

Summary
N6 methyladenosine (m6A) is the most abundant RNA modification, but little is known about its role in mammalian hematopoietic development. Here, we show that conditional deletion of the m6A writer METTL3 in murine fetal liver resulted in hematopoietic failure and perinatal lethality. Loss of METTL3 and m6A activated an aberrant innate immune response, mediated by the formation of endogenous double-stranded RNAs (dsRNAs). The aberrantly formed dsRNAs were long, highly m6A modified in their native state, characterized by low folding energies, and predominantly protein-coding. We identified coinciding activation of pattern recognition receptor pathways normally tasked with the detection of foreign dsRNAs. Disruption of the aberrant immune response via abrogation of downstream Mavs or Rnasel signaling partially rescued the observed hematopoietic defects in METTL3-deficient cells in vitro and in vivo. Our results suggest that m6A modification protects against endogenous dsRNA formation and a deleterious innate immune response during mammalian hematopoietic development.
Keywords: innate immune response, dsRNA, RNA modification, N6-methyladenosine, METTL3, hematopoietic development
Graphical Abstract
In Brief
Little is known about the role of N6-methyladenosine (m6A) modifications in mammalian hematopoietic development, Gao et al. find that m6A modification of endogenous transcripts preserves their recognition as self by preventing aberrant formation of double-stranded RNA. Deletion of m6A writer Mettl3 and loss of m6A activates pattern recognition receptor pathways, culminating in a deleterious innate immune response and hematopoietic failure.
Introduction
N6-methyladenosine (m6A) is an abundant modification found in RNAs which affects the stability, translation and conformation of the modified transcripts (Desrosiers et al., 1974; Frye et al., 2018; Liu et al., 2015). The effects of m6A are determined by m6A writers, erasers and readers (Meyer and Jaffrey, 2017; Patil et al., 2017). Many studies have explored the mechanisms by which m6A modification modulates transcript fates (Zhao et al., 2017a). For instance, m6A methylation promotes mRNA degradation and turnover (Batista et al., 2014; Wang et al., 2014a) while enhancing translational efficiency (Wang et al., 2015). In the cellular and the organismal contexts, m6A RNA methylation impacts key processes, such as maintenance of pluripotency in embryonic stem cells (Batista et al., 2014; Bertero et al., 2018; Geula et al., 2015), X chromosome inactivation (Patil et al., 2016), maternal to zygotic transition (Zhao et al., 2017b) and cytokine signaling (Li et al., 2017a; Tong et al., 2018). In embryonic stem cells, m6A reduces stability of key naïve pluripotency-promoting transcripts and is necessary for stem cell differentiation. Deletion of the m6A methyltransferase METTL3, the catalytic component of the m6A writer complex, enforces a naïve pluripotent state (Batista et al., 2014; Geula et al., 2015). This raises the question whether m6A RNA methylation similarly regulates self-renewal and differentiation in somatic stem cells.
Hematopoiesis affords unique insights into somatic stem cell function. Hematopoietic stem cells (HSCs) represent the most widely applied stem cell therapy in clinical practice, and native HSC pathways are coopted in hematopoietic malignancies such as leukemia (Orkin and Zon, 2008). During zebrafish embryogenesis, m6A is essential for the early specification of hematopoietic stem and progenitor cells (HSPCs) during the endothelial-to-hematopoietic transition (Zhang et al., 2017). Knockdown of METTL3 in human cord blood CD34+ HSPCs and primary acute myeloid leukemia (AML) in vitro promotes terminal myeloid differentiation (Vu et al., 2017), while inhibition of METTL14, which heterodimerizes with METTL3 in the methyltransferase complex, promotes myeloid differentiation of HSPCs and AML cells (Weng et al., 2018). These studies raise the possibility of targeting m6A RNA modification for therapeutic purposes, but little is known about the role of m6A in mammalian hematopoietic development.
Mx1-Cre is widely used for conditional deletion of floxed genes in a hematopoiesis-specific manner. However, interferon-responsive Mx1-mediated Cre expression requires induction of an interferon-mediated immune response with the double-stranded RNA (dsRNA) mimic polyinosinic acid: polycytidylic acid (pI:pC). Therefore Mx1-Cre-Mettl3fl/fl models, as used by other groups, are limited in their ability to discern the effects of Mettl3 deletion on innate immune signaling (Cheng et al., 2019; Lee et al., 2019). Previous work has shown the importance of RNA editing in protecting HSCs from aberrant immune activation (Hartner et al., 2009), and recent studies support a role of m6A in modulating the antiviral immune response (Chen et al., 2019; Winkler et al., 2019). To explore the impact of the m6A modification, its potential roles in immune activity and any corresponding effects in hematopoiesis, we generated hematopoietic-specific Vav-Cre+-Mettl3fl/fl (vcMettl3−/−) mice. This allows us to examine the role of m6A in fetal liver hematopoiesis without the need for exogenous Cre induction.
We found that loss of METTL3 and m6A in vcMettl3−/− mice resulted in hematopoietic failure with expansion of Lin−Sca-1+c-Kit+ (LSK) HSPCs that were defective in the production of progenitors and mature blood cells. Further analysis revealed that loss of m6A resulted in robust transcriptional upregulation of interferon stimulated genes (ISGs) and 2’,5’-oligoadenylate synthetase (Oas) genes, which function in the detection of dsRNA pathogen-associated molecular patterns. This suggested the induction of a dsRNA-mediated innate immune response upon depletion of m6A. Concordantly, we found downstream activation of the OAS-RNase L and PKR-eIF2α pathways, and upregulation of the dsRNA sensors MDA5 and RIG-I (Silverman, 2007; Zhou et al., 1993). Via J2 anti-dsRNA mediated RNA immunoprecipitation we directly demonstrated de novo formation of endogenous dsRNAs in vcMettl3−/− fetal liver cells, which were substantially enriched for long and highly m6A modified transcripts characterized by low folding energies, signifying higher propensity to form dsRNAs. The relevance of the dsRNA response to the observed hematopoietic phenotype is substantiated by partial rescue of hematopoietic colony formation and contribution to in vivo hematopoiesis upon knockdown of Rnasel or deletion of the common innate immune response mediator Mavs.
In summary, we show that loss of METTL3 and m6A results in aberrant activation of a deleterious dsRNA-induced innate immune response, contributing to hematopoietic failure.
Results
Hematopoietic deletion of Mettl3 results in bone marrow failure and embryonic lethality
To determine the role of METTL3 and RNA m6A modification during murine hematopoiesis, we developed a model of Vav-Cre mediated deletion of the Mettl3 gene. Vav is expressed exclusively in hematopoietic stem cells starting at E10.5-11.5, resulting in efficient deletion of the floxed gene by E13.5-E14.5 (Stadtfeld and Graf, 2005). Crossing of Mettl3 heterozygous mice (Vav-Cre+-Mettl3wt/fl; hereafter referred to as vcMettl3+/−) to Vav-Cre−-Mettl3fl/fl wildtype (vcMettl3+/+) mice failed to yield viable Vav-Cre+-Mettl3fl/fl (vcMettl3−/−) offspring (Figure 1A and S1A). While vcMettl3−/− pups were rarely observed at birth, typical Mendelian ratios were preserved at E14.5, when fetal liver is the predominant site of definitive hematopoiesis (Figure S1A). Careful tracking of survival at different stages of development demonstrated predominantly late embryonic and perinatal lethality (Figures S1A-S1D). Examination of rare vcMettl3−/− newborn mice revealed smaller size, pale appearance, and significant pancytopenia (Figures S1D and S1E), accompanied by significant reduction in bone marrow cellularity (Figure S1F). These experiments show that deletion of Mettl3 in fetal liver HSCs resulted in bone marrow failure and perinatal lethality.
Figure 1. Loss of METTL3 results in dysfunctional fetal liver hematopoietic stem cells.
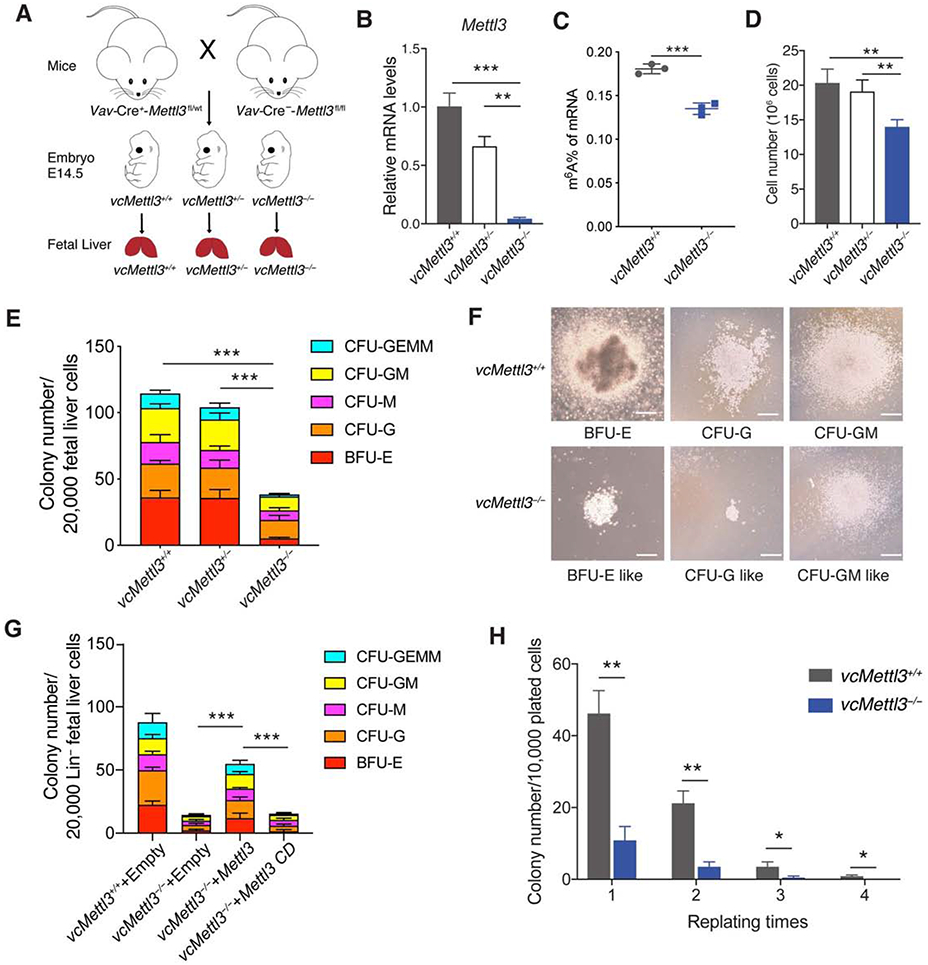
(A) Breeding scheme to obtain Vav-Cre-Mettl3fl/fl mice vcMettl3+/+, vcMettl3+/−, and vcMettl3−/− E14.5 fetal livers.
(B) Quantitation of Mettl3 expression via Q-RT-PCR, normalized to Gapdh (n=3 biological replicates per group).
(C) Quantitation of m6A of mRNA in vcMettl3+/+ and vcMettl3−/− E14.5 fetal livers via ELISA (n=3 biological replicates per group).
(D) Fetal liver cell numbers at E14.5 in vcMettl3+/+, vcMettl3+/− and vcMettl3−/− embryos (n=5 biological replicates per group), counted after mechanical dissociation and red blood cell lysis.
(E, F) Assessment of colony forming unit (CFU) potential of vcMettl3+/+, vcMettl3+/− and vcMettl3−/− E14.5 fetal liver cells (E) (n=3 biological replicates per group). Demonstration of typical colonies in vcMettl3+/+ and vcMettl3−/− cultures (F). CFU-GEMM, CFU-granulocyte, erythrocyte, monocyte, megakaryocyte; CFU-GM, CFU-granulocyte, macrophage; CFU-M, CFU-macrophage; CFU-G, CFU-granulocyte; BFU-E, burst-forming unit-erythroid. Scale bar, 100 μm.
(G) CFUs of E14.5 lineage depleted fetal liver cells transduced with empty, Mettl3 and catalytically dead Mettl3 (Mettl3 CD) expressing retroviral vectors (n=3 biological replicates per group).
(H) Serial colony replating assay enumerating colonies/10,000 plated cells (n=3 biological replicates per group).
Data are represented as mean ± SEM, and representative of at least two independent experiments; The P values were calculated using two-tailed Student's t test. * p<0.05, ** p<0.01, *** p<0.001.
See also Figure S1.
Loss of METTL3 results in an expanded but dysfunctional HSPC population
To explore the mechanism of hematopoietic failure in vcMettl3−/− mice, we evaluated hematopoiesis at E14.5 prior to fetal loss (Figure S1A). At E14.5, vcMettl3−/− embryos and fetal livers were grossly indistinguishable from their vcMettl3+/+ and vcMettl3+/− counterparts (Figure S1B). Efficient deletion and loss of Mettl3 expression in E14.5 vcMettl3−/− fetal livers was confirmed by Q-RT-PCR (Figure 1B) and western blot (Figure S1G), which also confirmed the known loss of METTL3’s binding partner, METTL14, at the protein level (Wang et al., 2014b). Deletion of Mettl3 resulted in a significant reduction of mRNA m6A modification as assayed by ELISA (Figure 1C), and verified for select transcripts, known to be highly m6A modified, via m6A RIP-PCR (Figure S1H). Deletion of Mettl3 was specific to hematopoietic tissue (Figure S1I).
We next analyzed the fetal liver hematopoiesis phenotypically and functionally. vcMettl3−/− fetal livers exhibited a significant reduction in total cellularity compared to vcMettl3+/+ and vcMettl3+/− fetal livers (Figure 1D) and reduction in mature erythroid progenitors by histology (Figure S2A). Flow cytometric analysis of lineage differentiation revealed delayed erythroid maturation (Figures S2B and S2C), as well as lineage skewing within the myeloid lineage from differentiated granulocytes to immature monocytes (Figures S2D and S2E).
Given the observed hematopoietic failure, we tested the ability of vcMettl3−/− fetal liver HSPCs to give rise to colonies in vitro. Colony forming unit (CFU) activity was severely compromised in vcMettl3−/− fetal liver cells, with a significant reduction in the formation of all colony types (Figure 1E), and abnormal colony size and morphology (Figure 1F) compared to vcMettl3+/+ fetal liver. Enforced over-expression of METTL3 in vcMettl3−/− fetal liver via retroviral transduction rescued colony formation ability, while over-expression of catalytically dead METTL3 or transduction with empty vector did not (Figure 1G); this confirms that the HSPC defect was indeed mediated by loss of the METTL3 m6A methyltransferase activity. Defective serial replating ability of vcMettl3−/− HSPCs was indicative of a self-renewal defect (Figure 1H).
To characterize these defects in HSPC activity, we analyzed the hierarchical composition of the HSPC compartments in E14.5 fetal livers. vcMettl3−/− fetal livers had an increased percentage of Lin−Sca-1+c-Kit+ (LSK) HSPCs (Figures 2A and 2B) at the expense of Lin−c-Kit+Sca-1− (LK) progenitors (Figures 2A and 2C). Despite decreased overall fetal liver cellularity, absolute numbers of LSK HSPCs were significantly increased in vcMettl3−/− fetal livers (Figure 2D). Analysis of subpopulations within the LSK compartment showed an increase in phenotypic long-term HSC (LT-HSC; Lin−Sca-1+c-Kit+CD34−Flt3−) and short-term HSC (ST-HSC; Lin−Sca-1+c-Kit+CD34+Flt3−) at the expense of Flt3+ multi-potent progenitors (MPP; Lin−Sca-1+c-Kit+CD34+Flt3+) (Figures 2E and 2F). These findings demonstrate that loss of METTL3 resulted in accumulation of phenotypically immature HSC populations that were functionally impaired. Indeed, analysis of the LK progenitor compartment in E14.5 fetal liver revealed a significant decrease of common myeloid progenitors (CMP; Lin−Sca-1−c-Kit+CD34+CD16/32−) and granulocyte-macrophage progenitors (GMP; Lin−Sca-1−c-Kit+CD34+CD16/32+) (Figures S2F and S2G). Annexin V staining showed no increase of apoptotic rate in vcMettl3−/− compared to vcMettl3+/+ fetal livers (Figures 2G and 2H), while BrdU and 7AAD labeling did reveal a significant proliferative defect with reduced BrdU uptake in vcMettl3−/− fetal liver cells (Figures 2I and 2J). Together, these data indicate that loss of RNA m6A methylation resulted in defective fetal hematopoietic progenitor proliferation, lineage commitment and maturation, with accumulation of immature HSPCs and resultant hematopoietic failure.
Figure 2. Mettl3-deficient fetal liver hematopoietic stem and progenitor cells show aberrant differentiation trajectory and impaired proliferation.
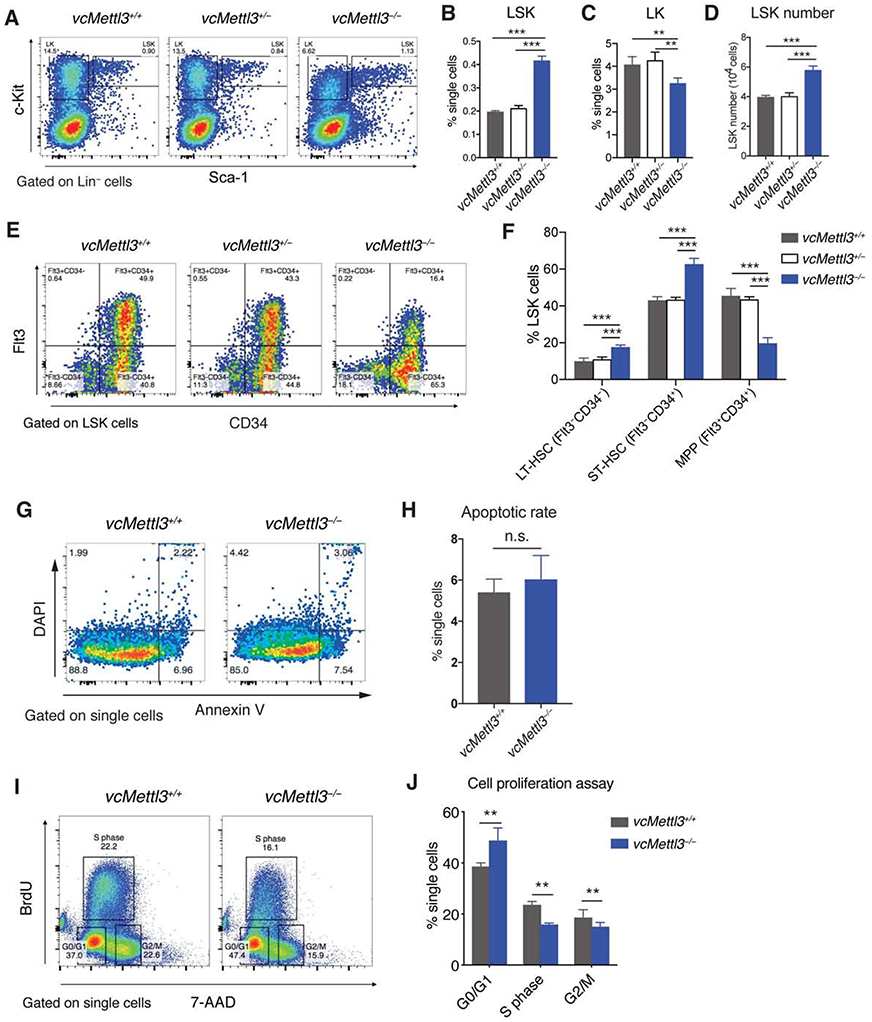
(A-D) Determination of LSK and LK cell distribution in vcMettl3+/+, vcMettl3+/− and vcMettl3−/− E14.5 fetal livers by flow cytometry (A). Quantitation relative to single cell population of LSK cells (B) and LK cells (C), and absolute LSK cell numbers per fetal liver (D; n=5 biological replicates per group).
(E and F) Determination (E) and quantification (F) of phenotypic hematopoietic stem cell subsets in vcMettl3+/+ and vcMettl3−/− E14.5 fetal livers via flow-cytometric identification of long-term (LT) (Flt3−CD34− LSK) and short-term (ST) (Flt3−CD34+ LSK) hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs) (Flt3+CD34+ LSK; n = 3 biological replicates per group).
(G and H) Determination (G) and quantification (H) of apoptotic rate in fetal livers via annexin V staining (n = 3 biological replicates per group).
(I and J) Assessment (I) and quantification (J) of fetal liver cell proliferation via BrdU uptake and 7AAD staining of DNA content (n = 3 biological replicates per group).
Data are represented as mean ± SEM and representative of at least three independent experiments; the p values in (B)–(D) and (H) were calculated using two-tailed Student’s t test. n.s., not statistically significant; **p < 0.01; ***p < 0.001. The p values in (F) and (J) were calculated using two-way ANOVA. **p < 0.01; ***p < 0.001.
See also Figures S2 and S3.
To determine whether the observed stem cell defects were cell-intrinsic, we performed fetal liver cell transplantation into lethally irradiated congenic recipients. While vcMettl3+/+ fetal liver cells fully rescued lethally irradiated Pep3b recipient mice, all recipients of vcMettl3−/− donor fetal liver cells died within two weeks post-transplant (Figure S3A). Flow cytometric evaluation 8 days post-transplantation prior to death from graft failure demonstrated nearly absent contribution of CD45.2+ vcMettl3−/− fetal liver cells to bone marrow or peripheral blood chimerism. By contrast, CD45.2+ vcMettl3+/+ fetal liver cells represented the majority of recovering cells in recipients of vcMettl3+/+ fetal liver cells (Figures S3B-S3D). As the prompt death of vcMettl3−/− fetal liver recipient mice precluded detailed analysis, we performed competitive transplantation assays (Figure S3E). We co-injected lethally irradiated CD45.1/2 recipient mice with either CD45.2+ vcMettl3+/+ or vcMettl3−/− fetal liver cells at a 1:1 ratio with CD45.1+ competitor bone marrow cells. Recipients of both vcMettl3+/+ and vcMettl3−/− competitive transplants survived. However, vcMettl3−/− fetal liver cells failed to contribute to recipient bone marrow and peripheral blood chimerism at 8 weeks post-transplant (Figures S3F-S3H). We ruled out a contributory homing defect by staining vcMettl3+/+ and vcMettl3−/− fetal liver cells with the cell tracer dye CFSE, followed by injection of labeled cells into lethally irradiated recipient mice. After 16 hours, homing efficiency to the bone marrow and spleen was unchanged between recipients of vcMettl3+/+ and vcMettl3−/− cells (Figures S3I and S3J). In summary, while vcMettl3−/− cells homed to recipient marrow, they failed to reconstitute lethally irradiated recipient mice, confirming a cell-intrinsic HSPC defect in vcMettl3−/− mice.
Loss of METTL3 elicits expression of innate immune response genes
To identify gene expression changes accounting for the observed phenotype in vcMettl3−/− fetal liver HSPCs, we performed bulk and single cell RNA sequencing on sorted LSK cells from independent littermate E14.5 vcMettl3+/+ and vcMettl3−/− fetal livers (Figure 3A).
Figure 3. Loss of METTL3 results in significant upregulation of genes of the innate immune response in LSK cells.
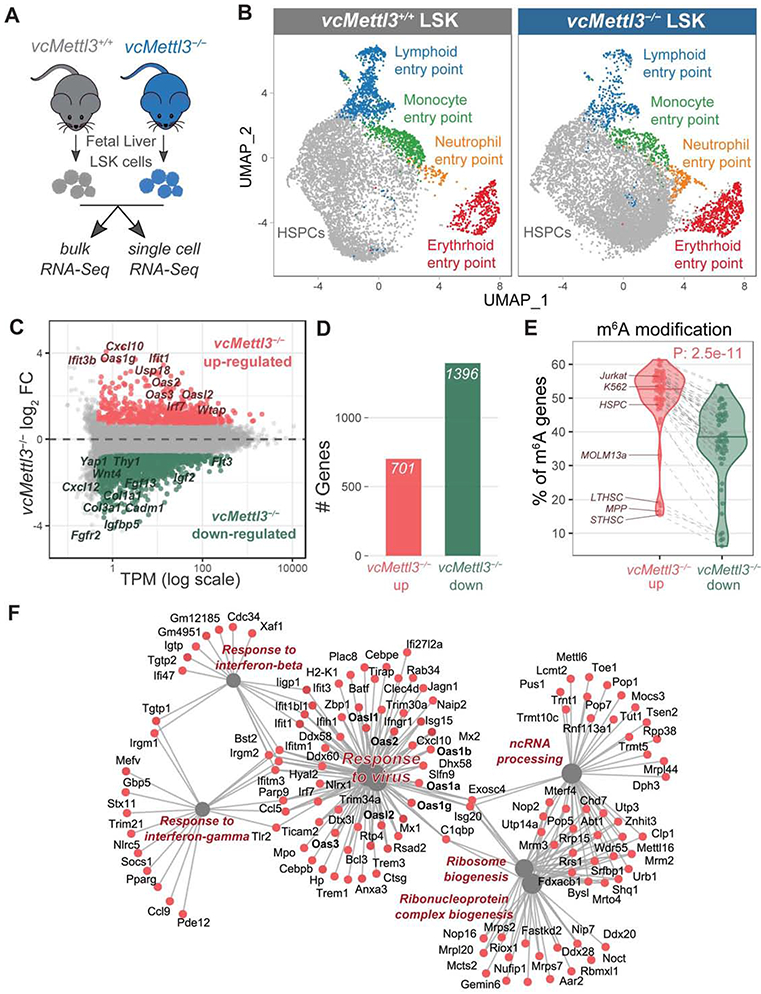
(A) Schematics of the experimental design for bulk and single cell RNA-Seq.
(B) UMAP (Uniform Manifold Approximation and Projection) representation of vcMettl3+/+ and vcMettl3−/− LSK cells, based on single-cell RNA sequencing (scRNA-seq). Clusters corresponding to entry points for differentiation toward mature cell types are highlighted (n = 2 biological replicates per group).
(C) Scatter plot of gene expression average levels (x) and fold changes (y) in vcMettl3−/− versus vcMettl3+/+ LSK cells, examined by bulk RNA-seq. Significantly upregulated and downregulated genes are highlighted in red and green, respectively (FC, fold change; TPM, transcript per million; n = 3 biological replicates per group).
(D) Number of upregulated and downregulated genes in vcMettl3−/− LSK cells (n=3 biological replicates per group).
(E) Percentage of genes with annotated m6A peaks among genes either upregulated or downregulated upon vcMettl3−/−. The analysis was performed with 52 different published m6A datasets, each corresponding to a dot in the violin plot. Hematopoietic datasets are highlighted.
(F) Gene-Concept network visualization of genes upregulated in vcMettl3−/− LSK cells (in red) and their enriched pathways (in grey).
The P value in (E) was calculated using two-tailed Wilcoxon rank-sum test.
Expression changes in phenotypically determined cell populations may be attributable to changes in cell population composition. To dissect changes within the LSK population at single cell resolution, we performed 3’ single cell RNA sequencing on vcMettl3+/+ and vcMettl3−/− LSK cells in duplicate using a microfluidic chip-based platform (Dura et al., 2018). A total of 6,621 vcMettl3+/+ and 8,326 vcMettl3−/− LSK cells were sequenced, with at least 500 genes per cell (Figure S4A). Based on expression markers previously identified in murine LSK populations (Dahlin et al., 2018), we identified a predominant HSPC cluster enriched in stem cells, as well as entry points to 4 different blood lineages (lymphoid, erythroid, neutrophil, monocyte) (Figures 3B and S4B). Importantly, all the identified clusters were populated by both vcMettl3+/+ and vcMettl3−/− cells, with minor differences in composition (Figures 3B and S4C). The major difference consisted in a shift within the HSPC cluster from the lymphoid towards the erythroid entry point, without major gain or loss of cell populations (Figure 3B). Given the relative preservation of cellular composition within the overall population, it is unlikely that changes in cell composition affected expression changes identified in bulk sequencing of vcMettl3+/+ vs vcMettl3−/− LSK cells.
Analysis of bulk RNA sequencing data revealed that loss of METTL3 in LSK resulted in upregulation of 701 genes and downregulation of 1396 genes (Figures 3C and 3D). Eight-fold downregulation of Mettl3 mRNA in vcMettl3−/− versus vcMettl3+/+ groups confirmed efficient deletion in E14.5 fetal liver LSK cells (Table S1).
The m6A modification has been associated with destabilization of RNA and enhanced transcript degradation. It would therefore be expected that loss of m6A modification results in upregulation of m6A modified transcripts (Batista et al., 2014; Geula et al., 2015; Wang et al., 2014a). We cross-referenced genes upregulated and downregulated in vcMettl3−/− fetal liver LSK to 52 m6A-Seq data sets obtained from murine and human tissues and cell lines (Barbieri et al., 2017; Geula et al., 2015; Li et al., 2018; Liu et al., 2020; Vu et al., 2017). This analysis included hematopoietic data sets, including m6A-Seq performed in murine LT-HSC, ST-HSC & MPP; human CD34+ umbilical cord blood stem cells (Li et al., 2018); and human MOLM-13 acute myeloid leukemia cell lines (Barbieri et al., 2017; Vu et al., 2017). Consistently across all the m6A data sets, genes that were upregulated in fetal liver LSK upon METTL3 loss were more likely to be m6A modified than genes that were downregulated (P = 2.5 e-11) (Figure 3E). The observed upregulation of expected m6A targets following Mettl3 deletion in fetal liver LSK was congruent with the predicted negative effects of m6A on transcript stability.
To understand which pathways could be contributing to the observed stem cell failure phenotype upon loss of METTL3, we performed pathway analysis on up- and downregulated genes. Upregulated genes were strongly enriched for pathways involved in antiviral and interferon response, as well as ribosome biogenesis and RNA processing and metabolism (Figure 3F and S4D). In contrast, downregulated genes were enriched for processes such as cell proliferation, cytokine signaling, cellular activation, cell adhesion and projection (Figure S4D). Network analysis of upregulated genes in vcMettl3−/− fetal liver LSK cells revealed that genes involved in “response to virus” played a central role in the upregulated pathways (Figure 3F).
In summary, we found that loss of m6A was associated with upregulation of expected m6A modified transcripts, while the majority of differentially expressed genes were downregulated and mostly not expected to be m6A modified. The most highly upregulated pathways were characteristic of an innate immune response.
Interferon-stimulated genes and Oas genes are transcriptionally activated upon loss of METTL3
Given the enrichment of innate immune response pathways amongst the most upregulated genes in vcMettl3−/− fetal liver LSK cells (Figures 3C, 3F and S4D), we sought to understand the mechanism of regulation underlying this response. Among the top 100 most highly upregulated genes, several interferon-stimulated genes, including most members of the oligoadenylate synthetase (Oas) family of genes, were highly enriched. OAS proteins sense double-stranded RNAs and are established detectors of exogenous dsRNA in the context of viral infection (Silverman, 2007; Son et al., 2015). When binding dsRNA, OAS proteins generate 5’-phosphorylated 2’-5’-linked oligoadenylate (2-5A) nucleotides that activate the endoribonuclease RNase L. RNase L amplifies the innate immune response by degrading both cellular and viral RNAs (Han et al., 2014; Silverman, 2007). Of the 12 known Oas family genes, 8 were detectably expressed in LSK cells. Of these, we found that 7 were significantly upregulated following Mettl3 deletion in LSK (Figure 4A and Table S1), as well as in total fetal liver cells (Figure S4E).
Figure 4. OAS family genes are highly upregulated via epigenetic regulation in response to Mettl3 loss in HSPCs.
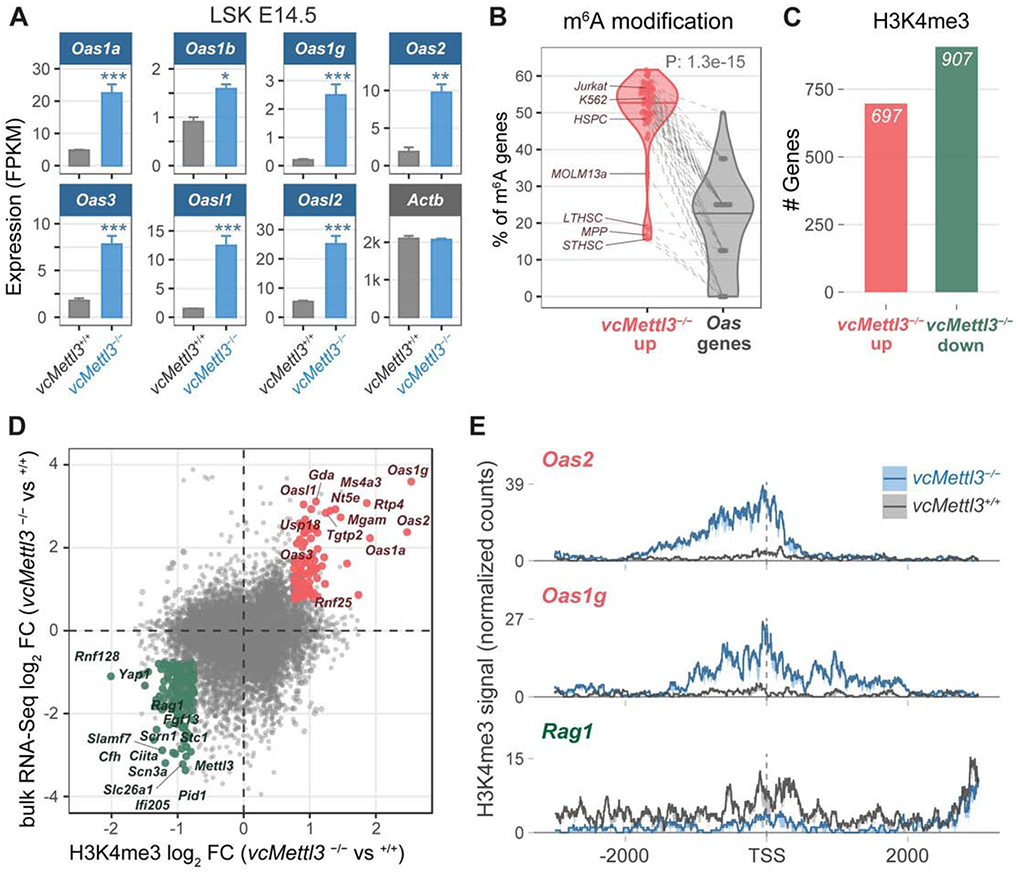
(A) Oas family gene expression in vcMettl3+/+ and vcMettl3−/− E14.5 fetal liver LSK cells as measured by RNA-seq (n=3 biological replicates per group).
(B) Percentage of genes with annotated m6A peaks in Oas genes compared to the remaining genes upregulated in vcMettl3−/− LSK. The meta-analysis was performed with 52 different published m6A datasets.
(C) Number of genes with significantly increased (red) versus decreased (green) H3K4me3 occupancy around the transcription start site (TSS) in vcMettl3−/− versus vcMettl3+/+ LSK cells (n=2 biological replicates per group).
(D) Scatterplot comparing gene expression changes (y-axis) versus H3K4me3 occupancy changes (x-axis) between vcMettl3−/− and vcMettl3+/+ LSK cells. Genes with consistent and significant epigenetic and gene expression changes are highlighted (red=up, green=down).
(E) H3K4me3 occupancy profiles of Oas1g, Oas2 and Rag1 (3k bp around the TSS) in vcMettl3+/+ and vcMettl3−/− LSK cells. The average normalized signal and standard error from 2 replicates is displayed.
The P value in (B) was calculated using two-tailed Wilcoxon rank-sum test.
See also Table S1.
As noted above, upregulated genes were more likely to be m6A modified than downregulated genes, in keeping with the expected effects of m6A on transcripts stability. By contrast, Oas transcripts are generally devoid of m6A modifications in all the 52 available human and murine m6A-Seq data sets that we examined (P = 1.3 e-15) (Figure 4B). Upregulation of the Oas genes following Mettl3 deletion was therefore not likely to be directly mediated by the effects of m6A on transcript stability, but by an alternative regulatory mechanism. To determine whether expression of Oas and other ISGs was transcriptionally induced, we performed cleavage under targets and release using nuclease (CUT&RUN) analysis for H3K4me3 histone modification (Skene et al., 2018), the key epigenetic mark at promoters of actively transcribed genes (Santos-Rosa et al., 2002). We applied CUT&RUN to two independent sets of vcMettl3+/+ and vcMettl3−/− E14.5 fetal liver LSK cells and determined H3K4me3 occupancy at transcription start sites (TSS).
Analysis of CUT&RUN data revealed altered H3K4me3 occupancy for numerous genes in vcMettl3−/− compared to vcMettl3+/+ LSK cells: 697 and 907 genes displayed increased and decreased H3K4me3 deposition at their transcription start sites, respectively (Figure 4C and Table S1). We found a strong positive correlation (Pearson coefficient 0.70) between significant gene expression changes and variations in H3K4me3 promoter occupancy, with 112 and 175 genes showing concordant increases and decreases, respectively (Figures 4D and 4E). Specifically, several Oas genes and other ISGs upregulated in vcMettl3−/− LSK showed increased H3K4me3 occupancy at their TSS, as illustrated for Oas2 and Oas1g (Figure 4E). Previous studies have identified widespread alterations in histone modifications following m6A depletion. Consequently, differential gene expression following Mettl3 deletion may be accounted for by transcriptional changes attributable to an altered histone profile (Wang et al., 2018). Of note, the expression of most histone modifying genes was not altered in vcMettl3−/− LSK cells (Table S1), evidence that ISG upregulation reflects a specific cellular response in LSK that is transcriptionally mediated and not secondary to global chromatin dysregulation.
In summary, these data highlight transcriptionally mediated upregulation of Oas and other ISGs upon loss of m6A underlying the observed anti-viral immune response gene expression signature.
Loss of METTL3-mediated RNA m6A modification results in formation of endogenous dsRNAs
Most viruses produce double-stranded RNAs upon viral infection (Son et al., 2015). Detection of foreign dsRNA triggers activation of the cellular innate immune response to inhibit viral replication and contain infection (Jensen and Thomsen, 2012). Structural studies have previously demonstrated that m6A destabilizes base-paired RNA duplexes (Liu et al., 2015; Roost et al., 2015). We hypothesized that loss of m6A modification might result in the aberrant formation of endogenously-derived dsRNA, and a consequent downstream aberrant innate immune response characterized by activation of Oas and other ISGs. To determine whether loss of METTL3 induced the formation of dsRNA, we stained E14.5 fetal liver lineage depleted (Lin−) cells with J2 antibody, specific for dsRNA of > 40bp length (Schönborn et al., 1991). vcMettl3−/− Lin− fetal liver cells showed a significant accumulation of dsRNAs compared to vcMettl3+/+ cells (Figures 5A and 5B).
Figure 5. Loss of METTL3 leads to aberrant dsRNA formation upon depletion of m6A.
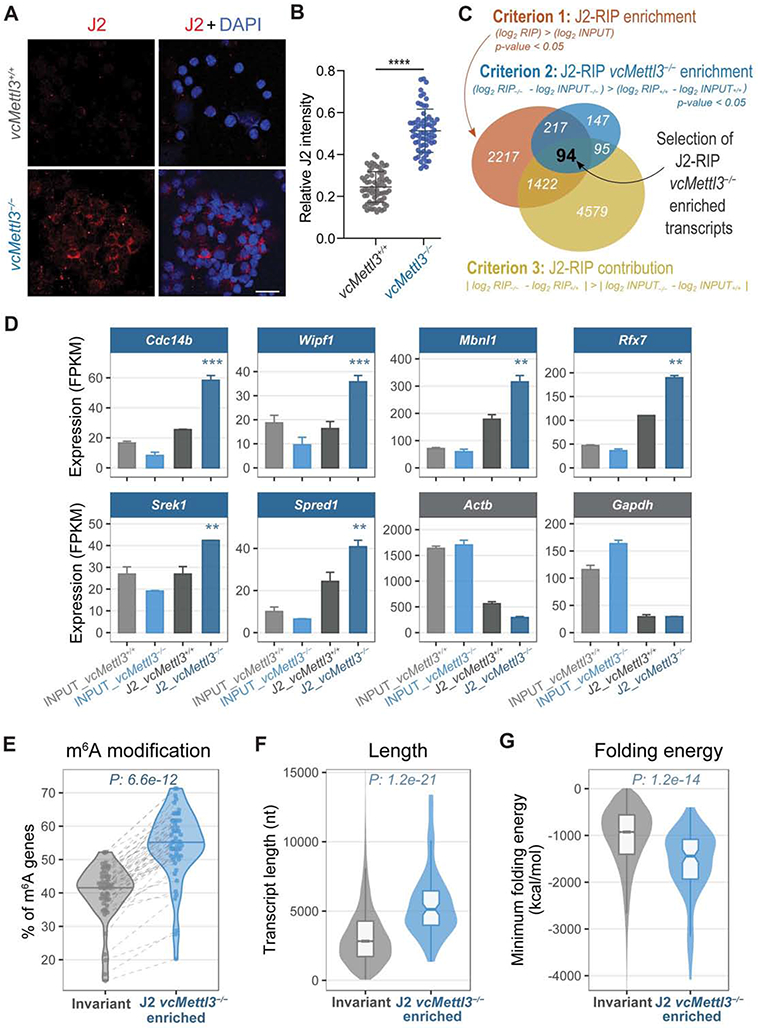
(A) Detection of dsRNA via immunocytochemistry in vcMettl3+/+ and vcMettl3−/− fetal liver cells using the dsRNA specific antibody J2. Scale bar, 50 μm. Data are representative of four independent experiments.
(B) Quantification of J2 intensity of each cell normalized to its DAPI signal (vcMettl3+/+ n=69, vcMettl3−/− n=66). Data are representative of four independent experiments.
(C) Schematics of the criteria used to identify the 94 genes significantly enriched in J2 RIP of vcMettl3−/− versus vcMettl3+/+ fetal liver cells.
(D) J2 RIP-Seq signals from six representative J2 vcMettl3−/− enriched genes, compared with Actb and Gapdh as negative controls (n=2 biological replicates per group).
(E) Percentage of genes with annotated m6A peaks among the 94 genes enriched in vcMettl3−/− J2 RIP compared with invariant genes. The meta-analysis was performed considering 52 different published m6A datasets.
(F) Distributions of the transcript length of vcMettl3−/− J2 RIP enriched genes compared with invariant genes.
(G) Distributions of the predicted folding energy of vcMettl3−/− J2 RIP enriched genes compared with invariant genes.
The P values in (E,F,G) were calculated using two-tailed Wilcoxon rank-sum test.
To characterize these dsRNAs, we performed J2 anti-dsRNA immunoprecipitation (Blango and Bass, 2016; Lybecker et al., 2014) followed by deep sequencing. To limit in vitro artifacts and capture dsRNAs differentially formed in vivo, we lysed red blood cell depleted vcMettl3+/+ and vcMettl3−/− fetal liver cells in dsRNA RIP buffer. Input and J2 antibody- versus isotype control (IgG)-bound RNAs were isolated and cDNA libraries generated. IgG control libraries could not be generated, indicative of the stringency of the assay (Figure S5A). Input J2 RIP sequencing samples clearly clustered according to their genotype (vcMettl3+/+ or vcMettl3−/−) and sample type (J2 or INPUT) (Figure S5B). We identified 94 RNAs significantly enriched in vcMettl3−/− versus vcMettl3+/+ fetal liver J2 RIP (Figure 5C and Table S2). These 94 genes were enriched in J2-RIP vs input signal, and in vcMettl3−/− over vcMettl3+/+ J2-RIP, after normalizing for input variations (see STAR methods and Figure 5C for a detailed description of the selection criteria). Although multiple families of retrotransposons, such as long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs), are known to form dsRNAs and are highly expressed, we did not identify significant enrichment in vcMettl3−/− J2-RIP for these elements (Figures S5C and S5D). Out of the 94 genes identified, 92 were protein coding and 2 were long non-coding RNAs, including Malat1, which has previously been shown to be highly m6A modified (Liu et al., 2015; Zhou et al., 2016). The enrichment profiles of 6 representative genes are displayed in Figure 5D, with Actb and Gapdh as negative controls. The 94 genes were also expressed in LSK cells (Figure S5E left) with a minor expression increase in vcMettl3−/− cells (Figure S5E right). Importantly, when compared to non-enriched RNAs, the 94 J2 vcMettl3−/− enriched genes were more highly m6A modified in their native state (P = 6.6 e-12, Figure 5E), longer (P = 1.2 e-21, Figures 5F and S5F) and marked by lower predicted folding energies (P = 1.2 e-14, Figures 5G and S5G). These transcripts are therefore predicted to have a higher propensity to adopt a dsRNA conformation upon loss of m6A. Overall, these experiments demonstrate that loss of the m6A modification resulted in aberrant formation of endogenously derived dsRNAs.
Loss of METTL3 induces an aberrant dsRNA innate immune response that compromises hematopoietic stem cell function
To determine whether endogenous dsRNA formation could indeed activate an innate immune response and contribute to hematopoietic failure, we assessed for concomitant activation of immune signaling pathways downstream of dsRNA detection (Figure 6A) (Li et al., 2017b). In the setting of viral infection, viral dsRNAs bind to and activate pattern recognition receptors (PRRs). These include two critical sensors of cytoplasmic RNA: retinoic acid inducible gene I (RIG-I, also known as Ddx58) and melanoma differentiation associated gene 5 (MDA5, also known as Ifih1), which mediate downstream transcriptional activation of interferons. Indeed, expression of both Ifih1 and Ddx58 were upregulated both in LSK (Table S1) and in total fetal liver cells (Figure 6B). We further identified activation of the protein kinase R (PKR) and eIF2α pathways via phosphorylation of PKR and eIF2α, respectively (Figures 6C and 6D). Both of these pathways are induced following detection of dsRNAs, and subsequently inhibit translation (Dabo and Meurs, 2012). An additional pathway involved in dsRNA response is mediated by the OAS proteins, which are known to activate the endoribonuclease RNase L. We found that RNase L was activated in vcMettl3−/− fetal livers, demonstrated by the significant increase of RNase L mediated cleavage of tRNA-His-36 (Donovan et al., 2017), which was detected by Q-RT-PCR (Figure 6E). As expected, tRNA cleavage detection by Q-RT-PCR was more sensitive, and visible earlier than rRNA specific cleavage, which was detectable via TapeStation in newborn bone marrow but not E14.5 fetal liver cells (Figures S6A and S6B). In addition to Oas genes, RIG-I and MDA5, several other ISGs, such as Usp18 and Rtp4, were upregulated upon Mettl3 deletion (Figure 6F). To further dissect the nature of the immune response we probed interferon (IFN) gene expression shortly after Mettl3 deletion at E12.5. Neither type I nor type III IFNs were upregulated by Q-RT-PCR in E12.5 fetal liver (Figure S6C). In E14.5 vcMettl3−/− whole fetal liver, Ifnl3, a Type III IFN, was upregulated, while Type I IFNs (Ifna4, Ifnb1) were not (Figure 6G).
Figure 6. Loss of METTL3 induces an aberrant dsRNA induced innate immune response.
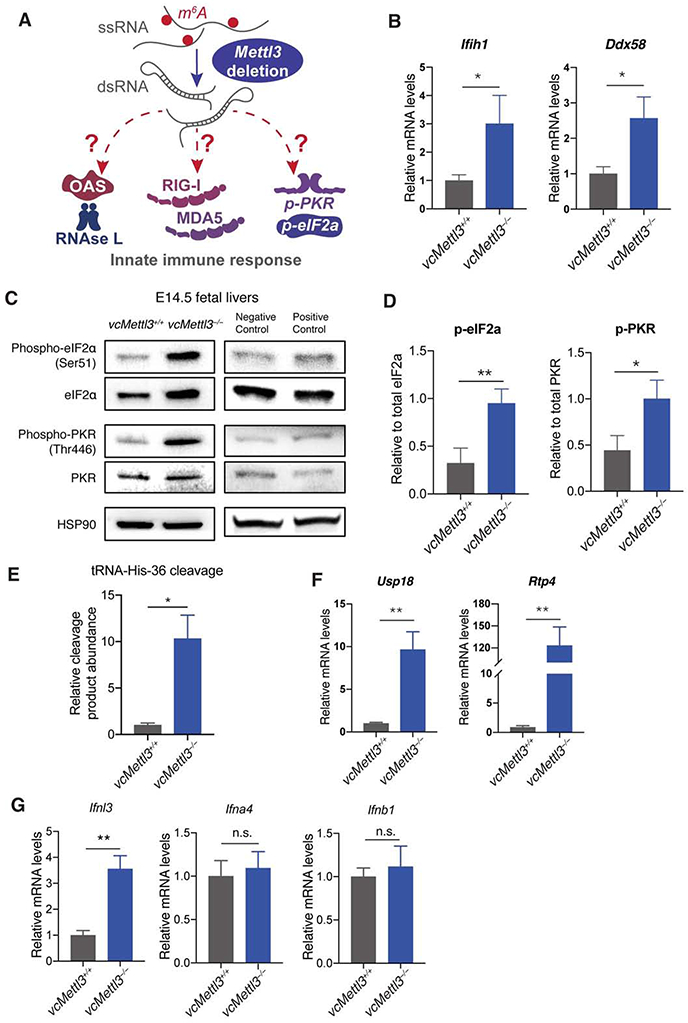
(A) Scheme depicting the dsRNA induced innate immune response pathways.
(B) Q-RT-PCR determination of expression of dsRNA sensor genes Ifih1 (MDA5) and Ddx58 (RIG-I) in E14.5 fetal livers, normalized to Gapdh (n=3 biological replicates per group).
(C-D) Detection of PKR phosphorylation and eIF2α phosphorylation in fetal liver in response to Mettl3 deletion via Western blot (C); bands from three representative blots were quantified via Image J, p-PKR and p-eIF2a protein expressions were normalized to PKR and eIF2a expression, respectively (D) (n=3 biological replicates per group).
(E) RtcB Q-RT-PCR analysis of tRNA specific cleavage by RNase L, normalized to U6 (n=3 biological replicates per group).
(F) Q-RT-PCR determination of expression of immune response genes downstream of interferon activation in E14.5 fetal livers, normalized to Gapdh (n=3 biological replicates per group).
(G) Q-RT-PCR determination of IFN III (Ifnl3) and I (Ifna4, Ifnb1) expression in E14.5 fetal livers, normalized to Gapdh (n=3 biological replicates per group).
Data are represented as mean ± SEM, and representative of at least three independent experiments; The P values were calculated using two-tailed Student's t test. * p<0.05, ** p<0.01.
See also Figure S6.
These data collectively suggest that PRR sensing of dsRNAs, Oas and ISG upregulation represented the initiating events of the aberrant innate immune activation in vcMettl3−/− fetal liver LSK, as opposed to direct m6A mediated effects on transcript stability.
Disruption of the innate immune response in part rescues fetal HSPC function
Since most PRR-related innate immune pathways converge on the MAVS-mediated IFN immune response, we sought to rescue progenitor proliferation and colony formation via deletion of Mavs in vcMettl3−/− fetal livers. We performed lentiviral transduction of vcMettl3−/− Lin− fetal liver cells with Mavs sgRNA or control sgRNA concurrently with the Cas9 enzyme, and tested their colony forming ability. Mavs deletion produced partial, but significant rescue of colony formation by vcMettl3−/− Lin− fetal liver cells (Figure 7A). Of note, while deletion of Mavs completely abrogated Ifnb induction by the positive control dsRNA pI:pC (Figure S7A), it only partially reduced Oas gene induction and the IFN immune response in vcMettl3−/− fetal liver cells (Figure 7B) and did not reduce the dsRNA formation in vcMettl3−/− fetal liver cells (Figure S7B). Furthermore, inhibition of RNase L activity via Rnasel knockdown also partially rescued hematopoietic colony formation (Figure S7C). Importantly, deletion of Mavs in lineage-depleted fetal liver cells partially rescued engraftment of vcMettl3−/− cells in vivo in competitive transplantation assays (Figure 7C and 7D). Previous studies have identified loss of MYC as the cause of hematopoietic failure (Cheng et al., 2019; Lee et al., 2019). In our model, Myc expression was upregulated in E12.5 and E14.5 fetal livers and only downregulated in bone marrow at birth suggesting that the innate immune response precedes dysregulation of MYC (Figure S7D).
Figure 7. Inhibition of the innate immune response partially rescues hematopoietic failure secondary to Mettl3 deletion.
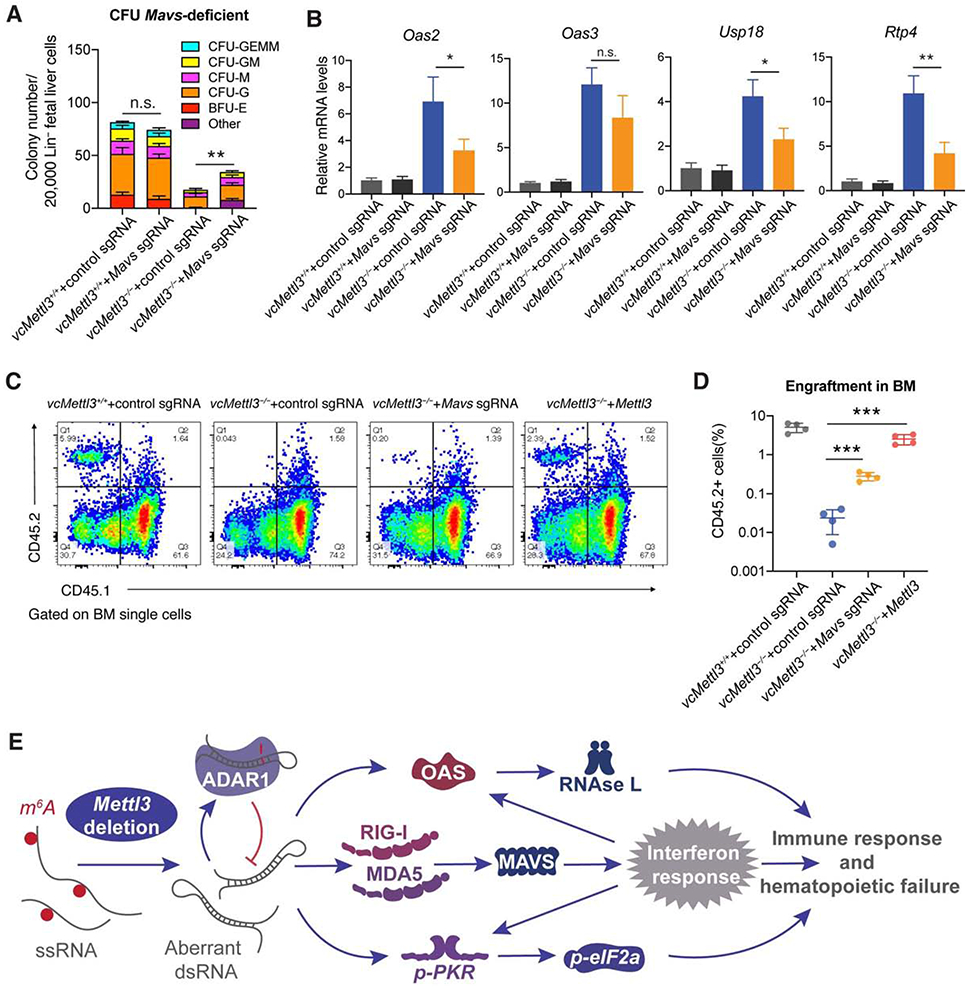
(A) Determination of the effect of CRISPR/Cas9 mediated Mavs deletion on colony formation in vcMettl3−/− compared to vcMettl3+/+ Lin− fetal liver cells (n=3 biological replicates per group).
(B) qRT-PCR measurement of Oas and interferon response gene expression secondary to control versus targeted sgRNA-mediated deletion of Mavs in vcMettl3−/− compared to vcMettl3+/+ fetal liver cells, normalized to Gapdh (n = 3 biological replicates per group).
(C and D) Determination (C) and quantification (D) of the engraftment of vcMettl3−/− cells with CRISPR-Cas9-mediated deletion of Mavs or overexpression of Mettl3 6 weeks post-transplantation via flow cytometric detection of CD45.2 fetal liver cells versus CD45.1 control cells (n = 4 separate mice per group). One independent experiment is shown.
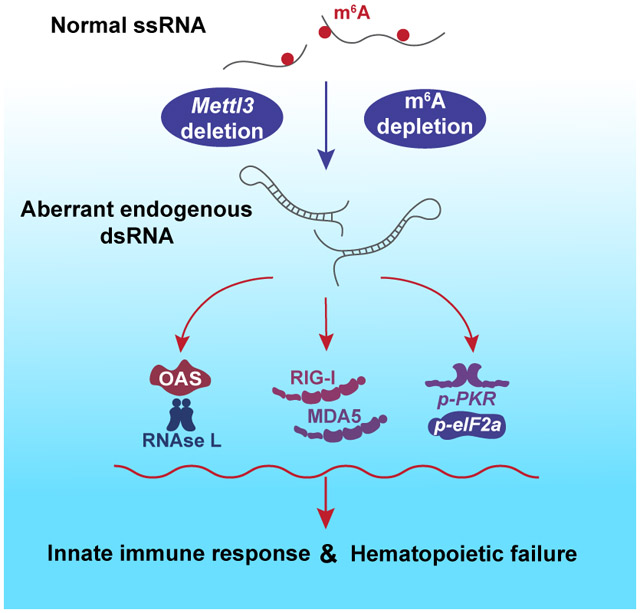
(E) Model of the Mettl3 deletion-induced dsRNA-mediated innate immune response.
Data are represented as mean ± SEM, and representative of at least two independent experiments unless stated otherwise; The P values were calculated using two-tailed Student's t test. n.s. not statistically significant, * p<0.05, ** p<0.01, *** p<0.001.
See also Figure S7.
Of note, we found that expression of adenosine deaminase acting on RNA 1 (Adar1) was significantly increased in vcMettl3−/− fetal livers (Figure S7E). ADAR1 is known to prevent endogenous dsRNA formation by A-to-I editing, which hinders dsRNA base pairing. ADAR1 thereby functions to prevent the detection of endogenous RNAs as non-self and induction of a dsRNA-mediated aberrant innate immune response (Liddicoat et al., 2015). The observed activation of Adar1 is congruent with a cellular response to aberrant dsRNA formation, albeit insufficient to quench induction of an innate immune response and rescue of the observed stem and progenitor cell defect in this context (Li et al., 2017b).
In summary, we present evidence revealing that depletion of m6A RNA modification promoted formation of endogenously derived dsRNA, which was associated with activation of the innate immune response via the OAS-RNase L, PKR-eIF2α and MDA5-RIG-I pathways. The resulting inflammatory response was deleterious to fetal liver HSPC proliferation and differentiation, contributing to bone marrow failure and embryonic lethality (Figure 7E).
Discussion
We here present an examination of the effects of Vav-Cre mediated deletion of the m6A writer METTL3 on murine fetal hematopoiesis. We found that depletion of m6A resulted in expansion of the hematopoietic stem and progenitor pool, with accumulation of dysfunctional immature cells defective in self-renewal and differentiation in vitro and in vivo. We identified upregulation of the MDA5-RIG-I, the PKR-eIF2α, and OAS-RNase L pathways in hematopoietic stem and progenitor cells, driven by increased formation of endogenously derived dsRNAs. We showed that this aberrantly triggered innate inflammatory response contributed to the observed HSPC defects via rescue experiments that disrupted RNase L or MAVS activity. These findings suggest that the presence of the m6A RNA modification prevents the formation of endogenously derived dsRNA, and preserves the integrity of the cellular innate immune response.
The role of m6A modification in hematopoiesis has been of great interest, with several studies evaluating the effects of m6A writer, reader and eraser functions in normal hematopoiesis and leukemia. Initial interest was driven by the finding that METTL3 is overexpressed in acute myeloid leukemia compared to other malignancies. Indeed, in studies in human MOLM-13 cell lines, CD34+ umbilical cord derived HSPCs, and murine leukemia cells, METTL3 knockdown promotes spontaneous differentiation along the myeloid lineage, and prevents engraftment in xenotransplantation models (Barbieri et al., 2017; Vu et al., 2017). This led to the hypothesis that METTL3 might be a viable therapeutic target for myeloid leukemia.
Our findings indicate that the effects of m6A depletion on native hematopoiesis might be limiting in the development of METTL3-targeted differentiation therapies. METTL3 function is essential for native hematopoiesis, as evidenced by early embryonic lethality and the hematopoietic failure occurring in vcMettl3−/− mice.
Previously, zebrafish and murine studies demonstrated the role of m6A during the endothelial to hematopoietic transition (EHT). In both systems, loss of METTL3 coincides with inhibition of the EHT, and impairs emergence of definitive hematopoiesis (Lv et al., 2018; Zhang et al., 2017). The authors of these studies argued that in murine models this phenotype was observed only when using a Vec-Cre promoter, but not when using a Vav-Cre promoter. However, Lv and colleagues restricted their analysis of Vav-Cre-Mettl3fl/fl hematopoiesis to E10.5 embryos prior to the onset of Vav expression (E11.5) and were therefore unlikely to detect the effects of Mettl3 deletion in fetal HSPCs.
Recently, two groups published analyses of Mx1-Cre driven Mettl3 excision in the adult mouse hematopoietic system (Cheng et al., 2019; Lee et al., 2019). While the reported phenotypes are similar to those presented here, our Vav-Cre driven model and derivative data provide specific, valuable insights. First, Vav-Cre mediated hematopoietic-specific deletion of Mettl3 in E14.5 fetal HSPCs permits analysis within 3 days after Mettl3 deletion, allowing for detection of direct, early effects of Mettl3 deletion. By contrast, Mx1 requires a prolonged induction phase before expression changes can be evaluated. Second, Mx1 is an interferon-responsive gene, and Cre-induction is mediated via injection of pI:pC, a strong inducer of the antiviral immune response (Gantier and Williams, 2007). The antiviral innate immune response induced by endogenous dsRNA formation following Mettl3 deletion as described here is likely to be masked by the exogenous induction of interferon signaling by pI:pC in the Mx1-Cre model.
Cheng et al. and Lee et al. observe reduction of MYC, whose mRNA is highly m6A modified, >10 days after pI:pC treatment, and propose that loss of MYC contributed to the observed phenotype. However, in our Vav-Cre-Mettl3fl/fl model, Myc expression was initially upregulated (E12.5 and E14.5) and only downregulated at birth at the time of the lethal bone marrow failure. Myc upregulation in vcMettl3−/− E14.5 fetal livers likely results from the increased mRNA stability of Myc mRNA after Mettl3 deletion and m6A depletion. Considering that upregulation of the innate immune response largely precedes Myc downregulation, it is conceivable that Myc downregulation was at least in part secondary to immune response activation, consistent with previous studies (Ramana et al., 2000; Raveh et al., 1996).
Our findings highlight inherent differences between ESCs and HSCs. Although Mettl3 deletion compromises differentiation in both, Mettl3-deficient murine ESCs persist in a state of naïve pluripotency, while Mettl3-deficient HSCs are eventually lost. Stem cells in general have been found to have a cell-intrinsic program of ISG expression that protects them against viral infection (Wu et al., 2018). Unlike HSCs, ESCs do not mount an innate immune response to dsRNA and are tolerant of exogenous, unmodified RNAs (Chen et al., 2010; Guo et al., 2015), supporting inherent differences between the ESC and HSC responses to loss of m6A mRNA modification. Indeed, ISG-induced resistance to RNA-mediated reprogramming of HSCs can be overcome by RNA modification (Warren et al., 2010). It is possible that the defect observed in fetal hematopoiesis in response to Mettl3 deletion is further shaped by the nature of immune regulation in fetal development under homeostasis and in response to infection (Yockey and Iwasaki, 2018).
In our current work, we established a link between m6A mRNA modification and innate immunity. We showed that loss of m6A coincides with enhanced formation of endogenously derived dsRNAs, suggesting that m6A normally functions to prevent dsRNA formation. Indeed, this can be expected based on previous structural studies (Roost et al., 2015). Via J2-RIP we identified the formation of 94 dsRNAs from endogenous protein coding and lncRNA transcripts. Aberrantly formed dsRNAs were likely sensed by PRRs resulting in induction of ISGs and specifically Oas genes, as evidenced by H3K4me3 enrichment at their promoter regions. OAS proteins are critical activators of RNase L activity, further amplifying the interferon response. Others have directly shown that nucleoside modification of RNA eliminates activation of 2'-5'-oligoadenylate synthetase and increases resistance to cleavage by RNase L (Anderson et al., 2011). Activation of the PKR-eIF2α pathway may contribute to hematopoietic failure through its known inhibitory function on translation and proliferation (Saelens et al., 2001). MDA5 and RIG-I, essential cytoplasmic PRRs upregulated in vcMettl3−/− fetal livers, recognize dsRNA and trigger the IFN response through MAVS (Besch et al., 2009). The overall gene expression profile we observed in vcMettl3−/− cells was characteristic of an interferon stimulated gene response. Importantly, PRRs can activate interferon response genes directly via interferon response factor (IRF) signaling (Grandvaux et al., 2002; Pulit-Penaloza et al., 2012). While Oas genes were the most highly upregulated class of ISGs, suggesting a dsRNA-induced positive feedback loop, the IFN response downstream of PRRs may be responsible for the prominent upregulation observed (Sadler and Williams, 2008). Which PRRs specifically recognize the newly formed dsRNAs following Mettl3 deletion and whether all or specific dsRNAs trigger the innate immune response remains to be shown.
Recently, Chen et al. showed that m6A modification of circular RNAs (circRNAs) prevents immune activation via recognition by the m6A reader YTHDF2, whereas unmodified circRNAs are recognized as non-self, similarly activating an anti-viral immune response (Chen et al., 2019). While circRNAs are not highly expressed in hematopoietic stem cells (Nicolet et al., 2018), it is conceivable that loss of m6A modification on circRNAs in fetal liver cells contributes to aberrant activation of the innate immune response. Future studies to isolate potential circRNAs may be of interest to quantitate their contribution. Another class of transcripts, retrotransposons, has a high propensity to form dsRNAs. Our results exclude a major contribution to the dsRNA pool by these elements: although LTR and SINE elements were enriched in J2-RIP, their signal was not specifically increased in vcMettl3−/− cells. Instead, depletion of m6A RNA modification resulted in the adoption of a dsRNA conformation by highly m6A modified transcripts, characterized by long sequence and low folding energy. Of note, Malat1, a long-noncoding RNA that is known to be highly m6A modified and highly expressed in HSCs and hematopoietic cells, was among the J2-RIP identified targets. It is of interest that MALAT1 function is regulated by an “m6A switch”, whereby m6A modification alters MALAT1 local conformation, affecting the accessibility of the transcript to RNA binding proteins (Liu et al., 2015; Zhou et al., 2016). Future studies that determine whether conformational changes in the identified dsRNAs are directly caused by the absence of m6A or mediated by the interaction of specific RNA binding proteins will be important.
Our studies reveal a critical regulatory function for METTL3 mediated m6A modification in assuring recognition of endogenous RNAs as self. This places METTL3 amongst other RNA editing enzymes, such as ADAR1 (Liddicoat et al., 2015; Xiang et al., 2018), as critical for the distinction between self and non-self RNAs, and for the regulation of the antiviral innate immune response. Extending this concept, our studies highlight the essential function of m6A RNA modification in the protection of cells from aberrant innate immune signaling in vivo.
While the specific targets of METTL3-mediated m6A mRNA modification are presumably context dependent and thus different at each stage within the hematopoietic differentiation hierarchy (Shi et al., 2019), the innate immune response to m6A depletion likely occurs in HSCs, progenitors, and mature cells. Future studies that determine a possible physiologic interplay between the METTL3 m6A transcriptome and innate immunity will be of significant interest.
Limitations of Study
In this study, we identified 94 transcripts that formed dsRNAs upon deletion of Mettl3 and loss of m6A RNA modification in fetal liver hematopoiesis. dsRNA formation induced a deleterious innate immune response that contributed to hematopoietic failure, but several questions remain to be addressed in future studies. It is of interest whether this mechanism is general or restricted to fetal hematopoiesis. In addition, it remains to be determined which dsRNA species are necessary or sufficient to induce innate immunity and whether this response serves a physiologic purpose. While we have identified downstream activation of MDA5, RIG-I, PKR-eIF2α and OAS-RNase L pathways, it will be of interest to define the direct sensors of the newly formed dsRNAs and dissect the hierarchy of events. In our study we deleted the m6A writer Mettl3, resulting in wide-spread loss of m6A; the question arises whether perturbation of m6A readers or erasers can similarly modify the innate immune response.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephanie Halene (stephanie.halene@yale.edu).
Materials Availability
Mettl3fl/fl mouse strain and newly generated plasmids in this paper are available under an MTA from the authors.
Data and Code Availability
Raw and analyzed sequencing data have been deposited under GEO: GSE148882 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148882)
Software used for the analyses are described and referenced in the individual Method Details subsections and are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| METTL3/MT-A70 Antibody | Bethyl lab | Cat# A301-567A; RRID: AB_1040004 |
| Anti-METTL14 antibody | SIGMA-ALDRICH | Cat# HPA038002; RRID: AB_10672401 |
| Anti-m6A antibody | Synaptic Systems | Cat# 202003; RRID: AB_2279214 |
| Anti-PKR antibody | SIGMA-ALDRICH | Cat# SAB3500326; RRID: AB_10646199 |
| Anti-phospho-PKR antibody | SIGMA-ALDRICH | Cat# SAB4504517; |
| eIF2α Antibody | Cell Signaling Technology | Cat# 9722S; RRID: AB_2230924 |
| Phospho-eIF2α (Ser51) Antibody | Cell Signaling Technology | Cat# 9721S; RRID: AB_330951 |
| CD34 Monoclonal Antibody (RAM34), FITC | eBioscience | Cat# 11-0341-85; RRID: AB_465021 |
| PE/Cy5 anti-mouse CD135 (Flt3) Antibody | Biolegend | Cat# 135312; RRID: AB_2263031 |
| Miltenyi lineage detection | Miltenyi | Cat# 130-092-613; RRID: AB_1103214 |
| APC-Cy7 Streptavidin | Biolegend | Cat# 405208 |
| PE anti-mouse Ly-6A/E (Sca-1) Antibody | Biolegend | Cat# 108108; RRID: AB_313345 |
| APC anti-mouse CD117 (c-Kit) Antibody | Biolegend | Cat# 105812; RRID: AB_313221 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block™) | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| CD16/CD32 Monoclonal Antibody, PE-Cy7 | eBioscience | Cat# 25-0161-82; RRID: AB_469597 |
| APC anti-mouse/human CD44 Antibody | Biolegend | Cat# 103012; RRID: AB_312963 |
| TER-119 Monoclonal Antibody (TER-119), PE | Biolegend | Cat# 12-5921-82; RRID: AB_466042 |
| Biotin anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat# 108404; RRID: AB_313369 |
| Biotin anti-mouse/human CD45R/B220 Antibody | Biolegend | Cat# 103204; RRID: AB_312989 |
| CD11b Monoclonal Antibody (M1/70), Biotin | eBioscience | Cat# 13-0112-82; RRID: AB_466359 |
| Streptavidin eFluor™ 450 | eBioscience | Cat# 48-4317-82; RRID: AB_10359737 |
| Pacific Blue™ anti-mouse/human CD11b | Biolegend | Cat# 101224; RRID: AB_755986 |
| APC anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat# 108411; RRID: AB_313376 |
| PE/Cy7 anti-mouse CD45.1 Antibody | Biolegend | Car# 110730; RRID: AB_1134168 |
| CD45.2 Monoclonal Antibody (104), FITC | eBioscience | Cat# 11-0454-85; RRID: AB_465060 |
| HSP90 Antibody | StressMarq Biosciences | Cat# SMC-107; RRID: AB_854214 |
| GAPDH (FL-335) Antibogy | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2031; RRID: AB_631737 |
| Histone H3K4me3 antibody (pAb) | Active motif | Cat# 39159; RRID: AB_2615077 |
| Normal Rabbit IgG | Millipore | Cat# 12-370; RRID: AB_145841 |
| Monoclonal Antibody SCICONS J2 | English & Scientific Consulting Kft. | Cat# 10010200; RRID: AB_2651015 |
| Cy3 Streptavidin | VECTOR LABORATORIES | Cat# SA-1300 |
| Cy5 Streptavidin | VECTOR LABORATORIES | Cat# SA-1500 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MethoCult™ GF M3434 | STEMCELL Technologies | Cat# 03434 |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | New England Biolabs | Cat# M0531L |
| RBC Lysis Buffer, 10X | Santa Cruze Biotechnology | Cat# sc-296258 |
| Lipofectamine™ 2000 Transfection Reagent | ThermoFisher SCIENTIFIC | Cat# 11668019 |
| Lipofectamine™ RNAiMAX Transfection Reagent | ThermoFisher SCIENTIFIC | Cat# 13778100 |
| Mouse Thrombopoietin (mTPO) | Gemini Bio-product | Cat# 300-351P |
| Mouse Stem Cell Factor (mSCF) | Gemini Bio-product | Cat# 300-348P |
| Mouse Flt-3 Ligand (mFlt3L) | Gemini Bio-product | Cat# 300-306P |
| Mouse Interleukin-3 (mIL3) | Gemini Bio-product | Cat# 300-324P |
| Benchmark Fetal Bovine Serum (FBS) | Gemini Bio-product | Cat# 100-106 |
| DMEM, high glucose | ThermoFisher SCIENTIFIC | Cat# 11965092 |
| Protein A–micrococcal nuclease (pA-MN) fusion protein | Henikoff lab | Skene and Henikoff, 2018 |
| Spermidine trihydrochloride | Sigma-Aldrich | Cat# S2501 |
| Con A-coated BioMag®Plus microparticles | Polysciences, Inc | Cat# 86057-3 |
| iQ™ SYBR® Green Supermix | BIO-RAD | Cat# 1708880 |
| iScript cDNA Synthesis Kit | BIO-RAD | Cat# 1708890 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836170001 |
| SUPERase• In™ RNase Inhibitor | ThermoFisher SCIENTIFIC | Cat# AM2696 |
| SPRIselect beads | Beckman Coulter | Cat# B23317 |
| RNaseA, Dnase and protease-free | ThermoFisher SCIENTIFIC | Cat# EN0531 |
| Maxima H Minus Reverse Transcriptase | ThermoFisher SCIENTIFIC | Cat# EP0752 |
| Exonuclease I | New England Biolabs | Cat# M0293S |
| Ampure XP beads | Beckman Coulter | Cat# A63880 |
| Fluorinert™ FC-40 | Sigma-Aldrich | Cat# F9755 |
| Critical Commercial Assays | ||
| EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric) | Epigentek | Cat# P-9005-96 |
| FITC BrdU Flow Kits | BD Pharmingen™ | Cat# 559619 |
| CellTrace CFSE Cell Proliferation Kit, for flow | Thermo Fisher | Cat# C34554 |
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen™ | Cat# 556547 |
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Magnetic mRNA Isolation Kit | New England Biolabs | Cat# S1550S |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat# E7500S |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | Cat# E7645S |
| Qubit dsDNA HS Assay Kit | Life Technologies | Cat# Q32851 |
| Vector M.O.M. Immunodetection Kit | VECTOR LABORATORIES | Cat# BMK-2202 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE148882 |
| Mouse genome and transcript annotation | Gencode M15 | https://www.gencodegenes.org/mouse/release_M15.html |
| Transcript sequences | Ensembl 90 | https://useast.ensembl.org/info/website/archives/index.html |
| Annotation of Transposable Elements | Hammell Lab | http://labshare.cshl.edu/shares/mhammelllab/www-data/TEtranscripts/TE_GTF/mm10_rmsk_TE.gtf.gz |
| Annotation of m6A genes | Barbieri et al., 2017 | DOI: 10.1038/nature24678 |
| Annotation of m6A genes | Geula et al., 2015 | DOI: 10.1126/science.1261417 |
| Annotation of m6A genes | Li et al., 2018 | DOI: 10.1038/s41422-018-0072-0 |
| Annotation of m6A genes | Liu et al., 2020 | DOI: 10.1016/j.molcel.2019.09.032 |
| Annotation of m6A genes | Vu et al., 2017 | DOI: 10.1038/nm.4416 |
| Experimental Models: Cell Lines | ||
| Human: HEK293GP cells | Clontech | Cat# 631458 |
| Human: HEK293T cells | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | In house colony | JAX: 002014 |
| Mouse: B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J | In house colony | JAX: 008610 |
| Mouse: C57BL/6J | In house colony | JAX: 000664 |
| Mouse: Mettl3f/f | Li et al., 2017 | N/A |
| Oligonucleotides | ||
| ON-TARGETplus Mouse Rnasel siRNA smartpool | Horizon Discovery | L-043480-00-0020 |
| ON-TARGETplus Non-targeting Control Pool | Horizon Discovery | D-001810-10-05 |
| See Table S3 | This paper | N/A |
| Recombinant DNA | ||
| pMSCV-IRES-GFP | Addgene | Plasmid #20672 |
| pMSCV-Mettl3-GFP | This paper | N/A |
| pMSCV-Mettl3CD-GFP | This paper | N/A |
| LentiCRISPRv2-mCherry | Addgene | Plasmid #99154 |
| LentiCRISPRv2- Mavs-mCherry | This paper | N/A |
| LentiCRISPRv2-Control-mCherry | This paper | N/A |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| psPAX2 | Addgene | Plasmid #12260 |
| Software and Algorithms | ||
| FlowJo v10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| Prism v8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Bowtie2 v2.2.9 | John Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Fiji | NIH | https://fiji.sc/ |
| Drop-seq tools v1.12 | Broad Institute | https://github.com/broadinstitute/Drop-seq |
| STAR v2.3.3a | N/A | https://github.com/alexdobin/STAR |
| edgeR | Bioconductor | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| clusterProfiler | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Picard Tools v2.9.0 | Broad Institute | https://broadinstitute.github.io/picard/ |
| Fastuniq v1.1 | N/A | https://sourceforge.net/projects/fastuniq/ |
| RNAfold v2.4.11 | ViennaRNA package | https://www.tbi.univie.ac.at/RNA/ |
| Rsubread | Bioconductor | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| Seurat v3.0 | N/A | https://satijalab.org/seurat/ |
| FastQC v0.11.5 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Other | ||
| Qubit fluorometer | ThermoFisher SCIENTIFIC | Cat# Q33216 |
| HiSeq 2500 Sequencing System | Illumina | Cat# SY–401–2501 |
| HemaTrue Veterinary Hematology Analyzer | Heska | N/A |
| Leica TCS SP5 | Leica | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were bred and maintained under specific-pathogen-free conditions at the animal facility of Yale University School of Medicine. Animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Yale University. Both female and male mice were used in experiments and their ages were described below.
Syngeneic (B6.SJL-Ptprca Pepcb/BoyJ; JAX: 002014) transplant recipient mice were 8-12 weeks of age. Male and female animals were represented in balanced proportion when in-house colony stock availability necessitated using mixed sex recipients.
We crossed floxed Mettl3 mice (Li et al., 2017a) with B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J (Vav-Cre) mice to obtain conditional Mettl3-deficient mice. The Vav-Cre mice were purchased from the Jackson laboratory. Mice were 8-16 weeks of age when they crossed with each other. Pregnant mice were sacrificed at E14.5 and the fetal livers of their embryos were analyzed.
Mettl3fl/fl mice without Vav-Cre (vcMettl3+/+) were used as wildtype control for Vav-Cre+-Mettl3fl/fl (vcMettl3−/−) mice. All of the vcMettl3−/− and vcMettl3+/+ mice were littermates and co-housed for any experiments described.
Exclusion criteria such as inadequate staining or low cell yield due to technical problems were pre-determined. Animals were assigned randomly to experimental groups. Each cage contained animals of all different experimental groups.
Cell lines
293T cells were cultured under 5% CO2 at 37 °C. The cells were derived from the kidney of a female embryo. We purchased the 293T cells from ATCC.
293GP cells were cultured under 5% CO2 at 37 °C. The cells were derived from the kidney of a female embryo and stably integrated with gag and pol. We purchased the 293GP cells from Clontech.
METHOD DETAILS
Mouse fetal liver isolation and transplantation
Hematopoietic cells were harvested from embryonic day E14.5 fetal liver (FL) from Vav-Cre-Mettl3fl/fl mice by gently dissociating into single cells in PBS with 5% BSA. The tail and feet from each embryo were digested with Proteinase K and used for genotyping. FL cells were lysed by 1x RBC lysis buffer and kept on ice until further use.
For non-competitive transplantation, FL cells were injected into lethally irradiated (850 cGy, XRAD 320) B6.SJL-Ptprca Pepcb/BoyJ (Pep3b) recipient mice at doses of 1 million cells per mouse via retro-orbital injection.
For competitive transplantation, FL cells were injected into lethally irradiated (850 cGy) CD45.1/2 recipient mice at doses of 1 million cells per mouse via retro-orbital injection along with 1 million competitor wildtype bone marrow cells from Pep3b mice.
Transplanted recipient mice were monitored daily for any signs of distress after transplantation. Sick mice were euthanized and analyzed for engraftment. All other mice were bled and sacrificed at assay endpoints (typically beyond eight weeks post-transplant), and engraftment was confirmed by flow cytometry.
To test the homing efficiency of FL cells, FL cells were incubated with 5 μM CellTrace™ CFSE for 20 min and transplanted into lethally irradiated (900 cGy) Pep3b mice (1.5 million cells per recipient mouse) via retro-orbital injection. After 16 hours, recipient mice were sacrificed and cell homing efficiency in their bone marrow and spleens was analyzed by flow cytometry.
Western Blot
Samples were boiled to denature proteins and separated in 12% mini-PROTEAN TGX Precast Protein gels. Proteins were then transferred to 0.45 μm PVDF membrane with a standard wet transfer system at 100V for 1 hr. Membranes were blocked with 5% skim milk for 30 min and incubated with primary antibodies overnight at 4°C. Excess antibody was washed away with TBST (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween 20) 3 times. Membranes were incubated with HRP-linked secondary antibody for 1 hr at room temperature. After 4 washes, membranes were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, 34080). Antibodies were used at the following dilutions in 5% skim milk: METTL3 1:2000, METTL14 1:1000, PKR 1:1000, phospho-PKR 1:1000, eIF2a 1:1000, phospho-eIF2a 1:1000, GAPDH 1:5000, HSP90 1: 2500, and HRP-linked Anti-rabbit IgG 1:10000, HRP-linked Anti-mouse IgG 1:5000.
RNA extraction, reverse-transcription and quantitative PCR
RNA was isolated using the RNeasy Mini Kit (QIAGEN) per vendor supplied protocol. Reverse-transcription of 1μg RNA was performed using the iScript cDNA Synthesis Kit (BIO-RAD) per standard protocol. Quantitative PCR was carried out in triplicate with target specific primers (see Table S3) using iQ™ SYBR® Green Supermix and quantitated using the CFX96 Real-Time System (BIO-RAD). Analysis was performed on at least 3 fetal livers per genotype to ensure statistical significance.
Measurement of m6A levels in fetal livers
Fetal livers were harvested from Vav-Cre-Mettl3fl/fl mice, and mRNA was isolated using the Magnetic mRNA Isolation Kit (New England Biolabs) following the standard provided protocol. 150 ng mRNA of each sample was used for the measurement of m6A levels using the EpiQuik m6A RNA Methylation Quantification Kit following the supplier provided protocol.
m6A-RIP PCR
Magnetic mRNA Isolation Kit (New England Biolabs) selected mRNAs from Vav-Cre-Mettl3fl/fl E14.5 fetal livers were incubated with m6A antibody (Synaptic Systems) in the lysis buffer and immunoprecipitated with Protein G Dynabeads (Thermo Fisher Scientific). The m6A enriched mRNAs were extracted by TRIzol reagent and reverse-transcribed using the iScript cDNA Synthesis Kit per standard protocol. Quantitative PCR was carried out in triplicate with target specific primers (see Table S3) using iQ™ SYBR® Green Supermix and quantitated using the CFX96 Real-Time System. Analysis was performed on at least 3 fetal livers per genotype to ensure statistical significance.
Flow Cytometry
Fetal liver single cell suspensions were blocked with rat anti-mouse CD16/32 antibody for 5 min (except when stained with CD16/32 antibody), followed by staining with antibodies (listed under reagents) in FACS buffer (5% BSA, 2 mM EDTA in PBS) in the dark at 4 °C for 30 min. Cells were washed with 1 ml FACS buffer and spun down at 500xg for 5 min. Flow cytometry analysis was performed on LSR Fortessa and LSR II (BD Biosciences) instruments, while sorting was performed on the FACS Aria instrument (BD Biosciences). Flow cytometry data were analyzed with FlowJo software (TreeStar).
Cell proliferation and apoptosis assay
To measure FL cell proliferation, RBC lysed FL cells were stained with BrdU and 7-AAD using the FITC BrdU Flow kit, and proliferation was quantified by flow cytometry.
To assay apoptosis, FL cells were stained with Annexin V and DAPI and analyzed by flow cytometry per standard protocol.
Histology and Immunohistochemistry
Bones and fetal livers were harvested and fixed in Bouin’s fixative solution overnight. Bones were decalcified, dehydrated and embedded in paraffin. Tissue sections, 5 μm in thickness, were air-dried at room temperature and stained with Hematoxylin & Eosin.
Immunofluorescent staining
E14.5 lineage depleted cells were spun onto glass slides and fixed with 4% Paraformaldehyde for 10 min at room temperature. Cells were stained with the dsRNA-specific antibody J2 and antibody binding detected with the M.O.M.™ (mouse on mouse) staining kit according to supplier’s recommendations. In brief, cells were permeabilized with 0.5% Triton X for 10 min and incubated for 1 hr in working solution of M.O.M.™ Mouse IgG Blocking Reagent. After two washes with PBS, slides were incubated for 5 minutes in working solution of M.O.M.™ Diluent, followed by 2 washes. Next, slides were incubated with mouse J2 anti-dsRNA antibody for 30 min, washed and subsequently incubated with M.O.M.™ Biotinylated Anti-Mouse IgG Reagent for 10 min at room temperature. After two washes, streptavidin-Cy3 detection antibody or strepotavidin-Cy5 detection antibody was applied for 20 min, slides washed three times with PBS and covered with DAPI containing vector shield. J2 antibody staining was analyzed via confocal imaging using Leica TCS SP5. For analysis of J2 intensity of each cell, J2 signal was quantified by Image J and normalized to the DAPI signal of each cell quantified by Image J.
Colony-Forming Unit (CFU) assay and serial re-plating
20,000 FL cells were plated in 1.1ml MethoCult (M3434, Stem Cell Technologies, Vancouver, CA) per 35 mm dish in triplicate per supplier’s protocol. Dishes were cultured at 37°C and 5% CO2 in humidified incubator chambers. Colonies were counted and morphologically classified 7 days after plating.
For serial replating, colonies were resuspended and re-plated (10,000 cells/dish in triplicates) for a total of four consecutive platings (7, 14, 21 and 28 days). Colonies were counted and evaluated 7 days after each plating.
Plasmids
METTL3 and catalytically dead METTL3 plasmids are described in previous work (Li et al., 2017a). Both cDNAs were cloned into pMSCV-IRES-GFP vectors for transduction of hematopoietic cells and confirmed by Sanger sequencing. sgRNA against Mavs and Control sgRNA were designed and cloned into LentiCRISPRv2-mCherry lentiviral vector, designated as LentiCRISPRv2-Mavs-mCherry (sgRNA: CCAGGATCGACTGCGGGCTTCCT) and LentiCRISPRv2-Control-mCherry (sgRNA: CACCGGCACTCACATCGCTACATCA). The sequences were verified by Sanger sequencing. The efficiency of sgRNA was determined via Q-RT-PCR and Western blot.
Virus transduction and rescue experiments
Retroviral supernatants were generated via co-transfection of 293GP cells with pCMV-VSVG and pMSCV-Mettl3-GFP / pMSCV-Mettl3 CD-GFP plasmids, followed by spin-concentration. Lentiviral supernatants were generated via co-transfection of 293T cells with pCMV-VSVG, psPAX2 and LentiCRISPRv2-Mavs-mCherry / LentiCRISPRv2-control-mCherry plasmids, followed by spin-concentration.
Bead lineage-depleted Mettl3-deficient FL cells were infected with viral supernatants at equivalent multiplicity of infection (MOI) via spinoculation (1000 xg for 1 hr at 25°C) with addition of 8 mg/ml polybrene (Sigma). After transduction, cells were cultured for two days in HSPC culture medium (DMEM+10% FBS, 50 ng/ml mSCF, 50 ng/ml mTPO, 50 ng/ml mFlt3L, 10ng/ml mIL3).
For CFU assays, virally transduced and sorted cells were plated into 35 mm dishes (20,000 cells/dish).
For competitive transplantation analysis, 0.1 million virally transduced and sorted positive cells were co-transplanted with 1 million competitor (CD45.1) bone marrow cells into irradiated Pep3b recipients (900 cGy). Engraftment was measured 6 weeks post transplantation.
Rnasel knockdown and rescue experiment
30 pmol Rnasel siRNA or non-targeting siRNA (Horizon Discovery) was transfected into vcMettl3+/+ or vcMettl3−/− E14.5 fetal liver cells separately with Lipofectamine™ RNAiMAX Transfection Reagent. After 2 days, siRNA transfected fetal liver cells were plated at 20,000 cells per 35 mm dish with MethoCult for CFU assay. Colonies were counted and morphologically classified 7 days post plating.
Ribosomal RNA (rRNA) cleavage assay
Fetal liver and bone marrow cells were harvested in RLT cell lysis buffer (RNeasy mini kit, QIAGEN). Total RNA was extracted and was resolved on RNA chips using an Agilent 2200 TapeStation to determine the integrity of 28S and 18S rRNA.
RtcB-ligase assisted Q-RT-PCR
RNA fragments generated by RNase L were detected as described (Donovan et al., 2017). Total RNA was purified by RNeasy mini kit (QIAGEN) and RNAs with 2’-3’ cyclic phosphate were ligated to the adapter 5’ rGrArUrCrGrUCGGACTGTAGAACTCTGAAC 3’ using RtcB RNA ligase (New England Biolabs). EDTA-quenched ligation reactions were reverse transcribed using Multiscribe reverse transcriptase (ThermoFisher) and the primer 5’ TCCCTATCAGTGATAGAGAGTTCAGAGTTCTACAGTCCG 3’. The resulting cDNAs were assessed for RNase L cleavage products by Q-RT-PCR for specific cleavage sites and normalized to U6, which has a naturally occurring 2’-3’ cyclic phosphate. Primers are listed in Table S3.
J2-RIP-seq
Fresh Vav-Cre-Mettl3fl/fl E14.5 fetal livers were harvested. One part of the fetal liver was extracted by TRIzol Reagent for input RNA, the other part of the fetal liver was lysed in dsRIP lysis buffer (100 mM NaCl, 50 mM Tirs-HCl pH 7.4, 3 mM MgCl2, 0.5% NP-40) with 1x Complete Proteinase Inhibitor and 30 U SUPERase• In™ RNase Inhibitor. The fetal liver lysate was incubated with 5 μg SCICONS J2 antibody for 2 hours at 4°C. Protein G Dynabeads were washed in dsRIP lysis buffer and added to the lysate to allow J2-RNA complex formation for 1 hour at 4°C. Beads were washed four times with dsRIP lysis buffer, and J2 antibody pull-down RNA was extracted with TRIzol Reagent. Isolated RNAs were rRNA depleted, fragmented, ligated, and sequenced on the NovaSeq System per standard protocol at the Yale Center for Genome Analysis (YCGA).
RNA-seq
E14.5 FL Lin−Sca-1+c-Kit+ (LSK) cells were sorted from 3 embryos per genotype. Total RNA was isolated using the RNeasy Plus Micro Kit (QIAGEN) and submitted for mRNA sequencing to the YCGA via mRNA selection, standard library preparation and sequencing on the Illumina HiSeq2500 system (1x75 bp).
Single cell RNA-sequencing (scRNA-seq)
E14.5 FL LSK cells from two mice per genotype were subjected to scRNA-seq as previously described (Dura et al., 2018). In brief, microfluidic devices were infused with PBS and pressurized to remove air bubbles inside the microwells using a manually operated syringe with a closed outlet. Single cell suspensions with 20,000 cells/50 μl in PBS + 1%BSA were pipetted on the inlet and drawn into the device. Once the channel was completely filled with cell solution, the fluid flow was stopped, and cells were allowed to settle by gravity. Excess cells were washed out with PBS, and mRNA capture beads at concentrations of 30,000–120,000 beads/50–150 μl were similarly loaded into the cell-loaded wells. Microwells were sealed with fluorinated oil (Fluorinert FC-40), the tubing at the outlet was disconnected, and the microfluidic devices were exposed to three freeze-thaw cycles with 5 min freezing (−80°C) alternating with 5 min thawing at room temperature. Following lysis, the microfluidic device was incubated for one hour inside a wet chamber for mRNA capture onto beads. After incubation, beads were collected into 6x saline-sodium citrate (SSC) buffer and washed twice with 6x SSC buffer prior to reverse transcription.
Single cell library preparation and sequencing
Library preparation steps follow the same steps as in the DropSeq and SeqWell methods. Briefly, the beads were washed with 6×SSC twice after retrieval, and the captured mRNA was reverse-transcribed using Maxima H Minus reverse transcriptase (ThermoFisher) with a custom template switching oligo (AAGCAGTGGTATCAACGCAGAGTGAATrGrGrG). The cDNA coated beads were treated with Exonuclease I (Exo I, NEB) at 37°C for 45 min to chew away any unbound mRNA capture probes. cDNA on coated beads was amplified using a half PCR reaction. The amplified DNA was purified using Ampure XP beads (Beckman Coulter) at 0.6x ratio, and the quality of the amplified DNA was assessed by Agilent BioAnalyzer using high sensitivity chips. Purified cDNA was pooled and input for standard Nextera tagmentation and amplification reactions (Nextera XT, Illumina) using a custom primer instead of the i5 index primer to amplify only those fragments that contain the cell barcodes and UMIs. The PCR product was further purified using Ampure XP beads at 0.6x ratio and library quality-verified on Agilent BioAnalyzer high sensitivity chips. The libraries were sequenced on HiSeq 2500 (Illumina) using a custom primer with 75 cycles on Read 1 and 75 cycles on Read 2. For Read 1, only the first 20 bases were used in analysis. PhiX libraries were used at 20% as spike-in controls.
CUT&RUN
CUT&RUN experiments were carried out as described (Skene et al., 2018) with modifications. Briefly, nuclei from 1x105 E14.5 FL LSK cells were isolated with NE buffer (20 mM HEPES-KOH, pH 7.9, 10 mM KCl, 0.5 mM Spermidine, 0.1% Triton X-100, 20% Glycerol and 1x protease inhibitor cocktails from Roche), captured with Bio-Mag Plus Concanavalin A coated beads (Polysciences) and incubated with primary antibody for 2 hours at 4°C. After washing away unbound antibody with wash buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine and 1x protease inhibitor cocktails from Roche), protein A-MNase was added to a final concentration of 700 ng/ml and incubated for 1 hour at 4 °C. The nuclei were washed again and placed in a metal block at 0°C. To activate protein A-MNase, CaCl2 was added to a final concentration of 2 mM. The reaction was carried out for 45 min and stopped by addition of an equal volume of 2XSTOP buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 mg/mL RNase A and 40 mg/mL glycogen). The protein-DNA complex was released by incubation for 10 min at 37 °C and centrifugation. DNA was extracted by QIAGEN PCR MinElute Kit, followed by Qubit fluorometer. Protein A-MNase (batch 6) was kindly provided by Dr. Steve Henikoff. The antibodies used were: H3K4me3 antibody, Cat #39159, Active motif; Normal rabbit IgG, Cat #12-370, Millipore.
Library Preparation and Sequencing for CUT&RUN
We followed the protocol of NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) to make the CUT&RUN DNA library for sequencing. The NEBNext End Prep was performed for 30 min at 20°C and followed by 30 min at 65°C. Adaptor ligation was carried out for 15 min at 20°C, followed by USER Enzyme treatment for 15 min at 37°C.
After adaptor ligation, SPRIselect beads were added to the reaction to purify the adaptor-ligated DNA. After PCR enrichment of adaptor-ligated DNA, the reaction was cleaned up with 0.9x volume of SPRIselect beads. 8 barcoded libraries were quantified and mixed at equal molar ratios. Samples were measured with a bioanalyzer and sequenced in the Illumina HiSeq 2500 system (2x75 bp).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell RNA-Seq analysis
Single cell expression measurements (digital counts) were obtained from raw sequence data (FASTQ) with Drop-seq tools v1.12 (https://github.com/broadinstitute/Drop-seq). Reads were aligned to the mouse genome (GRCm38.p5) with STAR (version 2.5.3a) using the Gencode M15 transcript annotation as transcriptome guide). The final digital expression matrix was obtained selecting cells with at least 500 identified genes.
Single cell expression data analysis was performed with Seurat 3.0 (https://satijalab.org/seurat/) following a standard comparison analysis workflow. In short, digital counts were first normalized with the “LogNormalize” method. Variable features were selected with the “vst method”. vcMettl3+/+ and vcMettl3−/− cells were integrated with the “FindIntegrationAnchors” and “IntegrateData” functions. Integrated data were then scaled. After Principal Component Analysis, cell clusters were identified with the “FindNeighbors” and “FindClusters” functions. Clusters were assigned to hematopoietic populations and entry points based on markers previously identified (Dahlin et al., 2018). Results were visualized with the UMAP dimension reduction technique.
Bulk RNA-Seq analysis
After quality control (FastQC), reads generated from each sample were aligned to the mouse genome (GRCm38.p5) with STAR (version 2.5.3a, –quantMode GeneCounts), using the Gencode M15 transcript annotation as transcriptome guide. Normalization with the TMM method and identification of differentially expressed genes were performed with the edgeR package in Bioconductor (https://bioconductor.org/). Differentially expressed genes were identified with the “glmQLFTest” function, using a double threshold on gene expression changes and associated statistical significances (absolute log2 fold change > 0.75, FDR < 0.05).
Functional annotation and enrichment analysis with Gene Ontology terms and KEGG or REACTOME pathways were performed with the clusterProfiler Bioconductor package (https://yulab-smu.github.io/clusterProfiler-book/).
The meta-analysis of m6A annotated genes was based on a collection of 52 m6A-Seq datasets obtained from murine and human tissues and cell lines (Barbieri et al., 2017; Geula et al., 2015; Li et al., 2018; Liu et al., 2020; Vu et al., 2017).
CUT&RUN analysis
Raw sequence data were aligned to the mouse genome (GRCm38.p5) with Bowtie2 v2.2.9, (--local --very-sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700). Duplicated reads were removed with Picard tools v2.9.0. Using the Gencode M15 annotation and bedtools coverage function (v2.26.0), the number of reads aligned around known transcription start sites (TSS) was determined for each sample, considering a window of 3k nucleotides. Differential changes in H3K4me3 occupancy between vcMettl3+/+ and vcMettl3−/− LSK cells were identified with edgeR (p-value < 0.05, log2 absolute log2 fold change > 0.75).
J2 RIP-Seq analysis
After quality control (FastQC) and removal duplicates removal with FastUniq (v1.1) (https://sourceforge.net/projects/fastuniq/), reads generated from J2 RIP and INPUT samples were aligned to the mouse genome (GRCm38.p5) with STAR (version 2.5.3a, –quantMode GeneCounts), using the Gencode M15 transcript annotation as transcriptome guide. Data were normalized with the TMM method in edgeR.
J2 vcMettl3−/− enriched genes were identified applying 3 criteria:
-
Enrichment of J2-RIP over INPUT, considering both vcMettl3+/+ and vcMettl3−/− samples. This criterion selects transcripts that are enriched in the J2-RIP samples versus the INPUT samples. These are the transcripts that are more likely to be in a dsRNA conformation.
The analysis is performed with generalized linear models implemented in edgeR (glmQLFTest function). The contrast used for this criterion is: (RIP+/+ + RIP−/−) − (INPUT+/+ + INPUT−/−).
After applying this contrast, considering average expression values among replicates, 3950 genes with (log2 RIP) > (log2 INPUT) and with p – value < 0.05 were selected.
-
Enrichment of J2 vcMettl3−/− over vcMettl3+/+, normalizing for INPUT. This criterion selects transcripts that are specifically enriched in the vcMettl3−/− J2-RIP, normalizing for the INPUT. These are the transcripts increasing their dsRNA conformation upon deletion of Mettl3.
The analysis is performed with generalized linear models implemented in edgeR (glmQLFTest function). The contrast used for this criterion is: (RIP−/− − RIP+/+) − (INPUT−/− − INPUT+/+).
This is also equivalent to the contrast: (RIP−/− − INPUT−/−) − (RIP+/+ − INPUT+/+).
After applying this contrast, considering average expression values among replicates, 553 genes with (log2 RIP−/− − log2 RIP+/+) > (log2 INPUT−/− − log2 INPUT+/+), equivalent to (log2 RIP−/− − log2 INPUT−/−) > (log2 RIP+/+ − log2 INPUT+/+), and with p – value < 0.05 were selected. The intersection between criterion 1 and criterion 2 results in 311 genes.
-
Contribution of J2 RIP changes over INPUT changes. This criterion selects transcripts for which the absolute change in J2 RIP levels (vcMettl3−/− vs vcMettl3+/+) is larger than the absolute change in INPUT levels. This criterion is important to exclude genes that, upon Mettl3 deletion, decrease in both J2 RIP and in the INPUT, but more in the latter. These genes would result enriched in criterion 2, but just because their decrease in J2 RIP is lower than their decrease in INPUT. Considering average expression values among replicates, genes with ∣log2 RIP−/− − log2 RIP+/+∣ > ∣log2 INPUT−/− − log2 INPUT+/+∣ were selected.
This criterion is not associated with an edgeR contrast, therefore it’s not dependent on a p-value.
This criterion, applied to the 311 genes selected before, results in the final set of 94 J2 vcMettl3−/− enriched genes.
RNA sequences used for length and folding analysis were downloaded from Ensembl v90, corresponding to the Gencode M15 annotation.
Folding minimum free energies were calculated with the RNAfold algorithm contained in the ViennaRNA package (v2.4.11).
The analysis of Transposable Elements (TE), including the classification of TE classes and families, was based on GTF files curated by the Hammell Lab: (http://labshare.cshl.edu/shares/mhammelllab/www-data/TEtranscripts/TE_GTF/mm10rmskTE.gtf.gz).
Reads mapped on transposable elements were identified and quantified with the Rsubread Bioconductor package.
Statistical Analysis
All the remaining statistical analyses were performed using GraphPad Prism 8 (GraphPad Software). Significance was calculated using two-tailed, unpaired Student’s t-tests or two-way ANOVA. Differences were considered to be statistically significant at the p < 0.05 level (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s. not significant). Unless otherwise noted, all error bars represent SEM.
Supplementary Material
Table S1. Bulk RNA-seq and H3K4me3 CUT&RUN data. Related to Figure 3 and Figure 4.
The file contains transcriptome-wide gene annotation, m6A annotation from a collection of external datasets, bulk RNA-seq and H3K4me3 CUT&RUN differential analysis results.
Table S2. J2-RIP-seq data. Related to Figure 5.
The file contains transcriptome-wide gene annotation and J2-RIP enrichment analysis results.
Table S3. Primers. Related to the STAR Methods.
Highlights.
Loss of METTL3 inhibits proliferation and differentiation of hematopoietic stem cells
Depletion of m6A results in aberrant dsRNA formation of long m6A modified transcripts
Loss of METTL3 induces deleterious innate immune responses in hematopoiesis
Mavs and Rnasel depletion partially rescue defects in Vav-Cre+-Mettl3fl/fl mice
Acknowledgements
We thank A. Brooks for research histology services. We thank Christopher Castaldi at the Yale Center for Genome Analysis (YCGA) for valuable advice and high throughput sequencing and the Yale University High Performance Computing Center for use of clusters to run bioinformatics analysis. We thank Jun Zhao at Department of Pathology, Yale University School of Medicine, for her help with the single cell RNA-Seq analysis. We thank Grace Chen, Department of Immunobiology, Yale University School of Medicine, for the fruitful discussion regarding circular RNAs. We thank Shirui Chen at the University of Tokyo, for her help with the J2 RIP-seq experiment. This study was supported in part by the State of Connecticut under the Regenerative Medicine Research Fund (to S.H.; its contents are solely the responsibility of the authors and do not necessarily represent the official views of the State of Connecticut or Connecticut Innovations, Incorporated), the NIH/NIDDK R01DK102792 (to S.H.), The Frederick A. Deluca Foundation (to S.H.), the National Natural Science Foundation of China (91753141 to H.-B.L.), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (to H.-B.L.), and the Howard Hughes Medical Institute (R.A.F). Y.G. was supported by the James Hudson Brown - Alexander Brown Coxe Postdoctoral Fellowships. R.V. was supported by the American Society of Hematology Physician Scientist Career Development Award and the William U. Gardner Memorial Student Research Fellowship. T.T. was supported by a pilot grant from the Yale Cooperative Center of Excellence in Hematology (YCCEH; NIDDK U54DK106857). G.V. was supported by CNR Short Term Mobility 2018. Y.S. was supported by the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 81800122). X.F. was supported by the National Natural Science Foundation of China (No. 81801588). This study was supported by the Animal Modeling Core of the Yale Cooperative Center of Excellence in Hematology (NIDDK U54DK106857).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, and Kariko K (2011). Nucleoside modifications in RNA limit activation of 2'-5'-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res 39, 9329–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, Sadee C, et al. (2018). The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA methylation in pluripotency. Nature 555, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, et al. (2009). Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 119, 2399–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blango MG, and Bass BL (2016). Identification of the long, edited dsRNAome of LPS-stimulated immune cells. Genome Res 26, 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-L, Yang L, and Carmichael GG (2010). Molecular basis for an attenuated cytoplasmic dsRNA response in human embryonic stem cells. Cell cycle (Georgetown, Tex) 9, 3552–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al. (2019). N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Bruning JC, Vu LP, et al. (2019). m(6)A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Rep 28, 1703–1716 e1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabo S, and Meurs EF (2012). dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4, 2598–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, Weinreb C, Wolock S, Hannah R, Diamanti E, et al. (2018). A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood 131, e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, and Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 71, 3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Rath S, Kolet-Mandrikov D, and Korennykh A (2017). Rapid RNase L-driven arrest of protein synthesis in the dsRNA response without degradation of translation machinery. Rna 23, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dura B, Choi JY, Zhang K, Damsky W, Thakral D, Bosenberg M, Craft J, and Fan R (2018). scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3' mRNA profiling. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Harada BT, Behm M, and He C (2018). RNA modifications modulate gene expression during development. Science 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, and Williams BR (2007). The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev 18, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, and Hiscott J (2002). Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76, 5532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y-L, Carmichael GG, Wang R, Hong X, Acharya D, Huang F, and Bai F (2015). Attenuated Innate Immunity in Embryonic Stem Cells and Its Implications in Developmental Biology and Regenerative Medicine. Stem cells (Dayton, Ohio) 33, 3165–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, and Korennykh A (2014). Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, and Orkin SH (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S, and Thomsen AR (2012). Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L (2019). Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017a). m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, and Weiss SR (2017b). Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. (2018). Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res 28, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li K, Cai J, Zhang M, Zhang X, Xiong X, Meng H, Xu X, Huang Z, Peng J, et al. (2020). Landscape and Regulation of m(6)A and m(6)Am Methylome across Human and Mouse Tissues. Mol Cell 77, 426–440 e426. [DOI] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, Yang YG, Zhou Q, and Liu F (2018). Endothelial-specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res 28, 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker M, Zimmermann B, Bilusic I, Tukhtubaeva N, and Schroeder R (2014). The double-stranded transcriptome of Escherichia coli. Proc Natl Acad Sci U S A 111, 3134–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol 33, 319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet BP, Engels S, Aglialoro F, van den Akker E, von Lindern M, and Wolkers MC (2018). Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res 46, 8168–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Pickering BF, and Jaffrey SR (2017). Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins. Trends Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulit-Penaloza JA, Scherbik SV, and Brinton MA (2012). Type 1 IFN-independent activation of a subset of interferon stimulated genes in West Nile virus Eg101-infected mouse cells. Virology 425, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, and Stark GR (2000). Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. Embo J 19, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh T, Hovanessian AG, Meurs EF, Sonenberg N, and Kimchi A (1996). Double-stranded RNA-dependent protein kinase mediates c-Myc suppression induced by type I interferons. J Biol Chem 271, 25479–25484. [DOI] [PubMed] [Google Scholar]
- Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, and Kool ET (2015). Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc 137, 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, and Williams BR (2008). Interferon-inducible antiviral effectors. Nat Rev Immunol 8, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens X, Kalai M, and Vandenabeele P (2001). Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem 276, 41620–41628. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, and Kouzarides T (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411. [DOI] [PubMed] [Google Scholar]
- Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, and Lukacs N (1991). Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic acids research 19, 2993–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, and He C (2019). Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RH (2007). Viral encounters with 2',5'-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81, 12720–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Son KN, Liang Z, and Lipton HL (2015). Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J Virol 89, 9383–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, and Graf T (2005). Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development 132, 203–213. [DOI] [PubMed] [Google Scholar]
- Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, Zhu S, Li H, Li B, Chen L, et al. (2018). m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res 28, 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014a). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014b). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue MH, Wang J, Kumar S, Wechsler-Reya RJ, Zhang ZL, Ogawa Y, Kellis M, Duester G, et al. (2018). N-6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci 21, 195–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205 e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai-Schmiedel J, Friedman N, Le-Trilling VTK, et al. (2019). m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol 20, 173–182. [DOI] [PubMed] [Google Scholar]
- Wu X, Dao Thi VL, Huang Y, Billerbeck E, Saha D, Hoffmann HH, Wang Y, Silva LAV, Sarbanes S, Sun T, et al. (2018). Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 172, 423–438.e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang JF, Yang Q, Liu CX, Wu M, Chen LL, and Yang L (2018). N(6)-Methyladenosines Modulate A-to-I RNA Editing. Mol Cell 69, 126–135 e126. [DOI] [PubMed] [Google Scholar]
- Yockey LJ, and Iwasaki A (2018). Interferons and Proinflammatory Cytokines in Pregnancy and Fetal Development. Immunity 49, 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y, et al. (2017). m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, and He C (2017a). Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, and He C (2017b). m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Hassel BA, and Silverman RH (1993). Expression cloning of 2–5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72, 753–765. [DOI] [PubMed] [Google Scholar]
- Zhou KI, Parisien M, Dai Q, Liu N, Diatchenko L, Sachleben JR, and Pan T (2016). N(6)-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J Mol Biol 428, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Bulk RNA-seq and H3K4me3 CUT&RUN data. Related to Figure 3 and Figure 4.
The file contains transcriptome-wide gene annotation, m6A annotation from a collection of external datasets, bulk RNA-seq and H3K4me3 CUT&RUN differential analysis results.
Table S2. J2-RIP-seq data. Related to Figure 5.
The file contains transcriptome-wide gene annotation and J2-RIP enrichment analysis results.
Table S3. Primers. Related to the STAR Methods.
Data Availability Statement
Raw and analyzed sequencing data have been deposited under GEO: GSE148882 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148882)
Software used for the analyses are described and referenced in the individual Method Details subsections and are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| METTL3/MT-A70 Antibody | Bethyl lab | Cat# A301-567A; RRID: AB_1040004 |
| Anti-METTL14 antibody | SIGMA-ALDRICH | Cat# HPA038002; RRID: AB_10672401 |
| Anti-m6A antibody | Synaptic Systems | Cat# 202003; RRID: AB_2279214 |
| Anti-PKR antibody | SIGMA-ALDRICH | Cat# SAB3500326; RRID: AB_10646199 |
| Anti-phospho-PKR antibody | SIGMA-ALDRICH | Cat# SAB4504517; |
| eIF2α Antibody | Cell Signaling Technology | Cat# 9722S; RRID: AB_2230924 |
| Phospho-eIF2α (Ser51) Antibody | Cell Signaling Technology | Cat# 9721S; RRID: AB_330951 |
| CD34 Monoclonal Antibody (RAM34), FITC | eBioscience | Cat# 11-0341-85; RRID: AB_465021 |
| PE/Cy5 anti-mouse CD135 (Flt3) Antibody | Biolegend | Cat# 135312; RRID: AB_2263031 |
| Miltenyi lineage detection | Miltenyi | Cat# 130-092-613; RRID: AB_1103214 |
| APC-Cy7 Streptavidin | Biolegend | Cat# 405208 |
| PE anti-mouse Ly-6A/E (Sca-1) Antibody | Biolegend | Cat# 108108; RRID: AB_313345 |
| APC anti-mouse CD117 (c-Kit) Antibody | Biolegend | Cat# 105812; RRID: AB_313221 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block™) | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| CD16/CD32 Monoclonal Antibody, PE-Cy7 | eBioscience | Cat# 25-0161-82; RRID: AB_469597 |
| APC anti-mouse/human CD44 Antibody | Biolegend | Cat# 103012; RRID: AB_312963 |
| TER-119 Monoclonal Antibody (TER-119), PE | Biolegend | Cat# 12-5921-82; RRID: AB_466042 |
| Biotin anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat# 108404; RRID: AB_313369 |
| Biotin anti-mouse/human CD45R/B220 Antibody | Biolegend | Cat# 103204; RRID: AB_312989 |
| CD11b Monoclonal Antibody (M1/70), Biotin | eBioscience | Cat# 13-0112-82; RRID: AB_466359 |
| Streptavidin eFluor™ 450 | eBioscience | Cat# 48-4317-82; RRID: AB_10359737 |
| Pacific Blue™ anti-mouse/human CD11b | Biolegend | Cat# 101224; RRID: AB_755986 |
| APC anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat# 108411; RRID: AB_313376 |
| PE/Cy7 anti-mouse CD45.1 Antibody | Biolegend | Car# 110730; RRID: AB_1134168 |
| CD45.2 Monoclonal Antibody (104), FITC | eBioscience | Cat# 11-0454-85; RRID: AB_465060 |
| HSP90 Antibody | StressMarq Biosciences | Cat# SMC-107; RRID: AB_854214 |
| GAPDH (FL-335) Antibogy | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2031; RRID: AB_631737 |
| Histone H3K4me3 antibody (pAb) | Active motif | Cat# 39159; RRID: AB_2615077 |
| Normal Rabbit IgG | Millipore | Cat# 12-370; RRID: AB_145841 |
| Monoclonal Antibody SCICONS J2 | English & Scientific Consulting Kft. | Cat# 10010200; RRID: AB_2651015 |
| Cy3 Streptavidin | VECTOR LABORATORIES | Cat# SA-1300 |
| Cy5 Streptavidin | VECTOR LABORATORIES | Cat# SA-1500 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MethoCult™ GF M3434 | STEMCELL Technologies | Cat# 03434 |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | New England Biolabs | Cat# M0531L |
| RBC Lysis Buffer, 10X | Santa Cruze Biotechnology | Cat# sc-296258 |
| Lipofectamine™ 2000 Transfection Reagent | ThermoFisher SCIENTIFIC | Cat# 11668019 |
| Lipofectamine™ RNAiMAX Transfection Reagent | ThermoFisher SCIENTIFIC | Cat# 13778100 |
| Mouse Thrombopoietin (mTPO) | Gemini Bio-product | Cat# 300-351P |
| Mouse Stem Cell Factor (mSCF) | Gemini Bio-product | Cat# 300-348P |
| Mouse Flt-3 Ligand (mFlt3L) | Gemini Bio-product | Cat# 300-306P |
| Mouse Interleukin-3 (mIL3) | Gemini Bio-product | Cat# 300-324P |
| Benchmark Fetal Bovine Serum (FBS) | Gemini Bio-product | Cat# 100-106 |
| DMEM, high glucose | ThermoFisher SCIENTIFIC | Cat# 11965092 |
| Protein A–micrococcal nuclease (pA-MN) fusion protein | Henikoff lab | Skene and Henikoff, 2018 |
| Spermidine trihydrochloride | Sigma-Aldrich | Cat# S2501 |
| Con A-coated BioMag®Plus microparticles | Polysciences, Inc | Cat# 86057-3 |
| iQ™ SYBR® Green Supermix | BIO-RAD | Cat# 1708880 |
| iScript cDNA Synthesis Kit | BIO-RAD | Cat# 1708890 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836170001 |
| SUPERase• In™ RNase Inhibitor | ThermoFisher SCIENTIFIC | Cat# AM2696 |
| SPRIselect beads | Beckman Coulter | Cat# B23317 |
| RNaseA, Dnase and protease-free | ThermoFisher SCIENTIFIC | Cat# EN0531 |
| Maxima H Minus Reverse Transcriptase | ThermoFisher SCIENTIFIC | Cat# EP0752 |
| Exonuclease I | New England Biolabs | Cat# M0293S |
| Ampure XP beads | Beckman Coulter | Cat# A63880 |
| Fluorinert™ FC-40 | Sigma-Aldrich | Cat# F9755 |
| Critical Commercial Assays | ||
| EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric) | Epigentek | Cat# P-9005-96 |
| FITC BrdU Flow Kits | BD Pharmingen™ | Cat# 559619 |
| CellTrace CFSE Cell Proliferation Kit, for flow | Thermo Fisher | Cat# C34554 |
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen™ | Cat# 556547 |
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Magnetic mRNA Isolation Kit | New England Biolabs | Cat# S1550S |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat# E7500S |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | Cat# E7645S |
| Qubit dsDNA HS Assay Kit | Life Technologies | Cat# Q32851 |
| Vector M.O.M. Immunodetection Kit | VECTOR LABORATORIES | Cat# BMK-2202 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE148882 |
| Mouse genome and transcript annotation | Gencode M15 | https://www.gencodegenes.org/mouse/release_M15.html |
| Transcript sequences | Ensembl 90 | https://useast.ensembl.org/info/website/archives/index.html |
| Annotation of Transposable Elements | Hammell Lab | http://labshare.cshl.edu/shares/mhammelllab/www-data/TEtranscripts/TE_GTF/mm10_rmsk_TE.gtf.gz |
| Annotation of m6A genes | Barbieri et al., 2017 | DOI: 10.1038/nature24678 |
| Annotation of m6A genes | Geula et al., 2015 | DOI: 10.1126/science.1261417 |
| Annotation of m6A genes | Li et al., 2018 | DOI: 10.1038/s41422-018-0072-0 |
| Annotation of m6A genes | Liu et al., 2020 | DOI: 10.1016/j.molcel.2019.09.032 |
| Annotation of m6A genes | Vu et al., 2017 | DOI: 10.1038/nm.4416 |
| Experimental Models: Cell Lines | ||
| Human: HEK293GP cells | Clontech | Cat# 631458 |
| Human: HEK293T cells | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | In house colony | JAX: 002014 |
| Mouse: B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J | In house colony | JAX: 008610 |
| Mouse: C57BL/6J | In house colony | JAX: 000664 |
| Mouse: Mettl3f/f | Li et al., 2017 | N/A |
| Oligonucleotides | ||
| ON-TARGETplus Mouse Rnasel siRNA smartpool | Horizon Discovery | L-043480-00-0020 |
| ON-TARGETplus Non-targeting Control Pool | Horizon Discovery | D-001810-10-05 |
| See Table S3 | This paper | N/A |
| Recombinant DNA | ||
| pMSCV-IRES-GFP | Addgene | Plasmid #20672 |
| pMSCV-Mettl3-GFP | This paper | N/A |
| pMSCV-Mettl3CD-GFP | This paper | N/A |
| LentiCRISPRv2-mCherry | Addgene | Plasmid #99154 |
| LentiCRISPRv2- Mavs-mCherry | This paper | N/A |
| LentiCRISPRv2-Control-mCherry | This paper | N/A |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| psPAX2 | Addgene | Plasmid #12260 |
| Software and Algorithms | ||
| FlowJo v10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| Prism v8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Bowtie2 v2.2.9 | John Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Fiji | NIH | https://fiji.sc/ |
| Drop-seq tools v1.12 | Broad Institute | https://github.com/broadinstitute/Drop-seq |
| STAR v2.3.3a | N/A | https://github.com/alexdobin/STAR |
| edgeR | Bioconductor | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| clusterProfiler | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Picard Tools v2.9.0 | Broad Institute | https://broadinstitute.github.io/picard/ |
| Fastuniq v1.1 | N/A | https://sourceforge.net/projects/fastuniq/ |
| RNAfold v2.4.11 | ViennaRNA package | https://www.tbi.univie.ac.at/RNA/ |
| Rsubread | Bioconductor | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| Seurat v3.0 | N/A | https://satijalab.org/seurat/ |
| FastQC v0.11.5 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Other | ||
| Qubit fluorometer | ThermoFisher SCIENTIFIC | Cat# Q33216 |
| HiSeq 2500 Sequencing System | Illumina | Cat# SY–401–2501 |
| HemaTrue Veterinary Hematology Analyzer | Heska | N/A |
| Leica TCS SP5 | Leica | N/A |