

Summary
PARP inhibitors (PARPi) cause synthetic lethality in BRCA-deficient tumors. Whether specific vulnerabilities to PARPi exist beyond BRCA mutations and related defects in homology-directed repair (HDR) is not well understood. Here, we identify the ubiquitin E3 ligase TRIP12 as negative regulator of PARPi sensitivity. We show that TRIP12 controls steady-state PARP1 levels and limits PARPi-induced cytotoxic PARP1 trapping. Upon loss of TRIP12, elevated PARPi-induced PARP1 trapping causes increased DNA replication stress, DNA damage, cell cycle arrest, and cell death. Mechanistically, we demonstrate that TRIP12 binds PARP1 via a central PAR-binding WWE domain and, using its carboxy-terminal HECT domain, catalyzes polyubiquitylation of PARP1, triggering proteasomal degradation and preventing supra-physiological PARP1 accumulation. Further, in cohorts of breast and ovarian cancer patients, PARP1 abundance is negatively correlated with TRIP12 expression. We thus propose TRIP12 as regulator of PARP1 stability and PARPi-induced PARP trapping, with potential implications for PARPi sensitivity and resistance.
Keywords: genome instability, endogenous DNA lesions, replication stress, cancer, BRCA mutations, PARP inhibitors, PAR-targeted protein ubiquitylation, HECT-type ubiquitin ligases, synthetic lethality, personalized cancer therapy
Graphical Abstract
Highlights
-
•
TRIP12 is a PAR-targeted ubiquitin ligase (PTUbL)
-
•
The HECT and WWE domains of TRIP12 cooperate to mediate PARP1 turnover
-
•
TRIP12 loss sensitizes cancer cells to PARP inhibitors in a PARP1-dependent manner
-
•
TRIP12 status may help to estimate how cancer cells respond to PARP inhibition
Gatti et al. demonstrate that the enzyme TRIP12 is a PAR-targeted ubiquitin ligase (PTUbL), which regulates PARP1 turnover. TRIP12 and PARP1 are inversely correlated in human cancers, and loss of TRIP12 leads to increased PARP inhibitor-induced PARP trapping, replication-associated DNA damage, and amplifies PARP inhibitor-induced cancer cell death.
Introduction
Poly(ADP-ribose) polymerase 1 (PARP1) is an abundant chromatin-associated enzyme, whose activity is strongly induced in response to genotoxic stress (Gibson and Kraus, 2012; Ray Chaudhuri and Nussenzweig, 2017; Teloni and Altmeyer, 2016). PARP1 has affinity for DNA single- and double-strand breaks (DSBs), for stalled and reversed replication forks, and for a range of atypical DNA secondary structures (Alemasova and Lavrik, 2019; Neelsen and Lopes, 2015). Upon DNA binding and allosteric activation, PARP1 uses the energy carrier nicotinamide adenine dinucleotide (NAD+) as substrate to generate poly(ADP-ribose) (PAR) chains (Gupte et al., 2017; Eustermann et al., 2015; Hottiger, 2015). PAR formation is important for the timely recruitment of PAR-binding repair factors and for rearranging the local chromatin structure in response to DNA damage and it induces liquid-liquid phase separation of intrinsically disordered PAR-binding proteins at chromosome break sites (Altmeyer et al., 2015; Dantuma and van Attikum, 2016; Pellegrino and Altmeyer, 2016). PAR signaling thus promotes DNA repair reactions and helps maintain genome stability. When genotoxic stress is too severe, however, sustained PARylation can deplete the cellular NAD+ pool and cause a PAR-dependent type of cell death known as PARthanatos (Tang et al., 2019; Wang et al., 2016; Yu et al., 2002).
While PARP inhibitors (PARPi) protect cells from PARP1-mediated NAD+ depletion, their clinical use is primarily associated with causing synthetic lethality in BRCA1/2-deficient tumors (Lord and Ashworth, 2017; O’Connor, 2015). The tumor suppressors BRCA1 and BRCA2 are critical components of the homology-directed repair (HDR) pathway, which promotes error-free repair of DNA double-strand breaks (DSBs) and emerges as an important contributor to replication fork stability (Chen et al., 2018). In a fraction of heritable and sporadic breast and ovarian cancers BRCA1/2 function is lost, and this situation generates a cancer-specific vulnerability to PARPi (Lord and Ashworth, 2017; O’Connor, 2015). Based on the promising pre-clinical and clinical results in BRCA-deficient contexts, current efforts aim at identifying signatures of HDR dysfunction, so that PARPi therapies could be used in HDR-defective settings and potentially beyond BRCA mutations (Davies et al., 2017; Lord and Ashworth, 2016).
Vulnerabilities to PARPi may, however, also exist in HDR-proficient cancer cells. In fact, by blocking access of NAD+ to the catalytic site, PARPi lock PARP1 in an inactive cytotoxic conformation, a situation generally referred to as PARP trapping (Murai et al., 2012; Pommier et al., 2016). Consistent with PARP trapping being an important cause of PARPi toxicity in BRCA-proficient cells, loss of PARP1 rescues them from PARPi-induced genotoxic stress and cell death (Michelena et al., 2018; Murai et al., 2012). We therefore reasoned that when PARPi are applied in conditions of BRCA1/2 and HDR proficiency, it is neither the impaired PARP1 function nor the inability to produce PAR, but rather the inactivated trapped PARP1 itself, which becomes detrimental for cell survival. However, in BRCA-deficient conditions, the response to PARPi is not uniform, and desired responses are only observed in a fraction of patients with germline BRCA1/2 mutations (Faraoni and Graziani, 2018). For instance, the overall response rate in BRCA-mutated ovarian cancer is <50% in multiple phase II clinical trials (Boussios et al., 2019), warranting identification of additional biomarkers for PARPi responses. As regulators of PARP1 may determine PARPi-induced PARP1 trapping and PARPi efficiency, uncovering such regulators and characterizing their molecular and cellular functions might help to better stratify cancer patients for PARPi therapy and pave the way for patients without a BRCAness signature to possibly benefit from PARPi.
Results
TRIP12 Counteracts PARP1 Abundance
To identify regulators of PARP1 and PARPi sensitivity, we focused on putative PAR-binding components of the ubiquitin-proteasome system. In human cells, only six ubiquitin E3 enzymes are known to combine within their structure a high-affinity PAR-binding domain of the WWE type, which recognizes iso-ADP-ribose, with a ubiquitin E3 ligase domain of either the homologous to the E6-AP carboxyl terminus (HECT) or really interesting new gene (RING) type (Figure 1A). Sequence alignment of the WWE domains confirms the presence of the eponymous tryptophan and glutamate residues in all six proteins (Figure 1B). Using a previously established high-content cell imaging system to assess multiple parameters of PARPi sensitivity and resistance (Michelena et al., 2018), we followed an RNAi approach with two independent small interfering RNAs (siRNAs) against each target, measuring nuclear PARP1 intensities in transgenic HeLa Kyoto cells stably expressing EGFP-tagged PARP1 from its natural promoter (Aleksandrov et al., 2018) as readout. This revealed elevated PARP1 levels particularly upon downregulation of the HECT domain ubiquitin E3 ligase TRIP12 (Figure 1C). TRIP12, a primarily nuclear protein of around 200 kDa, which is also known as ULF, is altered in around 4% of cancer patients, with a somatic mutation frequency of 2.8% according to The Cancer Genome Atlas (TCGA) PanCancer Atlas data on cBioPortal (Cerami et al., 2012; Gao et al., 2013). In support of our initial observation, individual validation experiments by both high-content microscopy and western blot confirmed elevated PARP1 abundance upon TRIP12 depletion, while steady-state PAR levels were only very mildly increased (Figures 1D and S1A). The cell cycle distribution remained largely unaffected under these conditions (Figure S1B). Similar results were obtained in human bone osteosarcoma epithelial cells (U-2 OS) cells (Figures S1C and S1D) and with three independent siRNAs against TRIP12 (Figure S1E). Elevated PARP1 levels upon TRIP12 depletion were also observed in hTERT-immortalized RPE-1 cells (Figure S1F), and in the triple-negative breast cancer cell line HCC1143 (Figure S1G). When the TRIP12 knockdown was enhanced through two consecutive rounds of siRNA transfection, the effect on PARP1 abundance was even more pronounced (Figure S1H). PARP1 mRNA levels, on the other hand, were only very mildly affected by TRIP12 loss (Figure S1I), suggesting that TRIP12 regulates PARP1 mainly at the protein rather than the gene expression level. In line with our knockdown experiments, overexpression of GFP-TRIP12 reduced ectopically expressed PARP1 in PARP1 knockout (KO) cells (Figures 1E and S1J), and GFP-TRIP12 expression was inversely correlated with nuclear abundance of endogenous PARP1 (Figures 1F and S1K). Collectively, these results reveal that nuclear PARP1 levels are negatively regulated by the ubiquitin ligase TRIP12.
Figure 1.

The HECT-Type Ubiquitin Ligase TRIP12 Controls PARP1 Abundance
(A) Schematic representation of WWE domain-containing ubiquitin E3 ligases. HECT, homologous to the E6-AP carboxyl terminus; RING, really interesting new gene.
(B) Multiple sequence alignment of WWE domains present in ubiquitin E3 ligases. While RNF146, HUWE1, and TRIP12 each contain a single WWE domain, DELTEX1, DELTEX2, and DELTEX4 each contain a tandem WWE domain. The tryptophan and glutamate residues typical of the WWE domain are marked in red.
(C) HeLa Kyoto PARP1-LAP cells expressing GFP-PARP1 from its natural promoter were transfected with negative control siRNA or with siRNAs against WWE-containing ubiquitin E3 ligases. Nuclear GFP-PARP1 levels were analyzed 72 h after siRNA transfection by high-content microscopy. Pooled results from two independent siRNAs against each target protein are shown. Mean (solid line) and standard deviation (SD) from the mean (dashed lines) are indicated. Representative single cell images are shown below. Scale bar, 10 μm.
(D) Western blot analysis of GFP-PARP1 levels in HeLa Kyoto PARP1-LAP cells transfected with negative control siRNA or siRNA against TRIP12. GFP-PARP1 and poly(ADP-ribose) band intensities were quantified in Fiji and the fold change is indicated below.
(E) Naive U-2 OS cells (first lane) and U-2 OS PARP1 knockout (KO) cells transfected with Myc-PARP1 and increasing amounts of GFP-TRIP12 (0, 1, 2, and 4 μg of plasmid, respectively) were analyzed for PARP1 levels by western blot.
(F) U-2 OS transfected with GFP-TRIP12 were analyzed by high-content microscopy for GFP-TRIP12 and endogenous PARP1 levels. Nuclear GFP-TRIP12 and PARP1 intensities per cell are plotted, color-coded by GFP-TRIP12 expression. Representative single cell images are shown to the right. Scale bar, 10 μm.
See also Figure S1.
TRIP12 Limits PARylation and Affects Outcomes of PAR Signaling
When we used short-term chemical inhibition of the PAR-degrading enzyme PARG to detect endogenous PAR formation as performed previously (Hanzlikova et al., 2018), we observed a moderate increase in PAR levels after TRIP12 depletion (Figures S2A and S2B). Moreover, when we induced DNA damage, PARP1 accumulation at sites of laser micro irradiation was more sustained (Figure 2A), and PAR formation at the laser stripe was more pronounced in absence of TRIP12 (Figure 2B). Consistent with elevated PAR formation at DNA break sites, recruitment of the PAR-binding protein XRCC1 to laser damage was enhanced upon TRIP12 depletion (Figure 2C). Similarly, XRCC1 chromatin association in response to H2O2-induced oxidative DNA damage was increased in TRIP12-depleted cells (Figure S2C). On the other hand, MDC1 recruitment, which is dependent on ataxia telangiectasia-mutated (ATM) activation and γH2AX generation rather than PAR, was not facilitated by TRIP12 depletion (Figure 2D). Collectively, these findings indicate that TRIP12 constrains PAR formation and PAR-dependent protein recruitment to sites of DNA damage.
Figure 2.

TRIP12 Limits PARylation and Affects Outcomes of PAR Signaling
(A) U-2 OS cells were transfected with the indicated siRNAs and localized DNA damage was induced by laser micro-irradiation. Cells were allowed to recover for the indicated time periods and stained for γH2AX as DNA damage marker and PARP1. Representative single cell images are shown and quantification of PARP1 recruitment to sites of DNA damage is depicted below as mean ± SD. ∗∗∗, p < 0.001; ∗∗, p < 0.01.
(B) U-2 OS cells were treated as in (A) and stained for γH2AX and PAR. Representative single cell images are shown and quantification of PAR formation at sites of DNA damage is depicted below as mean ± SD. ∗∗∗, p < 0.001; ∗∗, p < 0.01.
(C) U-2 OS cells were treated as in (A) and stained for γH2AX and XRCC1. Representative single cell images are shown and quantification of XRCC1 recruitment to sites of DNA damage is depicted below as mean ± SD. ∗∗, p < 0.01.
(D) U-2 OS cells were treated as in (A) and stained for γH2AX and MDC1. Representative single cell images are shown and quantification of MDC1 recruitment to sites of DNA damage is depicted below as mean ± SD.
See also Figure S2.
As hyper-activation of PARP1 under severe conditions of genotoxic stress can lead to PARthanatos, we aimed to test whether loss of TRIP12 would sensitize cells to PARP1-dependent cell death. Using AIF (apoptosis-inducing factor) translocation from the mitochondria to the nucleus as hallmark of PARthanatos upon exposure to oxidative damage in conjunction with inhibition of the NAD+ salvage pathway, we observed elevated nuclear AIF localization and condensed nuclei in TRIP12-depleted cells (Figures S2D and S2E). Inhibition of PARP activity under these conditions rescued the observed AIF translocation, demonstrating that AIF release was indeed PARylation dependent (Figure S2D). By limiting the cellular PARylation capacity, TRIP12 thus affects multiple outcomes of PAR formation in response to genotoxic stress.
Cooperation between a PAR-Binding WWE Domain and a HECT-Type Ubiquitin Ligase Domain Enables PAR-Targeted Ubiquitylation
Next, we aimed to elucidate the mechanism of PARP1 regulation by TRIP12. We found ectopically expressed TRIP12 to interact with PARP1, and this interaction was impaired by PARPi (Figure 3A). We also observed an interaction between endogenous TRIP12 and PARP1, although only upon treatment with PARGi to stabilize PARP1 auto-PARylation under otherwise unchallenged conditions (Figure S3A). Applying PARPi in conjunction with PARGi greatly reduced this interaction (Figure S3B). Consistently, a PARylation-defective mutant (PARP1 E988K) showed reduced binding to TRIP12 compared to wild-type PARP1 (Figure S3C). As TRIP12 contains a putative PAR-binding WWE domain, we expressed and purified the TRIP12 WWE domain (amino acids 797–911) as a GST-fusion protein and tested whether it would directly interact with auto-PARylated PARP1. As an additional specificity control, we generated a mutant version containing a single amino acid exchange (TRIP12 WWE R869A), which, based on sequence alignment with other WWE domains (Wang et al., 2012; see also Figure 1B), would be predicted to abolish PAR binding. These in vitro interaction experiments revealed that the TRIP12 WWE domain indeed bound PARP1, and that the interaction was strongly dependent on PARP1 auto-PARylation (Figure 3B). Consistently, the purified wild-type WWE domain of TRIP12, but not the R869A mutant, precipitated PARylated proteins, including PARP1, from H2O2-treated cell extracts (Figure 3C). Moreover, in co-immunoprecipitation experiments, the interaction between TRIP12 and PARP1 was reduced for the TRIP12 WWE domain mutant R869A (Figure S3D).
Figure 3.

TRIP12 Interacts with and Poly-Ubiquitylates PARP1 in a PAR- and WWE-Dependent Manner
(A) HEK293T cells were transfected with the indicated plasmids for co-immunoprecipitation (coIP) experiments, with or without prior PARPi treatment (olaparib: 10 μM, 1 h). FLAG-PARP1 was immunoprecipitated and the interaction with GFP-PARP1 was analyzed by western blot. IP samples were adjusted based on input levels of PARP1 to correct for the effect of TRIP12 on PARP1 abundance.
(B) In vitro interaction assay of purified GST, GST-TRIP12-WWE wild type (WT), and GST-TRIP12-WWE R869A mutant with PARP1 and auto-PARylated PARP1. Recombinant purified PARP1 was incubated or not with a low (20 μM, +) or high (200 μM, ++) concentration of NAD+ for 15 min at 30°C to induce PARP1 auto-PARylation prior to the GST interaction assay. PARP1 and PAR binding to the purified GST-fusion proteins was assessed by western blot.
(C) Ex vivo interaction assay of purified GST, GST-TRIP12-WWE WT, and GST-TRIP12-WWE R869A mutant with whole cell lysates from HeLa cells, which were exposed to 1-mM H2O2 for 15 min prior to cell lysis to induce PAR formation. PARG was depleted from these cells by siRNA to avoid the PARG-mediated rapid degradation of PAR. PARP1 and PAR binding to the purified GST-fusion proteins was assessed by western blot.
(D) In vitro ubiquitylation assay with purified E1 (UBE1), E2 (UBE2L3), E3 (FLAG-TRIP12 WT, WWE mutant (R869A), or HECT mutant (C2034A) purified from HEK293T cells and ubiquitin, using auto-PARylated PARP1 as a target protein. PARP1 ubiquitylation was assessed by western blot with anti-ubiquitin antibody. TRIP12 levels were assessed by running the supernatant from the in vitro reaction via western blot.
(E) In vivo ubiquitylation assay in HEK293T cells expressing FLAG-PARP1 and GFP-TRIP12 WT, WWE mutant (R869A), or HECT mutant (C2034A) together with HA-ubiquitin. FLAG-PARP1 was immunoprecipitated and PARP1 ubiquitylation was assessed by western blot. IP samples were adjusted based on input levels of PARP1 to correct for the effect of TRIP12 on PARP1 abundance.
(F) In vitro ubiquitylation assay to monitor TRIP12 activity in absence or presence of purified PAR chains. FLAG-TRIP12 WT or the PAR-binding-deficient WWE mutant (R869A) were purified from HEK293T cells and incubated with E1 (UBE1), E2 (UBE2L3), and ubiquitin with or without different amounts of purified PAR chains as indicated. Auto-ubiquitylation of TRIP12 was assessed by western blot.
See also Figure S3.
To directly test whether TRIP12 functions as PAR-targeted ubiquitin ligase (PTUbL; Pellegrino and Altmeyer, 2016) for PARP1, we reconstituted the ubiquitylation reaction in vitro using purified E1 and E2 enzymes, auto-PARylated PARP1, and immuno-purified full-length TRIP12. Although wild-type TRIP12 was indeed able to ubiquitylate PARP1, neither the TRIP12 WWE mutant (R869A), nor a catalytically inactive mutant containing a single amino acid exchange in the HECT ubiquitin ligase domain (C2034A), was able to modify PARP1 (Figure 3D). Importantly, also when expressed in cells, TRIP12 wild type, but not the WWE (R869A) and HECT domain (C2034A) mutants, triggered PARP1 ubiquitylation (Figure 3E). Thus, TRIP12 catalyzes PARP1 ubiquitylation in a PAR-dependent manner.
For the RING-type ubiquitin ligase RNF146/Iduna, an allosteric mechanism of PAR-dependent activation was described (DaRosa et al., 2015); we therefore reasoned that a similar mechanism might be at work for the HECT-type ubiquitin ligase TRIP12. Indeed, addition of purified PAR chains greatly stimulated the enzymatic activity of TRIP12, and this effect was abolished when the PAR-binding-deficient WWE mutant (R869A) was used (Figure 3F). Taken together, our in vitro and in vivo results thus provide biochemical support for an allosteric mechanism to activate TRIP12 in a PAR-binding- and WWE-domain-dependent manner. With analogous mechanisms being employed by both RING-type (RNF146/Iduna) and HECT-type (TRIP12) ubiquitin E3 ligases, PAR-binding-mediated allosteric activation seems to be a general mechanism for activation of and target protein recognition by WWE-containing ubiquitin E3 ligases.
In line with the ubiquitylation data, overexpression of wild-type TRIP12 reduced PARP1 levels, while overexpression of the WWE (R869A) and HECT domain (C2034A) mutants had no effect (Figure 4A). Conversely, depletion of TRIP12 resulted in reduced ubiquitylation of PARP1 in cells (Figure 4B). TRIP12 depletion also stabilized PARP1 in cycloheximide (CHX) experiments, an effect that was partially masked by reduced TRIP12 expression after prolonged CHX exposure (Figure 4C). Similarly, inhibition of PAR formation by the PARPi PJ-34 stabilized PARP1 levels, and additional depletion of TRIP12 did not result in a further increase in PARP1, consistent with PAR-dependent turnover of PARP1 by TRIP12 (Figures 4D and S4A). Reassuringly, the elevated PARP1 abundance upon TRIP12 depletion was rescued by siRNA-resistant wild-type TRIP12, but not by the catalytically inactive C2034A mutant or by the PAR binding-deficient R869A mutant (Figure 4E), despite all TRIP12 versions being expressed at similar levels (Figure S4B). Moreover, short-term inhibition of the proteasome stabilized PARP1 upon TRIP12 overexpression (Figure 4F), in agreement with PAR-dependent, proteasome-mediated degradation of PARP1. Finally, we also found TRIP12 to be recruited to sites of DNA damage in a PAR-, PARP1-, and WWE-domain-dependent manner (Figures S4C and S4D), indicating that TRIP12 not only controls basal PARP1 levels but also participates in PAR-triggered protein ubiquitylation in response to genotoxic stress. Taken together, our in vitro and in vivo data reveal that through its PAR-binding WWE domain and the carboxy-terminal HECT domain TRIP12 functions as PTUbL, which controls PARP1 abundance and activity.
Figure 4.

TRIP12 Controls PARP1 Stability and Proteasomal Degradation in a PAR-Dependent Manner
(A) U-2 OS PARP1 KO cells were transfected with the indicated plasmids and the effect of GFP-TRIP12 WT or mutant expression on Myc-PARP1 levels was assessed by western blot.
(B) U-2 OS cell were transfected with control siRNA or siRNA against TRIP12 for 48 h, followed by the indicated plasmid transfections for 24 h. Myc-PARP1 was immunoprecipitated and its ubiquitylation was assessed by western blot with an anti-HA antibody. Immunoprecipitation with a FLAG antibody was performed to detect unspecific binding. IP samples were adjusted based on input levels of PARP1 to correct for the effect of TRIP12 on PARP1 abundance.
(C) U-2 OS were transfected with control siRNA or siRNA against TRIP12 and exposed to cycloheximide (CHX, 50 μM) for the indicated time periods. TRIP12 and PARP1 levels were assessed by western blot. PARP1 band intensities were quantified in Fiji and the fold changes are indicated below.
(D) U-2 OS were transfected with control siRNA or siRNA against TRIP12 and exposed to a PARPi with low PARP1 trapping activity (PJ-34, 10 μM) as indicated for 16 h to block PARylation with minimal cytotoxicity. TRIP12 and PARP1 levels were assessed by western blot.
(E) U-2 OS cells were transfected with control siRNA or siRNA against TRIP12 for 48 h, followed by the indicated plasmid transfections for 24 h. All TRIP12 plasmids were rendered siRNA-resistant by introducing multiple silent mutations. High-content imaging-derived violin plot show nuclear PARP1 intensities. Median (solid lines) and quartiles (dashed lines) are indicated.
(F) U-2 OS PARP1 KO cells were transfected with plasmids for Myc-PARP1 and GFP-TRIP12 expression and exposed to the proteasome inhibitor MG132 (10 μM) as indicated. PARP1 levels were assessed by western blot.
See also Figure S4.
TRIP12 Constrains PARP1 Trapping and Extenuates PARPi-Induced Genotoxicity
As PARP1 is the major target of PARPi, and given that PARPi-induced cancer cell death has been associated with cytotoxic PARP1 trapping, entailing replication problems during S-phase progression and DNA damage (Michelena et al., 2018; Murai et al., 2012; Muvarak et al., 2016; Zimmermann et al., 2018), we wondered whether TRIP12 might impact this aspect of PARPi action. Loss of TRIP12 indeed resulted in enhanced chromatin association of PARP1 (in unchallenged conditions and more pronounced upon PARPi and when PARPi was combined with alkylation damage by methyl methanesulfonate [MMS]), both when quantifying the detergent-resistant pool of PARP1 in hundreds of individual cells by high-content microscopy and also in biochemical fractionation assays (Figures 5A, 5B, S5A, and S5B). Consistent with elevated PARP1 trapping upon impaired TRIP12 function, we observed an increase in DNA damage signaling marked by γH2AX, primarily in the S- and G2-phase of the cell cycle (Figures 5C and S5C). DNA damage signaling upon PARPi is associated with impaired S-phase progression and G2/M arrest (Michelena et al., 2018), and we observed a more pronounced PARPi-evoked accumulation of cells in G2/M upon TRIP12 depletion, compared to control cells (Figure 5D). Importantly, both the increased DNA damage signaling measured by γH2AX formation as well as the more pronounced cell cycle arrest were rescued by PARP1 co-depletion (Figures S5D and S5E), indicating that these signs of genotoxic stress were caused by catalytically inactivated PARP1. Also, additional markers of PARPi-induced genotoxic stress, such as replication-associated formation of 53BP1, RAD51 and RPA foci, were elevated in TRIP12-depleted cells, and PARP1 co-depletion alleviated these effects (Figures 5E–5G). Collectively, these results provide evidence that TRIP12, by limiting PARP1 abundance and PARP trapping, extenuates PARPi-induced DNA damage.
Figure 5.

TRIP12 Constrains PARP1 Trapping and Extenuates PARPi-Induced DNA Damage
(A) U-2 OS cells transfected with control siRNA or siRNA against TRIP12 and exposed to MMS (0.01%) and PARPi (olaparib, 10 μM) for 1 h as indicated. Cells were pre-extracted and chromatin-bound PARP1 was analyzed by high-content microscopy. Violin plots with median (solid lines) and quartiles (dashed lines) are shown; unpaired t test was used for statistical analysis. Representative single cell images of endogenous chromatin-bound PARP1 are shown below. Scale bar, 10 μm. ∗∗∗∗, p < 0.0001.
(B) U-2 OS cells transfected with control siRNA or siRNA against TRIP12 were treated with MMS (0.01%) and PARPi (olaparib, 10 μM) for 2 h as indicated. Cells were then subjected to chromatin fractionation to separate the soluble from the chromatin-bound fraction. Chromatin-bound PARP1 was analyzed by western blot. PARP1 band intensities were quantified in Fiji and the fold changes are indicated below.
(C) HeLa Kyoto PARP1-LAP cells were transfected with control siRNA or siRNA against TRIP12 and exposed to PARPi (olaparib, 10 μM) for 16 h. QIBC-derived cell cycle resolved γH2AX profiles are shown. Representative single-cell images of γH2AX staining are shown below. Scale bar, 10 μm.
(D) QIBC-derived EdU profiles are shown and percentages of cells in G1, S, and G2 are provided for the same cells analyzed in (C).
(E) U-2 OS cells were transfected with siRNAs and exposed to PARPi (olaparib, 10 μM) as indicated. QIBC-derived cell cycle resolved profiles of nuclear 53BP1 foci are shown. Representative single-cell images of 53BP1 foci are shown to the right. Scale bar, 10 μm.
(F) U-2 OS cells were treated as in (E) and QIBC-derived cell-cycle resolved profiles of nuclear RAD51 foci are shown. Representative single cell images of RAD51 foci are shown to the right. Scale bar, 10 μm.
(G) U-2 OS cells were treated as in (E) and QIBC-derived cell-cycle resolved profiles of nuclear RPA foci are shown. Representative images of RPA foci are shown to the right. Scale bar, 10 μm.
See also Figure S5.
Downregulation of TRIP12 Sensitizes Cancer Cells to PARPi and Is Associated with Elevated PARP1 Abundance in Cancer Patients
Finally, to directly test whether TRIP12 status determines cancer cell survival upon PARPi exposure, we performed colony formation assays in multiple cell lines. Remarkably, TRIP12 downregulation greatly sensitized BRCA-proficient cancer cells to the PARPi olaparib, and the hyper-sensitization was strictly dependent on the presence of PARP1, as PARP1 co-depletion rescued clonogenic survival in all cases (Figures 6A–6D). Similar results were obtained with a second and third siRNA against TRIP12 (Figures S6A and S6B) and with two different clinically approved PARPi, rucaparib and talazoparib (Figures S6C and S6D). Consistent with the hyper-sensitization to PARPi originating from supra-physiological PARP1 accumulation, TRIP12 depletion had no effect on the survival of PARP1 KO cells (Figure S6E). These results are consistent with the notion that PARP1, locked in a catalytically nonproductive state, is an important contributor to PARPi-induced cell death in BRCA-proficient cells, and suggest that variations in PARP1 protein levels impact PARPi sensitivity. In line with this, controlled overexpression of PARP1 in a stable doxycycline-inducible cell line resulted in reduced survival upon PARPi (Figure S6F).
Figure 6.

Downregulation of TRIP12 Sensitizes Cancer Cells to PARPi and Is Associated with Elevated PARP1 Abundance in Cancer Patients
(A) U-2 OS cells were transfected with siRNAs and exposed to PARPi olaparib as indicated for clonogenic survival assays. Relative clonogenic survival in response to increasing doses of PARPi olaparib is shown. Means ± SD are depicted.
(B) HeLa Kyoto PARP1-LAP cells were treated as in (A) and relative clonogenic survival in response to increasing doses of PARPi olaparib is shown. Means ± SD are depicted.
(C) HCC1143 cells were treated as in (A) and relative clonogenic survival in response to increasing doses of PARPi olaparib is shown. Means ± SD are depicted.
(D) MCF-7 cells were treated as in (A) and relative clonogenic survival in response to increasing doses of PARPi olaparib is shown. Means ± SD are depicted.
(E) Scatterplot of adjusted PARP1 protein levels against the relative TRIP12 RNA expression in breast cancer, with BRCA mutant samples highlighted in red. Upper right shows p values and r in Pearson’s product-moment correlation tests.
(F) As in (E) for the ovarian cancer samples. Upper right shows p values and r in Pearson’s product-moment correlation tests.
See also Figure S6.
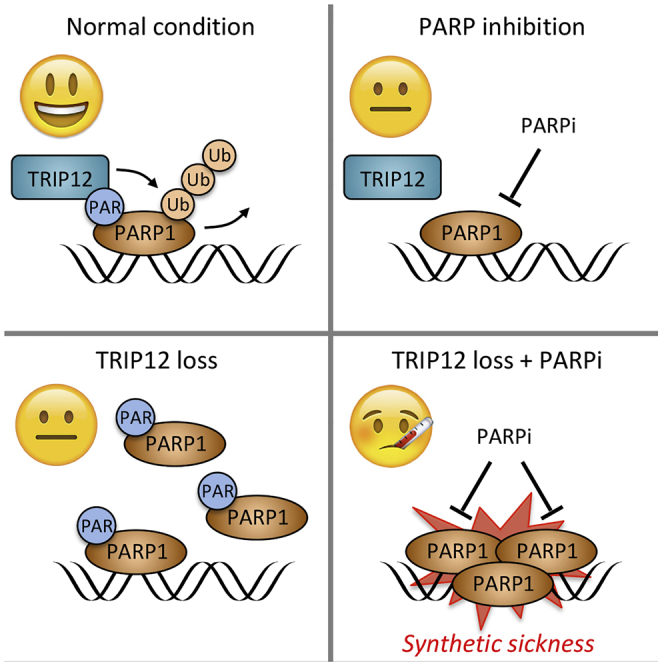
As our findings suggested that TRIP12 expression might serve as an indicator of PARP1 stability (and thus PARP1 abundance and consequently PARPi sensitivity), we explored the NCI/NIH CPTAC Data Portal (Edwards et al., 2015; Ellis et al., 2013) for studies providing matched genotyping, gene expression, and protein abundance data. We identified such matched multiomics data for cohorts of 102 breast cancer patients and 145 ovarian cancer patients. In both cohorts, TRIP12 expression showed a negative correlation with PARP1 protein abundance (Figures 6E, 6F, S7A, and S7B), consistent with our mechanistic and functional data. BRCA status did not provide information about PARP1 abundance in these patient cohorts (Figures 6E, 6F, S7C, and S7D), suggesting that the negative correlation between TRIP12 expression and PARP1 abundance is unrelated to BRCA function. In summary, our findings establish TRIP12 as a PTUbL, which constrains PARP1 abundance and PARPi efficiency, and suggest that TRIP12 measurements might be useful to evaluate PARPi sensitivity beyond BRCA mutations (Figures 7A and 7B).
Figure 7.

Model for PAR-Targeted Ubiquitylation of PARP1 by TRIP12 and the Ensuing Proteasomal Degradation of PARP1
(A) TRIP12 binds auto-modified PARP1 via its WWE domain and triggers PARP1 polyubiquitylation and proteasomal degradation.
(B) When TRIP12 expression or function is impaired, PARP1 accumulates to supra-physiological levels, resulting in enhanced PARP1 trapping and elevated PARPi susceptibility. See main text for further discussion.
Discussion
BRCA deficiency has emerged as paradigm for PARPi sensitivity, with impaired BRCA function leading to synthetic lethality in PARPi-exposed cancer cells. Here, we reveal a mechanistically distinct synthetic lethal/sick relationship between PARPi and impaired TRIP12 function. While BRCA defects entail HDR deficiency with a compromised capacity to protect stalled replication forks and deal with DNA double-stand breaks, TRIP12 dysfunction leads to supra-physiological PARP1 levels and causes hyper-sensitization to PARPi by elevated PARP1 trapping. Hence, concomitant loss of PARP1 reverses PARPi sensitivity under conditions of impaired TRIP12 function, in contrast to the synthetic lethality caused by BRCA deficiency. It was recently demonstrated that mutations in PARP1 that disrupt its interaction with DNA and alleviate PARP1 trapping provide protection from PARPi cytotoxicity and may underlie clinical cases of PARPi resistance (Pettitt et al., 2018). It will thus be important to evaluate whether additional cases of PARP1 mutations associated with PARPi resistance may emerge from currently ongoing PARPi trials, and whether upregulation of TRIP12 may provide an alternative means for cancer cells to escape PARPi therapy.
PARPi monotherapy has demonstrated promising results in specific patient cohorts, in particular in germline BRCA mutation-associated breast and ovarian cancer. Olaparib showed a response rate of 41% in germline BRCA mutated carriers with recurrent, advanced breast cancer (Tutt et al., 2010), and response rates of 31%–40% were reported in germline mutated BRCA ovarian cancer patients with recurrent disease (Fong et al., 2010). Recently, data presented from a randomized phase III trial, which compared single-agent olaparib with chemotherapy treatment of physician’s choice (TPC) in germline BRCA mutated platinum-sensitive relapsed ovarian cancer, showed favorable outcomes of olaparib over TPC with objective response rate of 72% versus 51% (Penson et al., 2020). However, predicting PARPi sensitivity has remained challenging due to the lack of good biomarkers, in particular beyond BRCA- and HDR-deficiencies. PARP1 and PARP activity measurements have been proposed as predictive tools (Coskun et al., 2019; Makvandi et al., 2016; Makvandi et al., 2018; Oplustilova et al., 2013; Thomas et al., 2018), but PARP1 gene expression does not necessarily reflect PARP1 protein abundance, and PARP activity measurements may not necessarily predict PARP1 trapping. Moreover, antibody-based detection of PAR and PARP1 has limitations in terms of sensitivity and specificity. Therefore, additional markers for PARPi efficiency are needed. Based on the molecular mechanism of TRIP12-mediated PARP1 turnover described here and the inverse correlation between TRIP12 expression and PARP1 abundance in cohorts of breast and ovarian cancer patients, we propose that evaluation of TRIP12 status could provide such an additional stratification marker for PARP1 abundance and PARPi sensitivity, and for PARPi single agent and combination therapies. Validation of TRIP12-mediated PARP1 turnover and its effect on PARPi efficiency in suitable animal models would help address this point further. Additionally, once data become available from cancer patients treated with PARPi that allow for comparison of TRIP12 status, PARP1 protein abundance, and PARPi response, a clinical evaluation of the relationship between TRIP12 status and PARPi response will be possible.
WWE-dependent PAR binding and PAR-targeted ubiquitylation has been primarily characterized for the RING domain ubiquitin ligase RNF146/Iduna (DaRosa et al., 2015; Wang et al., 2012; Zhang et al., 2011). A crystal structure of the RNF146/Iduna RING-WWE domains together with iso-ADPr revealed a mechanism of RING-type ubiquitin E3 ligase activation by PAR binding-induced allosteric conformational changes leading to a functional RING structure (DaRosa et al., 2015). Although structural information on the precise interplay between the WWE and HECT domains of TRIP12 is currently not available, our biochemical and functional data suggest that a similar allosteric mechanism, triggered by non-covalent PAR binding, might stimulate the catalytic activity of the HECT-type ubiquitin E3 ligase TRIP12. Further studies and dedicated protein structure illumination will be needed to elucidate the conformational changes underlying PAR-triggered activation of the two families of ubiquitin ligases, the RING domain, and the HECT domain ligases, and how they affect their respective reaction mechanism (direct ubiquitin transfer from E2 to the substrate by RING domain enzyme versus ubiquitin conjugation to an active-site cysteine by HECT domain enzymes). Of note, RNF146/Iduna was previously shown to bind and ubiquitylate PARylated PARP1 among other PARylated proteins (Kang et al., 2011), and the mitotic checkpoint factor and RING-type ubiquitin ligase CHFR targets PARP1 for degradation upon mitotic stress and DNA damage (Kashima et al., 2012; Liu et al., 2013). Also, RNF168 acts on PARP1 and regulates PARP1-dependent repair functions (Kim et al., 2018). It will thus be interesting to investigate how RING- and HECT-type ubiquitin ligases functionally cooperate to regulate PARP1 degradation, and how this in turn affects PARPi-induced PARP1 trapping in different cell cycle phases and at different types of DNA lesions. Our results show that TRIP12 regulates PARP1 steady-state levels in unchallenged conditions, indicating that basal PARP1 activity (which is hardly detectable by PAR antibodies), likely triggered by endogenous DNA lesions, is sufficient for TRIP12 to bind PARP1 and regulate its turnover. However, we also found TRIP12 to be recruited to sites of DNA damage in a PARP1- and PAR-dependent manner, suggesting that TRIP12, as PAR-targeted ubiquitin ligase (PTUbL), additionally controls the strength and duration of PAR signaling upon genotoxic insults.
A limited number of TRIP12 targets had been identified by previous studies; however, whether their ubiquitylation is stimulated by prior PARylation is currently not known. For instance, TRIP12 catalyzes amino-terminal ubiquitylation of several substrates of the ubiquitin fusion degradation (UFD) pathway (Park et al., 2009), and also controls the half-life and abundance of the tumor suppressor and p53 activator p14/ARF in non-cancer cells (Chen et al., 2010). Moreover, TRIP12 was previously identified as a negative regulator of the DNA damage response protein RNF168, which promotes regulatory chromatin-associated ubiquitylation events and coordinates the recruitment of downstream genome caretakers in response to DNA replication stress and chromosome breakage (Gudjonsson et al., 2012). Based on the emerging paradigm of PAR-dependent ubiquitylation, future studies may discern PAR-dependent and -independent functions of TRIP12 and aim for comprehensive identification of TRIP12 targets in different cells lines, to elucidate their roles for genome stability and chemotherapy sensitivity and resistance. Moreover, with the intensified development of ubiquitin E3 ligase inhibitors, small molecules targeting TRIP12 may soon become available to test combined inhibition of PARPs and TRIP12 in order to maximize PARPi-induced PARP trapping and PARPi efficiency.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H2AX Phospho S139 primary antibody (mouse) | Biolegend | Cat# 613401; RRID:AB_315794 |
| H2AX Phospho S139 primary antibody (rabbit) | Cell Signaling | Cat# 2344; RRID:AB_2118009 |
| TRIP12 primary antibody (rabbit) | Bethyl | Cat# A301-814A; RRID:AB_1264344 |
| PARP1 primary antibody (rabbit) | Abcam | Cat# ab227244; RRID:N/A |
| PARP1 (H-250) primary antibody (mouse) | Santa-Cruz | Cat# sc-7150; RRID:AB_2160738 |
| c-Myc primary antibody (mouse) | Santa-Cruz | Cat# sc-40; RRID:AB_627268 |
| Poly(ADP-ribose) primary antibody (rabbit) | Enzo Life Sciences | Cat# ALX-210-890-R100; RRID:AB_2052276 |
| RPA70 primary antibody (rabbit) | Abcam | Cat# ab79398; RRID:AB_1603759 |
| 53BP1 primary antibody (rabbit) | Santa-Cruz | Cat# sc-22760; RRID:AB_2256326 |
| 53BP1 primary antibody (mouse) | Millipore | Cat# MAB3802; RRID:AB_2206767 |
| AIF primary antibody (rabbit) | Santa-Cruz | Cat# sc-5586; RRID:AB_2224668 |
| FLAG primary antibody (mouse) | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Ubiquitin (P4D1) primary antibody (mouse) | Santa-Cruz | Cat# sc-8017; RRID:AB_628423 |
| HA.11 primary antibody (mouse) | BioLegend | Cat# 901513; RRID:AB_2565335 |
| RAD51 primary antibody (rabbit) | BioAcademia | Cat# 70-002 |
| XRCC1 primary antibody (mouse) | Novus Biologicals | Cat# H00007515-B01P; RRID:AB_1522704 |
| MDC1 primary antibody (rabbit) | Abcam | Cat# ab11171; RRID:AB_297810 |
| RNF168 primary antibody (rabbit) | Stewart et al., 2009 | N/A |
| Tubulin primary antibody (mouse) | Sigma-Aldrich | Cat# T6199; RRID:AB_477583 |
| Histone H3 primary antibody (rabbit) | Abcam | Cat# ab1791; RRID:AB_302613 |
| PARP2 primary antibody (rabbit) | Active Motif | Cat# 39743; RRID:AB_2793328 |
| GFP primary antibody (rabbit) | Torrey Pines Biolabs | Cat# TP401 071519; RRID:AB_10013661 |
| Alexa Fluor 488 goat-anti-Mouse | ThermoFisher | Cat# A-11029; RRID:AB_2534088 |
| Alexa Fluor 488 goat-anti-Rabbit | ThermoFisher | Cat# A-11034; RRID:AB_2576217 |
| Alexa Fluor 568 goat-anti-Mouse | ThermoFisher | Cat# A-11031; RRID:AB_144696 |
| Alexa Fluor 568 goat-anti-Rabbit | ThermoFisher | Cat# A-11036; RRID:AB_10563566 |
| Alexa Fluor 647 goat-anti-Mouse | ThermoFisher | Cat# A-21235; RRID:AB_2535804 |
| Alexa Fluor 647 goat-anti-Rabbit | ThermoFisher | Cat# A-21244; RRID:AB_2535812 |
| Anti-Mouse IgG (H+L), Peroxidase | AdipoGen Life Science | Cat# VC-PI-2000-M001 |
| Anti-Rabbit IgG (H+L), Peroxidase | AdipoGen Life Science | Cat# VC-PI-1000-M001 |
| Bacterial and Virus Strains | ||
| Library Efficiency DH5α Competent Cells | ThermoFisher | Cat# 18263012 |
| BL21 (DE3) competent cells | ThermoFisher | Cat# EC0114 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Q5 High-Fidelity DNA polymerase | New England Biolabs | Cat# M0491S |
| T5 Exonuclease | New England Biolabs | Cat# M0363S |
| Taq DNA ligase | New England Biolabs | Cat# M0208S |
| Phusion High-Fidelity polymerase | New England Biolabs | Cat# M0530S |
| β-Nicotinamide adenine dinucleotide hydrate | Sigma-Aldrich | Cat# N7004 |
| dATP | Sigma-Aldrich | Cat# D4788 |
| dCTP | Sigma-Aldrich | Cat# D4913 |
| dGTP | Sigma-Aldrich | Cat# D5038 |
| dTTP | Sigma-Aldrich | Cat# 11969048001 |
| Dulbecco’s modified Eagle’s medium (DMEM) | ThermoFisher | Cat# 61965-026 |
| RPMI-1640 medium | ThermoFisher | Cat# A1049101 |
| Ham’s F12 medium | ThermoFisher | Cat# 11765054 |
| OptiMEM | ThermoFisher | Cat# 31985070 |
| Tet system approved fetal bovine serum | Clontech | Cat# 631107 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A9418 |
| Penicillin-streptomycin | ThermoFisher | Cat# 15140-122 |
| Hygromycin B | ThermoFisher | Cat# 10687010 |
| Geneticin (G418 sulfate) | ThermoFisher | Cat# 10131027 |
| 5-bromo-2-deoxyuridine (BrdU) | Sigma-Aldrich | Cat# B5002 |
| PJ-34 | Enzo Life Sciences | Cat# ALX-270-289-M001 |
| Rucaparib | Selleckchem | Cat# S1098 |
| Talazoparib | Selleckchem | Cat# S1288 |
| Hydrogen peroxide | Sigma-Aldrich | Cat# H3410 |
| Olaparib | Selleckchem | Cat# S1060 |
| Methylmethanesulfunate | Sigma-Aldrich | Cat# 129925 |
| PARG inhibitor | Tocris | Cat# 5952 |
| FK866 | Sigma-Aldrich | Cat# F8557 |
| MG132 | Sigma-Aldrich | Cat# M7449 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| TRIzol reagent | ThermoFisher | Cat# 15596026 |
| Random hexamers | Roche | Cat# 11034731001 |
| Protease inhibitor cocktail cOmplete | Roche | Cat# 11873580001 |
| PhosSTOP | Roche | Cat# 4906837001 |
| S7 Nuclease | Sigma-Aldrich | Cat# 10107921001 |
| Benzonase | Merck | Cat# 71206-3 |
| 3x FLAG peptide | Sigma-Aldrich | Cat# F4799 |
| Glycine | Sigma-Aldrich | Cat# G8898 |
| UBE1 | Boston Biochem | Cat# E-305 |
| UBE2L3 | Boston Biochem | Cat# E2-640 |
| Ubiquitin | Boston Biochem | Cat# U-100H |
| hPARP1 | Michael Hottiger | N/A |
| hPARG | Michael Hottiger | N/A |
| PAR polymer | Trevigen | Cat# 4336-100-01 |
| TRIP12 WWE domain WT | This paper | N/A |
| TRIP12 WWE domain R869A | This paper | N/A |
| Protein G Sepharose 4 Fast Flow | GE Healthcare | Cat# 17-0618-01 |
| ProBond Nickel resin | ThermoFisher | Cat# R80101 |
| DAPI | ThermoFisher | Cat# D1306 |
| Mowiol 4.88 | Calbiochem | Cat# 475904 |
| Critical Commercial Assays | ||
| Rapid Ligation kit | Roche | Cat# 11635379001 |
| Fugene HD | Promega | Cat# E2311 |
| 5-ethynyl-2′-desoxyuridine (EdU) labeling kit | ThermoFisher | Cat# C10337 |
| Lipofectamine 3000 | ThermoFisher | Cat# L3000008 |
| MultiScribe Reverse Transcriptase | ThermoFisher | Cat# 4311235 |
| KAPA Biosystems Sybr Fast qPCR Kit | Roche | Cat# KK4600 |
| Lipofectamine RNAiMAX | ThermoFisher | Cat# 13778-150 |
| QIAGEN MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| Energy regeneration System | Boston Biochem | Cat# B-10 |
| Experimental Models: Cell Lines | ||
| U-2 OS (female origin, STR authenticated) | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| hTERT-RPE1 (female origin) | ATCC | Cat# CRL-4000; RRID:CVCL_4388 |
| HEK293T (female origin) | ATCC | N/A |
| MCF-7 (female origin) | Ross Chapman | N/A |
| U-2 OS PARP1 KO (female origin) | Keith Caldecott | N/A |
| HeLa (female origin) | Raffaella Santoro | N/A |
| HeLa Kyoto PARP1-LAP cells (female origin) | Stoyno S. Stoynov | N/A |
| HCC1143 (female origin) | Mohamed Bentires-Alj | N/A |
| U-2 OS Tet-ON (female origin) | Ana Zubiaga | N/A |
| U-2 OS Tet-ON HA-empty vector control cells | This paper | N/A |
| U-2 OS Tet-ON HA-PARP1 cells | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 | N/A | |
| Recombinant DNA | ||
| pcDNA-Myc-PARP1 WT | Laboratory of Michael Hottiger | N/A |
| pcDNA-Myc-PARP1 E988K | Laboratory of Michael Hottiger | N/A |
| pCMV-FLAG-PARP1 | Laboratory of Michael Hottiger | N/A |
| pcDNA-HA-Ubiquitin | Laboratory of Lorenza Penengo | N/A |
| pAc-GFP-C1 TRIP12 WT | This paper | N/A |
| pAc-GFP-C1 TRIP12 C2034A | This paper | N/A |
| pAc-GFP-C1 TRIP12 R869A | This paper | N/A |
| pAc-GFP-C1 TRIP12 WT siRNA resistant | This paper | N/A |
| pAc-GFP-C1 TRIP12 C2034A siRNA resistant | This paper | N/A |
| pAc-GFP-C1 TRIP12 R869A siRNA resistant | This paper | N/A |
| pcDNA 3.1 FLAG TRIP12 WT | This paper | N/A |
| pcDNA 3.1 FLAG TRIP12 C2034A | This paper | N/A |
| pcDNA 3.1 FLAG TRIP12 R869A | This paper | N/A |
| pGEX 6P-1 TRIP12 WWE WT | This paper | N/A |
| pGEX 6P-1 TRIP12 WWE R869A | This paper | N/A |
| pTRE2Hyg2-HA-PARP1 WT | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8.0 | GraphPad Software | https://www.graphpad.com/ |
| Olympus ScanR Image Analysis Software 3.0.1 | Olympus | https://www.olympus-lifescience.com/de/microscopes/inverted/scanr/#! |
| TIBCO Spotfire 7.0.1 | TIBCO Software | https://spotfire.tibco.com/ |
| Fiji 2.0.0 | Schindelin et al., 2012 | https://fiji.sc/ |
| Clustal Omega | Madeira et al., 2019 | https://www.ebi.ac.uk/Tools/msa/clustalo |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthias Altmeyer (matthias.altmeyer@uzh.ch).
Materials Availability
Materials generated in this study will be made available upon reasonable request to the Lead Contact.
Data and Code Availability
This study did not generate or analyze code or datasets (such as microarray, genomics, transcriptomics, protein structure data).
Experimental Model and Subject Details
Cell lines and culture conditions
Human U-2 OS cells (ATCC), RPE-1 (ATCC), HEK293T (ATCC), HeLa (kindly provided by Raffaella Santoro), MCF-7 (kindly provided by Ross Chapman), U-2 OS PARP1 KO cells (kindly provided by Keith Caldecott), HeLa Kyoto PARP1-LAP cells stably expressing EGFP-tagged PARP1 from its natural promoter (kindly provided by Stoyno S. Stoynov), and TetOn HA-PARP1 U-2 OS cells were grown under standard sterile cell culture conditions (37°C, humidified atmosphere, 5% CO2) in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific) containing 10% fetal bovine serum (GIBCO) and 1% of penicillin-streptomycin antibiotics. HCC1143 cells (kindly provided by Mohamed Bentires-Alj) were grown in RPMI-1640 medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (GIBCO) and 1% penicillin-streptomycin antibiotics. All cultured cells were routinely tested for mycoplasma contamination and scored negative. In addition, cells were regularly assessed by quantitative image-based cytometry (QIBC) for proper proliferation and absence of abnormal stress signals.
Bacterial strains
Cloning was done using chemically competent bacteria generated in-house, derived from Library Efficiency DH5α Competent Cells (ThermoFisher), and recombinant protein expression from BL21 (DE3) competent cells.
Method Details
Generation of doxycycline-inducible HA-PARP1 cell line
pTREHyg2-HA was linearized with Phusion High-Fidelity polymerase using primers pTRE2Hyg2_lin_frw and pTRE2Hyg2_lin_rev. PARP1 was amplified with Phusion polymerase using primers PARP1 GA_to_pTRE_fwr and PARP1 GA_to_pTRE_rev. The amplified products were DpnI digested and purified by gel extraction followed by isothermal Gibson assembly. U-2 OS TetOn cells (kindly provided by Ana Zubiaga) stably expressing PARP1 were generated by transfection of pTRE2Hyg2-HA containing the PARP1 sequence. Induction of PARP1 expression was achieved by Doxycycline treatment (1000ng/ml).
Drug treatment
The following compounds were used at the indicated final concentrations: PJ-34 (10 μM, ALX-270-289-M001, Enzo Life Science), Olaparib (1-10 μM, AZD-2281, Selleckchem), Rucaparib (1-8 μM, AG-014699, Selleckchem), Talazoparib (6.25-50 nM, BMN673, Selleckchem), β-Nicotinamide adenine dinucleotide hydrate (20-200 μM, N7004, Sigma-Aldrich), hydrogen peroxide (0.02-0.5 mM, H3420, Sigma-Aldrich), Methyl methanesulfunate, MMS (0.01%, 129925, Sigma-Aldrich), PARG inhibitor (10 μM, PDD00017273, Tocris), FK866 (1 μM, F8557, Sigma-Aldrich), MG132 (10 μM, M7449, Sigma-Aldrich), cycloheximide (50 μM, C7698, Sigma-Aldrich).
Edu labeling
For pulsed EdU (5-ethylnyl-2′-desoxyuridine, Thermo Fisher Scientific) incorporation, cells were incubated for 20 minutes in medium containing 10 μM EdU. The Click-iT EdU Alexa Fluor Imaging Kit (Thermo Fisher Scientific) was used for EdU detection.
siRNA and plasmid transfections
Transfections with siRNAs were performed for 72h, unless stated otherwise, with Ambion Silencer Select siRNA using Lipofectamine RNAi/MAX (Thermo Fisher Scientific). For individual transfections the siRNAs were used at a final concentration of 25nM. When multiple siRNAs were combined, the final concentration was kept constant in all samples of an experiment. The following Ambion Silencer Select siRNAs were used: TRIP12 (s17808, s17809, s17810), PARP1 (s1098), DTX1 (s4355, s4356), DTX2 (s41519, s223231), DTX4 (s23319, s23321), HUWE1 (s19596, s19597), RNF146 (s37821, s37822), PARG (s16158). Negative Silencer Select control Neg1 from Ambion was used as a non-targeting control (siControl). Plasmid transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific) or Fugene HD (Promega) according to the manufacturer’s recommendations. Calcium phosphate transfection was used for HEK293T cells. Unless stated otherwise, cells were analyzed 24h after transfection.
Antibodies
GFP (rabbit, TP401, Torrey Pines Biolabs; 1:1000 for WB, 1 μg for IP), TRIP12 (rabbit, A301-814A, Bethyl; 1:500 for WB, 1 μg for IP), Tubulin (mouse, T6199, Sigma; 1:5000 for WB), PARP1 (rabbit, ab227244, Abcam; 1:500 for IF, 1:1000 for WB), PARP1 (H-250) (rabbit, sc-7150, Santa Cruz; 1:500 for IF, 1:1000 for WB), PARP2 (rabbit, 39743, Active Motif, 1:500 for WB), c-Myc (mouse, sc-40, Santa Cruz; 1:500 for WB), Poly(ADP-ribose) (rabbit, ALX-210-890A-0100, Enzo; 1:1000 for IF, 1:2000 for WB), γH2AX (mouse, 613402, BiolLegend, 1:1000 for IF), γH2AX (rabbit, 9718, Cell Signaling, 1:1000 for IF), AIF (rabbit, sc-5586, Santa Cruz, 1:500 for IF), FLAG (mouse, F3165, Sigma; 1:1000 WB), Ub P4D1 (mouse, sc-8017, Santa Cruz; 1:1000 for WB), HA.11 (mouse, 901513, BioLegend; 1:1000 for WB), H3 (rabbit, ab1791, Abcam; 1:5000 for WB), 53BP1 (mouse, MAB3802 Merk Millipore; 1:1000 for IF), RAD51 (rabbit, 70-002, BioAcademia; 1:1000 for IF), XRCC1 (mouse, H00007515-B01P, Novus Biologicals; 1:500 for IF), MDC1 (rabbit, ab11171, Abcam, 1:1000 for IF), RPA70 (rabbit, ab79398, Abcam, IF 1:500 for IF), RNF168 (rabbit, 1:2000 for WB, kindly provided by Daniel Durocher).
Immunostaining
For standard immunofluorescence staining, high-content microscopy and QIBC analyses, cells were seeded on sterile 12 mm glass coverslips and allowed to proliferate until they reached a cell density of 70%–90%. Cells were then fixed in 3% formaldehyde for 15 minutes at room temperature, washed once in PBS, permeabilized for 5 minutes at room temperature in 0.2% Triton X-100 (Sigma-Aldrich) in PBS, washed twice in PBS and incubated in blocking solution (filtered DMEM containing 10% FBS and 0.02% Sodium Azide) for 15 minutes at room temperature. When the staining was combined with an EdU Click-iT reaction, this reaction was performed prior the incubation with primary antibody according to manufacturer’s recommendations (Thermo Fisher Scientific). Where indicated, cells were pre-extracted in 0.2% Triton X-100 in PBS for two minutes on ice prior to formaldehyde fixation. To preserve cellular PAR levels during the fixation step, the formaldehyde solution was supplemented with PARPi and PARGi to a final concentration of 10 μM each (Figures 5A, S2A, and S2B). All primary antibodies were diluted in blocking solution and incubated for 2h at room temperature. Secondary antibodies (Alexa Fluor 488, 568, 647 anti-mouse and anti-rabbit IgG from Thermo Fischer Scientific) were diluted 1:500 in blocking solution and incubated at room temperature for 1h. Cells were washed once with PBS and incubated for 10 minutes with 4’,6-Diamidino-2-Phenylindole Dihydrochloride (DAPI, 0.5 μg/ml) in PBS at room temperature. Following three washing steps in PBS, coverslips were briefly washed with distilled water and mounted on 5 μl Mowiol-based mounting media (Mowiol 4.88 (Calbiochem) in Glycerol/TRIS).
High-content microscopy and quantitative image-based cytometry (QIBC)
Automated multichannel wide-field microscopy for high-content imaging and quantitative image-based cytometry (QIBC) was performed using the Olympus ScanR System as described previously (Kilic et al., 2019; Teloni et al., 2019). Images were analyzed with the inbuilt Olympus ScanR Image Analysis Software Version 3.0.1, a dynamic background correction was applied, and nuclei segmentation was performed using an integrated intensity-based object detection module based on the DAPI signal. All downstream analyses were focused on properly detected nuclei containing a 2C-4C DNA content as measured by total and mean DAPI intensities. Fluorescence intensities were quantified and are depicted as arbitrary units. Color-coded scatterplots of asynchronous cell populations were generated with Spotfire data visualization software (TIBCO) and GraphPad Prism 8.0 was used for depicting data as violin plots. Within one experiment, similar cell numbers were compared for the different conditions. For visualizing discrete data in scatterplots, mild jittering (random displacement of data points along discrete data axes) was applied in order to demerge overlapping data points. Representative scatterplots and quantifications of independent experiments, typically containing several thousand cells each, are shown.
Laser micro-irradiation
Cells were grown on 12 mm coverslips and pre-sensitized with 10 μM 5′-bromo-2-deoxyuridine (BrdU) for 24 hours prior to laser micro-irradiation with a pulsed UV-A laser. Laser tracks were visualized on a Leica DMI 6000 inverted microscope using an HCX Plan APO DIC 63x oil objective (1.4-0.6 NA) and analyzed using Fiji.
RNA extraction, reverse transcription and quantitative PCR
RNA was purified with TRIzol reagent (Life Technologies), primed with random hexamers (11034731001, Roche) and reverse-transcribed to cDNA using a MultiScribe Reverse Transcriptase (4311235, Thermo Fisher). Quantitative PCR (qPCR) was performed with KAPA SYBR FAST qPCR Kit (KAPA Biosystems) on a Rotor Gene Q system (QIAGEN). Samples were run in triplicates, normalized to RPS12, and the results are depicted as relative fold changes. All primer sequences are provided in the Key Resources Table.
Plasmids and constructs
Cloning was done using chemically competent bacteria generated in-house, derived from Library Efficency™ DH5α™ Competent Cells (Thermo Fisher Scientific). Correct cloning, integration into target vectors and mutagenesis was confirmed by DNA sequencing.
Cloning of GFP-tagged TRIP12 plasmids
The TRIP12 cDNA was cloned into pAc-GFP1-C1 (Clontech) using the primers TRIP12 KpnI_to_pAc_fwd and TRIP12 XmaI_to_pAc_rev. The complete WWE domain (as encoded by TRIP12 transcript ENST 00000389044.8) was generated by insertion of a DNA fragment encoding for the TRIP12 amino acid sequence AAHQVGEDEISLSTLGRVYTIDFNSMQ, synthesized by Integrated DNA technologies (IDT) with 5′-PvuI and 3′-NruI restriction sites. The primers WWE domain PvuI_to_pAc_fwr and WWE domain NruI_to_pAc_rev were used to insert the fragment. The amino acid substitutions C2034A and R869A were generated by site-directed mutagenesis with the following primers: TRIP12 C2034A_fwr, TRIP12 C2034A_rev, TRIP12 R869A_fwr and TRIP12 R869A_rev.
The siRNA resistant versions of the pAc-GFP1-C1 TRIP12 constructs were generated by site-directed mutagenesis resulting in silent mutations using the following primers: TRIP12_siRES_fwr and TRIP12_siRES_rev.
Cloning of FLAG-tagged TRIP12 plasmids
The pcDNA 3.1 FLAG TRIP12 WT, C2034A and R869A plasmids were generated by Gibson assembly with the following homology primers TRIP12 GA_to_pcDNA_fwr and TRIP12 GA_to_pcDNA_rev. pcDNA 3.1 FLAG vector was linearized with Phusion High-Fidelity polymerase using primers pcDNA 3.1 FLAG_lin_fwr and pcDNA 3.1 FLAG_lin_rev.
Cloning of GST-TRIP12 WWE plasmids
GST-TRIP12 WWE plasmids were generated by PCR amplification of the WWE domain from full-length WT and R869A GFP-TRIP12 plasmids using primers WWE domain EcoRI_to_pGEX_fwr and WWE domain EagI_to_pGEX_rev.
The target vector pGEX 6P-1 was linearized with EcoRI and EagI for 2h at 37°C, followed by dephosphorylation with calf intestinal phosphatase (CIP, NEB) for 1 hour at 37°C. The PCR amplified TRIP12 WWE domain encoding sequences were digested with EcoRI and EagI for 2h at 37°C, followed by gel purification and ligation using the Rapid Ligation Kit (Roche).
PARP1 and ubiquitin expression plasmids
The pcDNA-Myc-PARP1 WT, pcDNA-Myc-PARP1 E998K, and the pCMV-FLAG-PARP1 plasmids were kindly provided by Michael Hottiger. The pcDNA-HA-Ubiquitin plasmid was kindly provided by Lorenza Penengo.
TRIP12 purification from HEK293T cells
HEK293T cells were transfected with plasmids encoding FLAG-TRIP12 WT or the point mutants R869A or C2034A. Cells were lysed in TNE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0,1% Igepal CA630, 1 mM EDTA) supplemented with 2 mM MgCl2, cOmplete inhibitor cocktail (Roche), phosphoSTOP (Roche) and 25 U/ml benzonase. One milligram of protein lysates was immunoprecipitated using protein G-Sepharose (GE Heltcare) coupled to 1 μg of anti-FLAG antibody for 3 hours at 4°C. After four washes with IP buffer the beads were first equilibrated in TE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl) supplemented with cOmplete inhibitor cocktail (Roche), 1 mM PMSF, and then re-suspended in 5 volumes of TE buffer with 0.3 mg/ml FLAG peptide (3x FLAG peptide, Sigma) for 1 hour at 4°C at low rpm. Samples were then centrifuged at 100 x g for two minutes and the supernatant containing the eluted protein was stored at −80°C.
In vitro ADP-ribosylation assay with recombinant proteins
To obtain auto-PARylated PARP1, human recombinant His-tagged PARP1 was incubated in reaction buffer RB (50 mM Tris-HCl pH 8.0, 4 mM MgCl2, 150 mM NaCl, 1x cOmplete inhibitor cocktail) supplemented with 20 μM - 200 μM of NAD+ and 200 nM of double-stranded annealed 40 bp long oligomer (40bp long oligomer_fwr and 40bp long oligomer_rev) for 15 minutes at 37°C. Auto-PARylation was stopped by addition of Olaparib to a final concentration of 10 μM.
In vitro ubiquitylation
Auto-PARylated PARP1 was bound to histidine beads (ProBond) for 45 minutes at 4°C. The beads were then equilibrated in reaction buffer (25 mM Tris-HCl pH 7.4, 100 mM NaCl) and equally distributed in reaction tubes. 20 μl of FLAG-TRIP12 WT or mutants were incubated with 0.3 μg human recombinant E1 Ubiquitin-activating enzyme (UBE1, E-305-025, Boston Biochem), 0.3 μg of human recombinant UBE2L3 (E2-640, Boston Biochem), and 1 μg of purified ubiquitin in 25 mM Tris-HCl pH 7.4, 100 mM NaCl at 30°C for 2h and 30 minutes at low rpm. Energy regeneration system was used to recycle ATP in the reaction (B-10, Boston Biochem). The supernatant was then collected by 100 x g centrifugation for 2 minutes and then boiled at 95°C for 5 minutes after the addition of 10x SDS-PAGE loading buffer. The remaining beads, containing the bound PARP1, were then boiled at 95°C for 5 minutes after the addition of 10x SDS-PAGE loading buffer. Supernatant and beads were run on two separate gels (Bolt 4%–12% Bis-Tris Plus, NW04122, Thermo Fisher Scientific) and transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare). Membranes were denatured using a solution containing 6 M guanidine hydrochloride, 20 mM Tris-HCl (pH 7.4), 1 mM PMSF, and 5 μM β-mercaptoethanol for 30 min at 4°C. After extensive washing in Tris-buffered saline (TBS), membranes were blocked in TBS buffer containing bovine serum albumin (BSA) (5%) overnight and then incubated with anti-Ub P4D1 antibody (Santa Cruz) for 1 h or with the indicated antibodies. For in vitro ubiquitylation assays to monitor TRIP12 activity in absence or presence of purified PAR chains, 20 μl of purified FLAG-TRIP12 WT or R869A mutant were incubated with 0.3 μg E1, 0.3 μg E2, 1 μg of ubiquitin, and purified PAR (4336-100-01, TREVIGEN) at 0, 0.2, or 1 μM final concentration in 25 mM Tris-HCl pH 7.4, 100 mM NaCl at 30°C for 2h and 30 minutes at low rpm. Energy regeneration system (B-10, Boston Biochem) was used in all reactions to recycle ATP. The reactions were stopped by adding 10x SDS-PAGE loading buffer and samples were boiled for 5 minutes at 95°C. Proteins were separated by SDS-PAGE (Bolt 4%–12% Bis-Tris Plus, NW04122, Thermo Fisher Scientific) and transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare). Membranes were denatured using a solution containing 6 M guanidine hydrochloride, 20 mM Tris-HCl (pH 7.4), 1 mM PMSF, and 5 μM β-mercaptoethanol for 30 min at 4°C. After extensive washing in Tris-buffered saline (TBS), membranes were blocked in TBS buffer containing bovine serum albumin (BSA) (5%) overnight and then incubated with anti-Ub P4D1 antibody (Santa Cruz) for 1 h.
GST pull-down assays
Expression of recombinant GST-fusion proteins was performed in BL21 (DE3) cells from pGEX-6P-1 encoding GST, GST-TRIP12-WWE WT or GST-TRIP12-WWE R869A. 4 mL of bacterial pre-culture from a single colony was inoculated into 400 mL of LB medium with 100 μg/ml ampicillin and grown at 37°C and 230 rpm until reaching OD600 of 0.6. The expression was induced with 0.5 mM IPTG for 2 hours at 37°C and 230 rpm. Bacteria were harvested by centrifugation at 5′000 x g for 20 min and resuspended in 20 mL binding buffer (50 mM Tris pH 7.4, 200 mM NaCl, 0.05% NP-40, 1 mM EDTA, 1 mM DTT, 10% glycerol, 1 mM PMSF, pepstatin, bestatin, and leupeptin 1 μg/ml each) followed by cell lysis by two passages through a French press. The supernatant was recovered by centrifugation at 25’000 x g for 30 min. The supernatant was loaded onto a pre-equilibrated 1 mL GSTrap HF column (GE Healthcare Life Sciences) and washed with 10 column volumes of binding buffer. Bound proteins were eluted with 10 column volumes of elution buffer (50 mM Tris pH 7.4, 15 0mM NaCl, 0.05% NP-40, 1 mM EDTA, 1 mM DTT, 10% glycerol, 40 mM glutathione, and 1 tablet cOmplete protease inhibitor tablet (Roche) per 50 mL buffer). Eluted fractions were analyzed by 10% SDS-PAGE. Peak fractions with the highest concentration were pooled and dialyzed against dialysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 20% glycerol) and the protein concentration was determined by Bradford assay.
For in vitro interactions with purified proteins, 2 μg of human recombinant PARP1 was incubated in reaction buffer RB (50 mM Tris-HCl pH 8.0, 4 mM MgCl2, 150 mM NaCl, 1x cOmplete inhibitor cocktail) supplemented with 20 μM - 200 μM NAD+ and 200 nM of annealed double-strand break-mimicking oligomer (40bp long oligomer_fwr and 40bp long oligomer_rev) for 15 minutes at 37°C. Meanwhile, purified GST, GST-TRIP12 WWE WT and GST-TRIP12 WWE R869A (4 μg each) were bound to glutathione Sepharose beads (GE Healtcare) in PBS for 45 minutes at 4°C. The beads were then equilibrated in TNE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0,1% Igepal CA630, 1 mM EDTA) supplemented with cOmplete inhibitor cocktail (Roche) and phosphoSTOP (Roche) and incubated with unmodified or auto-PARylated PARP1 for 2 hours at 4°C. The beads were collected by centrifugation, washed four times with TNE buffer and eluted by boiling in 10x SDS-PAGE sample buffer. Samples were run on SDS-PAGE and subjected to immunoblotting.
For in vitro interactions with whole cell extracts, HeLa cells were transfected with siPARG and treated with 1 mM H2O2 for 15 minutes. Cells were collected after two washes with cold PBS and lysates were prepared with TNE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0,1% Igepal CA630, 1 mM EDTA) supplemented with cOmplete inhibitor cocktail (Roche) and phosphoSTOP (Roche). 5 μg of GST, GST-TRIP12 WWE WT and GST-TRIP12 WWE R869A were bound to glutathione Sepharose beads (GE Healtcare) in PBS supplemented with 0.05% Igepal for 1 hour at 4°C. The GST-proteins were then washed twice in PBS and combined with 500 μg of HeLa cell lysates for 2 hours at 4°C. The beads were collected by centrifugation, washed three times with PBS and eluted by boiling in 10x SDS-PAGE sample buffer.
In vivo ubiquitylation
HEK293T or U-2 OS cells transfected with the indicated plasmids and siRNAs were washed twice with cold PBS and directly lysed on ice with RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Igepal CA630, 0.1% SDS, 0.1% Na-deoxycholic acid), supplemented with 2 mM MgCl2, cOmplete inhibitor cocktail (Roche), phosphoSTOP (Roche), 25 U/ml benzonase, 1 mM phenylmethylsulfonylfluoride (PMSF), and 10 mM N-ethylmaleimide (NEM). Lysates were incubated for 5 minutes at room temperature and then centrifuged at 15’000 x g for 15 minutes. 500 μg of lysates were incubated with protein G-Sepharose (GE Healtcare) coupled to 1 μg of anti-Myc or anti-FLAG antibody for 3 hours at 4°C. Beads were collected by centrifugation, washed four times with RIPA buffer and eluted by boiling in 10x SDS-PAGE sample buffer. Samples were subjected to SDS-PAGE for immunoblotting. For PARP1 ubiquitylation upon overexpression of TRIP12 wild-type or mutant and upon TRIP12 depletion, IP samples were adjusted based on input levels of PARP1 to correct for the effect of TRIP12 on PARP1 abundance and allow comparisons between samples containing similar PARP1 amounts.
Co-immunoprecipitation
HEK293T cells were treated as indicated and the cells were washed twice with cold PBS and directly lysed on ice in 500 μl of TNE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0,1% Igepal CA630, 1 mM EDTA) supplemented with 1 mM DTT, 2 mM MgCl2, cOmplete inhibitor cocktail (Roche), phosphoSTOP (Roche) and 25 U/ml benzonase. Cell lysates were incubated for 5 minutes at room temperature and then centrifuged at 15’000 x g for 15 minutes. 800 μg of lysates were then incubated with 1 μg of rabbit anti-TRIP12 antibody (A301-814A, Bethyl) for 3 hours at 4°C. Protein G-Sepharose beads (20 μl slurry, 17-0618-01, GE Healthcare) were added for an additional hour. Beads were then collected by centrifugation, washed four times with TNE buffer and eluted by boiling in 10x SDS-PAGE sample buffer. Samples were run on SDS-PAGE and subjected to immunoblotting.
HEK293T cells were transfected as indicated and cells were washed twice with cold PBS and directly lysed on ice in 500 μl TNE buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0,1% Igepal CA630, 1 mM EDTA) supplemented with 2 mM MgCl2, cOmplete inhibitor cocktail (Roche), phosphoSTOP (Roche) and 25 U/ml benzonase. Cell lysates were incubated for 5 minutes at room temperature and then centrifuged at 15’000 x g for 15 minutes. 500 μg of lysate was then incubated with protein G-Sepharose coupled to 1 μl of anti-FLAG or anti-GFP antibody for 12 hours at 4°C. Beads were collected by centrifugation, washed four times with TNE buffer and eluted by boiling in 10x SDS-PAGE sample buffer. Samples were subjected to SDS-PAGE for immunoblotting. For PARP1 interactions upon loss or gain of TRIP12 function, IP samples were adjusted based on PARP1 input signals to correct for the effect of TRIP12 on PARP1 abundance and allow comparisons between samples containing similar PARP1 amounts.
Chromatin fractionation
Cell pellets were collected by scraping the cells in 1x PBS supplemented with 1x protease inhibitor cocktail (cOmplete, Roche) and split in two equal fractions: (A) for the total proteome, cells were resuspended in 1x MNase buffer (0.3 M Sucrose, 50 mM Tris pH 7.5, 30 mM KCl, 7.5 mM NaCl, 4 mM MgCl2, 1 mM CaCl2, 0.125% NP-40, 0.25% Na-Deoxycholate) supplemented with 1x protease inhibitor cocktail and 10 U of MNase for every 5 million cells. Cells were incubated for 30 min at 37°C, boiled in 1x SDS-loading buffer for 5 min, spun down at 16’000 x g for 5 min and the supernatant was collected for immunochemical assays. (B) For chromatin-bound and soluble fractions, cells were resuspended in chromatin extraction buffer (10 mM HEPES pH 7.6, 3 mM MgCl2, 0.5% Triton X-100, 1 mM DTT) supplemented with 1x protease inhibitor cocktail. Cells were rotated for 30 min at room temperature and spun down at 1’300 x g for 10 min at 4°C. The pellet was kept and the supernatant was centrifuged at 16’000 x g for 5 min at 4°C and collected again (soluble fraction). The supernatant was boiled in 1x SDS-loading buffer for 5 min, spun down at 16’000 x g for 5 min and used for immunochemical assays. The pellet of the chromatin extraction step (chromatin-bound fraction) was resuspended in MNase buffer supplemented with 1x protease inhibitor cocktail and 10 U of MNase for every 5 million cells. Cells were incubated for 30 min at 37°C, boiled in 1x SDS-loading buffer for 5 min, spun down at 16’000 x g for 5 min and the supernatant was collected for immunochemical assays. Protein amounts were quantified using the standard Bradford method before addition of the SDS-PAGE loading buffer.
Immunoblotting
Proteins were separated by standard SDS-PAGE and transferred onto PVDF membranes. To reduce auto-PARylation of PARP1 interfering with PARP1 abundance measurements by western blot, cell extracts were treated with purified PARG to degrade PAR for 15 minutes at 37°C after cell lysis (Figures 1D, 1E, 4C, 4D, 4F, 1F, 1G, 1H, and 3B). Membranes were blocked with 5% milk in PBS-T (PBS + 0.1% Tween-20) for one hour at room temperature and incubated with primary antibodies over night at 4°C. Membranes were then washed three times with PBS-T and incubated with HRP-conjugated secondary antibodies for one hour at room temperature, washed again three times with PBS-T, and protein signals were detected using ECL™ Western Blotting Detection Reagent (Amersham™).
Clonogenic survival assay
Cells transfected with the indicated siRNAs or induced by addition of doxycycline were seeded at single cell density and exposed to PARPi at the indicated final concentration. All conditions were performed in triplicates. Cells were then incubated for 10 days and the number of colonies with more than 50 cells was counted after staining with crystal violet (0.5% crystal violet in 20% ethanol). GraphPad Prism 8.0 was used to display clonogenic survival data.
Cancer genomics analyses
A total of 102 breast cancer samples and 145 ovarian cancer samples, for which both transcriptomics and proteomics data are publicly available, were analyzed for correlations between transcript and protein levels (Goncalves et al., 2017). Proteomics data were generated by the NCI/NIH Clinical Proteomic Tumor Analysis Consortium (CPTAC) (Edwards et al., 2015; Ellis et al., 2013). Transcriptomics data were generated by the TCGA Research Network: https://www.cancer.gov/tcga (Grossman et al., 2016). As copy number loss (CNL) is a primary confounding factor for protein expression, samples with PARP1 CNL were excluded from the analyses. For transcriptomics analyses, raw TRIP12 and PARP1 transcript signals were normalized to the total RNA signals per sample. For proteomics analyses, the unshared log ratio was used as raw value. The ovarian cancer proteome data were generated in two batches, which were merged with z-score normalization within each batch. For BRCA1/2 mutation records, we matched rsID from reported somatic mutations with the Ensembl SNP database (retrieved from R BioMart library, Ensembl Variation 99) with missense variants and labeled the samples as BRCA mutated if either BRCA1 or BRCA2 contained such a mutation. Samples without such mutations were considered as BRCA wild-type (WT). To determine the effect of TRIP12 transcript levels on PARP1 protein levels, a two-step linear regression was used: First, a linear model was built with PARP1 protein levels as response variable and PARP1 transcript levels as predictor. The residual PARP1 protein levels from the first model were then used in the second linear model for TRIP12 transcript association.
Protein sequence alignments
Multiple sequence alignment of WWE domains was performed analogous to previous work (Wang et al., 2012) using Clustal Omega:
Quantification and Statistical Analysis
For QIBC analysis, between 9 and 20 images per condition, depending on the microscope objective used and the cell confluence (i.e., 9 images with a 10x objective and 20 images with a 20x objective) were acquired in an unbiased fashion from asynchronous cell populations grown on glass coverslips. Typically, between 1000 and 5000 cells per condition were analyzed, and representative single cell data of cell cohorts of comparable size are shown as one- or two-dimensional cell cycle-resolved scatterplots. Fluorescence intensities were quantified and are depicted as arbitrary units. Color-coded scatterplots of asynchronous cell populations were generated with Spotfire data visualization software (TIBCO). Within one experiment, similar cell numbers were compared for the different conditions. For visualizing discrete data in scatterplots, mild jittering (random displacement of data points along discrete data axes) was applied in order to demerge overlapping data points. Representative scatterplots and quantifications of independent experiments are shown. GraphPad Prism 8.0 was used for depicting data as violin plots and unpaired t test was used for statistical analysis. qPCRs to confirm knockdown efficiencies were performed in technical triplicates with results depicted as means ± SD. Fiji was used for quantification of Western Blot band intensities. For laser micro-irradiation, the average pixel intensity of laser tracks was measured within the locally irradiated area (Idamage), in the nucleoplasm outside the locally irradiated area (Inucleoplasm) and in a region not containing cells in the same field of view (Ibackground). The level of protein accumulation relative to the protein level in the nucleoplasm was calculated as follows: (Idamage - Ibackground)/(Inucleoplasm - Ibackground). The data are presented as means ± SD obtained from 15 to 30 cells per condition. GraphPad Prism 8.0 was used to display clonogenic survival data, represented as means ± SD. ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ∗∗, p < 0.01; ∗, p < 0.05; p ≥ 0.05 was considered not significant.
Acknowledgments
We acknowledge the University of Zurich Center for Microscopy and Image Analysis for microscopy support and are indebted to the following researchers for scientific input and for providing reagents and technical advice: Mohamed Bentires-Alj, Keith Caldecott, Daniel Durocher, Michael Hottiger, Sarah Charlotte Larsen, Jiri Lukas, Michael Lund Nielsen, Lorenza Penengo, Sara Przetocka, Sven Rottenberg, Raffaella Santoro, Alessandro Sartori, Stoyno Stoynov, and Maries van den Broek. Cancer patient data used in this publication were generated by the Clinical Proteomic Tumor Analysis Consortium (NCI/NIH). We thank DMMD, IMCR, and IMLS members for helpful discussions, and our lab members for feedback on the manuscript. Research in the lab of M.A. is supported by the Swiss National Science Foundation (grants 150690 and 179057), the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation program (ERC-2016-STG 714326), the Swiss Cancer Research Foundation (KFS-4406-02-2018), and the UZH Candoc and Postdoc programs.
Author Contributions
M.G. performed most of the experiments and developed the project, helped by R.I.; Q.H. and M.B. performed human cancer data analyses; M.A. conceived and supervised the project and wrote the manuscript draft with help from M.G.; and all authors read and edited the manuscript.
Declaration of Interests
M.G. and M.A. are listed as inventors on a UZH patent application for determining PARPi responsiveness.
Published: August 4, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107985.
Supplemental Information
References
- Aleksandrov R., Dotchev A., Poser I., Krastev D., Georgiev G., Panova G., Babukov Y., Danovski G., Dyankova T., Hubatsch L. Protein dynamics in complex DNA lesions. Molecular Cell. 2018;69:1046–1061 e1045. doi: 10.1016/j.molcel.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Alemasova E.E., Lavrik O.I. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019;47:3811–3827. doi: 10.1093/nar/gkz120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M., Neelsen K.J., Teloni F., Pozdnyakova I., Pellegrino S., Grøfte M., Rask M.D., Streicher W., Jungmichel S., Nielsen M.L., Lukas J. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose) Nat. Commun. 2015;6:8088. doi: 10.1038/ncomms9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussios S., Karihtala P., Moschetta M., Karathanasi A., Sadauskaite A., Rassy E., Pavlidis N. Combined strategies with poly (ADP-Ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer: a literature review. Diagnostics (Basel) 2019;9:9. doi: 10.3390/diagnostics9030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Shan J., Zhu W.G., Qin J., Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464:624–627. doi: 10.1038/nature08820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.C., Feng W., Lim P.X., Kass E.M., Jasin M. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu. Rev. Cancer Biol. 2018;2:313–336. doi: 10.1146/annurev-cancerbio-030617-050502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun E., Tuna G., Jaruga P., Tona A., Erdem O., Dizdaroglu M. Identification and quantification of DNA repair protein poly(ADP ribose) polymerase 1 (PARP1) in human tissues and cultured cells by liquid chromatography/isotope-dilution tandem mass spectrometry. DNA Repair (Amst.) 2019;75:48–59. doi: 10.1016/j.dnarep.2019.01.008. [DOI] [PubMed] [Google Scholar]
- Dantuma N.P., van Attikum H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016;35:6–23. doi: 10.15252/embj.201592595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRosa P.A., Wang Z., Jiang X., Pruneda J.N., Cong F., Klevit R.E., Xu W. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature. 2015;517:223–226. doi: 10.1038/nature13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H., Glodzik D., Morganella S., Yates L.R., Staaf J., Zou X., Ramakrishna M., Martin S., Boyault S., Sieuwerts A.M. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards N.J., Oberti M., Thangudu R.R., Cai S., McGarvey P.B., Jacob S., Madhavan S., Ketchum K.A. The CPTAC data portal: a resource for cancer proteomics research. J. Proteome Res. 2015;14:2707–2713. doi: 10.1021/pr501254j. [DOI] [PubMed] [Google Scholar]
- Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., Townsend R.R., Kinsinger C., Mesri M., Rodriguez H., Liebler D.C., Clinical Proteomic Tumor Analysis Consortium (CPTAC) Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustermann S., Wu W.F., Langelier M.F., Yang J.C., Easton L.E., Riccio A.A., Pascal J.M., Neuhaus D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell. 2015;60:742–754. doi: 10.1016/j.molcel.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraoni I., Graziani G. Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancers (Basel) 2018;10:487. doi: 10.3390/cancers10120487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong P.C., Yap T.A., Boss D.S., Carden C.P., Mergui-Roelvink M., Gourley C., De Greve J., Lubinski J., Shanley S., Messiou C. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010;28:2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B.A., Kraus W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Goncalves E., Fragoulis A., Garcia-Alonso L., Cramer T., Saez-Rodriguez J., Beltrao P. Widespread post-transcriptional attenuation of genomic copy-number variation in cancer. Cell Systems. 2017;5:386–398 e384. doi: 10.1016/j.cels.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson T., Altmeyer M., Savic V., Toledo L., Dinant C., Grøfte M., Bartkova J., Poulsen M., Oka Y., Bekker-Jensen S. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012;150:697–709. doi: 10.1016/j.cell.2012.06.039. [DOI] [PubMed] [Google Scholar]
- Gupte R., Liu Z., Kraus W.L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017;31:101–126. doi: 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H., Kalasova I., Demin A.A., Pennicott L.E., Cihlarova Z., Caldecott K.W. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. Cell. 2018;71:319–331.e3. doi: 10.1016/j.molcel.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M.O. Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu. Rev. Biochem. 2015;84:227–263. doi: 10.1146/annurev-biochem-060614-034506. [DOI] [PubMed] [Google Scholar]
- Kang H.C., Lee Y.I., Shin J.H., Andrabi S.A., Chi Z., Gagné J.P., Lee Y., Ko H.S., Lee B.D., Poirier G.G. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc. Natl. Acad. Sci. USA. 2011;108:14103–14108. doi: 10.1073/pnas.1108799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima L., Idogawa M., Mita H., Shitashige M., Yamada T., Ogi K., Suzuki H., Toyota M., Ariga H., Sasaki Y., Tokino T. CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J. Biol. Chem. 2012;287:12975–12984. doi: 10.1074/jbc.M111.321828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019;38:e101379. doi: 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.J., Lee S.Y., Kim S., Chung J.M., Kwon M., Yoon J.H., Park S., Hwang Y., Park D., Lee J.S., Kang H.C. A novel reciprocal crosstalk between RNF168 and PARP1 to regulate DNA repair processes. Mol. Cells. 2018;41:799–807. doi: 10.14348/molcells.2018.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Wu J., Paudyal S.C., You Z., Yu X. CHFR is important for the first wave of ubiquitination at DNA damage sites. Nucleic Acids Res. 2013;41:1698–1710. doi: 10.1093/nar/gks1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C.J., Ashworth A. BRCAness revisited. Nat. Rev. Cancer. 2016;16:110–120. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- Lord C.J., Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D., Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makvandi M., Xu K., Lieberman B.P., Anderson R.C., Effron S.S., Winters H.D., Zeng C., McDonald E.S., Pryma D.A., Greenberg R.A., Mach R.H. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy. Cancer Res. 2016;76:4516–4524. doi: 10.1158/0008-5472.CAN-16-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makvandi M., Pantel A., Schwartz L., Schubert E., Xu K., Hsieh C.J., Hou C., Kim H., Weng C.C., Winters H. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin. Invest. 2018;128:2116–2126. doi: 10.1172/JCI97992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelena J., Lezaja A., Teloni F., Schmid T., Imhof R., Altmeyer M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018;9:2678. doi: 10.1038/s41467-018-05031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J., Huang S.Y., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muvarak N.E., Chowdhury K., Xia L., Robert C., Choi E.Y., Cai Y., Bellani M., Zou Y., Singh Z.N., Duong V.H. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents—a potential therapy for cancer. Cancer Cell. 2016;30:637–650. doi: 10.1016/j.ccell.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen K.J., Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- O’Connor M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Oplustilova L., Wolanin K., Mistrik M., Korinkova G., Simkova D., Bouchal J., Lenobel R., Bartkova J., Lau A., O’Connor M.J. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2013;11:3837–3850. doi: 10.4161/cc.22026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Yoon S.K., Yoon J.B. The HECT domain of TRIP12 ubiquitinates substrates of the ubiquitin fusion degradation pathway. J. Biol. Chem. 2009;284:1540–1549. doi: 10.1074/jbc.M807554200. [DOI] [PubMed] [Google Scholar]
- Pellegrino S., Altmeyer M. Interplay between ubiquitin, SUMO, and poly(ADP-ribose) in the cellular response to genotoxic stress. Front. Genet. 2016;7:63. doi: 10.3389/fgene.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penson R.T., Valencia R.V., Cibula D., Colombo N., Leath C.A., 3rd, Bidziński M., Kim J.W., Nam J.H., Madry R., Hernández C. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): a randomized Phase III trial. J. Clin. Oncol. 2020;38:1164–1174. doi: 10.1200/JCO.19.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt S.J., Krastev D.B., Brandsma I., Dréan A., Song F., Aleksandrov R., Harrell M.I., Menon M., Brough R., Campbell J. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018;9:1849. doi: 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y., O’Connor M.J., de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016;8:362ps17. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A., Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017;18:610–621. doi: 10.1038/nrm.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- Tang D., Kang R., Berghe T.V., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teloni F., Altmeyer M. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 2016;44:993–1006. doi: 10.1093/nar/gkv1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teloni F., Michelena J., Lezaja A., Kilic S., Ambrosi C., Menon S., Dobrovolna J., Imhof R., Janscak P., Baubec T., Altmeyer M. Efficient Pre-mRNA cleavage prevents replication-stress-associated genome instability. Mol. Cell. 2019;73:670–683.e12. doi: 10.1016/j.molcel.2018.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A., Murai J., Pommier Y. The evolving landscape of predictive biomarkers of response to PARP inhibitors. J. Clin. Invest. 2018;128:1727–1730. doi: 10.1172/JCI120388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutt A., Robson M., Garber J.E., Domchek S.M., Audeh M.W., Weitzel J.N., Friedlander M., Arun B., Loman N., Schmutzler R.K. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- Wang Z., Michaud G.A., Cheng Z., Zhang Y., Hinds T.R., Fan E., Cong F., Xu W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., An R., Umanah G.K., Park H., Nambiar K., Eacker S.M., Kim B., Bao L., Harraz M.M., Chang C. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;354:354. doi: 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.W., Wang H., Poitras M.F., Coombs C., Bowers W.J., Federoff H.J., Poirier G.G., Dawson T.M., Dawson V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu S., Mickanin C., Feng Y., Charlat O., Michaud G.A., Schirle M., Shi X., Hild M., Bauer A. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011;13:623–629. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- Zimmermann M., Murina O., Reijns M.A.M., Agathanggelou A., Challis R., Tarnauskaitė Ž., Muir M., Fluteau A., Aregger M., McEwan A. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 2018;559:285–289. doi: 10.1038/s41586-018-0291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze code or datasets (such as microarray, genomics, transcriptomics, protein structure data).