

Summary
Arginyltransferase ATE1 mediates posttranslational arginylation and plays key roles in multiple physiological processes. ATE1 utilizes arginyl (Arg)-tRNAArg as the donor of Arg, putting this reaction into a direct competition with the protein synthesis machinery. Here, we address the question of ATE1- Arg-tRNAArg specificity as a potential mechanism enabling this competition in vivo. Using in vitro arginylation assays and ATE1 knockout models, we find that, in addition to full length tRNA, ATE1 is also able to utilize short tRNAArg fragments that bear structural resemblance to tRNA-derived fragments (tRF), a recently discovered class of small regulatory non-coding RNAs with global emerging biological role. ATE1 knockout cells show a decrease in tRFArg generation and a significant increase in the ratio of tRNAArg:tRFArg compared to wild type, suggesting a functional link between tRFArg and arginylation. We propose that generation of physiologically important tRFs can serve as a switch between translation and protein arginylation.
Graphical Abstract

eTOC Blurb
Avcilar-Kucukgoze et al. dissect the tRNA-dependent mechanisms of regulation of arginylation and suggest a role for tRNA-derived fragments, a type of small noncoding RNAs, in arginylation.
Introduction
Protein arginylation mediated by arginyltransferase ATE1 is a posttranslational modification of emerging importance that is essential for multiple physiological processes and targets many proteins in vivo (Saha and Kashina, 2011). ATE1 gene is functionally conserved in all eukaryotes, and it regulates shoot and leaf development in Arabidopsis (Graciet et al., 2009), migratory behavior in Dictyostelium (Batsios et al., 2019), and multiple other processes relevant to the functioning of many physiological systems (Dissmeyer, 2019; Eldeeb et al., 2018; Ji et al., 2019; Kashina, 2015; Wang et al., 2017b). Deletion of ATE1 leads to embryonic lethality in flies and mice (Kwon et al., 2002; Saha and Kashina, 2011). Global proteomics screens have identified hundreds of putative arginylated targets of ATE1, suggesting that this single enzyme can potentially mediate a vast variety of intracellular reactions. Despite this complexity and high physiological relevance (Kashina and Yates, 2015; Wong et al., 2007), the molecular mechanisms of ATE1 action and regulation in vivo are poorly understood.
Biochemical studies and structural predictions suggest that ATE1 is a globular monomeric enzyme that can exist in a soluble form in the cytosol, and can potentially associate with the endoplasmic reticulum and the protein synthesis machinery (Carpio et al., 2013; Ciechanover et al., 1988; Galiano et al., 2016; Ji and Kwon, 2017; Lopez Sambrooks et al., 2012; Wang et al., 2011). A recent study has identified a sequence context around the target site on proteins that can potentially facilitate arginylation (Wang et al., 2018). None of these studies, however, provide any explanation on how ATE1 prioritizes among its wide variety of targets, and what upstream mechanisms can mediate its regulation. ATE1 utilizes Arg-tRNAArg as a donor of Arg in the reaction. tRNAsArg belong to the moderately abundant tRNA isoacceptors (i.e., tRNAs reading different codons but carrying the same amino acid) in human cells (Kirchner et al., 2017). Hence, arginylation directly competes with ribosomes and protein synthesis in the cell for Arg-tRNAArg.
Arginyl-tRNA-synthetase (RARS) in mammalian cells exists in two variants, a ~72 kDa form that is largely in a ribosome-associated multi-synthetase complex, which also includes other aminoacyl-tRNA synthetases, and a ~60 kDa soluble form that is present freely in the cytosol (Cirakoglu and Waller, 1985; Deutscher and Ni, 1982; Kyriacou and Deutscher, 2008; Vellekamp et al., 1985). It has been previously proposed that the longer form is dedicated to translation, while the shorter form is utilized largely for arginylation (Sivaram and Deutscher, 1990). In support, an earlier study proposed that arginylation activity can partially co-fractionate with the activity of RARS (Ciechanover et al., 1988), although this study did not differentiate between the long and short RARS forms. While this hypothesis regarding a dedicated RARS for translation and arginylation is compelling, it has not been fully investigated beyond those initial studies. Furthermore, since lower eukaryotes contain only one RARS form, this mechanism cannot fully explain the ability of ATE1 to successfully compete with the protein synthesis machinery for mediation of protein arginylation.
Arg in eukaryotes is encoded by six different codons, and tRNAArg species in mammals are expressed from 24-28 different genes encoding 5 isoacceptors, most of which are moderately to low abundant in mammalian tissues (Dittmar et al., 2006; Ishimura et al., 2014; Kirchner et al., 2017). Therefore, the abundance among tRNAArg isoacceptors differs and does not seem to correlate with the Arg codon usage (Kirchner et al., 2017). It appears possible that one or few of these tRNAArg isoacceptors may be dedicated to arginylation.
Here we characterized tRNAArg specificity of ATE1 to determine whether this enzyme shows selectivity toward any particular tRNAArg isoacceptors, which may serve as a potential mechanism of mediating the balance between translation and arginylation. Our results show that, while all the tRNAArg isoacceptors can be utilized by ATE1, the acceptor stem of tRNAArg carrying the charged Arg moiety is competent for arginylation. This finding has led us to a surprising discovery that tRNA-derived fragments (tRF) generated from Arg-tRNAArg, can independently mediate arginylation in vitro with efficiencies similar to that of the full length tRNAArg. tRFs are a class of small non-coding RNAs that play major roles in normal physiology and disease (Anderson and Ivanov, 2014; Fu et al., 2015; Li et al., 2018; Shen et al., 2018; Sobala and Hutvagner, 2011; Zhu et al., 2018). We found that in cultured cells, lack of ATE1 shifts the balance between the levels of tRNAArg and tRFArg. Collectively these data provide biochemical evidence that tRFArg may be involved in arginylation. We propose that tRFArg generation provides a potential physiological mechanism that may regulate the partitioning of tRNAsArg and the balance between protein translation and arginylation.
Results
ATE1-mediated arginylation is highly specific toward arginyl-tRNAArg.
Our previous results show that ATE1 rapidly and efficiently mediates arginylation of protein and peptide substrates in an in vitro reaction containing E. coli tRNAArg, radioactively labeled Arg, and an RARS enzyme, in addition to ATE1 (Wang et al., 2011; Wang et al., 2014; Wang and Kashina, 2015). To test the specificity of this reaction for each component, we first tested whether ATE1 can transfer amino acids other than arginine, and whether it can utilize other tRNAs in the reaction. For these experiments, we initially used a total E. coli tRNA mixture containing all tRNA isoacceptors. We added radioactively labeled Met and Gln (chosen as the amino acids most structurally distinct from Arg), and the cognate aminoacyl-tRNA synthetases (ARSs) to aminoacylate tRNAMet and tRNAGln, and tested the ability of this mixture to support ATE1-mediated transfer of amino acids to a standard protein substrate (BSA, (Wang et al., 2011)). We reasoned that if ATE1 specificity to the aminoacyl-tRNA is low, it would be able to at least partially utilize these heterologous amino acids. However, despite reasonably high charging efficiency, we did not observe any detectable incorporation with any of the four isoforms of ATE1 (Fig. S1). While this result does not exclude the possibility of potential binding of these aminoacyl-tRNAs to ATE1, it clearly shows the inability of ATE1 to transfer Met or Gln onto BSA.
To further investigate ATE1 specificity for a particular tRNA-amino acid combination, we next tested whether ATE1 can recognize a non-cognate amino acid (i.e. other than Arg) charged to tRNAArg, or whether charging Arg to a different tRNA can support arginylation. In this experiment we used E. coli tRNAArg CCG (where CCG is the anticodon), a common substrate in our in vitro reactions, and E. coli tRNALys UUU, which carries the physicochemically closest amino acid to Arg, i.e. same charge of their side chains. To bypass the ARS specificity, we used flexizyme (Murakami et al., 2006) to charge these two tRNAs with either Arg (the native ATE1 amino acid substrate), Lys (a proteinogenic amino acid with positively charged side chain), or the non-proteinogenic citrulline (Ctr) (a structurally similar analog of Arg with no charge). We tested the ability of these molecules to mediate arginylation in a standard arginylation assay using angiotensin II, a highly efficient ATE1 peptide substrate (Wang et al., 2014). Notably, of all combinations, ATE1 only catalyzed arginylation only with Arg-tRNAArg (Fig. 1). Thus, ATE1 is highly specific to Arg-tRNAArg.
Figure 1. ATE1-mediated arginylation is highly specific toward Arg-tRNAArg.
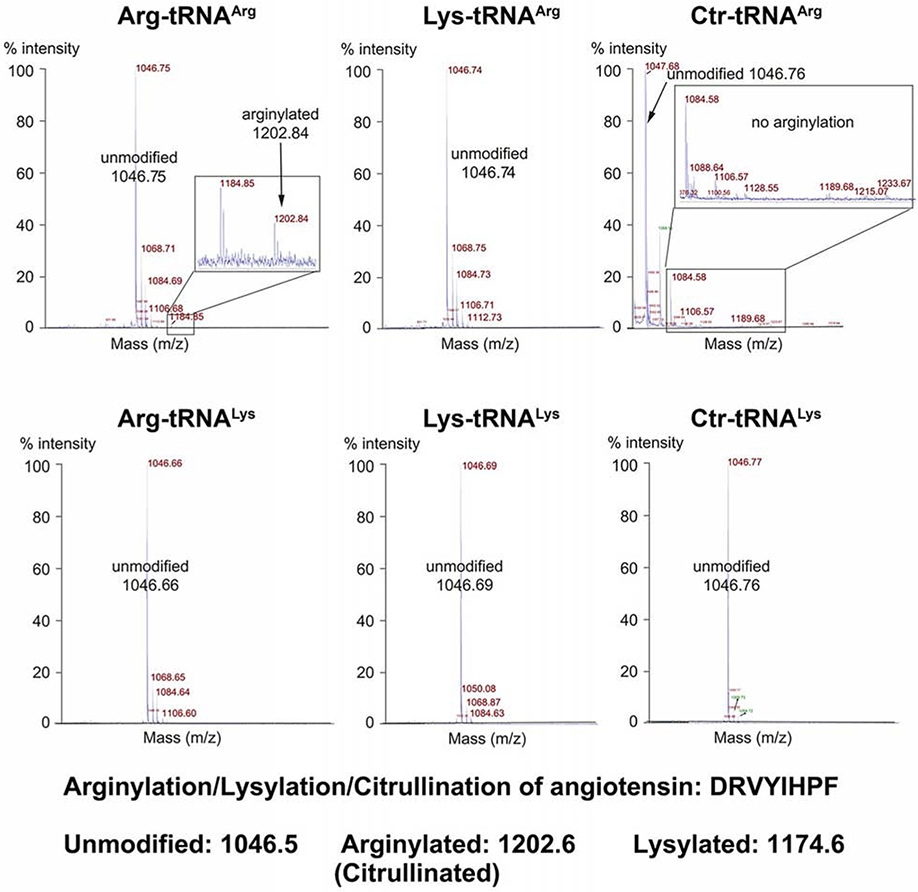
MALDI-TOF chromatograms of angiotensin II. Masses of unmodified and arginylated angiotensin are marked. Only tRNAArg charged with Arg is able to mediate arginylation.
ATE1 selectively binds to tRNA.
While many prior studies have demonstrated the essential role of tRNAArg in the arginylation reaction, it is not known whether ATE1 has any specific RNA-binding activity or whether ATE1-tRNA interaction occurs only transiently during the reaction. To address this question and test whether ATE1 can specifically bind any class(es) of intracellular RNA, we isolated total RNA from mouse liver and mixed it in vitro with a recombinant His-tagged ATE1 that is normally used in our arginylation assays. We then pulled down ATE1 by the His tag using Ni-NTA agarose beads, and examined the composition of RNAs bound to ATE1 in the pulldown.
Gel fractionation of the RNA before and after the pulldown showed that only the low molecular weight RNA were associated with ATE1, while the higher molecular weight RNAs appeared to have no interaction with ATE1 (Fig. 2A, top). Further size fractionation revealed that RNAs of the tRNA size are of the major components, and that a smaller amount of 5S and 5.8S ribosomal RNA was also present. Control pulldowns using empty Ni-NTA beads, or His-tagged fragment of gelsolin (an actin-binding protein with no known RNA binding activity), which was truncated to mimic ATE1 in molecular weight, did not result in any visible tRNA binding, or any RNA bands from the tRNA-depleted leftover sample (Fig. 2A, middle and bottom). Thus, ATE1 low molecular weight RNAs.
Figure 2. ATE1 selectively binds to tRNA.
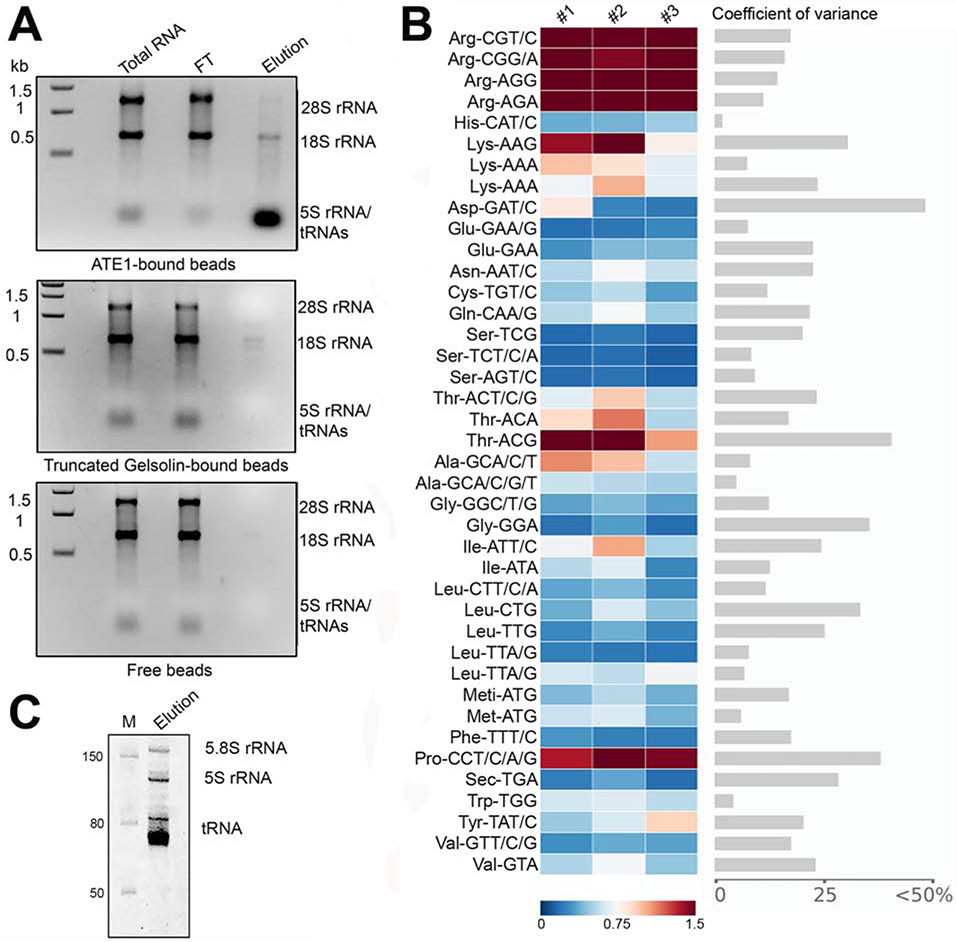
A, Gels of total RNA used as pulldown input, flow through (FT) and RNA eluate from His-tagged ATE1 immobilized on Ni-NTA beads (top), His-tagged gelsolin fragment immobilized on Ni-NTA beads (middle) and Ni-NTA beads alone (bottom). B, Comparative microarrays of tRNAs bound to ATE1 vs total tRNA of mouse liver summary of three biological replicates. Arrays are highly reproducible (Fig. S2). tRNA probes are depicted with their codon and the corresponding amino acid; Meti-ATG, initiator tRNAMet. Two different probes recognizing different tRNALeu (Leu-UAA) and tRNALys (Lys-UUU) isodecoders that pair to the same codon TTA/G-Leu or AAA-Lys codon but differ in their sequence outside the anticodon were used on the arrays. Data are shown as fold enrichment (gradient ruler at the bottom). Significantly enriched tRNAs in the ATE1 pull down compared to the total liver tRNAs were selected based on the confidence intervals of covariance analysis (an example between replicate 1 and 2 is included). C, Eluted RNA sample in denaturing 10% polyacrylamide gel. M, RNA marker.
To test whether any specific tRNAs in this mixture are bound to ATE1 with higher affinities than others, we used tRNA-tailored microarray (Beckert et al., 2018) in which tRNAs are visualized by ligation with a fluorophore Cy3-labeled stem-loop structure of a DNA/RNA oligonucleotide hairpin that is paired to the intact CCA-tails. We compared the ATE1-bound tRNA pool to the total mouse liver tRNA, which was labeled with a hairpin DNA/RNA oligonucleotide bearing the fluorophore ATTO647 on the same array. For this experiment, the ATE1-bound aminoacyl-tRNAs were extracted at alkaline conditions to hydrolyze the ester bond between the amino acid and tRNA and to allow the CCA ends for labeling. We found that all of the tRNAArg isoacceptor groups tested by each probe were enriched in the ATE1-bound fraction (Fig. 2B and S2), indicating that ATE1 has a higher affinity for tRNAArg species but does not strongly distinguish among different tRNAArg isoacceptors within these groups. Compared to tRNAArg, the majority of the other cellular tRNA species were under-represented in the pulldown. A notable exception included tRNAThr CGU, tRNALys CUU, and tRNAsPro, which showed enrichment in the ATE1-pulldown samples, although these tRNAs showed much higher variability among the replicates (Fig. 2B and S2). To test whether these tRNA species have any activity in the ATE1-mediated reaction, we used radioactively labeled Pro and Lys, along with total mouse tRNA and a mixture of aminoacyl tRNA synthetases, in an in vitro arginylation assay. We observed no incorporation of these amino acids into the test peptide substrate (Fig. S3). Thus, the binding of these tRNAs to ATE1 is likely unrelated to amino acid transfer.
ATE1 is able to utilize all mouse tRNAArg species.
One of the potential ways to enable ATE1 to successfully compete with the ribosome could involve selectivity for specific tRNAArg species. Mouse genome contains 25 different tRNAArg genes encoding 20 unique tRNAArg sequences for 5 different tRNAArg isoacceptors (Juhling et al., 2009). Thus, each family of tRNAArg isoacceptor contains multiple isodecoders (i.e., tRNA species with the same anticodon but with differences in the primary sequence). The microarray experiment presented above cannot distinguish among individual tRNAArg species, due to the necessity to maintain a common hybridization temperature across the array. This limitation constrained the design of the probes to recognize tRNA isoacceptors that may differ by as many as 8 nucleotides (nts) (Dittmar et al., 2006). In the ATE1-pulldown, the five tRNAArg isoacceptors and the entire repertoire of tRNAArg isodecoders were detected with four probes, of which only one probe was designed to detect tRNAArg CCG and tRNAArg UCG (Fig. 2B and S2). This raised the possibility that, of the two classes of tRNAArg isoacceptors pulled down by ATE1, the enzyme may exhibit specificity for individual isodecoders of each class.
To test this possibility more broadly and to determine the ATE1 specificity for tRNAArg isodecoders, we enzymatically synthesized all unique sequences of isodecoders of mouse tRNAArg by in vitro transcription. Each isodecoder was heat-denatured, annealed, and charged in vitro with radioactively labeled Arg by a recombinant human RARS. The charging efficiency of each was determined (Fig. S4A). Separately, we determined the linear tRNA concentration range in the ATE1-catalyzed arginylation reaction, using a range of concentrations of E. coli Arg-tRNAArg in excess of the peptide substrate (Fig. 3A). To confirm that the choice of the linear range was not affected by the tRNAArg source species, we compared arginylation efficiency of angiotensin II using the same amount of Arg-tRNAArg from E. coli or mouse (the ACG 1-1 isodecoder), and found that the two Arg-tRNAArg supported a similar arginylation level (Fig. S4B). Finally, we compared the efficiency of arginylation between angiotensin II and α-synuclein (a protein, rather than peptide substrate), and found that angiotensin II is much more efficient than α-synuclein (Fig. S4C). Based on these tests, we chose 0.25 μM Arg-tRNAArg and angiotensin II as the test substrate and tested the ability of each Arg-tRNAArg for the large-scale experiment to compare arginylation efficiency of different tRNAArg.
Figure 3. ATE1 is able to utilize all mouse tRNAArg species.
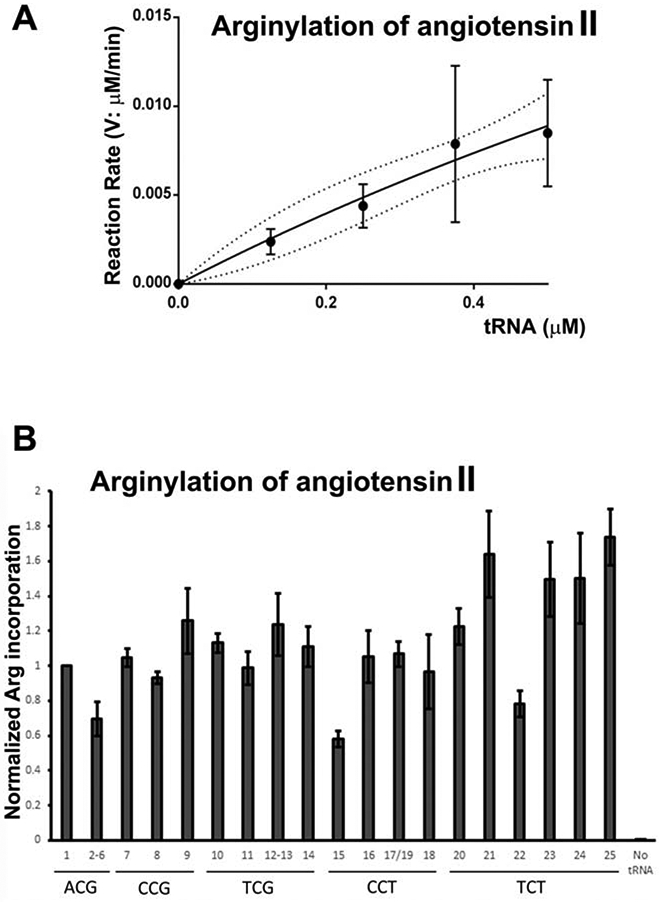
A, Rate of the arginylation reaction as a function of E. coli Arg-tRNAArg in the presence of excess of angiotensin II substrate B, Normalized Arg incorporation into angiotensin II using various pre-charged mouse Arg-tRNAArg isoacceptors and isodecoders. No tRNA, control without adding tRNA (see Table S1 for tRNA gene names and sequences, corresponding to the numbers on the chart).
All of the unique tRNAArg isodecoders were able to mediate arginylation of angiotensin II (Fig. 3B). The efficiency of this arginylation varied between isodecoders (potentially compounded by their different charging efficiencies, which were not accounted for in the plot in Fig 3B). However, it is clear from this result that ATE1 is able to transfer Arg from all of the isodecoders for tRNAArg species. Thus, it appears unlikely that ATE1 exhibits strong selectivity for specific tRNAArg to affect the balance between arginylation and translation.
tRNAArg acceptor stem-like structure charged with Arg can mediate arginylation.
Since all mouse tRNAArg isoacceptors are capable of mediating arginylation, while Arg-tRNALys previously tested in our assays is not (Fig. 1), it appears likely that tRNAArg species share some common structural features that make them better substrates for ATE1 than other tRNAs. Notably, ATE1 is highly specific to tRNAArg charged only with Arg (Fig. 1), suggesting that the combination of the acceptor stem and Arg moiety may be among the key determinants of the reaction. To test this, we developed a test based on previous work showing that the acceptor-T stem or the acceptor stem of tRNA, termed respectively as the minihelix and microhelix of tRNA, can be a substrate for charging (Hamann and Hou, 1995). We made derivatives of E. coli tRNAArg, containing the 12-base pair minihelix or the 7-base pair microhelix, each transcribed in vitro and charged with Arg using flexizyme (see inset structures in Fig. 4). Both Arg-charged mini- and microhelices were able to mediate arginylation (Fig. 4), suggesting that ATE1 requires only a small portion of the tRNAArg, the acceptor stem conjugated to Arg, for mediating arginylation.
Figure 4. tRNAArg acceptor stem-like structures charged with Arg can mediate arginylation.
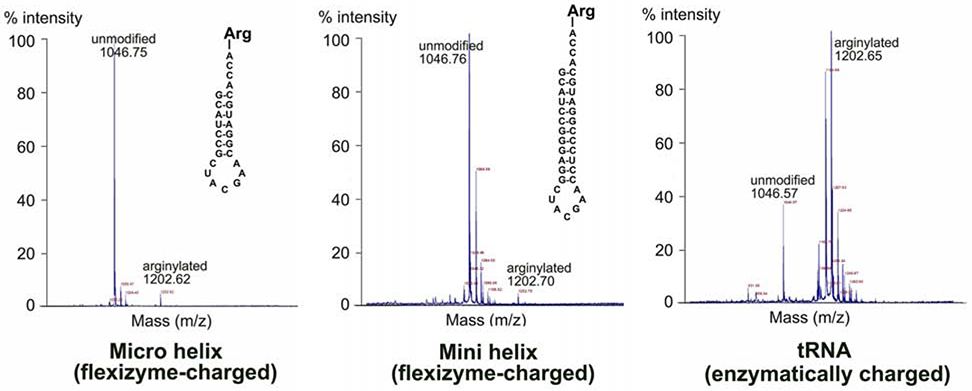
MALDI-TOF chromatograms of angiotensin II, arginylated in vitro using flexizyme-charged Arg-mini- and microhelix (left and middle panel), or RARS-charged Arg-tRNAArg. Masses of unmodified and arginylated angiotensin are marked. Structures of micro- and minihelix tRNAArg used in the reaction are shown on each chromatogram. The higher arginylated peak intensity in the tRNA-mediated reaction is likely due to the fact that this is a fully enzymatic reaction, where RARS present in the mixture continually charges tRNA for the arginylation reaction. Notably, the peak height/intensity in this method does not directly reflect the abundance of the peptide (Szajli et al., 2008).
tRNAArg-derived fragments can mediate arginylation.
tRNA-derived fragments (tRFs) can be generated in cells by several different mechanisms. Cleavage by angiogenin at the anticodon loop during cell stress releases two tRNA halves, which are believed to play a role in mediating stress response (Emara et al., 2010). Cleavage of tRNA at T and D loops occurs during normal physiology and produces three classes of smaller tRFs – tRF-3, tRF-5, and tRF-i (corresponding to the 3’ fragment, 5’ fragment, and internal fragment, respectively) (Shen et al., 2018). The production of these fragments raises the possibility that this type of normally occurring tRNA cleavage would likely release the anticodon loop but should not, in principle, affect the tRNA acceptor stem. If such cleavage, hypothetically speaking, happens after the tRNA charging, it could conceivably generate an amino acid-charged tRNA acceptor stem, which is incapable of participating in translation but can potentially still serve as an amino acid carrier. If true, this kind of molecule would bear marked similarities with the amino acid-charged mini- and microhelices tested above. Following this reasoning, we asked whether Arg-charged tRFArgs can be generated by the enzymes normally responsible for tRF production, and whether these charged tRFArg species can mediate arginylation.
The enzymes that produce shorter tRFs in normal conditions are not fully characterized. Based mostly on structural considerations, it was initially proposed that these tRFs are generated by tRNA cleavage via endonuclease Dicer (Cole et al., 2009), however it has been later found that Dicer ablation does not abrogate intracellular tRFs (Kuscu et al., 2018). A recent report suggests that in plants tRF generation depends on the activity of RNases T2 rather than Dicer (Megel et al., 2019), even though open questions remain about the subcellular RNase T2 localization – primarily in compartments targeted for secretion – which may be incompatible with tRF generation in cells. In search for an enzyme that can generate tRFs in vitro for use in arginylation assays, we tested both of these enzymes for their ability to cleave tRNAArg into tRF-sized fragments. In vitro transcribed mouse tRNAArgACG 1-1 isodecoder was incubated with Dicer or RNase T2, and the resulting cleavage products were analyzed by Northern blotting using 32P-labeled DNA probe complementary to 3’ end of tRNAArgACG 1-1, detecting the corresponding tRF-3.
Dicer in these assays acted considerably slower and less efficiently than RNase T2, and formed only one product, approximately 50 nts in length, which was larger than the size of tRF-3 described in vivo (28 nts) (Fig. 5A, left) . To make sure that this cleavage pattern is universal among tRNAArg isoacceptors, we tested Dicer with total RNA from mouse liver and detected the resulting products with Northern blot probes corresponding to different tRNAArg isoacceptors (Table S1). In all of these reactions, the product of 50 nts seemed to be the only one produced by Dicer cleavage, and the digestion never went far enough to deplete the intact tRNA (Fig. S5A). In contrast, treatment with RNase T2 resulted in rapid and efficient generation of a major fragment of the expected size (just below 30 nts on the gel, Fig. 5A, right). This reaction occurred in minutes and quickly led to the complete cleavage of the full-length tRNA and its full conversion to lower molecular weight fragments. We also tested whether ATE1 itself contains any tRNA cleaving activity, but found none (Fig. S5B). Based on this, we used RNase T2 in the subsequent tests.
Figure 5. tRNAArg-derived fragments mediate arginylation.
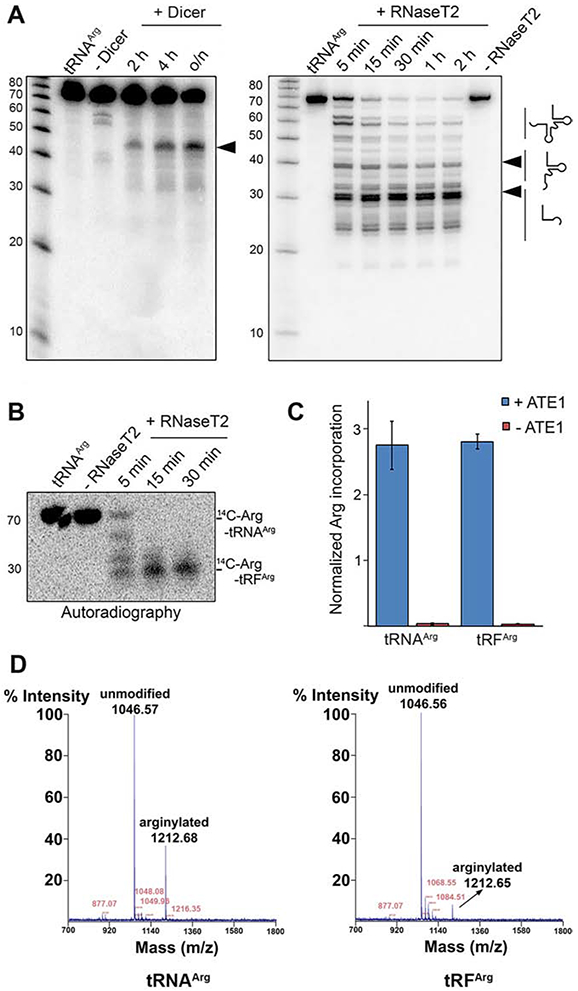
A, tRNA cleavage pattens by Dicer and RNase T2. The arrows show the cleavage products. B, Autoradiogram of 14C-Arg-tRNAArg cleaved with RNase T2. C, normalized incorporation of 3H-Arg into angiotensin II performed with 3H-Arg-tRNAArg or 3H-Arg-tRFArg. Data are mean ± SEM of three biological replicates. D. MALDI-TOF chromatogram of angiotensin II arginylated using purified pre-charged 13C15N-Arg-tRNAArg or 13C15N-Arg-tRFArg. Notably, the peak height/intensity in this method does not directly reflect the abundance of the peptide (Szajli et al., 2008).
Aminoacylated tRNA is normally labile, easily losing the charged amino acid under neutral pH, however the ester bond between amino acid and tRNA can be maintained under acidic conditions. Fortunately, RNase T2 has been shown to work better at acidic pH (Campomenosi et al., 2006), making it ideal for in vitro generation of charged tRFArg. To determine whether tRFArg generated by RNase T2 cleavage retain the aminoacyl moiety, we treated 14C-Arg-tRNAArg with RNase T2 under acidic conditions, and ran the products in an acidic denaturing gel to prevent hydrolysis of the arginyl group. While prior to cleavage, the 14C-Arg signal was detected only in the full length tRNA band on the gel, following cleavage with RNase T2 all the 14C-Arg signal associated with the lower molecular weight tRFArg band, without any detectable signal from the full-length 14C-Arg-tRNAArg, indicating that the aminoacyl moiety on the tRNA was retained in the tRF (Fig. 5B).
To test whether the RNase T2-generated Arg-tRFArg can mediate arginylation, we used this product in an in vitro arginylation reaction with ATE1, using angiotensin II as a substrate. Strikingly, Arg-tRFArg successfully mediated arginylation with the efficiency comparable to that of the full length Arg-tRNAArg (Fig. 5C). Arginine incorporation into the test peptide substrate after this reaction was also confirmed by MALDI-TOF mass spectrometry (Fig. 5D). Thus, Arg-tRFArg generated after RNase T2 cleavage of the full-length Arg-tRNAArg were able to mediate arginylation similarly to full-length tRNAArg.
ATE1 deletion alters the ratio of tRNAArg to tRFArg in cells.
Our finding that charged tRFArg can mediate arginylation in vitro points to a possibility that tRFArg generation in vivo may be functionally linked to arginylation. This hypothesis would predict that in cells lacking ATE1, there would be reduced generation of tRFArg, due to the lack of demand for tRFArg for arginylation. To test this possibility, we performed parallel tRNA- and tRF- sequencing in total RNA samples isolated from wild type and Ate1 knockout mouse embryonic fibroblasts and analyzed the composition and relative quantities of tRNA and tRF in each of these cell types. This analysis revealed several trends. First, while the level of tRNAArg was somewhat increased in Ate1 knockout cells, that of tRFArg was significantly decreased in these cells compared to wild type (Fig. 6A). In contrast to the change in the level of tRNAArg, the rest of tRNA species showed no change in abundance, with the exception of tRNAGly which was significantly decreased in Ate1 knockout cells (Dataset 1, Chart 1, bottom left and Chart 4). In the same samples, many tRF reads, in addition to tRFArg, were lower in Ate1 knockout compared to wild type (Dataset 1, Chart 1, bottom right and Chart 3), suggesting that Ate1 may be globally linked to tRF generation.
Figure 6. ATE1 deletion alters the ratio of tRNAArg to tRFArg in cells.
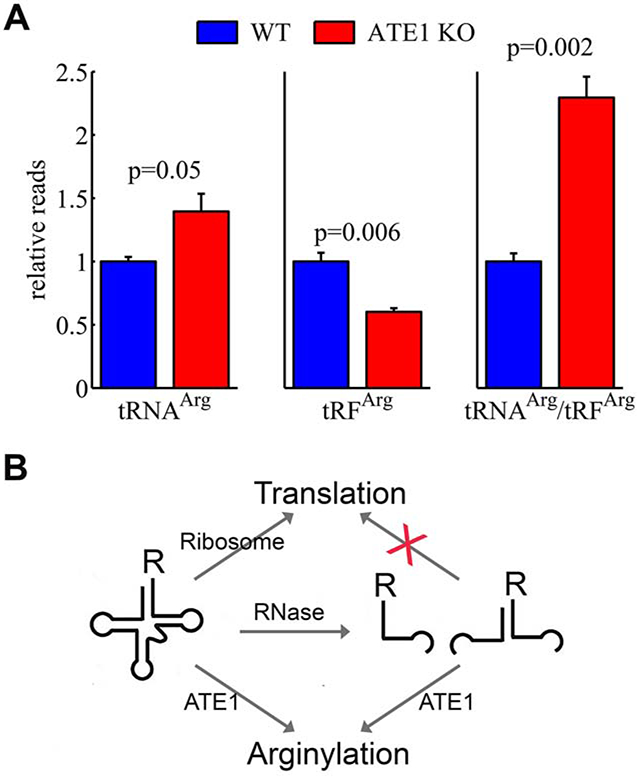
A. Ratios of tRNAArg/tRFArg in the total RNA preparations from wild type (WT) and ATE1 knockout (ATE1 KO) mouse embryonic fibroblasts, analyzed in 3 biological replicates. Error bars represent SEM, Welch’s one-tailed T-test was used for calculating statistical significance. B, Hypothetical model of the arginylation and translation dependent tRNAArg/tRFArg balance.
Analysis of individual tRNAArg and tRFArg reads (normalized to total mapped reads of nuclear tRNA) showed that both the increase in levels of tRNAArg and the corresponding decrease in tRFArg was not completely uniform (Dataset 1, Chart 1, top panels). Overall, these changes amounted to a nearly 2.5 fold statistically significant increase in tRNAArg/tRFArg ratio in Ate1 knockout cells compared to wild type (Fig. 6A, see also Supplemental Datasets 2-3). Some other tRNA/tRF ratios also showed changes, including those for amino acids with hydrophobic side chains (an increase) and Ser (a decrease) (Dataset 1, Chart 2), but none of these changes were nearly as pronounced as those for tRNAArg/tRFArg.
Discussion
Our study constitutes the first analysis of tRNA specificity for arginylation. We find that, in addition to numerous tRNAArg isoacceptors, ATE1 is also able to efficiently utilize the translation-incompetent Arg-charged 3’-tRFArg for Arg transfer. Strikingly, the fraction of tRFArg in cells is reduced in the absence of ATE1, accompanied by an increase in tRNAArg/ tRFArg ratio. This effect is likely the result of two factors – a specific relation between ATE1 and tRNAsArg, as well as an overall global decrease in tRF production in the Ate1 knockout, which suggests a potential functional interaction between ATE1 and the tRF-generating enzyme in cells (Fig. 6B). These findings have potentially global implications for the overall role of intracellular tRFs in balancing translation with arginylation and other pathways that involve the use of amino acids.
Despite numerous insights into ATE1’s important physiological role, relatively little is known about the structure of this enzyme and the mechanisms of its action. Our work shows for the first time that ATE1 can bind tRNA. This binding in principle should be able to aid ATE1 in sequestering some tRNA species of the pool to ensure that they are successfully protected from the activity of the protein synthesis machinery. While ATE1 binding to tRNA appears to lack strict specificity, the arginylation reaction is specific to Arg-tRNAArg and does not occur if the same tRNA is charged with Lys or Ctr. Thus, it is not the charge, or the side chain structure, but a combination of both that appears to drive the specificity of ATE1 to Arg.
While ATE1 structure is unknown, the closest related enzyme in bacteria, L/F transferase from E. coli, has been shown by structural and mutagenesis studies to contain a stretch of positively charged residues that facilitate its interaction with tRNA (Suto et al., 2006). Guided by this information, we searched ATE1 protein sequence for stretches of evolutionarily conserved positively charged amino acid residues that could potentially facilitate the interaction with the negatively charged RNA. This analysis revealed several sequence stretches rich in positively charged K/R (Dataset 4). Remarkably, our previous study showed that a mutation of K417 within one of these stretches abolishes ATE1 activity in yeast complementation assays (Rai et al., 2006), indicating a role in ATE1-mediated catalysis. Our present study suggests that, rather than inhibiting ATE1’s catalytic center, as previously suggested, this mutation may actually interfere with its binding to tRNA.
In addition to tRNA, ATE1 pulldowns also contain a prominent fraction of small ribosomal RNAs (5S and 5.8S). While these RNAs are not as abundant in the pulldown as the tRNA fraction, their binding to ATE1 appears to be at least partially specific, potentially due to their tendency to form stable hairpins and loops that lead to structural resemblance of tRNAs (Barciszewska et al., 1996). Since ATE1 has been previously found to be partially associated with the ribosomes, this binding may enable tethering a fraction of ATE1 to the ribosomes to facilitate ATE1’s proposed co-translational activity, as well as, potentially, its previously demonstrated role in regulating mRNA stability in response to heat shock (Deka et al., 2016). These possibilities require further investigation.
Our data raise the possibility that, while different tRNAArg isodecoders can all participate in arginylation, they show individual differences in their efficiency toward the angiotensin II substrate, ranging around 20-40% overall. It is difficult to assess whether these differences are sufficient to amount to any tRNA-dependent regulation of arginylation in the context of the cellular environment, where high local concentrations of tRNA in the proximity of the ribosomes would likely negate any effects related to this relatively small change. A prominent exception to this is tRNAArg #20, which shows charging efficiency around 20% in vitro or less compared to others, but mediates arginylation similarly to the rest. Potentially, the charged fraction of this tRNA could be well over 5-fold more efficient in cells, enabling it to play a role in facilitating or inhibiting arginylation.
Our study shows that ATE1 can utilize the Arg-charged to the tRNA acceptor stem as the amino acid donor in the arginylation reaction. Notably, previous studies show the importance of the acceptor stem for the recognition of L/F transferase, the closest analog of ATE1 in bacteira that uses aminoacylated tRNAs to transfer Leu and Phe to protein substrates in bacterial cells (Fung et al., 2014). Thus, our result uncovers yet another parallel between ATE1 and L/F transferase, the two enzymes that share very little sequence homology but utilize a similar mechanism of tRNA-dependent protein modifications.
Our finding that Arg-tRFArg can remain charged with Arg after cleavage from Arg-tRNAArg, is actually not unprecedented. A previous study has identified a case where the charged amino acid to tRNAAsp is retained after the tRNA is cleaved to a tRF, and that this aminoacyl-tRF has been proposed to play a role in cell proliferation and cancer (Honda et al., 2015). tRFArg are virtually unexplored at the molecular level, but are reported to be abundant in some human tissues, suggesting that they may play selective roles in different cell types (Torres et al., 2019). It is possible that aminoacyl-tRFs are more common than we currently realize, and that they carry currently unknown functions in different intracellular pathways that involve transfer of aminoacyl groups and and the activity of aa-tRNAs.
Our results suggest an interesting molecular link between ATE1 and the intracellular tRF-generating machinery. At present, the key enzymes involved in the intracellular generation of short tRFs are not fully known. Dicer has been originally believed to be the main player (reviewed in (Shen et al., 2018)), however recent reports also show that tRFs can be produced in the absence of Dicer (see, e.g. (Kuscu et al., 2018; Shen et al., 2018)). A more recent study in Arabidopsis thaliana implicates RNasesT2, rather than Dicer, in tRF generation (Megel et al., 2019). A potential complication in this mechanism relates to the fact that RNase T2 has been characterized as a secreted enzyme that is found in the ER, lysosomes, and extracellular space. It is possible that RNase T2 also exists in the cytosol, but if not, this compartmentalization would present a spacial constraint if this enzyme’s major activity were to be associated with the purely cytosolic tRFs. Recent evidence revealed RNase T2 presence in mitochondria (Huang et al., 2018; Liu et al., 2017) and stress granules (Vidalino et al., 2012), suggesting that its subcellular distribution is potentially broader than previously known. Different studies implicate this enzyme in the processing of cytosolic RNAs (Andersen and Collins, 2012; Megel et al., 2019; Thompson and Parker, 2009), and propose that this enzyme can exist in two forms, a glycosylated secreted form (distinguishable by a larger molecular weight), and a non-glycosylated intracellular form (Campomenosi et al., 2006). Overall, these questions require further investigation.
RNase T2 acts better at acidic pH (Thorn et al., 2012) and has been previously associated with lysosomal degradation (Fujiwara et al., 2017). At the same time, ATE1 has been long known to play a role in protein degradation (Varshavsky, 1992) and has been recently shown to be involved in autophagy and global regulation of protein flux between different degradation pathways (Jiang et al., 2016). It seems possible that in these pathways, a global increase in ATE1 activity may be linked to activation of RNase T2 during cellular stress and global degradation-dependent cellular rearrangements that ultimately lead to lysosomal and autophagy dependent protein breakdown. However, it is also possible that in cells ATE1 actually utilizes the tRFArg produced by another, as yet unknown enzyme.
tRFs are a recently discovered class of small non-coding RNAs that have been previously believed to constitute the products of tRNA decay, but have been recently demonstrated to play independent regulatory roles in multiple physiological pathways. Our study suggests that tRFArg generation in vivo may constitute a potential mechanism to regulate the flux of tRNAs charged with Arg between translation and arginylation (Fig. 6b, S6). Based on this possibility, we propose a role for tRFArg as molecules involved in arginylation, and potentially regulating the balance between intracellular protein synthesis and translation-independent utilization of amino acids.
Significance
Our study constitutes the first analysis of tRNA specificity for arginylation. We find that, in addition to numerous tRNAArg isoacceptors, ATE1 is also able to efficiently utilize the translation-incompetent Arg-charged 3’-tRFArg for Arg transfer. Strikingly, the fraction of tRFArg in cells is reduced in the absence of ATE1, accompanied by an increase in tRNAArg/ tRFArg ratio. This effect is likely the result of two factors – a specific relation between ATE1 and tRNAArg, as well as an overall global decrease in tRF production in the Ate1 knockout, which suggests a potential functional interaction between ATE1 and the tRF-generating enzyme in cells. These findings have potentially global implications for the overall role of intracellular tRFs in balancing translation with arginylation and other pathways that involve the use of amino acids.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Anna Kashina (akashina@upenn.edu).
Materials Availability
This study did not generate new unique reagents.
Data Availability
Original/source data for Fig. 6A and supplemental datasets 1-3 in the paper is available in the GEO database (Accession # GSE146344). Original/source data for Fig. 2B and S2 in the paper is available in Supplemental Dataset 5.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Growth Conditions
The cell lines used for tRNA- and tRF-Seq were wild type and ATE−/− immortalized mouse embryonic fibroblasts (Karakozova et al., 2006). The sex of both cell lines is female. Cells were maintained at 37°C in a humidified atmosphere at 5% CO2 in DMEM high glucose with Glutamax media supplemented with 10% of FBS and 1% of penicillin/streptomycin.
HEK293 cells were used for RNase T2 purification and cultivated as a suspension culture. While we did not determine the sex of HEK293 in this study, this cell line is parental to the HEK293T cells, which are derived from HEK293 by transfecting the large T antigen, and are female (https://www.atcc.org/products/all/CRL-11268G-1.aspx). Therefore, we assume that the sex of parental HEK293 cells used in this study is also female. Cells were maintained at 37°C in a humidified atmosphere at 5% CO2 in 293 SFM II media supplemented with 2% of Glutamax.
Bacterial Cells and Growth Conditions
The bacterial cell used in this study is BL21-CodonPlus (DE3)-RIL competent cells. The cells were transformed with pET29a-ATE1 construct according to manufacturer’s protocol and grown in LB supplemented with 100 μg/ml kanamycin and 50 μg/ml chloramphenicol at 37°C at 200 rpm.
METHOD DETAILS
RNA cleavage assay and Northern blotting
Mouse tRNAArg ACG 1-1 was in vitro transcribed by MEGAshortscipt T7 Transcription Kit according to manufacturer’s protocol. Total RNA was extracted from pulverized mouse liver with TRIzol according to manufacturer’s instruction. For Dicer cleavage experiments, in vitro transcribed mouse tRNAArg ACG 1-1 or total RNA was treated with commercially available Dicer enzyme according to the manufacturer’s instructions. For RNase T2 cleavage, 4 μM of mouse tRNAArg ACG 1-1 was treated with 0.24 ng/μl of human RNase T2 (purified as below) in 200 mM NaOAc pH 4.5 and 50 mM NaCl at 37°C. The RNA samples were directly mixed with 2xRNA loading dye and were fractionated in 15% urea polyacrylamide gel and electroblotted onto a nylon membrane (GE Healthcare, Amersham Hybond-N+). Hybridization was performed with a 5’-32P labeled DNA probe (see Key Resources Table the probe sequences). The autoradiography was visualized by phosphorimager (Molecular Dynamics, Storm 860).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Cell Strains | ||
| BL21-CodonPlus (DE3)-RIL competent cells | Stratagene | Cat# 230245 |
| Chemicals, and Recombinant Proteins | ||
| L-[2,3,4-3H]-Arg | PerkinElmer | Cat# NET1123001MC |
| L-[14C(U)]-Arg | Moravek Biochemicals | Cat# MC137 |
| L-[13C, 15N]-Arg | Pierce | Cat# 89990 |
| L-[2,3,4,5-3H]-Pro | PerkinElmer | Cat# NET483250UC |
| L-[4,5-3H(N)]-Lys | PerkinElmer | Cat# NET376250UC |
| Aminoacyl-tRNA synthetase mix | Sigma | Cat# A3646 |
| Recombinant Dicer enzyme | Genlantis | Cat# T510002 |
| ZR small-RNA PAGE recovery kit | Zymo Research | Cat# R1070 |
| C18 spin column | The Nest Group, Inc | Cat# HUM S18V |
| Ni-NTA agarose beads | Qiagen | Cat# 30210 |
| TRIzol | Ambion | Cat# 15596026 |
| T4 polynucleotide kinase | New England BioLabs | Cat# M0201 |
| Calf intestinal phosphatase | New England BioLabs | Cat# M0525 |
| MEGAshortscript Kit | Invitrogen | Cat# AM1354 |
| AutoFluor | National Diagnostics | Cat# LS-315 |
| Critical Commercial Assays | ||
| NEBNext Multiplex Small RNA Library Prep Set for Illumina | New England BioLabs | Cat# 7300 Cat# 7580 |
| NextSeq 500/550 V2 Kit | Illumina | Cat# (#FC-404-2005) |
| Deposited Data | ||
| Raw data for the tRNA- and tRF-seq | GEO database | Accession # GSE146344 |
| Experimental Models: Cell Lines | ||
| Wild type MEF cells | Karakozova et al., 2006 | N/A |
| Ate1−/− cells | Karakozova et al., 2006 | N/A |
| HEK293 cells | Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures GmbH | N/A |
| Oligonucleotides | ||
| For tRNAArgACG: 5’-TGGCGAGCCAGCCAGGAGTCGA-3’ | This paper | N/A |
| For tRNAArgTCT: 5’-TGGCGACTCTGGTGGGACTCGA-3’ | This paper | N/A |
| For tRNAArgCCG-TCG: 5’-TGGCGACCACGAAGGGACTCGA-3’ | This paper | N/A |
| tRNAArgCCT: 5’- TGGTACCCCAGGTGGGACTCGA | This paper | N/A |
| Recombinant DNA | ||
| pET29a-ATE1 plasmid | Wang et al., 2011 | N/A |
| Software and Algorithms | ||
| Clustal W | Kyoto University Bioinformatics Center Clustal W | https://www.genome.jp/tools-bin/clustalw |
| FastQC | FastQC | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR | Dobin et al., 2013 | http://code.google.com/p/rna-star/ |
| BioRender | BioRender | https://biorender.com |
| MATLAB | MathWorks | https://www.mathworks.com |
| Other | ||
| HisTrap FF crude purification column | Cytiva | Cat# 17528601 |
To test the conjugation of L-[14C(U)]-Arg to mouse tRNAArg ACG after RNAse T2 treatment, we used 8% acidic denaturing gel (8% polyacrylamide, 7.5 M urea, 0.1 M NaOAc/AcOH pH 5.0) (Avcilar-Kucukgoze et al., 2016). Electrophoresis at 100-150 V (~12 V/cm) was performed in a cold room. The gel was treated with AutoFluor according to manufacturer’s instructions and dried at 80°C for 2 h (Hoefer Slab Gel Dryer, Model GD 2000).
tRNA charging and arginyl-tRF generation
The charging reaction was performed by mixing 8 μM of in vitro transcribed tRNAArgACG 1-1, 1.5 μM RARS, 3.3 mM ATP, 15 μM L-[2,3,4-3H]-Arg (L-[14C(U)]-Arg for autoradiography or [13C, 15N]-labeled Arg for mass spectrometry), 50 mM HEPES, 25 mM KCl, 15 mM MgCl2, 0.1 mM DTT, pH 7.5. After 2 h incubation at 37°C, the reaction was extracted with acid phenol:chloroform (5:1) and precipitated with the same volume of isopropanol. The pellet was washed with 70% ethanol 2-3 times anddissolved into 10 mM NaOAc, 1 mM EDTA pH 4.8 to preserve tRNA charging.
For in vitro tRF generation, 4 μM of Arg-tRNAArg were treated with human RNase T2 as described above. After acid phenol:chloroform (5:1) extraction, Arg-tRFArg were precipitated with the same volume of isopropanol. In the meantime, the same amount of Arg-tRNAArg were extracted with phenol:chloroform (5:1) and precipitated with the same volume of isopropanol, for use as a positive control in the arginylation assay. The cpm values were measured for both Arg-tRNAArg and Arg-tRFArg samples before their use in arginylation assays.
In vitro arginylation assay
Arginylation assay was performed as described in (Wang et al., 2011; Wang et al., 2014). For all experiments except the assays shown in Fig. S1, a typical reaction was performed in 50 μl volume, containing 5 μM Arg-tRNAArg (or Arg-tRFArg), 15 μM the peptide substrate (angiotensin II), 3 μM ATE1, 50 mM HEPES, 25 mM KCl, 15 mM MgCl2, 0.1 mM DTT, pH 7.5. For the experiments shown in Fig. S3 and S4, the arginylation reaction was performed in 50 μl volume, containing 5 μM tRNAArg (or total tRNA), 15 μM substrate (angiotensin II or α-synuclein), 0.5 μM ATE1, 5 μM L-[2,3,4-3H]-Arg (L-[2,3,4,5-3H]-Pro or L-[4,5-3H(N)]-Lys), 2 mM ATP, 1 μM RARS (or 250 units of aminoacyl-tRNA synthetase mix), 50 mM HEPES, 25 mM KCl, 15 mM MgCl2, 0.1 mM DTT, pH 7.5. Total tRNA was extracted by ZR small-RNA PAGE recovery kit. After mixing, the reaction was incubated at 37°C for 5 min and then heated at 95°C for 15 min. Angiotensin II peptide was purified using C18 spin columns and analyzed on a scintillation counter (Beckman Coulter LS 6500) to measure Arg incorporation. For the tRNA/tRF assay shown in Fig. 5C the cpm count from the arginylated peptide in each sample was divided by the cpm counts measured in an aliquot of purified Arg-tRNAArg (or Arg-tRFArg) added to the reaction. For the assay shown in Fig. S4C, TCA precipitation of α-synuclein was performed as described in (Wang et al., 2017a).
For the protein-based assay shown in Figure S1, bovine serum albumin (BSA) was used as a substrate and the assay and measurements were performed as described in (Wang et al., 2011) using total E.coli tRNA. A typical reaction was performed in 50 μl volume, containing 50 mM HEPES, pH 7.5, 25 mM KCl, 15 mM MgCl2, 0.1 mM DTT, 2.5 mM ATP, 12.5 μM [3H]-Arginine, 40 μM tRNAArg, 2 μg RARS, 1 μg ATE1 and 8.3 μM BSA. The reaction was incubated at 37°C, and a 10 μl aliquot was taken out at each time point. For scintillation counting, samples at each time point were immediately quenched into 40 μl of 20% Trichloroacetic acid (TCA) containing 1 mM of unlabeled Arg, followed by heating at 95°C for 15 min, cooling down on ice for 20 min, and spinning at 13,000 rpm for 30 min at room temperature. Pellets were washed 3x with 5% cold TCA and once with cold acetone, air dried, and counted in a liquid scintillation counter.
MALDI-TOF mass spectrometry analysis was performed at the Wistar Proteomics Facility, University of Pennsylvania. All data were collected on an ABI/PerSeptive (Framingham, MA) Voyager DE-PRO MALDI-TOF instrument in positive-ion mode.
RNase T2 purification
C-terminal His-tagged human RNase T2 construct was transfected to HEK293 cells (Thorn et al., 2012). RNase T2 was purified from the cell culture supernatant (100 ml total volume). The medium was cleared by centrifugation at 3000 x g and 4°C for 60 min and then filtered with a 0.2 μm pore membrane. After adding 20 mM K2HPO4, pH 7.5, 0.5 M NaCl and 40 mM imidazole, the crude solution was loaded on a HisTrap FF purification column. Bound RNase T2 was eluted with an imidazole gradient and was typically released at 70-90 mM imidazole (Thorn et al., 2012).
ATE1 pull-down assay
His-tagged ATE1 was overexpressed in E. coli as described in (Wang et al., 2011). Bacteria harboring pET29a-ATE1 were grown in LB medium supplemented with 100 μg/ml kanamycin and 50 μg/ml chloramphenicol until OD600 reached 0.4-0.5. The culture was cold shocked on ice for 30 min, then induced with 0.4 mM IPTG for 18 h at 16°C. Cells were collected by centrifuging at 5,000 x g for 30 min and resuspended in lysis buffer 500 mM NaCl, 1 mM MgCl2, 50 mM Tris, 10 mM β-mercaptoethanol, 5 mM imidazole, 1 mM PMSF, pH 7.5. The cells were sonicated at level 5, 6x10s with 1 min interval (Fisher Scientific, Model 550 Sonic Dismembrator). The lysate was centrifuged at 30,000 x g for 30 min at 4°C and supernatant was loaded to the Ni-NTA agarose column equilibrated with lysis buffer, followed by washing in 1 M NaCl, 1 mM MgCl2, 50 mM Tris, 10 mM β-mercaptoethanol, 25 mM imidazole, pH 7.5. 100 μl of Ni-NTA beads with immobilized ATE1 were treated with 300 μg of total RNA extracted from mouse liver. The ATE1-bound beads and RNA mixture was gently rotated at room temperature for 1 h in 50 mM NaCl, 1 mM MgCl2, 50 mM Tris, 10 mM β-mercaptoethanol, 25 mM imidazole, pH 7.5. The mixture was centrifuged at 1200 x g for 1 min and the supernatant was collected as “flow-through”. The ATE1-bound beads were washed with the same buffer for 4-5 times and RNA was extracted with phenol/chloroform (5:1). After precipitation, 1 μg of eluate and flow-through were analyzed on 1% agarose gel. As negative controls, free beads and truncated gelsolin protein-bound beads have been treated as described above.
tRNA- and tRF-Sequencing
The tRNA- and tRF-Seq were performed by Arraystar Inc. using total TRIzol lysates from cultured wild type and Ate1 knockout mouse embryonic fibroblasts. Three biological replicates were analyzed for each condition.
Library preparation:
Agarose electrophoresis was used to check the integrity of total RNA samples, and NanoDrop ND-1000 instrument was used to measure the concentration of the samples. NEBNext® Multiplex Small RNA Library Prep Set for Illumina® reagents were used in library preparation. tRNAs were purified from total RNA samples and pretreated according to Hydro-tRNAseq method: a) over 2 μg total RNA was resolved on urea-polyacrylamide gels and recovered within a size window of 60-100 nt for tRNA; b) purified tRNA was subjected to limited alkaline hydrolysis by carbonate buffer; c) the partial hydrolyzed tRNA was dephosphorylated with calf intestinal phosphatase (CIP) and then re-phosphorylated with T4 polynucleotide kinase (T4 PNK). Partially hydrolyzed and re-phosphorylated tRNAs of each sample was used to construct Hydro-tRNAseq library. Library preparation procedures included: 1) 3'-adapter ligation; 2) 5'-adapter ligation; 3) cDNA synthesis; 4) PCR amplification; 5) size selection of ~140-155 bp PCR amplified fragments (corresponding to ~19-35 nt tRNA fragments). tRF were extracted from total RNA samples and pretreated as following to remove some RNA modifications that interfere with small RNA-seq library construction: 3-aminoacyl (charged) deacylation to 3-OH for 3 adaptor ligation, 3-cP (2,3-cyclic phosphate) removal to 3-OH for 3 adaptor ligation, 5-OH (hydroxyl group) phosphorylation to 5-P for 5-adaptor ligation, m1A and m3C demethylation for efficient reverse transcription. Pretreated total RNA of each sample was taken for tRF & tiRNA-seq library preparation. Library preparation procedures included: 1) 3'-adapter ligation; 2) 5'-adapter ligation; 3) cDNA synthesis; 4) PCR amplification; 5) size selection of ~135-160 bp PCR amplified fragments (corresponding to ~15-40 nt small RNAs).
The completed libraries were quantified by Agilent 2100 Bioanalyzer. Libraries were mixed in equal amounts according to the quantification results, and used for sequencing on the instrument.
Sequencing:
The DNA fragments in well mixed libraries were denatured with 0.1M NaOH to generate single-stranded DNA molecules, and loaded onto the reagent cartridge at 1.8 pM concentration. For tRNA, the sequencing run was performed on Illumina NextSeq 500 system and carried out by running 75 cycles. For tRF, the sequencing run was performed on Illumina NextSeq system using NextSeq 500/550 V2 kit and carried out by running 50 cycles.
tRNA-based microarray
tRNAs bound to ATE1 were determined by tRNA-tailored microarrays as described earlier (Beckert et al., 2018). Briefly, RNA was isolated with TRIzol which alkaline pH simultaneously deacylates all tRNAs and the intact 3’ NCCA ends were hybridized with Cy3-labeled RNA/DNA stem-loop oligonucleotide. In parallel, tfhe total tRNA from mouse liver was extracted by TRIzol and labeled with Atto647-labeled RNA/DNA stem-loop oligonucleotide. Both samples were hybridized on arrays containing the full-length mouse tDNAs (Dittmar et al., 2006). The arrays were normalized to spike-in standards (Kirchner et al., 2017), processed and quantified with in-house python scripts. See (https://www.protocols.io/view/microarray-based-quantification-of-cellular-trnas-hfcb3iw) for the detailed protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Analysis of tRNA- and tRF-Seq
Sequencing quality was examined by FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed reads (pass Illumina quality filter, trimmed 3-adaptor bases by cutadapt (Martin, 2011)) were aligned to cytoplasmic mature-tRNA sequence getting from GtRNAdb and also including the mitochondrial tRNA sequences from mitotRNAdb using STAR (Dobin et al., 2013) software. The transcript abundances for each sample was estimated with unique matched reads and the count value for tRNAs level were calculated using MATLAB software (MathWorks, Natick, MA, USA). The differentially expressed tRNAs were screened based on the count value using the R (3.2.1) and the MATLAB routines developed in our lab (Dong et al., 2015; Rai et al., 2016).
Supplementary Material
Supplemental Table 1, related to Fig. 3 and S4. Sequences and gene names of mouse tRNAArg, numbered according to the nomenclature in the main text and supplemental figures.
Supplemental Dataset 5, related to Fig. 2. Raw data for the microarray experiment shown in Fig. 2 of the main text.
Highlights.
ATE1-mediated arginylation is highly specific toward arginyl-tRNAArg.
ATE1 selectively binds to tRNA and can utilize all mouse tRNAArg species.
tRNAArg-derived fragments can mediate arginylation.
ATE1 deletion alters the ratio of tRNAArg to tRFArg in cells.
Acknowledgements.
We thank Dr. Junling Wang for his technical contributions to this paper, including the experiments shown in Figures 1, 3, 4, S1, and S4, as well as for critical reading and valuable comments on the manuscript. We also thank members of Kashina’s lab for helpful discussions and Arraystar, Inc. for the tRNA/tRF sequencing. This work was supported by NIH R35GM122505 and R01NS102435 to A.K., R01GM126210, AI139202, and HG010824 to Y-M.H., FOR1805 (Deutsche Forschungsgemeinschaft, DFG) to Z.I., Research Funding of the Lower Saxony Ministry for Science and Culture (MWK) ZN 2938 to R. K., and R21DA047936 and P20GM109091 to M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing interests.
References
- Andersen KL, and Collins K (2012). Several RNase T2 enzymes function in induced tRNA and rRNA turnover in the ciliate Tetrahymena. Molecular biology of the cell 23, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, and Ivanov P (2014). tRNA fragments in human health and disease. FEBS letters 588, 4297–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barciszewska MZ, Erdmann VA, and Barciszewski J (1996). Ribosomal 5S RNA: tertiary structure and interactions with proteins. Biological reviews of the Cambridge Philosophical Society 71, 1–25. [DOI] [PubMed] [Google Scholar]
- Batsios P, Ishikawa-Ankerhold HC, Roth H, Schleicher M, Wong CCL, and Muller-Taubenberger A (2019). Ate1-mediated posttranslational arginylation affects substrate adhesion and cell migration in Dictyostelium discoideum. Molecular biology of the cell 30, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckert B, Turk M, Czech A, Berninghausen O, Beckmann R, Ignatova Z, Plitzko JM, and Wilson DN (2018). Structure of a hibernating 100S ribosome reveals an inactive conformation of the ribosomal protein S1. Nature microbiology 3, 1115–1121. [DOI] [PubMed] [Google Scholar]
- Campomenosi P, Salis S, Lindqvist C, Mariani D, Nordstrom T, Acquati F, and Taramelli R (2006). Characterization of RNASET2, the first human member of the Rh/T2/S family of glycoproteins. Arch Biochem Biophys 449, 17–26. [DOI] [PubMed] [Google Scholar]
- Carpio MA, Decca MB, Lopez Sambrooks C, Durand ES, Montich GG, and Hallak ME (2013). Calreticulin-dimerization induced by post-translational arginylation is critical for stress granules scaffolding. The international journal of biochemistry & cell biology 45, 1223–1235. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Ferber S, Ganoth D, Elias S, Hershko A, and Arfin S (1988). Purification and characterization of arginyl-tRNA-protein transferase from rabbit reticulocytes. Its involvement in post-translational modification and degradation of acidic NH2 termini substrates of the ubiquitin pathway. The Journal of biological chemistry 263, 11155–11167. [PubMed] [Google Scholar]
- Cirakoglu B, and Waller JP (1985). Multiple forms of arginyl- and lysyl-tRNA synthetases in rat liver: a re-evaluation. Biochimica et biophysica acta 829, 173–179. [DOI] [PubMed] [Google Scholar]
- Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, and Hutvagner G (2009). Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 15, 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka K, Singh A, Chakraborty S, Mukhopadhyay R, and Saha S (2016). Protein arginylation regulates cellular stress response by stabilizing HSP70 and HSP40 transcripts. Cell death discovery 2, 16074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher MP, and Ni RC (1982). Purification of a low molecular weight form of rat liver arginyl-tRNA synthetase. The Journal of biological chemistry 257, 6003–6006. [PubMed] [Google Scholar]
- Dissmeyer N (2019). Conditional Protein Function via N-Degron Pathway-Mediated Proteostasis in Stress Physiology. Annual review of plant biology 70, 83–117. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Goodenbour JM, and Pan T (2006). Tissue-specific differences in human transfer RNA expression. PLoS genetics 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong DW, Srinivasan S, Guha M, and Avadhani NG (2015). Defects in cytochrome c oxidase expression induce a metabolic shift to glycolysis and carcinogenesis. Genomics data 6, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldeeb MA, Fahlman RP, Esmaili M, and Ragheb MA (2018). Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells. International journal of molecular sciences 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu GF, and Anderson P (2010). Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. The Journal of biological chemistry 285, 10959–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Lee I, Lee YS, and Bao X (2015). Small Non-coding Transfer RNA-Derived RNA Fragments (tRFs): Their Biogenesis, Function and Implication in Human Diseases. Genomics & informatics 13, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Wada K, and Kabuta T (2017). Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. Journal of biochemistry 161, 145–154. [DOI] [PubMed] [Google Scholar]
- Fung AW, Leung CC, and Fahlman RP (2014). The determination of tRNALeu recognition nucleotides for Escherichia coli L/F transferase. RNA 20, 1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MR, Goitea VE, and Hallak ME (2016). Post-translational protein arginylation in the normal nervous system and in neurodegeneration. Journal of neurochemistry 138, 506–517. [DOI] [PubMed] [Google Scholar]
- Graciet E, Walter F, O'Maoileidigh DS, Pollmann S, Meyerowitz EM, Varshavsky A, and Wellmer F (2009). The N-end rule pathway controls multiple functions during Arabidopsis shoot and leaf development. Proceedings of the National Academy of Sciences of the United States of America 106, 13618–13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann CS, and Hou YM (1995). Enzymatic aminoacylation of tRNA acceptor stem helices with cysteine is dependent on a single nucleotide. Biochemistry 34, 6527–6532. [DOI] [PubMed] [Google Scholar]
- Honda S, Loher P, Shigematsu M, Palazzo JP, Suzuki R, Imoto I, Rigoutsos I, and Kirino Y (2015). Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proceedings of the National Academy of Sciences of the United States of America 112, E3816–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu P, and Wang G (2018). Regulation of mitochondrion-associated cytosolic ribosomes by mammalian mitochondrial ribonuclease T2 (RNASET2). The Journal of biological chemistry 293, 19633–19644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, and Ackerman SL (2014). RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji CH, Kim HY, Heo AJ, Lee SH, Lee MJ, Kim SB, Srinivasrao G, Mun SR, Cha-Molstad H, Ciechanover A, et al. (2019). The N-Degron Pathway Mediates ER-phagy. Molecular cell 75, 1058–1072 e1059. [DOI] [PubMed] [Google Scholar]
- Ji CH, and Kwon YT (2017). Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Molecules and cells 40, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee J, Lee JH, Lee JW, Kim JH, Choi WH, Yoo YD, Cha-Molstad H, Kim BY, Kwon YT, et al. (2016). The arginylation branch of the N-end rule pathway positively regulates cellular autophagic flux and clearance of proteotoxic proteins. Autophagy 12, 2197–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, and Putz J (2009). tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic acids research 37, D159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR 3rd, Mogilner A, Zebroski H, and Kashina A (2006). Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science 313, 192–196. [DOI] [PubMed] [Google Scholar]
- Kashina AS (2015). Protein Arginylation: Over 50 Years of Discovery. Methods Mol Biol 1337, 1–11. [DOI] [PubMed] [Google Scholar]
- Kashina AS, and Yates JR 3rd (2015). Identification of Arginylated Proteins by Mass Spectrometry. Methods Mol Biol 1337, 93–104. [DOI] [PubMed] [Google Scholar]
- Kirchner S, Cai Z, Rauscher R, Kastelic N, Anding M, Czech A, Kleizen B, Ostedgaard LS, Braakman I, Sheppard DN, et al. (2017). Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS biology 15, e2000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Kumar P, Kiran M, Su Z, Malik A, and Dutta A (2018). tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 24, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, and Varshavsky A (2002). An essential role of N-terminal arginylation in cardiovascular development. Science 297, 96–99. [DOI] [PubMed] [Google Scholar]
- Kyriacou SV, and Deutscher MP (2008). An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Molecular cell 29, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xu Z, and Sheng J (2018). tRNA-Derived Small RNA: A Novel Regulatory Small Non-Coding RNA. Genes 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Huang J, Zheng Q, Xie L, Lu X, Jin J, and Wang G (2017). Mammalian mitochondrial RNAs are degraded in the mitochondrial intermembrane space by RNASET2. Protein & cell 8, 735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Sambrooks C, Carpio MA, and Hallak ME (2012). Arginylated calreticulin at plasma membrane increases susceptibility of cells to apoptosis. The Journal of biological chemistry 287, 22043–22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Megel C, Hummel G, Lalande S, Ubrig E, Cognat V, Morelle G, Salinas-Giege T, Duchene AM, and Marechal-Drouard L (2019). Plant RNases T2, but not Dicer-like proteins, are major players of tRNA-derived fragments biogenesis. Nucleic acids research 47, 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Ohta A, Ashigai H, and Suga H (2006). A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nature methods 3, 357–359. [DOI] [PubMed] [Google Scholar]
- Rai R, Mushegian A, Makarova K, and Kashina A (2006). Molecular dissection of arginyltransferases guided by similarity to bacterial peptidoglycan synthases. EMBO Rep 7, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Zhang F, Colavita K, Leu NA, Kurosaka S, Kumar A, Birnbaum MD, Gyorffy B, Dong DW, Shtutman Mv et al. (2016). Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene 35, 4058–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, and Kashina A (2011). Posttranslational arginylation as a global biological regulator. Developmental biology 358, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yu X, Zhu L, Li T, Yan Z, and Guo J (2018). Transfer RNA-derived fragments and tRNA halves: biogenesis, biological functions and their roles in diseases. J Mol Med (Berl) 96, 1167–1176. [DOI] [PubMed] [Google Scholar]
- Sivaram P, and Deutscher MP (1990). Existence of two forms of rat liver arginyl-tRNA synthetase suggests channeling of aminoacyl-tRNA for protein synthesis. Proceedings of the National Academy of Sciences of the United States of America 87, 3665–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A, and Hutvagner G (2011). Transfer RNA-derived fragments: origins, processing, and functions. Wiley interdisciplinary reviews RNA 2, 853–862. [DOI] [PubMed] [Google Scholar]
- Suto K, Shimizu Y, Watanabe K, Ueda T, Fukai S, Nureki O, and Tomita K (2006). Crystal structures of leucyl/phenylalanyl-tRNA-protein transferase and its complex with an aminoacyl-tRNA analog. The EMBO journal 25, 5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajli E, Feher T, and Medzihradszky KF (2008). Investigating the quantitative nature of MALDI-TOF MS. Molecular & cellular proteomics : MCP 7, 2410–2418. [DOI] [PubMed] [Google Scholar]
- Thompson DM, and Parker R (2009). The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. The Journal of cell biology 185, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn A, Steinfeld R, Ziegenbein M, Grapp M, Hsiao HH, Urlaub H, Sheldrick GM, Gartner J, and Kratzner R (2012). Structure and activity of the only human RNase T2. Nucleic acids research 40, 8733–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Reina O, Stephan-Otto Attolini C, and Ribas de Pouplana L (2019). Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proceedings of the National Academy of Sciences of the United States of America 116, 8451–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (1992). The N-end rule. Cell 69, 725–735. [DOI] [PubMed] [Google Scholar]
- Vellekamp G, Sihag RK, and Deutscher MP (1985). Comparison of the complexed and free forms of rat liver arginyl-tRNA synthetase and origin of the free form. The Journal of biological chemistry 260, 9843–9847. [PubMed] [Google Scholar]
- Vidalino L, Monti L, Haase A, Moro A, Acquati F, Taramelli R, and Macchi P (2012). Intracellular trafficking of RNASET2, a novel component of P-bodies. Biology of the cell / under the auspices of the European Cell Biology Organization 104, 13–21. [DOI] [PubMed] [Google Scholar]
- Wang J, Han X, Leu NA, Sterling S, Kurosaka S, Fina M, Lee VM, Dong DW, Yates JR 3rd, and Kashina A (2017a). Protein arginylation targets alpha synuclein, facilitates normal brain health, and prevents neurodegeneration. Scientific reports 7, 11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Han X, Saha S, Xu T, Rai R, Zhang F, Wolf YI, Wolfson A, Yates JR 3rd, and Kashina A (2011). Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chemistry & biology 18, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Han X, Wong CC, Cheng H, Aslanian A, Xu T, Leavis P, Roder H, Hedstrom L, Yates JR 3rd, et al. (2014). Arginyltransferase ATE1 catalyzes midchain arginylation of proteins at side chain carboxylates in vivo. Chemistry & biology 21, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Kashina AS (2015). Assaying ATE1 Activity In Vitro. Methods Mol Biol 1337, 73–77. [DOI] [PubMed] [Google Scholar]
- Wang J, Pavlyk I, Vedula P, Sterling S, Leu NA, Dong DW, and Kashina A (2017b). Arginyltransferase ATE1 is targeted to the neuronal growth cones and regulates neurite outgrowth during brain development. Dev Biol 430, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pejaver VR, Dann GP, Wolf MY, Kellis M, Huang Y, Garcia BA, Radivojac P, and Kashina A (2018). Target site specificity and in vivo complexity of the mammalian arginylome. Scientific reports 8, 16177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Xu T, Rai R, Bailey AO, Yates JR 3rd, Wolf YI, Zebroski H, and Kashina A (2007). Global analysis of posttranslational protein arginylation. PLoS biology 5, e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Liu X, Pu W, and Peng Y (2018). tRNA-derived small non-coding RNAs in human disease. Cancer letters 419, 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1, related to Fig. 3 and S4. Sequences and gene names of mouse tRNAArg, numbered according to the nomenclature in the main text and supplemental figures.
Supplemental Dataset 5, related to Fig. 2. Raw data for the microarray experiment shown in Fig. 2 of the main text.
Data Availability Statement
Original/source data for Fig. 6A and supplemental datasets 1-3 in the paper is available in the GEO database (Accession # GSE146344). Original/source data for Fig. 2B and S2 in the paper is available in Supplemental Dataset 5.