

Abstract
The resolution of inflammation is an active response involving the interaction of pro-resolving mediators with specific receptors, such as N-formyl peptide receptor 2 (FPR2). FPRs represent potentially important therapeutic targets for the treatment of some pathologies, including asthma and rheumatoid arthritis. Previously, we identified selective or mixed FPR agonists with a pyridazin-3(2H)-one scaffold, all containing a 4-bromophenylacetamide fragment at N-2. The most effective compounds in this series were EC3, a potent mixed FPR1/FPR2/FPR3 agonist, and EC10, which had a preference for FPR1. We report here a new series of pyridinone and pyrimidindione derivatives containing the 4-(bromophenyl)acetamide substituent that was essential for activity in the pyridazinone series. All new compounds were evaluated for FPR agonist activity in HL60 cells transfected with FPR1 or FPR2 and in human neutrophils. While most of the pyridinone derivatives had reasonable FPR agonist activity in the submicromolar/micromolar range, the pyrimidindione derivatives were less active. Compound 2a (N-(4-bromophenyl)-2-[3-cyano-5-(3-methoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]acetamide) was the most active pyridinone derivative and had a 10-fold preference for FPR2 (EC50 = 120 nM) versus FPR1 (EC50 = 1.6 μM). To assess their therapeutic activity, compounds 2a, EC3, and EC10 were evaluated in vivo using a rat model of rheumatoid arthritis. All three compounds increased the pain threshold and reduced pain hypersensitivity in the treated rats versus control rats, although 2a and EC10 were much more effective than EC3. Thus, these FPR agonists represent potential leads to develop for the treatment of inflammatory diseases such as rheumatoid arthritis.
Keywords: Formyl peptide receptor (FPR), Agonist, Rheumatoid arthritis, Inflammation, Neutrophil
1. Introduction
Inflammation is a protective response of the immune system and helps to prevent infection by invading microbial pathogens. This complex and multifactorial host response is mediated by the action of many cell types with distinct functions and is essential for host survival [1]. The infiltration of leukocytes and their activation are characteristic events of inflammation, as these cells can generate inflammatory mediators to recruit additional phagocytic cells, phagocytose foreign agents, produce reactive oxygen species, release proteases, and destroy invading pathogens [2,3]. However, deregulation of these responses or an excessive activation of leukocytes can lead to tissue damage and acute or chronic inflammation. Pro-inflammatory mediators provoke the classical signs of inflammation, inducing a sufficiently strong response to the injury while limiting damage to host tissue. Subsequently, endogenous anti-inflammatory mediators ensure resolution of the inflammatory response. The resolution of inflammation was originally considered as a passive process, but recently it was demonstrated that resolution of inflammation is an active response carried out by specific pro-resolving mediators (SPMs), such as protectins, maresins, resolvins, and lipoxins [4–7].
SPMs perform a dual role by combining pro-resolving and anti-inflammatory activities, both mediated by interactions with specific receptors [6,8]. N-formyl peptide receptor 2 (FPR2) has been reported to be a target for SPMs, and its activation by these pro-resolving mediators can contribute to the resolution of inflammation [9,10]. FPR2 is a G-protein-coupled chemoattractant receptor that belongs to the FPR family, which in humans includes two other isoforms, FPR1 and FPR3, that exhibit a high level of amino acid homology with FPR2 [11]. The three isoforms are expressed at different levels in various cells: FPR1 and FPR2 are present in many immune cells, including monocytes, neutrophils, T lymphocytes, and macrophages, but also in endothelial cells, hepatocytes, astrocytes [11,12], whereas FPR3 is found primarily in monocytes, macrophages, and dendritic cells, but not in neutrophils [12,13]. Activation of FPRs can induce a variety of pro- or anti-inflammatory responses, depending on the cell type and receptor subtype involved [11]. The multifaceted role of FPRs has stimulated interest in targeting FPRs for the treatment of some pathologies, including asthma, chronic pulmonary disease, rheumatoid arthritis, Alzheimer disease, and cancer [14–23]. For example, Amgen published the results of an in vivo study of the mixed FPR1/FPR2 agonist Cpd43 [24] (Fig. 1). This compound was administered at 30 mg/kg (i.p.) in mice to investigate two animal models of rheumatoid arthritis, collagen-induced arthritis and antigen-induced arthritis. In both models, Cpd43 treatment significantly reduced disease severity, as demonstrated by decreased joint swelling and by the reduction of tissue inflammation, synovitis, and cartilage damage [25]. In addition, pre-treatment of the animals with FPR antagonists Boc-MLF and WRW4 resulted in complete reversal of the effects of Cpd43 treatment, demonstrating a direct correlation between FPR activation by Cpd43 and reduction of arthritis severity.
Fig. 1.
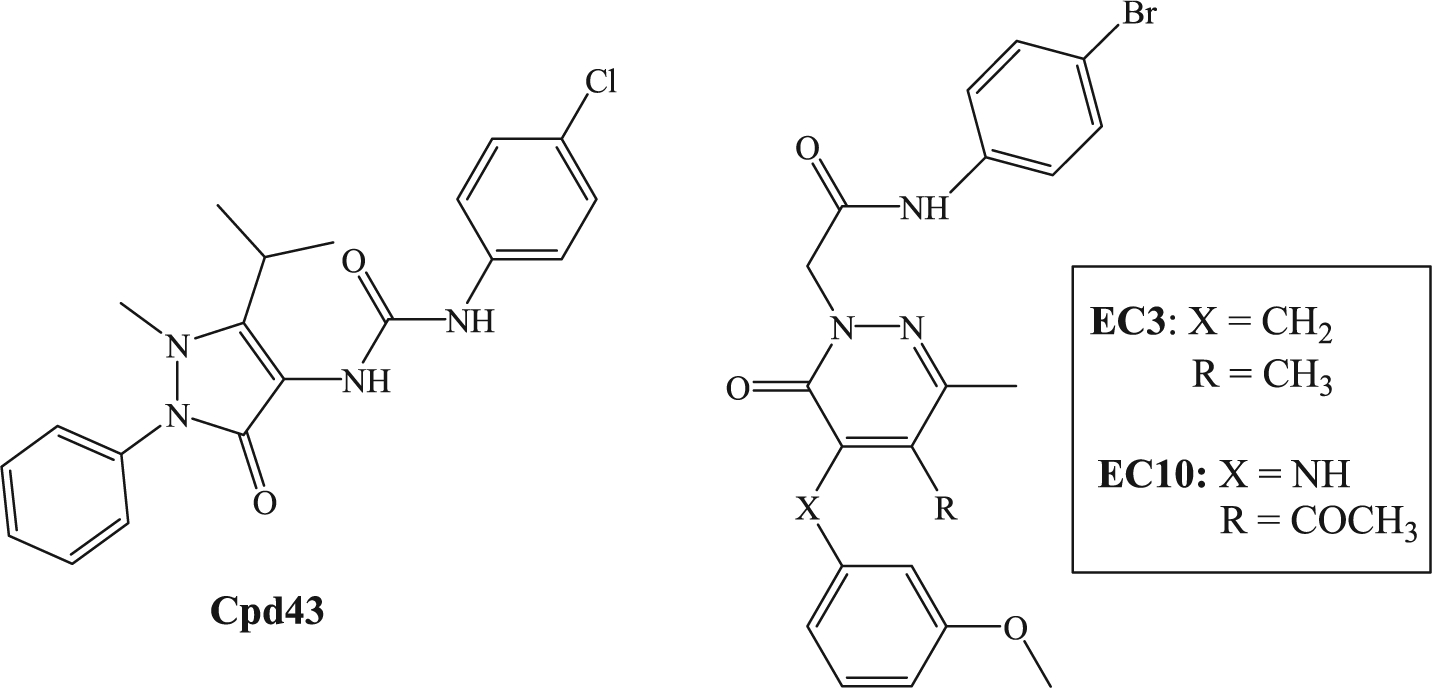
FPRs agonists.
Previously, we synthesized a number of FPR agonists with different scaffolds and activity/selectivity profiles [26–32], all displaying a common N-(4-bromophenyl)acetamide fragment. Among these compounds, the pyridazin-3(2H)-one derivatives EC3 [32] and EC10 [31] (Fig. 1) were the most potent, exhibiting EC50 values in the nanomolar range. In particular, EC3 was a potent mixed FPR1/FPR2/FPR3 agonist (EC50 = 19 nM for FPR1, 43 nM for FPR2, and 40 nM for FPR3), while EC10 was also a mixed agonist but with a preference for FPR1 (EC50 = 45 nM for FPR1, 170 nM for FPR2, and 210 nM for FPR3). Based on these results, we expanded our research to other nitrogen heterocyclic scaffolds and synthesized a new series of pyridinone and pyrimidindione derivatives containing a 4-bromophenylacetamide fragment (Fig. 2, compounds A and B). We also performed modifications at the level of 4-bromophenylacetamide chain in order to confirm the importance of this fragment for this series. All new compounds were tested as FPR agonists. Moreover, the most potent derivative (2a), as well the previously reported potent FPR agonists EC3 and EC10, were evaluated in vivo in a rat model of rheumatoid arthritis.
Fig. 2.
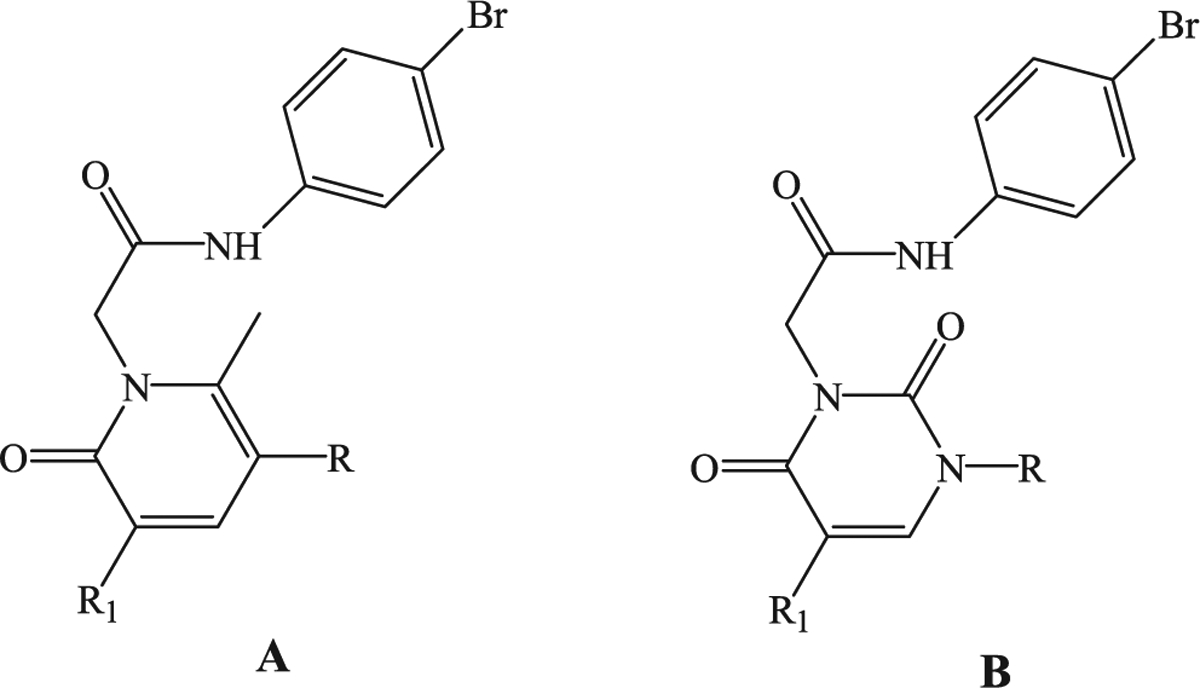
General structures of new potential FPR1/FPR2 agonists.
2. Results and discussion
2.1. Chemistry
All compounds were synthesized according to Schemes 1–3, and the structures were unequivocally confirmed on the basis of analytic and spectral data. Scheme 1 shows the synthetic pathways used to obtain the final compounds of type 2 and 3. Pyridinones 2a-d, containing a 4-bromophenylacetamide chain at N-1 and different substituents at position 5 of the heterocycle, were synthesized starting from the appropriate precursors 1a-d [33,34] by alkylation with N-(4-bromophenyl)-2-chloroacetamide [35] in anhydrous CH3CN and K2CO3. Similarly, compounds 3a-c were obtained by alkylation of precursor 1a with the suitable benzyl bromide in DMF (3a,b) or with 4-bromobenzoyl chloride and NaH in anhydrous THF (compound 3c).
Scheme 1. Reagents and conditions:
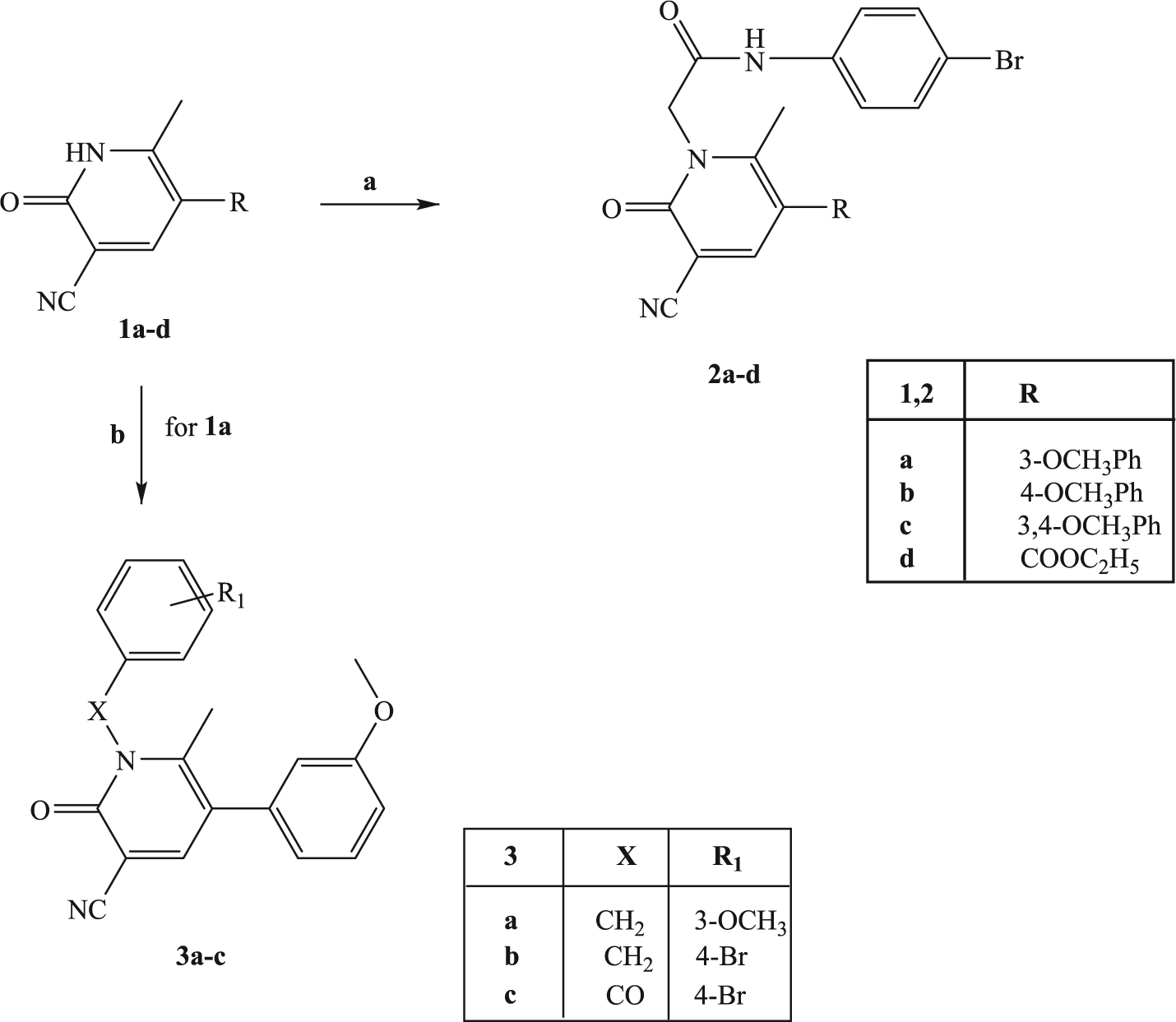
(a) N-(4-bromophenyl)-2-chloroacetamide, K2CO3, anhydrous CH3CN, reflux, 2–8 h; (b) for 3a,b: suitable benzylbromide, K2CO3, anhydrous DMF, reflux, 3 h; for 3c: 4-Br-PhCOCl, NaH, anhydrous THF, t.a., 24 h.
Scheme 3. Reagents and conditions:
(a) N-(4-bromophenyl)-2-chloroacetamide, K2CO3, anhydrous CH3CN, reflux, 2–6 h; (b) Br2, glacial CH3COOH, r.t. 90 min; (c) 3-methoxyphenylboronic acid, tetrakis, anhydrous toluene, 2N Na2CO3, 80 °C, 4–7 h.
Further elaborations of the pyridinone scaffold by synthesis of compounds of type 6 and 9 are illustrated in Scheme 2. To obtain compounds 6a-c, intermediates 4a,b [36] were directly alkylated with N-(4-bromophenyl)-2-chloroacetamide under standard conditions, followed by a coupling reaction with the appropriate aryl boronic acid using anhydrous copper(II)acetate and Et3N in the presence of activated molecular sieves.
Scheme 2. Reagents and conditions:
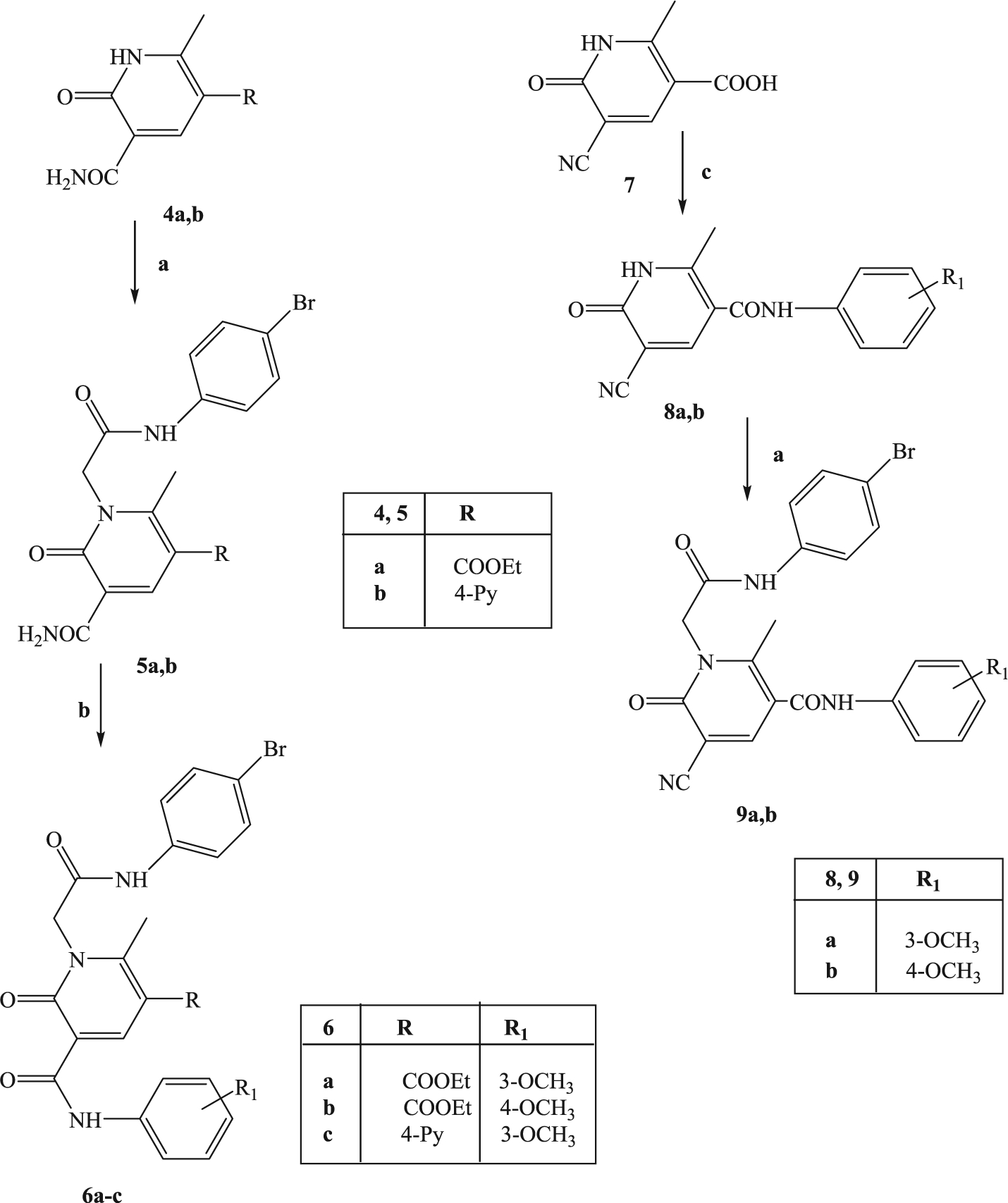
(a) N-(4-bromophenyl)-2-chloroacetamide, K2CO3, anhydrous CH3CN, reflux, 2–8 h; (b) arylboronic acid, anhydrous (COOCH3)Cu, Et3N, activated molecular sieves, anhydrous CH2Cl2, r.t. 48–52 h; (c) SOCl2, Et3N, t.a., 5 h and then arylamine, diethylether, r.t., 3 h.
For synthesis of 9a,b, the intermediate 7 [37] was treated with SOCl2 and the appropriate 3- or 4-methoxy aniline in diethyl ether to obtain derivatives 8a,b which, in turn, were alkylated under the same conditions previously described.
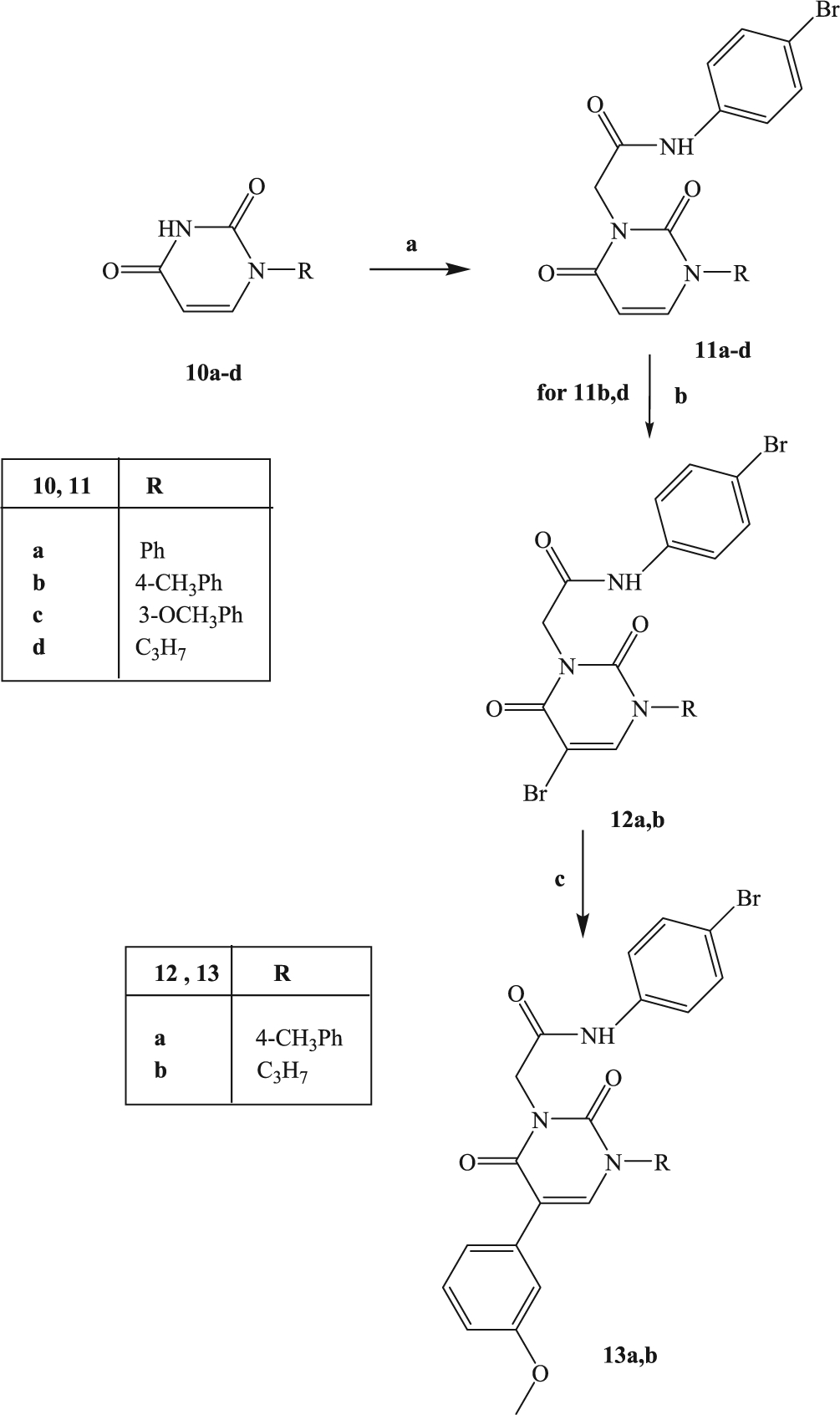
The synthetic procedure shown in Scheme 3 summarizes synthesis of the novel pyrimidine-2,4-diones 11a-d and 13a,b. Alkylation of the known pyrimidine-2,4-diones 10a-d [38–41] with N-(4-bromophenyl)-2-chloroacetamide resulted in 11a-d. Compounds 11b and 11d were then converted into the corresponding 5-bromine derivatives 12a,b, by reaction with bromine in acetic acid and finally transformed through a cross-coupling reaction with 3-methoxyphenylboronic acid, palladium-tetrakistriphenylphosphine (Tetrakis), and Na2CO3 in anhydrous toluene into the final compounds 13a,b.
2.2. Biological evaluation and structure-activity relationship (SAR) analysis
All newly synthesized compounds were evaluated for their ability to induce intracellular Ca2+ flux in human neutrophils and in human HL-60 cells transfected with either FPR1 or FPR2, and the results are reported as EC50 values in Tables 1 and 2 using as reference compounds fMLF (FPR1 agonist) and WKYMVm (FPR2 agonist). All compounds were also evaluated in non-transfected HL-60 wild-type cells and were found to be inactive.
Table 1.
Effect of compounds 2a-d, 3a-c, 6a-c, and 9a-b on Ca2+ mobilization in FPR-transfected HL60 cells and human neutrophils.
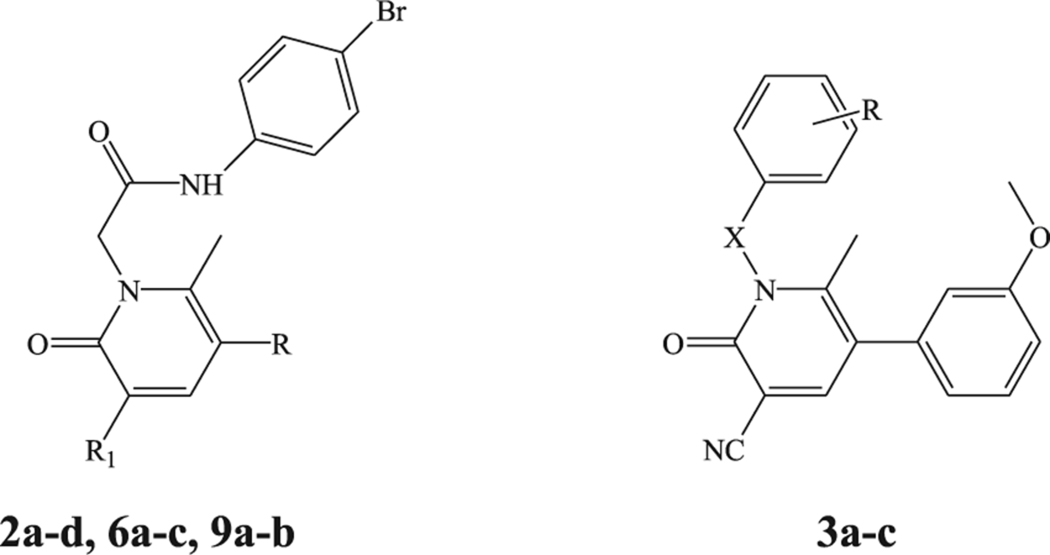 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | X | FPRl-HL60a EC50 (μM)a | FPR2-HL60a EC50 (μM) | hPMNa EC50 (μM) |
| 2a | 3-(OCH3)Ph | CN | 1.6 ± 0.5 | 0.12 ± 0.02 | 0.39 ± 0.17 | |
| 2b | 4-(OCH3)Ph | CN | - | 1.0 ± 0.2 | 0.48 ± 0.08 | 0.66 ± 0.21 |
| 2c | 3,4-(OCH3)2Ph | CN | - | 33.2 ± 4.9 | 0.60 ± 0.2 | 14.6 ± 2.2 |
| 2d | COOEt | CN | - | 3.0 ± 0.4 | 0.38 ± 0.09 | 0.83 ± 0.24 |
| 9a | CONH-(3-OCH3)Ph | CN | - | 3.9 ± 1.2 | 0.31 ± 0.09 | 2.5 ± 0.7 |
| 9b | CONH-(4-OCH3)Ph | CN | - | 0.96 ± 0.39 | 0.44 ± 0.12 | 1.2 ± 0.3 |
| 6a | COOEt | CONH-(3-OCH3)Ph | - | 0.4 ± 0.1 | 28.9 ± 2.7 | 0.35 ± 0.12 |
| 6b | COOEt | CONH-(4-OCH3)Ph | - | 7.9 ± 1.4 | 16.4 ± 3.3 | 2.3 ± 0.8 |
| 6c | 4-Py | CONH-(3-OCH3)Ph | - | 1.4 ± 0.4 | 1.8 ± 0.21 | 1.7 ± 0.3 |
| 3a | 3-OCH3 | - | CH2 | N.A.b | N.A.b | N.A.b |
| 3b | 4-Br | - | CH2 | N.A.b | N.A.b | N.A.b |
| 3c | 4-Br | - | CO | N.A.b | N.A.b | N.A.b |
| fMLF | 0.01 | |||||
| WKYMVm | 0.001 | |||||
EC50 values represent the mean of three independent experiments and were determined by nonlinear regression analysis of the concentration-response curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p < 0.05).
N.A., no activity (no response was observed during the first 2 min after addition of the compounds under investigation) considering the limits of efficacy < 20% and EC50 < 50 μM.
Table 2.
Effect of compounds 11a-d and 13a-b on Ca2+ mobilization in FPR-transfected HL60 cells and human neutrophils.
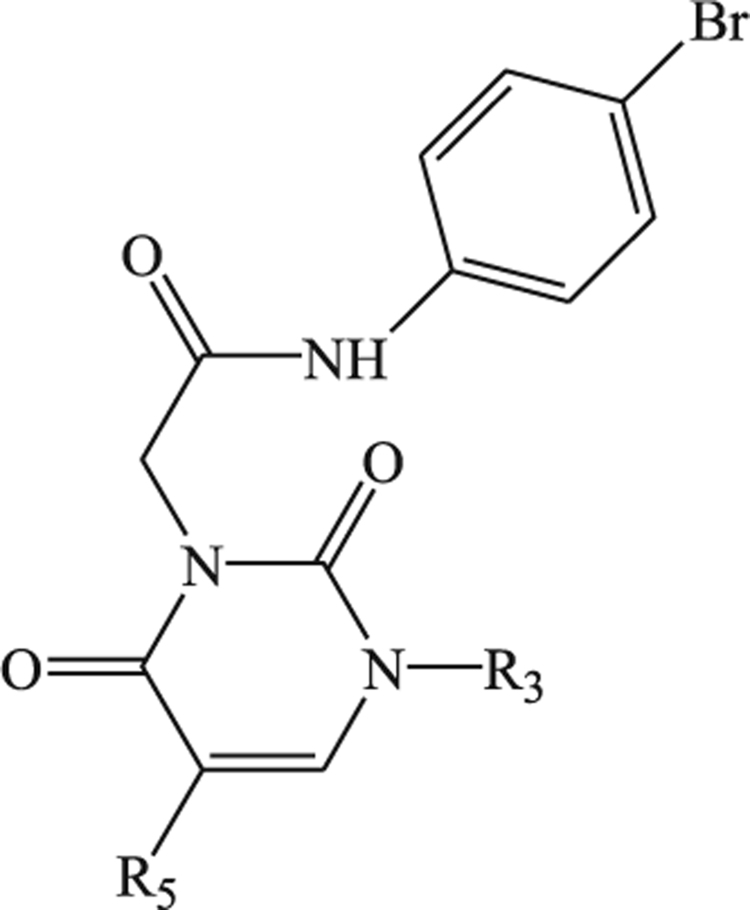 | |||||
|---|---|---|---|---|---|
| Compound | HL60-FPR1a EC50 (MM) | HL60-FPR2a EC50 (MM) | hPMNa EC50 (MM) | ||
| 11a |  |
H | 12.5 ± 2.8 | 8.9 ± 3.8 | 4.5 ± 2.1 |
| 11b | 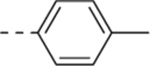 |
H | 13.6 ± 3.2 | 14.9 ± 4.2 | 6.2 ± 1.9 |
| 11c | 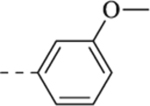 |
H | 4.3 ± 1.1 | 3.6 ± 1.4 | 3.1 ± 1.5 |
| 11d | -C3H7 | H | 5.5 ± 1.6 | 4.1 ± 1.3 | 5.0 ± 1.6 |
| 13a | 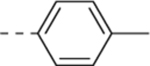 |
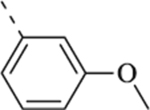 |
N.A.b | N.A.b | N.A.b |
| 13b | -C3H7 | 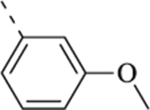 |
3.3 ± 0.3 | 2.4 ± 0.5 | 4.1 ± 0.7 |
| fMLF | 0.01 | ||||
| WKYMVm | 0.001 | ||||
EC50 values represent the mean of three independent experiments and were determined by nonlinear regression analysis of the concentration-response curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p < 0.05).
N.A., no activity (no response was observed during first the 2 min after addition of the compounds under investigation) considering the limits of efficacy <20% and EC50 < 50 μM.
Most compounds in the pyridinone series (Table 1) exhibited FPR agonist activity in the submicromolar/micromolar range, with variations in activity dependent on the R1 substituent. When R1 was a CN group (compounds of types 2 and 9), a moderate preference for FPR2 was observed as when 3- or 4-(OCH3)Ph (R) was directly connected (compounds 2a,b) or spaced from the pyridine ring by a CONH group (compounds 9a,b). In contrast, insertion of an additional OCH3 group to the phenyl ring (R = 3,4-(OCH3)Ph, compound 2c) was detrimental for FPR1 agonist activity, probably due to steric hindrance, but had less effect on FPR2 agonist activity. Interestingly, when the methoxyphenyl ring at position 5 of the pyridinone scaffold was replaced with an ethoxycarbonyl group (compound 2d), activity and selectivity for FPR2 were preserved and were comparable to those discussed above (EC50 = 3.00 μM, FPR1 and 0.38 μM, FPR2). This result offers a new perspective on active compound requirements, since the methoxyphenyl group was essential for activity in all of the compounds we have previously synthesized. Compounds 2a, 2d, and 9a had the best selectivity, with EC50 values for FPR2 being one order of magnitude lower compared to FPR1 (EC50 = 0.12, 0.38, 0.31 μM for FPR2 versus 1.6, 3.0, 3.9 μM for FPR1, respectively).
Maintaining the ethoxycarbonyl group as R and moving the CONH-(OCH3)Ph fragment to position 3 of the pyridinone scaffold (compounds 6a,b) resulted in a reversal of selectivity, which was particularly evident for 6a which exhibited a preference for FPR1 of about 72-fold (EC50 = 0.4 μM and 28.9 μM for FPR1 and FPR2, respectively). In contrast, compound 6c containing a pyridine at position 3 is a mixed FPR1/FPR2 agonist, with EC50 in the low micromolar range.
Finally, the modifications performed at the level of the 4-bromophenylacetamide chain (compounds 3a-c) led to completely inactive compounds, thus confirming that also for this series this chain is fundamental for the interaction with the receptor.
The pyrimidine-2,4-dione derivatives 11a-d and 13a,b reported in Table 2 had lower FPR agonist activity that the pyridinone compounds. They had EC50 values in the micromolar range, and no selectivity was observed toward the two FPR isoforms. From these data, it appears that position 3 tolerates as a (substituted)phenyl (11a-c) as the propyl chain (11d). The introduction of a substituent at position 5 was permitted only when a propyl group was present at position 3 (compound 13b, R3 = propyl, R5 = m-OCH3Ph), while the simultaneous presence at positions 3 and 5 of two phenyl rings (compound 13a) resulted in a completely inactive compound, which is also likely due to the steric hindrance.
Compounds 2a, 2c and 2d of this new series of derivatives were also evaluated for their chemoattractant activity toward human neutrophils. As shown in Table 3, compounds 2a, 2c, and 2d directly induced neutrophil chemotaxis and also inhibited WKYMVm-induced chemotaxis via desensitisation. Because resolution of inflammation involves receptor desensitization [42], we also tested compound 17b (Table 3), a previously reported FPR1 agonist with a pyridazin-3(2H)-one scaffold [26]. Compound 17b has been reported to exhibit pro-resolving/anti-inflammatory activity and was shown to protect against myocardial ischaemia–reperfusion injury in mice [43]. As with active compounds 2a,c,d, 17b also induced chemotaxis [26] and desensitized the neutrophil chemotactic response to WKYMVm (Table 3), suggesting that our compounds 2a,c,d have similar biological properties as this pro-resolving compound.
Table 3.
Effect of selected compounds on human neutrophil chemotaxis
| Compound | Chemotaxis | |
|---|---|---|
| EC50 (μM) | IC50 (μM)a | |
| 2a | 0.02 ± 0.004 | 6.1 ± 1.1 |
| 2c | 0.06 ± 0.015 | 18.8 ± 3.2 |
| 2d | 0.013 ± 0.001 | 8.4 ± 1.2 |
| 17bb | 0.6 ± 0.4b | 5.2 ± 1.6 |
 |
||
Desensitization of human neutrophil chemotaxis toward WKYMVm.
As reported previously [26].
The effectiveness of compounds EC3, EC10, and 2a was also evaluated using an in vivo model of rheumatoid arthritis (Fig. 3). Rats were treated intra-articularly (left paw, ipsilateral) with complete Freund’s adjuvant (CFA) on day 1. On day 14, when the CFA painful effect peaked [44], EC3 (10 and 30 mg kg−1), EC10 (3, 10 and 30 mg kg−1), and 2a (3, 10 and 30 mg kg−1) were administered p.o., and behavioral tests were performed over time (0–75 min) after administration. The effects of compounds herein reported were compared with ibuprofen (100 mg kg−1), a prototype of propionic acid derivative of NSAIDs group, administered at the active dosage usually employed in this model [45]. CFA-treated rats had a reduced pain threshold to noxious mechanical stimulus (paw pressure test) in comparison with sham + vehicle animals (36.6 ± 1 g for CFA + vehicle vs. 68.3 ± 1 g for sham + vehicle) (Fig. 3a–c). Ibuprofen administration (100 mg kg−1) significantly increased the pain threshold, reaching a peak between 15 and 30 min after treatment (65.3 ± 2.4 g for CFA + Ibuprofen vs 37.5 ± 1 g for CFA + vehicle, Fig. 3). EC3 at the higher dose (30 mg/kg) significantly increased the pain threshold from 30 to 60 min after injection (53.5 ± 2.3 g for CFA + 30 mg/kg EC3 vs 37.5 ± 1 g for CFA + vehicle), whereas 10 mg/kg was effective only 45 min after administration (Fig. 3a). As shown in Fig. 3b, EC10 dosed at 10 and 30 mg/kg increased the pain threshold, with a similar efficacy as EC3 between 15 and 60 min after administration. Compound 2a was significantly active when administered at 30 mg/kg from 15 to 45 min. In particular, 2a was able to completely control pain hypersensitivity at these times (63.3 ± 2 g for CFA + 30 mg/kg 2a vs. 37.5 ± 1 g for CFA + vehicle), as well as ibuprofen 15 min after administration. The lower dose of 10 mg/kg was effective between 30 and 60 min (Fig. 3c). CFA-induced hypersensitivity was also evaluated by an incapacitance test, where the change in hind limb weight bearing was measured as the difference (Δ Weight) between the weight burden on the contralateral (vehicle-treated) and the ipsilateral (CFA-treated) limbs. On day 14, CFA-treated rats had an increased postural unbalance in comparison with sham + vehicle animals (62.3 ± 1.3 g for CFA + vehicle vs −0.7 ± 1.5 g for sham + vehicle) (Fig. 3d–f). Ibuprofen administration (100 mg kg−1) was significantly active in reducing spontaneous pain from 15 to 30 min after treatment (34.7 ± 6.1 g for CFA + Ibuprofen vs 62.6 ± 2.5 g for CFA + vehicle, Fig. 3). EC3 at the higher dose (30 mg/kg) significantly reduced the postural unbalance from 15 to 60 min after administration (48.1 ± 1.05 g for CFA + 30 mg/kg EC3 vs. 62.8 ± 3.1 g for CFA + vehicle), while the lower dose (10 mg/kg) was effective only 30 min after administration (Fig. 3d). EC10 dosed at 10 and 30 mg/kg was significantly active in reducing spontaneous pain from 15 to 60 min (40.2 ± 1.1 g for CFA + EC10 vs. 62.8 ± 3.1 g CFA + vehicle) (Fig. 3e). Compound 2a significantly reduced the postural unbalance when dosed at 10 and 30 mg/kg between 15 and 60 min after administration, with similar efficacy as EC10 and ibuprofen, but with a more lasting effect than the latter (Fig. 3f).
Fig. 3.
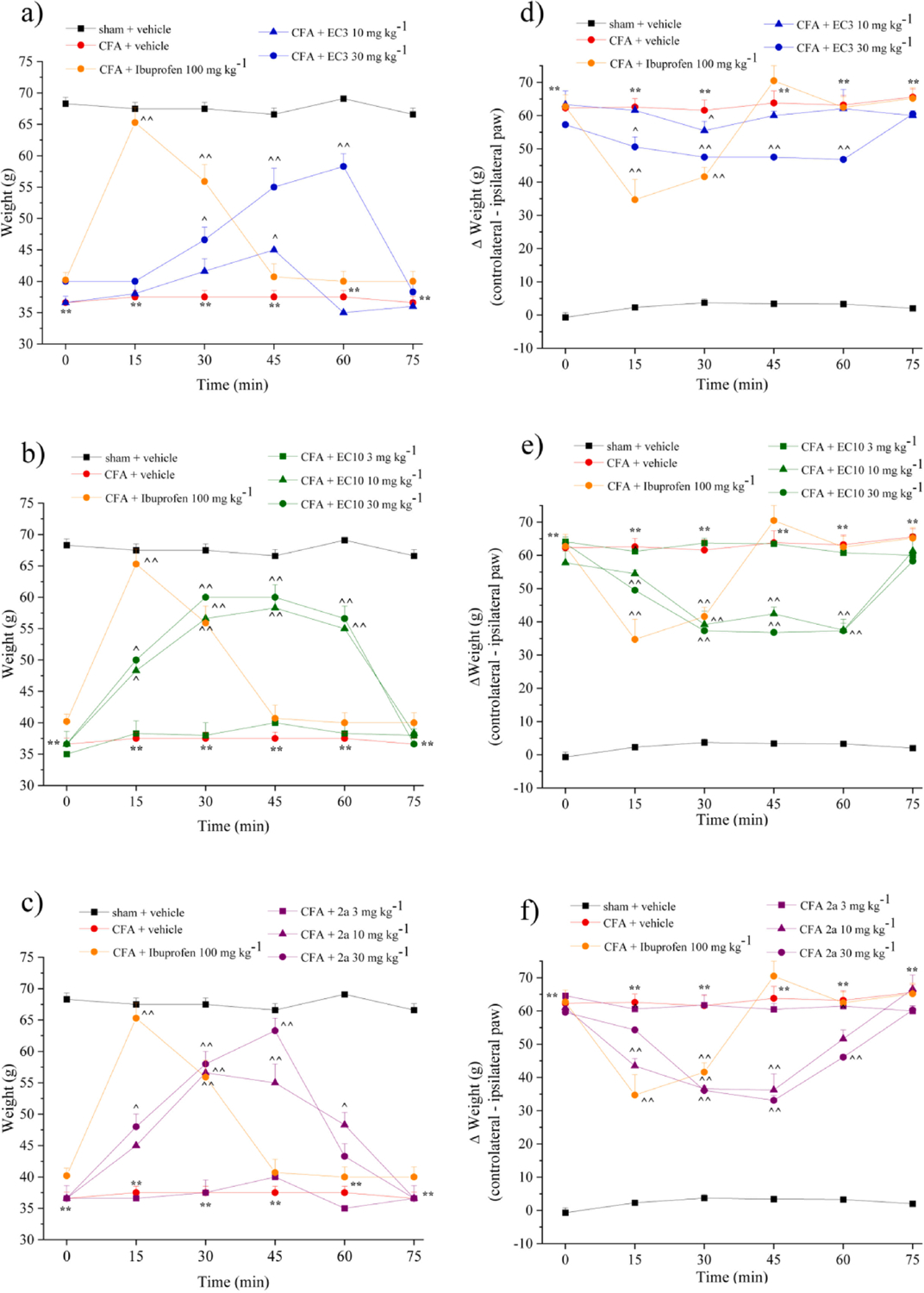
Effect of a single administration of selected FPR agonists on CFA-induced rheumatoid arthritis in rats.
Rheumatoid arthritis was induced by complete Freund’s adjuvant (CFA) injection into the left knee joint. On day 14 after rheumatoid arthritis induction, EC3 (10 and 30 mg/kg), EC10 (3, 10, and 30 mg/kg), and 2a (3, 10, and 30 mg/kg) and ibuprofen (100 mg kg−1) were suspended in CMC 1% and administered p.o. Behavioral tests were performed 15, 30, 45, 60, and 75 min after treatments. The response to a noxious mechanical stimulus was measured by the Paw pressure test (ipsilateral paw) (a), (b), (c). Spontaneous pain was assessed measuring the hind limb weight bearing alterations using the Incapacitance test; data are expressed as the difference (Δ weight) between the weight applied on the limb contralateral to the injury and the weight applied on the ipsilateral limb (d), (e), (f). Each value represents the mean ± SEM of five rats per group. **P < 0.01 vs sham + vehicle treated animals; ^P < 0.05 and ^^P < 0.01 vs CFA + vehicle treated animals.
3. Conclusion
We have identified a new series of FPR agonists with EC50 values in the submicromolar/micromolar range. These pyridinone compounds were effective FPR agonists, with the majority of compounds being mixed FPR1/FPR2 agonists that had a moderate preference for FPR2. The most active derivative (2a), as well as our previously reported FPR agonists EC3 and EC10, were also evaluated in vivo using a rat model of rheumatoid arthritis, and the results showed that all three compounds were able to increase the pain threshold using two different assays. Notably, compound 2a was highly active when administered at 30 mg/kg p.o. and, after 45 min, was able to completely control pain hypersensitivity in this model.
4. Experimental section
4.1. Material and methods
Reagents and starting materials were obtained from commercial sources. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. All reactions were monitored by thin layer chromatography (TLC) using commercial plates precoated with Merck silica gel 60F-254. Visualization was performed by UV fluorescence (λmax = 254 nm) or by staining with iodine or potassium permanganate. Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck), flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. Compounds were named following IUPAC rules, as applied by Beilstein-Institut AutoNom 2000 (4.01.305) or CA Index Name. All melting points were determined on a microscope hot stage Büchi apparatus and are uncorrected. The identity and purity of intermediates and final compounds was ascertained through NMR and TLC chromatography. 1H NMR and 13C NMR spectra were recorded with an Avance 400 instrument (Bruker Biospin Version 002 with SGU). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm (for 1H NMR) or 0.1 ppm (for 13C NMR), using the solvent as the internal standard. Coupling constants (J values) of 1H NMR are given in Hz and were calculated using “TopSpin 1.3” software rounded to the nearest 0.1 Hz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Bruker Micro TOF), and the reported mass values are within the error limits of ± 5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin-Elmer 260 elemental analyzer for C, H, and N and were within ± 0.4% of the theoretical values.
4.2. Chemistry
4.2.1. General procedure for 2a-d
To a mixture of the appropriate compound 1a-d [33,34] (0.21 mmol) and 0.42 mmol of K2CO3 in 5 mL of anhydrous CH3CN, 0.21–0.32 mmol of N-(4-bromophenyl)-2-chloroacetamide [35] was added, and the mixture was refluxed under stirring for 2–8 h. For compound 2d, it was necessary to reflux for 25 h. After cooling, the suspension was concentrated in vacuo, 5–10 mL of ice-cold H2O was added, and the precipitate was recovered by vacuum filtration. The final compounds were purified by flash chromatography using cyclohexane/ethyl acetate 1:4 (2a and 2c), cyclohexane/ethyl acetate 1:2 (2b), and cyclohexane/ethyl acetate 1:1 (2d) as eluents.
N-(4-Bromophenyl)-2-[3-cyano-5-(3-methoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]acetamide (2a). Yield = 32%; mp = 260–263 °C dec. (EtOH). 1H NMR (400 MHz, CDCl3) δ 2.56 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 4.98 (s, 2H, NCH2CO), 6.72 (s, 1H, Ar), 6.77 (d, 1H, Ar, J = 7.6 Hz), 6.94 (d, 1H, Ar, J = 7.6 Hz), 7.35 (t, 1H, Ar, J = 8.0 Hz), 7.40 (s, 4H, Ar), 7.80 (s, 1H, H-pyridinone), 9.19 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 19.4 (CH3), 50.6 (CH2), 55.4 (CH3), 101.5 (C), 113.8 (CH), 115.3 (CH), 115.5 (C), 117.3 (C), 121.2 (CH), 121.7 (CH), 122.2 (C), 130.2 (CH), 131.9 (CH), 136.5 (C), 137.8 (C), 148.4 (CH), 151.7 (C), 159.9 (C), 161.1 (C), 164.2 (C). ESI-MS calcd. for C22H18BrN3O3, 452.31; found: m/z 453.11[M+H]+. Anal. C22H18BrN3O3 (C, H, N).
N-(4-Bromophenyl)-2-[3-cyano-5-(4-methoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]acetamide (2b). Yield = 21%; mp = 144–146 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 2.44 (s, 3H, CH3), 3.86 (s, 3H, OCH3), 5.11 (s, 2H, NCH2CO), 6.96 (d, 2H, Ar, J = 8.4 Hz), 7.14 (d, 2H, Ar, J = 8.4 Hz), 7.28 (d, 2H, Ar, J = 8.4 Hz), 7.40 (d, 2H, Ar, J = 8.4 Hz), 7.78 (s, 1H, H-pyridinone), 9.73 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 19.3 (CH3), 50.5 (CH2), 55.4 (CH3), 101.5 (C), 115.5 (C), 116.3 (CH) 117.2 (C), 121.2 (CH), 122.1 (C), 127.5 (CH), 131.2 (CH), 136.5 (C), 137.7 (C), 148.4 (CH), 151.7 (C), 159.9 (C), 161.1 (C), 164.2 (C). ESI-MS calcd. for C22H18BrN3O3, 452.31; found: m/z 453.15[M+H]+. Anal. C22H18BrN3O3 (C, H, N).
N-(4-Bromophenyl)-2-[3-cyano-5-(3,4-dimethoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]acetamide (2c). Yield = 28%; mp = 236–238 °C dec. (EtOH). 1H NMR (400 MHz, CDCl3) δ 2.55 (s, 3H, CH3), 3.87 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 5.16 (s, 2H, NCH2CO), 6.55 (s, 1H, Ar), 6.74 (d, 1H, Ar, J = 8.0 Hz), 6.91 (d, 1H, Ar, J = 8.4 Hz), 7.39–7.51 (m, 4H, Ar), 7.80 (s, 1H, H-pyridinone), 9.26 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 19.3 (CH3), 49.6 (CH2), 56.0 (CH3), 56.2 (CH3) 101.0 (C), 111.4 (CH), 112.4 (CH), 115.9 (C), 117.0 (C), 120.9 (CH), 122.1 (CH), 122.1 (C), 128.6 (C), 131.7 (CH), 136.9 (C), 148.6 (CH), 149.2 (C), 151.8 (C), 160.9 (C), 164.4 (C). ESI-MS calcd. for C23H20BrN3O4, 482.33; found: m/z 483.08 [M+H]+. Anal. C23H20BrN3O4 (C, H, N).
1-[(4-Bromophenylcarbamoyl)-methyl]-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid ethyl ester (2d). Yield = 19%; mp = 238–240 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.38 (t, 3H, CH2CH3, J = 7.2 Hz), 2.87 (s, 3H, CH3), 4.35 (q, 2H, CH2CH3, J = 7.2 Hz), 5.03 (s, 2H, NCH2CO), 7.30 (m, 2H, Ar), 7.35 (m, 2H, Ar), 8.37 (s, 1H, H-pyridinone), 9.34 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 18.5 (CH3), 49.1 (CH2), 58.3 (CH2), 102.1 (C), 109.1 (C), 117.9 (C), 118.7 (C), 122.6 (CH), 131.1 (CH), 139.8 (C), 140.7 (C), 149.8 (CH), 161.4 (C), 165.2 (C), 168.2 (C). ESI-MS calcd. for C18H16BrN3O4, 418.24; found: m/z 418.97. [M+H]+. Anal. C18H16BrN3O4 (C, H, N).
4.2.2. General procedure for 3a,b
To a suspension of 1a (0.42 mmol) and K2CO3 (0.84 mmol) in an hydrous DMF (1.5 mL), 0.63 mmol of suitable benzylbromide (3-methoxybenzyl bromide for 3a and 4-bromobenzyl bromide for 3b) were added and the mixture was refluxed for 1 h. After cooling ice-cold H2O was added, and the suspension was extracted with ethyl acetate (3 × 10 mL) and dried on anhydrous sodium sulfate. After evaporation of the solvent, final compounds were purified by flash column chromatography using cyclohexane/ethyl acetate 2:1 as eluent.
1-(3-Methoxybenzyl)-5-(3-methoxyphenyl)-6-methyl-2-oxo-1,2-dihydro-pyridine-3-carbonitrile (3a). Yield = 13%; oil. 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H, CH3), 3.83 (s, 6H, 2 × OCH3), 5.52 (s, 2H, NCH2CO), 6.77 (s, 1H, Ar), 6.82 (m, 1H, Ar), 6.92 (d, 1H, Ar, J = 8.4 Hz), 7.08 (s, 1H, Ar), 7.10 (s, 1H, Ar), 7.29 (t, 2H, Ar, J = 7.8 Hz), 7.34 (t, 1H, Ar, J = 8.0 Hz), 7.71 (s, 1H, H-pyridinone). 13C NMR (100 MHz, CDCl3) δ 23.7 (CH3), 55.3 (CH3), 55.3 (CH3), 68.2 (CH2), 93.8 (C), 113.2 (CH), 113.3 (CH), 113.7 (CH), 114.9 (CH), 115.4 (C), 120.2 (CH), 121.5 (CH), 129.5 (CH), 129.7 (CH), 130.4 (C), 137.9 (C), 139.1 (C), 143.6 (CH), 159.3 (C), 159.7 (C), 161.7 (C). ESI-MS calcd. for C22H20N2O3, 360.41; found: m/z 361.13 [M+H]+. Anal. C22H20N2O3 (C, H, N).
1-(4-Bromobenzyl)-5-(3-methoxyphenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (3b). Yield = 29%; oil. 1H NMR (400 MHz, CDCl3) δ 2.44 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 5.47 (s, 2H, CH2), 6.76 (s, 1H, Ar), 6.81 (d, 1H, Ar, J = 7.6 Hz), 6.92 (dd, 1H, Ar, J = 8.4 Hz, J = 2 Hz), 7.33 (t, 1H, Ar, J = 8 Hz), 7.38 (d, 2H, Ar, J = 8 Hz), 7.50 (d, 2H, Ar, J = 8 Hz), 7.70 (s, 1H, H-pyridinone). 13C NMR (100 MHz, CDCl3) δ 23.7 (CH3), 55.4 (CH3), 67.6 (CH2), 93.7 (C), 113.2 (CH), 114.9 (CH), 115.3 (C), 116.8 (C), 121.4 (CH), 122.0 (C), 129.7 (CH), 130.5 (C), 131.6 (CH), 135.4 (C), 138.9 (C), 143.7 (CH), 159.3 (C), 161.5 (C). ESI-MS calcd. for C21H17BrN2O2, 409.28; found: m/z 410.01 [M+H]+. Anal. C21H17BrN2O2 (C, H, N).
1-(4-Bromobenzoyl)-5-(3-methoxyphenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (3c). To a solution of 1a (0.21 mmol) and 0.42 mmol of NaH in anhydrous THF (1.5 mL), 0.25 mmol of 4-bromobenzoyl chloride was added. The mixture was stirred at room temperature for 18 h, ice-cold H2O was added, and the suspension was extracted with ethyl acetate (3 × 10 mL). After evaporation of the solvent, the final compound was purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 as eluent. Yield = 76%; oil. 1H NMR (400 MHz, CDCl3) δ 2.56 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 6.83 (m, 1H, Ar), 6.88 (d, 1H, Ar, J = 7.6 Hz), 6.98 (dd, 1H, Ar, J = 8 Hz, J = 2.4 Hz), 7.39 (t, 1H, Ar, J = 8 Hz), 7.68 (d, 2H, Ar, J = 8.4 Hz), 7.91 (s, 1H, H-pyridinone), 8.10 (d, 2H, Ar, J = 8.4 Hz). 13C NMR (100 MHz, CDCl3) δ 23.7 (CH3), 55.4 (CH3), 100.5 (C), 113.9 (CH), 114.7 (CH), 121.2 (CH), 122.7 (C), 126.9 (C), 129.9 (C), 130.0 (CH), 132.1 (CH), 132.2 (CH), 136.2 (C), 137.9 (C), 143.3 (CH), 156.7 (C), 159.8 (C), 161.2 (C), 163.4 (C). ESI-MS calcd. for C21H15BrN2O3, 423.26; found: m/z 423.98 [M+H]+. Anal. C21H15BrN2O3 (C, H, N).
4.2.3. General procedure for 5a,b
Compounds 5a,b were obtained starting from intermediates 4a,b [36] following the same procedure described for 2a-d. The final compounds were purified by crystallization from ethanol.
1-[(4-Bromophenylcarbamoyl)methyl]-5-carbamoyl-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid ethyl ester (5a). Yield = 75%; mp = 289–291 °C dec. (EtOH). 1H NMR (400 MHz, DMSO-d6)δ 1.30(t, 3H, CH2CH3, J = 7.2 Hz), 2.75 (s, 3H, CH3), 4.28 (q, 2H, CH2CH3, J = 7.2 Hz), 5.05 (s, 2H, NCH2CO), 7.47–7.54 (m, 4H, Ar), 7.70 (exch br s, 1H, NH2), 8.58 (exch br s, 1H, NH2), 8.73 (s, 1H, H-pyridinone), 10.63 (exch br s, 1H, NH). ESI-MS calcd. for C18H18BrN3O5, 436.26; found: m/z 437.05 [M+H]+. Anal. C18H18BrN3O5 (C, H, N).
1-[(4-Bromophenylcarbamoyl)methyl]-2-methyl-6-oxo-1,6-dihydro-[3,4′]bipyridinyl-5-carboxylic acid amide (5b). Yield = 44%; mp = > 300 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 2.39 (s, 3H, CH3), 5.06 (s, 2H, NCH2CO), 7.39 (m, 2H, Ar), 7.48 (d, 2H, Ar, J = 7.6 Hz), 7.54 (d, 2H, Ar, J = 8.4 Hz), 7.60 (exch br s, 1H, NH2), 8.21 (s, 1H, H-pyridinone), 8.64 (m, 2H, Ar), 8.79 (exch br s, 1H, NH2), 10.63 (exch br s, 1H, NH). ESI-MS calcd. for C20H17BrN4O3, 441.28; found: m/z 442.03. [M+H]+. Anal. C20H17BrN4O3 (C, H, N).
4.2.4. General procedure for 6a-c
A mixture of intermediate 5a or 5b (0.23 mmol), 0.69–1.15 mmol of the appropriate arylboronic acid, 0.35 mmol of anhydrous cupric acetate, 0.46 mmol of triethylamine, and activated molecular sieves (800 mg, 4 Å) in 8 mL of dry dichloromethane was stirred at room temperature for 48–52 h. The suspension was filtered to remove the molecular sieves and the organic layer was washed with 15% aqueous ammonia (3 × 10 mL), then with 10 mL of H2O and finally dried over sodium sulfate. After evaporation of the solvent under reduced pressure, the final compounds were purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 for (6a), cyclohexane/ethyl acetate 1:1 (6b), and dichloromethane/methanol 9:1 (6c) as eluents.
1-[(4-Bromophenylcarbamoyl)methyl]-5-(3-methoxyphenylcarbamoyl)-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid ethyl ester (6a). Yield = 20%; mp = 248–251 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.32 (t, 3H, CH2CH3, J = 7.2 Hz), 2.79 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 4.29 (q, 2H, CH2CH3, J = 7.2 Hz), 5.13 (s, 2H, NCH2CO), 6.66 (d, 1H, Ar, J = 7.6 Hz), 7.16 (m, 1H, Ar), 7.22 (m, 1H, Ar), 7.38 (s, 1H, Ar), 7.48–7.55 (m, 4H, Ar), 8.85 (s, 1H, H-pyridinone), 10.70 (exch br s, 1H, NH), 11.48 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.8(CH3), 18.2 (CH3), 49.3 (CH2), 56.1 (CH3), 59.0 (CH2), 106.2 (CH), 109.1 (C), 110.0 (CH), 112.7 (CH), 118.7 (C), 121.9 (CH), 129.7 (CH), 131.3 (C), 132.2 (CH), 139.2 (C), 139.8 (C), 148.5 (CH), 161.4 (C), 162.2 (C), 163.8 (C), 165.1 (C), 168.2 (C). ESI-MS calcd. for C25H24BrN3O6, 542.38; found: m/z 543.11 [M+H]+. Anal. C25H24BrN3O6 (C, H, N).
1-[(4-Bromophenylcarbamoyl)methyl]-5-(4-methoxyphenylcarbamoyl)-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid ethyl ester (6b). Yield = 18%; mp = 284–286 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.30 (t, 3H, CH2CH3, J = 7.2 Hz), 2.78 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 4.30 (q, 2H, CH2CH3, J = 7.2 Hz), 5.12 (s, 2H, NCH2CO), 6.89 (d, 1H, Ar, J = 8.8 Hz), 7.49 (m, 2H, Ar), 7.54 (m, 2H, Ar), 7.58 (d, 2H, Ar, J = 8.8 Hz), 8.83 (s, 1H, H-pyridinone), 10.68 (exch br s, 1H, NH), 11.34 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 13.8(CH3), 18.2 (CH3), 49.4 (CH2), 56.1 (CH3), 58.9 (CH2), 106.3 (CH), 109.1 (C), 110.0 (CH), 113.0 (CH), 118.7 (C), 121.9 (CH), 129.4 (CH), 131.2 (C), 131.9 (CH), 139.1 (C), 139.8 (C), 148.4 (CH), 161.5 (C), 162.3 (C), 163.8 (C), 165.1 (C), 167.9 (C). ESI-MS calcd. for C25H24BrN3O6, 542.38; found: m/z 543.18 [M+H]+. Anal. C25H24BrN3O6 (C, H, N).
1-[(4-Bromophenylcarbamoyl)methyl]-2-methyl-6-oxo-1,6-dihydro-[3,4′]bipyridinyl-5-carboxylic acid (3-methoxyphenyl) amide (6c). Yield = 30%; mp = 278–280 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.43 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 5.14 (s, 2H, NCH2CO), 6.66 (d, 1H, Ar, J = 7.6 Hz), 7.16 (d, 1H, Ar, J = 8.4 Hz), 7.23 (t, 1H, Ar, J = 8.4 Hz), 7.38 (s, 1H, Ar), 7.43 (m, 2H, Ar), 7.49 (m, 2H, Ar), 7.56 (m, 2H, Ar), 8.34 (s, 1H, H-pyridinone), 8.66 (m, 2H, Ar), 10.69 (exch br s, 1H, NH), 11.79 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 19.5 (CH3), 49.4 (CH2), 56.1 (CH3), 105.8 (CH), 110.1 (CH), 112.4 (CH), 115.7 (C), 117.1 (C), 118.7 (C), 121.5 (CH), 124.9 (CH), 130.3 (CH), 132.2 (CH), 138.4 (C), 139.8 (C), 144.5 (CH), 145.9 (C), 150.5 (CH), 152.1 (C), 160.2 (C), 161.5 (C), 162.2 (C), 165.5 (C). ESI-MS calcd. for C27H23BrN4O4, 547.40; found: m/z 548.12 [M+H]+. Anal. C27H23BrN4O4 (C, H, N).
4.2.5. General procedure for 8a,b
A mixture of compound 7 [37] (1.12 mmol), 4.35 mL of SOCl2, and 0.05 mL of Et3N was stirred at room temperature for 5 h. After cooling, the excess of SOCl2 was removed under vacuum, the residue was dissolved in 2–3 mL of anhydrous diethyl ether, and 4–4.5 mmol of the appropriate arylamine was added. The mixture was stirred for 3 h at room temperature. Finally, the precipitate was recovered by vacuum filtration, washed with H2O, and recrystallized from ethanol.
5-Cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid (3-methoxyphenyl) amide (8a). Yield = 82%; mp = 204–205 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.43 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 6.65 (d, 1H, Ar, J = 8.1 Hz), 7.17 (d, 2H, Ar, J = 7.6 Hz), 7.21 (s, 1H, Ar), 8.30 (s, 1H, H-pyridinone), 10.16 (exch br s, 1H, NH), 11.79 (exch br s, 1H, NH). ESI-MS calcd. for C15H13N3O3, 283.29; found: m/z 284.06 [M+H]+. Anal. C15H13N3O3 (C, H, N).
5-Cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid (4-methoxyphenyl) amide (8b). Yield = 69%; mp = > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.45 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 6.88 (d, 2H, Ar, J = 8.8 Hz), 7.53 (d, 2H, Ar, J = 8.8 Hz), 8.33 (s, 1H, H-pyridinone), 10.02 (exch br s, 1H, NH), 12.82 (exch br s, 1H, NH). ESI-MS calcd. for C15H13N3O3, 283.29; found: m/z 284.08 [M+H]+. Anal. C15H13N3O3 (C, H, N).
4.2.6. General procedure for 9a,b
Compounds 9a,b were obtained by starting from intermediates 8a,b and following the same procedure described for 2a-d. The final compounds were purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 (9a) and dichloromethane/methanol 9:1 (9b) as eluents.
1-[(4-Bromophenylcarbamoyl)methyl]-5-cyano-2-methyl-6-oxo-1,6-dihydro-pyridine-3-carboxylic acid (3-methoxyphenyl) amide (9a). Yield = 12%; mp = > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.43 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 5.00 (s, 2H, N-CH2), 6.67 (m, 1H, Ar), 7.22 (m, 2H, Ar), 7.33 (m, 1H, Ar), 7.51 (m, 4H, Ar), 8.39 (s, 1H, H-pyridinone), 10.41 (exch br s, 1H, NH), 10.64 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 18.8 (CH3), 48.7 (CH2), 56.1 (CH3), 101.8 (C), 106.0 (CH), 109.7 (CH), 111.1 (C), 112.7 (CH), 117.2 (C), 120.1 (C), 121.6 (CH), 129.7 (CH), 131.3 (CH), 138.8 (C), 139.5 (C), 140.1 (C), 150.1 (CH), 159.1 (C), 162.2 (C), 163.1 (C), 168.2 (C). ESI-MS calcd. for C23H19BrN4O4, 495.33; found: m/z 496.06 [M+H]+. Anal. C23H19N4O4 (C, H, N).
1-[(4-Bromophenylcarbamoyl)methyl]-5-cyano-2-methyl-6-oxo-1,6-dihydro-pyridine-3-carboxylic acid (4-methoxyphenyl) amide (9b). Yield = 11%; mp = 292–293 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.53 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 4.99 (s, 2H, N-CH2), 6.90 (d, 1H, Ar, J = 8.1 Hz), 7.50 (m, 2H, Ar), 7.33 (m, 4H, Ar); 7.56 (d, 2H, Ar, J = 8.8 Hz), 8.37 (s, 1H, H-pyridinone), 10.29 (exch br s, 1H, NH), 10.64 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 19.6 (CH3), 49.1 (CH2), 56.1 (CH3), 101.8 (C), 106.4 (CH), 110.0 (CH), 111.6 (C), 112.7 (CH), 117.2 (C), 121.0 (C), 121.8 (CH), 130.0 (CH), 131.3 (CH), 139.0 (C), 139.8 (C), 141.1 (C), 150.8 (CH), 159.1 (C), 162.3 (C), 163.1 (C), 167.9 (C). ESI-MS calcd. for C23H19BrN4O4, 495.33; found: m/z 496.11 [M+H]+. Anal. C23H19N4O4 (C, H, N).
4.2.7. General procedure for 11a-d
Compounds 11a-d were obtained by starting from intermediates 10a-d [38–41] and following the same procedure described for 2a-d. The final compounds 11b and 11d were purified by crystallization from ethanol, whereas compounds 11a and 11c were purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 as eluent.
N-(4-Bromophenyl)-2-(2,6-dioxo-3-phenyl-3,6-dihydropyrimidin-1(2H)-yl)acetamide (11a). Yield = 32%; mp = 108–110 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 4.63 (s, 2H, N-CH2), 5.87 (d, 1H, CH = CH, J = 8.0 Hz), 7.41 (m, 3H, Ar), 7.52 (m, 6H, Ar), 7.82 (d, 1H, CH = CH, J = 8.0 Hz), 10.38 (exch br s, 1H, NH).13C NMR (100 MHz, DMSO-d6) δ 43.9 (CH2), 101.3 (CH), 115.4 (C), 121.5 (CH), 127.9 (CH), 128.9 (CH), 129.7 (CH), 132.1 (CH), 138.5 (C), 139.6 (C), 144.9 (CH), 151.1 (C), 162.6 (C), 165.7 (C). ESI-MS calcd. for C18H14BrN3O3, 400.23; found: m/z 400.98 [M+H]+. Anal. C18H14BrN3O3 (C, H, N).
N-(4-Bromophenyl)-2-(2,6-dioxo-3-p-tolyl-3,6-dihydropyrimidin-1(2H)-yl)acetamide (11b). Yield = 68%; mp = 122–124 °C (EtOH/H2O 1:5). 1H NMR (400 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3), 4.61 (s, 2H, N-CH2CO), 5.84 (d, 1H, CH = CH, J = 8.0 Hz), 7.25–7.30 (m, 4H, Ar), 7.48 (d, 2H, Ar, J = 8.8 Hz), 7.50 (d, 2H, Ar, J = 8.8 Hz), 7.78 (d, 1H, CH = CH, J = 8.0 Hz), 10.39 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 21.08 (CH3), 43.9 (CH2), 101.1 (CH), 115.4 (C), 121.4 (CH), 126.9 (CH), 130.1 (CH), 132.1 (CH), 137.1 (C), 138.5 (C), 138.6 (C), 145.0 (CH), 151.1 (C), 162.6 (C), 165.9 (C). ESI-MS calcd. for C19H16BrN3O3, 414.25; found: m/z 415.02 [M+H]+. Anal. C19H16BrN3O3 (C, H, N).
N-(4-Bromophenyl)-2-[3-(3-methoxyphenyl)-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetamide (11c). Yield = 31%; mp = 218–221 °C (EtOH/H2O 1:5). 1H NMR (400 MHz, DMSO-d6) δ 3.76 (s, 3H, OCH3), 4.61 (s, 2H, N-CH2CO), 5.85 (d, 1H, CH = CH, J = 8.0 Hz), 6.96–7.01 (m, 3H, Ar), 7.40 (m, 1H, Ar), 7.45–7.51 (m, 4H, Ar), 7.81 (d, 1H, CH = CH, J = 8.0 Hz), 10.37 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 43.9 (CH2), 55.9 (CH3), 101.2 (CH), 113.0 (CH), 114.8 (CH), 115.4 (C), 119.3 (CH), 121.5 (CH), 130.5 (CH), 132.1 (CH), 138.5 (C), 140.6 (C), 144.9 (CH), 150.9 (C), 160.2 (C), 162.6 (C), 165.9 (C). ESI-MS calcd. for C19H16BrN3O4, 430.25; found: m/z 430.98 [M+H]+. Anal. C19H16BrN3O4 (C, H, N).
N-(4-Bromophenyl)-2-(2,6-dioxo-3-propyl-3,6-dihydropyrimidin-1(2H)-yl)acetamide (11d). Yield = 78%; mp = 117–118 °C dec (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 0.83 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.56–1.61 (m, 2H, CH2CH2CH3), 3.67 (t, 2H, CH2CH2CH3, J = 7.2 Hz), 4.56 (s, 2H, N-CH2), 5.72 (d, 1H, CH = CH, J = 7.6 Hz), 7.44–7.51 (m, 4H, Ar), 7.74 (d, 1H, CH = CH, J = 8.0 Hz), 10.37 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 10.9 (CH3), 22.3 (CH2), 44.4 (CH2), 51.6 (CH2), 101.2 (CH), 116.0 (C), 121.2 (CH), 131.7 (CH), 137.1 (C), 143.2 (CH), 151.4 (C), 163.0 (C), 165.4 (C). ESI-MS calcd. for C15H16BrN3O3, 366.21; found: m/z 367.01 [M+H]+. Anal. C15H16BrN3O3 (C, H, N).
4.2.8. General procedure for 12a,b
To a solution of 11b or 11d (0.14 mmol) in 1.3 mL of glacial acetic acid, a solution of 0.14 mmol of Br2 in the same solvent was slowly added. The mixture was stirred at room temperature for 90 min. After evaporation of the solvent, 5 mL of cold H2O was added, and the precipitate was recovered by vacuum filtration and purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 as eluent.
2-(5-Bromo-2,6-dioxo-3-p-tolyl-3,6-dihydropyrimidin-1(2H)-yl)-N-(4-bromophenyl)acetamide (12a). Yield = 56%; mp = 166–168 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3), 4.67 (s, 2H, NCH2CO), 7.30 (m, 4H, Ar), 7.45–7.52 (m, 4H, Ar), 8.36 (s, 1H, CH), 10.39 (exch br s, 1H, NH). ESI-MS calcd. for C19H15Br2N3O3, 493.15; found: m/z 493.91 [M+H]+. Anal. C19H15Br2N3O3 (C, H, N).
2-(5-Bromo-2,6-dioxo-3-propyl-3,6-dihydropyrimidin-1(2H)-yl)-N-(4-bromophenyl)acetamide (12b). Yield = 29%; mp = 219–220 °C (EtOH), 1H NMR (400 MHz, DMSO-d6) δ 0.83 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.56–1.65 (m, 2H, CH2CH2CH3), 3.70 (t, 2H, CH2CH2CH3, J = 7.2 Hz), 4.62 (s, 2H, N-CH2CO), 7.44–7.50 (m, 4H, Ar), 8.36 (s, 1H, CH), 10.37 (exch br s, 1H, NH). ESI-MS calcd. for C15H15Br2N3O3, 445.11; found: m/z 445.95 [M+H]+.Anal.C15H15Br2N3O3 (C,H, N).
4.2.9. General procedure for 13a,b
To a suspension of the appropriate intermediate 12a,b (0.27 mol) in 3–4 mL of anhydrous toluene, 0.03 mmol of Tetrakis, 0.54–1.08 mmol of 3-methoxyphenylboronic acid in 1 mL of ethanol, and 2.1 mL of Na2CO3 2 N solution were added. The mixture was stirred at 80 °C for 4–7 h. After evaporation of the solvent, 5–10 mL of cold H2O was added, and the precipitate was recovered by vacuum filtration. The desired final compounds were purified by flash column chromatography using hexane/ethyl acetate 3:2 (13a) and cyclohexane/ethyl acetate1:2 (13b) as eluents.
N-(4-Bromophenyl)-2-[5-(3-methoxyphenyl)-2,6-dioxo-3-p-tolyl-3,6-dihydropyrimidin-1(2H)-yl]acetamide (13a). Yield = 27%; mp = 168–170 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 4.70 (s, 2H, N-CH2CO), 6.86 (d, 1H, Ar, J = 8.8 Hz), 7.16 (m, 2H, Ar), 7.27 (m, 4H, Ar), 7.36 (m, 1H, Ar), 7.48 (m, 4H, Ar), 7.95 (s, 1H, CH), 10.44 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 21.1 (CH3), 44.5 (CH2), 55.6 (CH3), 112.9 (C), 113.7 (CH), 114.3 (CH), 115.5 (C), 121.9 (CH), 121.5 (CH), 127.1 (CH), 129.5 (CH), 130.1 (CH), 130.4 (CH), 132.1 (CH), 134.5 (C), 137.1 (C), 138.5 (C), 142.5 (CH), 150.6 (C), 159.5 (C), 161.7 (C), 165.95 (C). ESI-MS calcd. for C26H22BrN3O4, 520.37; found: m/z 521.16 [M+H]+. Anal. C26H22BrN3O4 (C, H, N).
N-(4-Bromophenyl)-2-[5-(3-methoxyphenyl)-2,6-dioxo-3-propyl-3,6-dihydropyrimidin-1(2H)-yl]acetamide (13b). Yield = 10%; mp = 120–121 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 0.87 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.63–1.69 (m, 2H, CH2CH2CH3), 3.74 (s, 3H, OCH3), 3.75 (m, 2H, CH2CH2CH3), 4.66 (s, 2H, N-CH2CO), 7.15 (s, 3H, Ar), 7.28 (t, 1H, Ar, J = 8.2 Hz), 7.46 (d, 2H, Ar, J = 8.8 Hz), 7.51 (d, 2H, Ar, J = 9.2 Hz), 8.08 (s, 1H, CH), 10.40 (exch br s, 1H, NH).13C NMR (100 MHz, DMSO-d6) δ 11.1 (CH3), 22.2 (CH2), 44.3 (CH2), 50.9 (CH2), 55.6 (CH3), 111.9 (C), 113.2 (CH), 114.2 (CH), 115.4 (C), 120.8 (CH), 121.4 (CH), 129.6 (CH), 132.1 (CH), 134.9 (C), 138.6 (C), 143.1 (CH), 150.9 (C), 159.5 (C), 161.0 (C), 166.1 (C). ESI-MS calcd. for C22H22BrN3O4, 472.33; found: m/z 473.12 [M+H]+. Anal. C22H22BrN3O4 (C, H, N).
4.3. Pharmacology
4.3.1. Cell culture
Human promyelocytic leukemia HL60 cells stably transfected with FPR1 (FPR1-HL60 cells) or FPR2 (FPR2-HL60 cells) (kind gifts from Dr. Marie‐Josephe Rabiet, INSERM, Grenoble, France) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg/ml streptomycin, 100 U/ml penicillin, and G418 (1 mg/mL). Although stable cell lines are cultured under G418 selection pressure, G418 may affect some assays, so it was removed in the last round of culture before assays were performed.
4.3.2. Isolation of human neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells. Isolated neutrophils were washed twice and resuspended in HBSS−. Neutrophil preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion.
4.3.3. Ca2+ mobilization assay
Changes in intracellular Ca2+ were measured with a FlexStation III scanning fluorometer (Molecular Devices, Sunnyvale, CA, USA). The cells, suspended in Hank’s balanced salt solution without Ca2+ and Mg2+ but with 10 mM HEPES (HBSS−), were loaded with 1.25 μg/mL Fluo-4 AM dye and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS− containing 10 mM HEPES, resuspended in HBSS+ containing Ca2+, Mg2+, and 10 mM HEPES (HBSS+), and aliquoted into the wells of flat-bottom, half-area-well black microtiter plates (2 × 105 cells/well). For evaluation of direct agonist activity, compounds of interest were added from a source plate containing dilutions of test compounds in HBSS+, and changes in fluorescence were monitored (λex = 485 nm, λem = 538 nm) every 5 s for 240 s at room temperature after automated addition of compounds. Maximum change in fluorescence during the first 3 min, expressed in arbitrary units over baseline, was used to determine a response. Responses for FPR1 agonists were normalized to the response induced by 5 nM fMLF for FPR1-HL60 cells and neutrophils, or 5 nM WKYMVm for FPR2-HL60 cells, which were assigned a value of 100%. Curve fitting (5–6 points) and calculation of median effective inhibitory concentrations (IC50) were performed by nonlinear regression analysis of the dose–response curves generated using Prism 7 (GraphPad Software, Inc., San Diego, CA, USA).
4.3.4. Chemotaxis assay
Human neutrophils were resuspended in HBSS+ containing 2% (v/v) heat-inactivated FBS (2 × 106 cells/ml), and chemotaxis was analyzed in 96-well ChemoTx chemotaxis chambers (Neuroprobe, Gaithersburg, MD, USA), as described previously [26]. The compounds under investigation, 2 nM WKYMVm (positive control), or DMSO alone (negative control) were added into the lower wells of the chemotaxis chambers containing 30 μL of HBSS+ containing 2% (v/v) heat-inactivated FBS. To analyze desensitization of chemotaxis by the test compounds, neutrophils were preincubated with the indicated compounds or control DMSO for 30 min at room temperature and added to the upper wells of the chemotaxis chambers. The lower wells were loaded with 30 μL of HBSS+ containing 2% (v/v) heat-inactivated FBS with the same concentrations of test compounds plus 2 nM WKYMVm or control DMSO. In both cases (direct chemotaxis or chemotaxis desensitization), neutrophils in the upper chamber were allowed to migrate through the 5.0-μm pore polycarbonate membrane filter for 60 min at 37 °C and 5% CO2. The number of migrated cells was determined by measuring ATP in the lysates of transmigrated cells using a luminescence-based assay (CellTiter-Glo; Promega, Madison, WI, USA) and a Fluoroscan Ascent FL microplate reader. Luminescence measurements were converted to absolute cell numbers by comparison of the values with standard curves obtained with known numbers of neutrophils. Curve fitting (at least eight to nine points) and calculation of median effective concentration values (EC50 and IC50) were performed by nonlinear regression analysis of the dose–response curves generated using Prism software.
4.3.5. Animals
Male Sprague Dawley rats (Envigo, Varese, Italy) weighing approximately 200–250 g at the beginning of the experimental procedure were used. Animals were housed in Ce.S.A.L. (Centro Stabulazione Animali da Laboratorio, University of Florence) and used at least one week after their arrival. Four rats were housed per cage (size 26 × 41 cm2); animals were fed with standard laboratory diet and tap water ad libitum, kept at 23 ± 1 °C with a 12 h light/dark cycle, light at 7 a.m. All animal manipulations were carried out according to the Directive 2010/63/EU of the European parliament and of the European Union council (22 September 2010) on the protection of animals used for scientific purposes. The ethical policy of the University of Florence complies with the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH Publication No. 85–23, revised 1996; University of Florence assurance number: A5278–01). Formal approval to conduct the experiments described was obtained from the Italian Ministry of Health (No. 517/2017, 06/04/2017) and from the Animal Subjects Review Board of the University of Florence. Experiments involving animals have been reported according to ARRIVE guidelines [46]. All efforts were made to minimize animal suffering and to reduce the number of animals used.
4.3.6. Complete Freund’s Adjuvant-Induced inflammatory arthritis
Articular damage was induced by injection of CFA (Sigma-Aldrich) into the tibiotarsal joint [47,48]. Briefly, the rats were lightly anesthetized by 2% isoflurane. After sterilizing the left leg skin with 75% ethyl alcohol, the lateral malleolus was located by palpation and a 28-gauge needle was inserted vertically to penetrate the skin and turned distally for insertion into the articular cavity at the gap between the tibiofibular and tarsal bone until a distinct loss of resistance was felt. A volume of 50 μL of CFA was then injected (left paw, ipsilateral). Control rats received 50 μL of saline solution in the tibiotarsal joint. The paw pressure and the incapacitance tests (see below) were performed 14 days after CFA injection.
4.3.7. Drug administration
EC3(10 and 30 mg/kg), EC10 (3, 10, and 30 mg/kg), 2a (3, 10, and 30 mg/kg) and ibuprofen (100 mg kg−1) were suspended in CMC 1% and administered p.o. 14 days after CFA injection. Behavioral measurements were conducted at 15, 30, 45, 60, and 75 min after treatment.
4.3.8. Paw pressure test
Nociceptive threshold was determined with an analgesimeter (Ugo Basile, Varese, Italy). Briefly, a constantly increasing pressure was applied to a small area of the dorsal surface of the hind paw using a blunt conical mechanical probe. Mechanical pressure was increased until vocalization or a withdrawal reflex occurred while rats were lightly restrained. Vocalization or withdrawal reflex thresholds were expressed in grams. An arbitrary cut-off value of 100 g was adopted. The data were collected by an observer who was blinded to the protocol [49].
4.3.9. Incapacitance test
Weight bearing changes were measured using an Incapacitance Apparatus (Linton Instrumentation, UK) detecting changes in postural equilibrium after a hind limb injury. As described by Di Cesare Mannelli et al. [50], rats were trained to stand on their hind paws in a box with an inclined plane (65° from horizontal). This box was placed above the incapacitance apparatus. This allowed us to independently measure the weight that the animal applied on each hind limb. The value reported for each animal is the mean of five consecutive measurements. In the absence of hind limb injury, rats applied an equal weight on both hind limbs, indicating postural equilibrium, whereas an unequal distribution of weight on the hind limbs indicated a monolateral decreased pain threshold. Data are expressed as the difference between the weight applied to the limb contralateral to the injury and the weight applied to the ipsilateral one (Δ weight).
4.3.10. Statistical analysis
Each value represents the mean ± SEM of five rats per group. Groups were as shown in the figures, and different groups were used for acute treatments. The analysis of variance was performed by ANOVA. A Bonferroni’s significant difference procedure was used as post hoc comparison. P values of less than 0.05 or 0.01 were considered significant. Data were analysed using the ‘Origin 8.1′ software.
Acknowledgements:
This research was supported in part by National Institutes of Health IDeA Program Grants GM110732, GM115371, and GM103474; USDA National Institute of Food and Agriculture Hatch project 1009546; and the Montana State University Agricultural Experiment Station.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Beutler B, Innate immunity: an overview, Mol. Immunol 40 (2004) 845–859, 10.1016/j.molimm.2003.10.005. [DOI] [PubMed] [Google Scholar]
- [2].Brinkmann V, Zychlinsky A, Neutrophil extracellular traps: is immunity the second function of chromatin? J. Cell. Biol 198 (2012) 773–783, 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kolaczkowska E, Kubes P, Neutrophil recruitment and function in health and inflammation, Nat. Rev. Immunol 13 (2013) 159–175, 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- [4].Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL, Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals, J. Exp. Med 196 (2002) 1025–1037, 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Serhan CN, Levy B, Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators, J. Clin. Invest 128 (2018) 2657–2669, 10.1172/JCI97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N, Novel anti-inflammatorypro-resolving mediators and their receptors, Curr. Top. Med. Chem 11 (2011) 629–647, 10.2174/1568026611109060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serhan CN, Chiang N, Resolution phase lipid mediators of inflammation: agonists of resolution, Curr. Opin. Pharmacol 13 (2013) 632–640, 10.1016/j.coph.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Serhan CN, Chiang N, Dalli J, The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution, Semin. Immunol 27 (2015) 200–215, 10.1016/j.smim.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C, The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo, Pharmacol. Rev 58 (2006) 463–487, 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- [10].Stama ML, Slusarczyk J, Lacivita E, Kirpotina LN, Schepetkin IA, Chamera K, Riganti C, Perrone R, Quinn MT, Basta-Kaim A, Leopoldo M, Novel ureidopropanamide based N-formyl peptide 2 (FPR2) agonists with potential application for central nervous system disorders characterized by neuroinflammation, Eur. J. Med. Chem 141 (2017) 703–720, 10.1016/j.ejmech.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM, International union of basic and clinical pharmacology: LXXIII. Nomenclature for the formyl peptide receptor (FPR) family, Pharmacol. Rev 61 (2009) 119–161, 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Migeotte I, Communi D, Parmentier M, Formyl peptide receptors: a promiscuous sub family of G protein-coupled receptors controlling immune responses, Cytokine Grow Factor Rev. 17 (2006) 501–519, 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [13].Migeotte I, Riboldi E, Fransenn JD, Gregoire F, Loison C, Wittamer V, Detheux M, Robberecht P, Costagliola S, Vassart G, Identification and characterization of an endogenous chemotactic ligand specific for FPRL2, J. Exp. Med 201 (2005) 83–93, 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Perretti M, Leroy X, Bland EJ, Montero-Melendez T, Resolution pharmacology: opportunities for therapeutic innovation in inflammation, Trends Pharmacol. Sci 36 (2015) 737–755, 10.1016/j.tips.2015.07.007. [DOI] [PubMed] [Google Scholar]
- [15].Romano M, Cianci E, Simiele F, Recchiuti A, Lipoxins and aspirin-triggered lipoxins in resolution of inflammation, Eur. J. Pharmacol 760 (2015) 49–63, 10.1016/j.ejphar.2015.03.083. [DOI] [PubMed] [Google Scholar]
- [16].Kao W, Gu R, Jia Y, Wei X, Fan H, Harris J, Zhang Z, Quinn J, Morand EF, Yang YH, A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis, Br. J. Pharmacol 171 (2014) 4087–4096, 10.1111/bph.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bozinovski S, Anthony D, Anderson GA, Irving LB, Levy BD, Vlahos R, Treating neutrophilic inflammation in COPD by treating ALX/FPR2 resolution pathways, Pharmacol. Ther 140 (2013) 280–289, 10.1016/j.pharmthera.2013.07.007. [DOI] [PubMed] [Google Scholar]
- [18].Chen Y-C, Lin M-C, Lee CH, Liu S-F, Wang C-C, Fang W-F, Chao T-Y, Wu C-C, Wei Y-F, Chang H-C, Tsen C-C, Chen H-C, Defective formyl peptide receptor 2/3 and annexin A1 expressions associated with M2a polarization of blood immune cells in patients with chronic obstructive, J. Transl. Med 16 (2018) 69–81, 10.1186/s12967-018-1435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ries M, Loiola R, Shah NU, Gentleman MS, Solito E, Sastre M, The anti-inflammatory Annexin A1 induces the clearance and degradation of amyloid-β peptide, J. Neuroinflam 13 (2016) 234–248, 10.1186/s12974-016-0692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Prevete N, Liotti F, Marone G, Melillo RM, De Paulis A, Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth, Pharm. Res 102 (2015) 184–191, 10.1016/j.phrs.2015.09.017. [DOI] [PubMed] [Google Scholar]
- [21].Maris JM, Recent advances in neuroblastoma, N. Engl. J. Med 362 (2010) 2202–2211, 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Snapkov I, Oqvist CO, Figenschau Y, Kogner P, Johnsen JI, Sveinbjornsson B, The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis, BMC Cancer 16 (2016) 490–501, 10.1186/s12885-016-2545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu J, Li J, Zeng X, Rao Z, Gao J, Zhang B, Zhao Y, Yang B, Wang Z, Yu L, Wang W, Formyl peptide receptor suppresses melanoma development and promotes NK cell migration, Inflammation 37 (2014) 984–992, 10.1007/s10753-014-9819-z. [DOI] [PubMed] [Google Scholar]
- [24].Bürli RW, Xu H, Zou X, Muller K, Golden J, Frohn M, Adlam M, Plant MH, Wong M, McElvain M, Regal K, Viswanadhan VN, Tagari P, Hungate R, Potent hFPRL1 (ALXR) agonist as potential anti-inflammatory agents, Bioorg. Med. Chem. Lett 16 (2006) 3713–3718, 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- [25].Odobasic D, Jia Y, Kao W, Fan H, Wei X, Gu R, Ngo D, Kitching AR, Holdsworth SR, Morand EF, Yang YH, Formyl peptide receptor activation inhibits the expansion of effector T cells and synovial fibroblast and attenuates joint injury in models of rheumatoid arthritis, Intern. Immunopharm 61 (2018) 140–149, 10.1016/j.intimp.2018.05.028. [DOI] [PubMed] [Google Scholar]
- [26].Cilibrizzi A, Quinn MT, Kirpotina LN, Schepetkin IA, Holderness J, Ye RD, Rabiet MJ, Biancalani C, Cesari N, Graziano A, Vergelli C, Pieretti S, Dal Piaz V, Giovannoni MP, 6-Methyl-2,4-disubstituted pyridazin-3(2H)-ones: a novel class of small-molecule agonists for formyl peptide receptors, J. Med. Chem 52 (2009) 5044–5057, 10.1021/jm900592h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cilibrizzi A, Schepetkin IA, Bartolucci G, Crocetti L, Dal Piaz V, Giovannoni MP, Graziano A, Kirpotina LN, Quinn MT, Vergelli C, Synthesis enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists, Bioorg. Med. Chem 20 (2012) 3781–3792, 10.1016/j.bmc.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cilibrizzi A, Crocetti L, Giovannoni MP, Graziano A, Vergelli C, Bartolucci G, Soldani G, Quinn MT, Schepetkin IA, Faggi C, Synthesis HPLC enantioresolutio and X-ray analysis of a new series of C5-methyl pyridazines as N-formyl peptide receptor (FPR) agonists, Chirality 25 (2013) 400–408, 10.1002/chir.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Giovannoni MP, Schepetkin IA, Cilibrizzi A, Crocetti L, Khlebnikov AI, Dahlgren C, Graziano A, Dal Piaz V, Kirpotina LN, Zerbinati S, Vergelli C, Quinn MT, Further studies on 2-arylacetamide pyridazin-3(2H)-ones: design, synthesis and evaluation of 4,6-disubstituted analogs as formyl peptide receptors (FPRs) agonists, Eur. J. Med. Chem 64 (2013) 512–528, 10.1016/j.ejmech.2013.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Crocetti L, Vergelli C, Cilibrizzi A, Graziano A, Khlebnikov AI, Kirpotina LN, Schepetkin IA, Quinn MT, Giovannoni MP, Synthesis and pharmacological evaluation of new pyridazin-based thioderivatives as formyl peptide receptor (FPR) agonists, Drug Dev. Res 74 (2013) 259–271, 10.1002/ddr.21076. [DOI] [Google Scholar]
- [31].Vergelli C, Schepetkin IA, Ciciani G, Cilibrizzi A, Crocetti L, Giovannoni MP, Guerrini G, Iacovone A, Kirpotina LN, Khlebnikov AI, Ye RD, Quinn MT, 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists, Bioorg. Med. Chem 24 (2016) 2530–2543, 10.1016/j.bmc.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vergelli C, Schepetkin IA, Ciciani G, Cilibrizzi A, Crocetti L, Giovannoni MP, Guerrini G, Iacovone A, Kirpotina LN, Ye RD, Quinn MT, Synthesis of five- and six-membered N-phenylacetamido substituted heterocycles as formyl peptide receptor agonists, Drug Dev. Res 78 (2017) 49–62, 10.1002/ddr.21370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lesher GY, Opalka CJ Jr., Page DF, 3-Amino-5-(substituted)-2(1H)-pyridinones and 3-cyano compounds useful as cardiotonics, Sterling Drug Inc., USA, 1982, EP 0061774 A2. [Google Scholar]
- [34].Andersen SM, Aurell CJ, Zetteberg F, Bollmark M, Ehrl R, Schuisky P, Witt A, Development of a multi-kilogram-scale synthesis of AZD1283: a selective and reversible antagonist of the P2Y12 receptor, Org. Proc. Rese. Develop 17 (2013) 1543–1551, 10.1021/op400288v. [DOI] [Google Scholar]
- [35].Baraldi PG, Preti D, Tabrizi MA, Fruttarolo F, Saponaro G, Baraldi S, Romagnoli AR, Gessi S, Varani K, Borea PA, N6-[(Hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5’-N-ethylcarboxamido-adenosines: the first example of adenosine-related structures with potent agonist activity at human A2B adenosine receptor, Bioorg. Med. Chem 15 (2007) 2514–2547, 10.1016/j.bmc.2007.01.055. [DOI] [PubMed] [Google Scholar]
- [36].Abu-Shanab FA, Aly FM, Wakefield BJ, Synthesis of substituted nicotinamides from enamines derived from N,N-dimethylformammide dimethyl acetal, Synthesis. 8 (1995) 923–925, 10.1055/s-1995-4039. [DOI] [Google Scholar]
- [37].Fossa P, Boggia R, Lo Presti E, Mosti L, Dorigo P, Floreani M, Inotropic agents. Synthesis and structure-activity relationships of new milrinone related cAMP PDE III inhibitors, Farmaco 52 (1997) 523–530. [PubMed] [Google Scholar]
- [38].Winckelmann IB, Larsen EH, Improved one-step procedure for the preparation of 1-substituted and 1,3-disubstituted uracils and 2-thiouracils, Synthesis 12 (1986) 1041–1043, 10.1055/s-1986-31867. [DOI] [Google Scholar]
- [39].Robin A, Julienne K, Meslin J-C, Deniaud D, Straightforward pyrimidine ring construction: a versatile tool for the synthesis of nucleobase and nucleoside analogues, Eur. J. Org. Chem 3 (2006) 634–643, 10.1002/ejoc.200500556. [DOI] [Google Scholar]
- [40].Tao L, Yue Y, Zhang J, Chen S-Y, Yu X-Q, A mild and efficient method for N-arylnucleobase synthesis via the cross-coupling reactions of nucleobases with arylboronic acids catalyzed by simple copper salts, Hel. Chi. Ac 91 (6) (2008) 1008–1014, 10.1002/hlca.200890107. [DOI] [Google Scholar]
- [41].Browne DT, Eisinger J, Nelson JL, Synthetic spectroscopic models related to coenzymes and base pairs. II. Evidence for intermolecular base-base interactions in dinucleotide analogs, J. Am. Chem. Soc 90 (26) (1968) 7302–7309, 10.1021/ja01028a023. [DOI] [PubMed] [Google Scholar]
- [42].Gougerot-Pocidalo MA, El Benna J, Elbim C, Chollet-Martin S, Dang MC, Regulation of human neutrophil oxidative burst by pro- and anti-inflammatory cytokines, J. Soc. Biol 196 (1) (2002) 37–46. [PubMed] [Google Scholar]
- [43].Qin CX, May LT, Li R, Cao N, Rosli S, Deo M, Alexander AE, Horlock D, Bourke JE, Yang YH, Stewart AG, Kaye DM, Du XJ, Sexton PM, Christopoulos A, Gao XM, Ritchie RH, Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice, Nat. Commun 8 (2017) 14232, 10.1038/ncomms14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Di Cesare L, Mannelli L, Cinci L, Maresca M, Vergelli C, Pacini A, Quinn MT, Giovannoni MP, Ghelardini C, Effects of the neutrophil elastase inhibitor EL-17 in rat adjuvant-induced arthritis, Rheumatology 55 (7) (2016) 1285–1294, 10.1093/rheumatology/kew055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Di Cesare Mannelli L, Bani D, Bencini A, Brandi ML, Calosi L, Cantore M, Carossino AM, Ghelardini C, Valtancolli B, Failli P, Therapeutic effects of the superoxide dismutase mimetic compound MnIIMe2DO2A on experimental articular pain in rats, Mediators Inflamm. 905360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McGrath JC, Lilley E, Implementing guidelines on reporting research using animals: New requirements for publication in BJP, Br. J. Pharmacol 172 (2015) 3189–3193, 10.1111/bph.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Butler SH, Godefroy F, Besson JM, Weil-Fugazza J, A limited arthritic model for chronic pain studies in the rat, Pain 48 (1992) 73–81, 10.1016/0304-3959(92)90133-v. [DOI] [PubMed] [Google Scholar]
- [48].Micheli L, Ghelardini C, Lucarini E, Parisio C, Trallori E, Cinci L, Di Cesare Mannelli L, Intra-articular mucilages: behavioural and histological evaluations for a new model of articular pain, J. Pharm. Pharmacol 71 (2019) 971–981, 10.1111/jphp.13078. [DOI] [PubMed] [Google Scholar]
- [49].Bird MF, Cerlesi MC, Brown M, Malfacini D, Vezzi V, Molinari P, Micheli L, Di Cesare Mannelli L, Ghelardini C, Guerrini R, Calò DG, Lambert G. Characterisation of the novel mixed Mu-NOP peptide ligand dermorphin-N/OFQ (DeNo). PLoS One. 11 (2016) e0156897 10.1371/journal.pone.0156897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Di Cesare Mannelli L, Micheli L, Maresca M, Gravotto G, Bellumori M, Innocenti M, Mulinacci N, Ghelardini C. Anti-neuropathic effects of Rosmarinus officinalis L. terpenoid fraction: relevance of nicotinic receptors, Sci Rep. 6 (2016) 34832–34847, 10.1038/srep34832. [DOI] [PMC free article] [PubMed] [Google Scholar]