 2005, 7, 1023–1026. 10.1021/ol0475360. [DOI] [PubMed] [Google Scholar]
- Ollevier T.; Lavie-Compin G. An efficient method for the ring opening of epoxides with aromatic amines catalyzed by bismuth trichloride. Tetrahedron Lett. 2002, 43, 7891–7893. 10.1016/S0040-4039(02)01896-8. [DOI] [Google Scholar]
- Devan N.; Sridhar P. R.; Prabhu K. R.; Chandrasekaran S. Tetrathiomolybdate Assisted Epoxide Ring Opening with Masked Thiolates and Selenoates: Multistep Reactions in One Pot. J. Org. Chem. 2002, 67, 9417–9420. 10.1021/jo0263418. [DOI] [PubMed] [Google Scholar]
- Singh M. C.; Peddinti R. K. Antimony(III) chloride-catalyzed ring opening of epoxides with anilines. Tetrahedron Lett. 2007, 48, 7354–7357. 10.1016/j.tetlet.2007.08.023. [DOI] [Google Scholar]
- Moghadam M.; Tangestaninejad S.; Mirkhani V.; Mohammadpoor-Baltork I.; Gorjipoor S.; Yazdani P. Highly Efficient Aminolysis of Epoxides Catalyzed by Reusable Zirconyl Triflate, ZrO(OTf)2. Synth. Commun. 2009, 39, 552–561. 10.1080/00397910802406729. [DOI] [Google Scholar]
- Beaton M.; Gani D. Synthesis of 6-amino-3,5-deoxyinositol 1-phosphates via (1R,2R,4R,6S)-1,6-Epoxy-2,4-bis-benzyloxycyclohexane aminolysis in aqueous ytterbium triflate solution. Tetrahedron Lett. 1998, 39, 8549–8552. 10.1016/S0040-4039(98)01909-1. [DOI] [Google Scholar]
- Du Z.; Zhang W.; Zhang Y.; Wei X. Microwave-Enhanced Catalyst-Free Aminolysis of Epoxides with Anilines in Aqueous Phase: Efficient Synthesis of β-amino Secondary Alcohols. J. Chem. Res. 2011, 35, 726–728. 10.3184/174751911X13237015498335. [DOI] [Google Scholar]
- Singh A. K.; Rathore S.; Tang Y.; Goldfarb N. E.; Dunn B. M.; Rajendran V.; Ghosh P. C.; Singh N.; Latha N.; Singh B. K.; Rawat M.; Rathi B. Hydroxyethylamine based phthalimides as new class of plasmepsin hits: design, synthesis and antimalarial evaluation. PLoS One 2015, 10, e0139347 10.1371/journal.pone.0139347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Rajendran V.; Pant A.; Ghosh P. C.; Singh N.; Latha N.; Garg S.; Pandey K. C.; Singh B. K.; Rathi B. Design, synthesis and biological evaluation of functionalized phthalimides: A new class of antimalarials and inhibitors of falcipain-2, a major hemoglobinase of malaria parasite. Bioorg. Med. Chem. 2015, 23, 1817–1827. 10.1016/j.bmc.2015.02.029. [DOI] [PubMed] [Google Scholar]
- Rathi B.; Singh A. K.; Kishan R.; Singh N.; Latha N.; Srinivasan S.; Pandey K. C.; Tiwari H. K.; Singh B. K. Functionalized hydroxyethylamine based peptide nanostructures as potential inhibitors of falcipain-3, an essential proteases of Plasmodium falciparum. Bioorg. Med. Chem. 2013, 21, 5503–5509. 10.1016/j.bmc.2013.05.052. [DOI] [PubMed] [Google Scholar]
- Velcicky J.; Mathison C. J. N.; Nikulin V.; Pflieger D.; Epple R.; Azimioara M.; Cow C.; Michellys P.-Y.; Rigollier P.; Beisner D. R.; Bodendorf U.; Guerini D.; Liu B.; Wen B.; Zaharevitz S.; Brandl T. Discovery of Orally Active Hydroxyethylamine Based SPPL2a Inhibitors. ACS Med. Chem. Lett. 2019, 10, 887–892. 10.1021/acsmedchemlett.9b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanlokun J. O.; Olotu A. F.; David O. M.; Idowu T. O.; Soliman E. M.; Olorunsogo O. O. A novel compound purified from Alstonia boonei inhibits Plasmodium falciparum lactate dehydrogenase and plasmepsin II. J. Biomol. Struct. Dyn. 2018, 37, 2193–2200. 10.1080/07391102.2018.1483840. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Williams J. N.; Kovela S.; Takayama J.; Simpson H. M.; Walters D. E.; Hattori S.-i.; Aoki M.; Mitsuya H. Potent HIV-1 protease inhibitors incorporating squaramide-derived P2 ligands: Design, synthesis, and biological evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 2565–2570. 10.1016/j.bmcl.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S. A.; Adhikari N.; Bhargava S.; Jha T.; Gayen S. Structural exploration of hydroxyethylamines as HIV-1 protease inhibitors: new features identified. SAR QSAR Environ. Res. 2018, 29, 385–408. 10.1080/1062936X.2018.1447511. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Rajendran V.; Singh S.; Kumar P.; Kumar Y.; Singh A.; Miller W.; Potemkin V.; Poonam; Grishina M.; Gupta N.; Kempaiah P.; Durvasula R.; Singh B. K.; Dunn B. M.; Rathi B. Antiplasmodial activity of hydroxyethylamine analogs: Synthesis, biological activity and structure activity relationship of plasmepsin inhibitors. Bioorg. Med. Chem. 2018, 26, 3837–3844. 10.1016/j.bmc.2018.06.037. [DOI] [PubMed] [Google Scholar]
- Naeimi H.; Karshenas A. Highly regioselective conversion of epoxides to β-hydroxy nitriles using metal(II) Schiff base complexes as new catalysts under mild conditions. Polyhedron 2013, 49, 234–238. 10.1016/j.poly.2012.10.019. [DOI] [Google Scholar]
- Mohseni S.; Bakavoli M.; Morsali A. Theoretical and Experimental Studies on the Regioselectivity of Epoxide Ring Opening by Nucleophiles in Nitromethane without any Catalyst: Nucleophilic-Chain Attack Mechanism. Prog. React. Kinet. Mech. 2014, 39, 89–102. 10.3184/97809059274714X13874723178403. [DOI] [Google Scholar]
- Mayr H.; Patz M. Scales of Nucleophilicity and Electrophilicity: A System for Ordering Polar Organic and Organometallic Reactions. Angew. Chem., Int. Ed. 1994, 33, 938–957. 10.1002/anie.199409381. [DOI] [Google Scholar]
- Phan T. B.; Breugst M.; Mayr H. Towards a General Scale of Nucleophilicity?. Angew. Chem., Int. Ed. 2006, 45, 3869–3874. 10.1002/anie.200600542. [DOI] [PubMed] [Google Scholar]
- Mayr H.; Bug T.; Gotta M. F.; Hering N.; Irrgang B.; Janker B.; Kempf B.; Loos R.; Ofial A. R.; Remennikov G.; Schimmel H. Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles. J. Am. Chem. Soc. 2001, 123, 9500–9512. 10.1021/ja010890y. [DOI] [PubMed] [Google Scholar]
- Sloop J. C. 19-Fluorine nuclear magnetic resonance chemical shift variability in trifluoroacetyl species. Rep. Org. Chem. 2013, 3, 1–12. 10.2147/ROC.538495. [DOI] [Google Scholar]
- Potemkin V.; Potemkin A.; Grishina M. Internet Resources for Drug Discovery and Design. Curr. Top. Med. Chem. 2018, 18, 1955–1975. 10.2174/1568026619666181129142127. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Wu L.; Sharma N.; Ayushee; Kaushik K.; Grishina M.; Chhikara B. S.; Poonam; Potemkin V.; Rathi B. Theoretical and experimental studies of an oseltamivir–triazole-based thermoresponsive organogel. RSC Adv. 2019, 9, 21031–21041. 10.1039/C9RA02463H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poonam; Kumari P.; Grishina M.; Potemkin V.; Verma A.; Rathi B. Oxygen mediated highly efficient cobalt(ii) porphyrin-catalyzed reduction of functional chromones: experimental and computational studies. New J. Chem. 2019, 43, 5228–5238. 10.1039/C9NJ00266A. [DOI] [Google Scholar]
- Heaton N. J.; Bello P.; Herradón B.; del Campo A.; Jiménez-Barbero J.; Heaton N. J.; Bello P.; Herradón B.; del Campo A.; Jiménez-Barbero J. NMR Study of Intramolecular Interactions between Aromatic Groups: Van der Waals, Charge-Transfer, or Quadrupolar Interactions?. J. Am. Chem. Soc. 1998, 120, 12371–12384. 10.1021/ja985529z. [DOI] [Google Scholar]
- Manakov A. Y.; Likhacheva A. Y.; Potemkin V. A.; Ogienko A. G.; Kurnosov A. V.; Ancharov A. I. Compressibility of Gas Hydrates. ChemPhysChem 2011, 12, 2476–2484. 10.1002/cphc.201100126. [DOI] [PubMed] [Google Scholar]
- Avdin V. V.; Lymar A. A.; Batist A. V.; Nikitin E. A.; Belkanova M. Y.; Potemkin V. A. Structure formation in heavy metal oxyhydrates at low rates of gel formation. J. Struct. Chem. 2007, 48, 747–752. 10.1007/s10947-007-0114-9. [DOI] [Google Scholar]
- Sukharev Y. I.; Potemkin V. A.; Markov B. A. Autowave processes of forming gels as a cause of the coloring of oxyhydrate gels (the chromatic effect) of some rare earth metals (yttrium, gadolinium). Colloids Surf. 2001, 194, 75–84. 10.1016/S0927-7757(01)00757-9. [DOI] [Google Scholar]
- Kr̵íž J.; Dybal J.; Tuzar Z.; Kadlec P. Hydration Modes of an Amphiphilic Molecule: NMR, FTIR, and Theoretical Study of the Interactions in the Water–Lutidine System. J. Phys. Chem. B 2009, 113, 11950–11958. 10.1021/jp905436x. [DOI] [PubMed] [Google Scholar]
- Dračínskž M.; Bour̵ P. Computational Analysis of Solvent Effects in NMR Spectroscopy. J. Chem. Theory Comput. 2010, 6, 288–299. 10.1021/ct900498b. [DOI] [PubMed] [Google Scholar]
- Potemkin V. A.; Bartashevich E. V.; Belik A. V. A New approach to predicting the thermodynamic parameters of substances from molecular characteristics. Russ. J. Phys. Chem. A 1996, 70, 411–415. [Google Scholar]
- Potemkin V. A.; Pogrebnoy A. A.; Grishina M. A. Technique for Energy Decomposition in the Study of “Receptor-Ligand” Complexes. J. Chem. Inf. Model. 2009, 49, 1389–1406. 10.1021/ci800405n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.