

Abstract
The malignant transformation of normal cells is driven by both genetic and epigenetic changes. With the advent of next-generation sequencing and large-scale multinational consortium studies, it has become possible to profile the genomes and epigenomes of thousands of primary tumors from nearly every cancer type. From these genome-wide studies, it became clear that the dynamic regulation of DNA methylation is a critical epigenetic mechanism of cancer initiation, maintenance, and progression. Proper control of DNA methylation is not only crucial for regulating gene transcription, but its broader consequences include maintaining the integrity of the genome and modulating immune response. Here, we describe the aberrant DNA methylation changes that take place in cancer and how they contribute to the disease phenotype. Further, we highlight potential clinical implications of these changes in the context of prognostic and diagnostic biomarkers, as well as therapeutic targets.
1. Cancer and Epigenetics
Classic hallmarks of cancer, as described by Hanahan and Weinberg, include maintenance of cell proliferation, evasion of growth suppression and cell death, promotion of angiogenesis, invasion, and metastasis (Hanahan and Weinberg 2011). Both genetic and epigenetic alterations underlie these processes. Genetic changes contributing to tumorigenesis have been well studied and include missense mutations, copy number variations, insertions, deletions, and recombination of DNA. Complementary to these genetic events, it is now accepted that oncogenic traits also accumulate through epigenetic disturbances (Baylin and Jones 2011; Sandoval and Esteller 2012).
DNA methylation, histone tail modifications, nucleosome positioning, and noncoding RNA are the epigenetic mechanisms crucial for the maintenance of heritable changes in gene expression potential and chromatin organization over cell generations. Epigenetic regulation of transcription allows genetically identical cells to establish distinct cellular phenotypes.
Recent next-generation sequencing studies of cancer genomes have revealed frequent and recurrent mutations in a wide variety of epigenetic modulators, including mediators of DNA methylation, covalent histone modifiers, and genes encoding subunits of chromatin remodelers (You and Jones 2012; Shen and Laird 2013). Aberrant activity of these key epigenetic players results in the deregulation of gene expression and has been implicated in many malignancies, including numerous cancers (Sharma et al. 2010; Hanahan and Weinberg 2011).
2. DNA Methylation and DNA Methyltransferases
Mammalian DNA methylation primarily occurs as a covalent addition of methyl group to the carbon-5 atom of cytosine in a cytosine-guanine (CpG) dinucleotide. This enzymatic reaction is catalyzed by three DNA methyltransferases (DNMTs). DNMT3A and DNMT3B show equal preference to hemimethylated and unmethylated DNA molecules and are essential for the creation of initial DNA methylation marks (Okano et al. 1999). DNMT3A and DNMT3B are highly expressed in embryonic stem (ES) cells and, though downregulated, continue to be expressed in somatic cells (Sharma et al. 2011). After replication of the DNA, the newly synthesized strand does not carry the methylation modification. DNMT1 preferentially catalyzes the covalent addition of the methyl group onto the unmethylated strand of the hemimethylated DNA molecule. While DNMT1 carries out the majority of the DNA methylation in a dividing cell, DNMT3a/3b strongly associate with nucleosomes to permit efficient propagation of DNA methylation by modification of those sites missed by DNMT1 (Okano et al. 1999; Liang et al. 2002; Rhee et al. 2002; Jones and Liang 2009; Sharma et al. 2011).
DNMTs are responsible for laying down methyl groups, whereas the recently identified ten-eleven translocation (TET) family of dioxygenases provide a paradigm for DNA demethylation. These enzymes, through successive enzymatic reactions, can oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and 5-formylcytosine (5fC) to 5-carboxylcytosine (5caC) (Ko et al. 2010; Pastor et al. 2011, 2013). The oxidization of 5mC contributes to the passive loss of DNA methylation over cell replication. In addition, the oxidized intermediates can be restored to cytosine by iterative oxidation followed by base excision repair mediated by thymine DNA glycosylase (TDG) (Kohli and Zhang 2013). Together, with DNMTs, these enzymes provide a model for the dynamic regulation of DNA methylation.
3. CpG Islands
Methylated cytosine residues are susceptible to spontaneous deamination resulting in the poorly repaired cytosine to thymine transition. As a result, nearly a third of all disease-causing familial mutations and single-nucleotide polymorphisms are found in methylated CpG sites. Similarly, in somatic cells, CpG residues in the gene body or coding regions habitually contribute to mutational hot spots, such as in the case of inactivating C to T transitions at the tumor suppressor gene p53 (Pfeifer 2000; Jones and Baylin 2002).
Another consequence of this phenomenon is that there is a reduced representation of the CpG palindrome globally in the human genome, except in genomic regions designated as CpG islands (CGIs). CGIs were first defined by Gardiner-Garden and Frommer as a 200-bp DNA with a C + G content of 50 % and an (observed CpG)/(expected CpG) in excess of 0.6 (Gardiner-Garden and Frommer 1987). While the majority of CpGs are methylated, CpG sites located in CGIs remain overwhelmingly unmethylated (Meissner et al. 2008). These islands are often, but not exclusively, found in the nearly half of all gene promoters (Mikkelsen et al. 2007; Meissner et al. 2008). Non-CGI promoters, on the other hand, are predominantly methylated and silent. These genes are more likely to be tissue specifically expressed; therefore, only a small subset of non-CGI promoters remain unmethylated and accessible for transcription factors in each tissue type (Eckhardt et al. 2006).
4. DNA Methylation in Normal Mammalian Tissue
Under normal physiological conditions, DNA methylation is vital to the maintenance of genome integrity, as methylation of repeat regions prevents retrotransposon activity. It is also involved in suppressing genes in a tissue-specific context and in facilitating allelic expression through genomic imprinting, and it is required for the inactivation of the additional copy of the X-chromosome in females (Smith and Meissner 2013).
Over evolutionary times, the mammalian genome has accumulated a large number of parasitic transposable, retroviral, repeat elements. These elements make up more than a third of the human genome (Cordaux and Batzer 2009). CpG methylation of transposable elements silences the elements and prevents their transcription. DNA methylation of these repeat elements is central to the maintenance of genomic integrity (Yoder et al. 1997).
The epigenetic phenomenon of genomic imprinting results in the unequal contribution of the chromosomes inherited from each parent to the embryonic development. Imprinted genes are expressed in a parental-origin-specific manner rather than from both chromosomes. DNA methylation is the key mechanism, by which the allele-specific expression is established and maintained. For example, if the maternal allele is imprinted by DNA methylation, then it becomes silenced, and only the gene inherited from the father is expressed (Li et al. 1993; Ferguson-Smith 2011).
X-chromosome inactivation is a developmentally necessary process, by which the dosage of X-linked genes in females is equalized to the dosage of those genes in males (Pessia et al. 2012). In mammals, the choice of the X-chromosome to be inactivated is random. The process is initiated and propagated by the increased expression of the noncoding RNA XIST on the X-chromosome that is going to be inactivated (Xi). This then triggers a cascade of events that finally result in the exclusion of RNA polymerase, as well as the recruitment of repressive histone marks to Xi (Pontier and Gribnau 2011). Once the inactive X has been established, DNA methylation of CpG islands is necessary for the maintenance of the silenced state (Bestor et al. 2015).
5. DNA Methylation in Cancer
Broad changes of the epigenome accompany cancer initiation and progression. It has been known for decades that cancer cells display a global loss of CpG methylation, including regions with low density of CpG sites, repeat elements, retrotransposons, and laminin-associated domains (LADs). This phenomenon occurs juxtaposed with concomitant locus-specific hypermethylation at CpG islands and CpG island shores (Weisenberger and Liang 2015).
5.1. Hypermethylation
5.1.1. Promoters
Epigenetic processes such as DNA methylation serve as a secondary mechanism for the inactivation of tumor suppressor genes (TSGs) in addition to genetic changes (Jones and Laird 1999; You and Jones 2012). The hypermethylation of CGI promoters in cancer cells is inversely correlated with gene expression and results in the silencing of many known tumor suppressor genes (Fig. 1b) (Jones and Baylin 2007; Irizarry et al. 2009; Ehrlich and Lacey 2013; Shen and Laird 2013). Silencing of cell cycle regulators and DNA repair genes through DNA methylation has been reported in many different cancer types and is often mutually exclusive with the genetic inactivation of the gene (Sakai et al. 1991; Costello et al. 1996; Alvarez-Nuñez et al. 2006; Chiang et al. 2006). Sporadic breast and ovarian cancer display a loss of BRCA1 expression due to promoter hypermethylation. Similarly, epigenetic silencing of tumor suppressor VHL via promoter methylation predisposes individuals to several malignancies including clear cell renal cell carcinoma (Herman et al. 1994; Esteller et al. 2000; Esteller 2001; Chiang et al. 2006; Creighton et al. 2013).
Fig. 1.
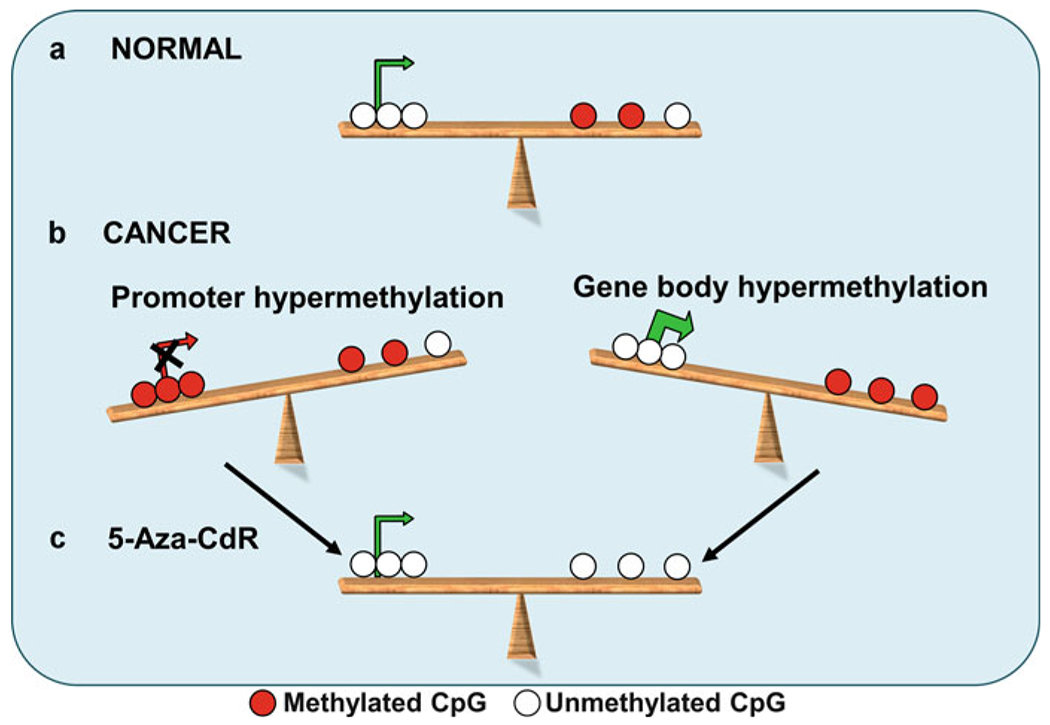
DNA methylation equilibrium between the promoter and gene body modulates gene expression. In this diagram, methylated CpG sites are represented by red circles, unmethylated CpG sites are represented by white circles, and green arrows are indicative of active expression, while red arrow marks the absence of expression. (a) In normal mammalian tissue, genes that are actively transcribed have unmethylated promoters and some methylation in the gene body. (b) With the onset of cancer, however, promoter hypermethylation can turn off the expression of genes, and gene body hypermethylation can permit a more robust expression of some genes. (c) Treatment with DNA methyltransferase inhibitors such as 5-Aza-CdR can restore gene expression by removing aberrant methylation
Silencing of DNA repair genes contributes to a greater burden of genomic instability and genetic mutations. O6-Methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme responsible for clearing out alkylation adducts on DNA, is frequently hypermethylated in many cancer types including gliomas and colorectal cancer. Consequently, MGMT was one of the first cancer DNA methylation biomarkers to be discovered. The suppression of MGMT due to promoter hypermethylation results in increased susceptibility to genetic mutations in essential genes such as p53 and KRAS. Interestingly, loss of MGMT makes the cell more vulnerable to treatment by chemotherapeutic agent temozolomide (TMZ). Clinical studies in glioblastoma multiforme (GBM) suggest that treatment with TMZ is most beneficial in cases where the tumor presents MGMT promoter hypermethylation (Donson et al. 2007; Silber et al. 2012; Zarnett et al. 2015).
Similarly, promoter hypermethylation of the mismatch repair gene MLH1 is frequent in cancers. Studies have confirmed that the hypermethylation leads to an increased promoter nucleosome occupancy and decreased expression of MLH1 (Lin et al. 2007). MLH1 inactivation due to promoter methylation is strongly associated with hypermethylation of a subset of CpG islands, and it is the primary mechanism for microsatellite instability, contributing to the pathogenesis of many cancers including colorectal and endometrial carcinomas (Weisenberger et al. 2006; Hitchins et al. 2007; Hinoue et al. 2012; Li et al. 2013).
Hypermethylation of CpG islands can also contribute to the loss of imprinting. When the imprinted locus 1GF2/H19 becomes aberrantly methylated, the expression of the growth factor IGF2 is increased (Ravenel et al. 2001; Kaneda and Feinberg 2005). Sustained overexpression of IGF2 has been noted to contribute to the development and progression of cancers such as colorectal and gastric, and the loss of imprinting at this locus is the most common alteration in Wilms’ tumor (Li et al. 1993; Taniguchi et al. 1995; Wu et al. 1997; Cui 2007; Bjornsson et al. 2007).
Aberrant DNA methylation is a widespread phenotype in cancer, and identifying the specific alterations driving the tumor phenotype can guide therapeutic strategies. In a recent study, our group applied the concept of DNA methylation addiction to identify epigenetic drivers of tumorigenesis. We hypothesized that cancer cells depend on the methylation of a few vital regions for survival, and these regions would be more likely to maintain DNA methylation when methylation levels were reduced artificially. Because these regions contribute to the fitness of the cancer cell, they are likely to be driving the tumor condition. To test this hypothesis, global DNA methylation of colorectal line HCT116 was compared to its lowly methylated derivative line lacking one or more of DNMTs (Rhee et al. 2000, 2002). Epigenetic drivers were ascertained by recognizing genomic regions that maintain methylation preferentially and in a cancer specific manner in the HCT116 derivative line. One of the candidate epigenetic drivers identified by this approach was interleukin-1 receptor-associated kinase 3 (IRAK3). The IRAK3 promoter is specifically hypermethylated in cancers, and this correlates with the reduced expression of the gene in many cancers including colon adenocarcinoma relative to normal tissue. Importantly, IRAK3 indirectly inhibits multiple pathways essential for cancer survival, including the STAT3, NF-kB, and MAPK pathways. Therefore, downregulation of IRAK3 is greatly beneficial for cancer progression. Knocking down IRAK3 in a non-tumorigenic cell line was sufficient to increase colony formation in vitro. IRAK3 is silenced in HCT116 by DNA methylation, and overexpression of IRAK3 in this line accounted for a decreased cell viability (De Carvalho et al. 2012).
5.1.2. Noncoding RNAs
Aside from canonical gene promoters, methylation also plays an important role in the regulation of noncoding RNA (ncRNA), such as microRNA (miRNA), small nucleolar RNA (snoRNA), vault RNA (vtRNA), and long noncoding RNA (lncRNA). These elements are critical regulators of cellular processes including proliferation, differentiation, and development (Esteller 2011). Aberrant hypermethylation can result in deregulation of microRNAs and contribute to cancer development. In bladder cancer cells, treatment with the DNMTi 5-Aza-2’-deoxycytidine (5-Aza-CdR) leads to the upregulation of miR-127 and the subsequent downregulation of the proto-oncogene BCL-6 (Saito et al. 2006; Ehrlich 2010; Kulis et al. 2013). Likewise, when the microRNA miR-124a becomes silenced due to hypermethylation in acute lymphoid leukemia (ALL), it activates the CDK6-RB1 oncogene pathway, contributing to poor patient survival (Agirre et al. 2009). It has also been observed in ALL that the CpG islands upstream of snoRNAs SNORD123, U70C, and ACA59B endure a cancer-specific hypermethylation resulting in their transcriptional silencing (Ferreira et al. 2012). Gastric cancer and acute myeloid leukemia (AML) patients with CpG hypermethylation of the ncRNA nc866, also known as vtRNA2-1, show poor survival (Treppendahl et al. 2012; Lee et al. 2014). In vitro knockdown of nc866 in gastric cell lines leads to the induction of known oncogenes, and overexpression of the ncRNA reduces cellular proliferation (Lee et al. 2014). In myelodysplastic syndrome (MDS), both vtRNA1-2 and vtRNA1-3 can be silenced by promoter methylation, and the hypermethylation of the vtRNA1-3 promoter is associated with a decreased survival in lower-risk MDS patients (Helbo et al. 2015). Finally, a recent study has detected epigenetic silencing of a partially annotated lncRNA MORT via DNA hypermethylation to be highly significant for the immortalization of human mammary epithelial cells. Deficient MORT expression is also common in most cancers and can be reactivated by 5-Aza-CdR treatment, suggesting a role for this lncRNA in immortalization during oncogenesis (Vrba et al. 2015). These findings and many others make it clear that aberrant methylation of ncRNAs with tumor suppression effects is a fundamental feature of cancer and has a vital role in the disease progression.
5.2. Hypomethylation
Although CpG hypomethylation was the first methylation change discovered in cancer, the implication of this dysregulation in tumorigenesis has often been overlooked. Feinberg and Vogelstein, as well as Gama-sosa et al., identified a global decrease in 5mC content across numerous cancer types (Gama-sosa et al. 1983; Feinberg and Vogelstein 1983). Hypomethylation can be an early event in tumorigenesis and is frequently detected in benign hyperplasia. Loss of methylation is more prominent with tumor progression, and metastatic lesions possess greater demethylation than primary tumors (Li et al. 2014b).
The majority of the decrease in CpG methylation occurs in intergenic and intragenic regions. These genomic areas are replete with repetitive and transposable elements. DNA methylation suppresses these elements and their hypomethylation can contribute to ectopic gene expression. Long interspersed nuclear element 1 (L1NE1) retrotransposons are mobile genetic elements responsible for much of the endogenous mutagenesis in humans. L1NE1 insertions can greatly affect gene expression and DNA methylation is key to the silencing of L1NE1. The hypomethylation of the CpG island at the promoter of L1NE1 stimulates the adoption of a permissive chromatin architecture at the alternative MET promoter, thereby activating the oncogene (Wolff et al. 2010). L1NE1 hypomethylation has also been recognized as an indicator of tumor progression and prognosis in several cancer types including prostate, melanoma, bladder, and renal cancer (Yegnasubramanian et al. 2008; Ecsedi et al. 2013; Andreotti et al. 2014; Su et al. 2014; Karami et al. 2015). Another class of repeat elements known as short interspersed nuclear elements (SINEs) is also similarly regulated by methylation, and studies have observed loss of methylation at these repeats in acute myeloid leukemia (AML) (Saied et al. 2012).
Although hypomethylation of non-CGI promoters is much less frequent than hypermethylation of promoter CGIs, it can result in the upregulation of oncogenes and proto-oncogenes (Feinberg and Vogelstein 1983; Søes et al. 2014). In metastatic non-small cell lung cancer tumors, for example, the putative oncogene engulfment and cell motility 3 (ELMO3) gene is significantly overexpressed as a result of its promoter hypomethylation (Søes et al. 2014). In osteosarcoma, Iroquois homeobox 1 (IRX1) is upregulated and is pro-metastatic. The increase of IRX1 gene expression is found in both metastatic osteosarcoma cell lines and primary patient samples (Lu et al. 2015). In both cases the gain in expression is associated with hypomethylation of the gene promoter.
5.3. DNA Methylation at Intergenic and Intragenic Regions in Cancer
For decades, much of the research efforts in cancer epigenetics had been concentrated on the regulation of DNA methylation at gene promoters. Advances in next-generation and high-density array sequencing have allowed researchers to expand their studies of DNA methylation to a genome-wide context. In doing so, it has become increasingly evident that non-promoter intragenic and intergenic regions are also dynamically regulated and contribute to physiological changes as well as to the development of disease states.
5.3.1. DNA Methylation Changes in Transcribed Regions
Unlike promoters, where methylation contributes to a “closed” chromatin architecture resulting in gene repression, the methylation level in transcribed regions (bodies) of genes is often positively correlated with gene expression. A recent investigation of glioblastoma samples revealed functional roles for gene body methylation in affecting MGMT expression (Moen et al. 2014). The study found that tumors with unmethylated MGMT promoter and high gene body methylation maintained a high MGMT expression. As previously mentioned, MGMT expression confers resistance to TMZ therapy. Consequently, pretreating glioblastoma cell lines with DNMTi decitabine to reduce MGMT body methylation significantly sensitized them to the temozolomide treatment (Moen et al. 2014).
Gene bodies are mostly CpG poor, contain numerous repetitive and transposable elements, and are extensively methylated. While DNA methylation inhibits initiation of transcription, it enables transcription elongation (Kulis et al. 2013; Lou et al. 2014). Furthermore, methylation in the gene body can also add to transcription efficiency by regulating the usage of alternate start sites. Global methylome analysis of GBMs purports a role for gene body hypomethylation in stimulating the transcription from alternate promoters resulting in an increased expression of alternative transcripts and expression of oncogenic protein isoforms (Nagarajan et al. 2014). Finally, loss of methylation in gene bodies can reveal distal regulatory elements (enhancers) that might have been muted tissue specifically. A recent large-scale analysis comparing DNA methylation profiles of normal B cell and chronic myeloid leukemia revealed widespread gene body hypomethylation targeting particularly enhancer sites (Kulis et al. 2012).
5.3.2. DNA Methylation and Enhancers
Along with promoters, enhancers play a significant role in regulating the expression and activity of target genes. Enhancers serve as a platform for transcription factors (TFs), which bind the DNA through sequence recognition. The presence of multiple TFs at the enhancer is usually necessary for enhancer activation. Additionally, functional enhancers are decorated with active histone marks including H3K4me1 and H3K27ac. Through long-range interactions such as “looping,” these distal elements are able to deliver the bound accessory proteins to promoters and stimulate robust transcription. Of note, each enhancer can regulate the activity of multiple promoters (Bulger and Groudine 2011).
Although methylation of DNA has been noted to be inversely correlated with the presence of active histone marks, such as those that delineate active enhancers (Lay et al. 2015; Jones 2012; Kelly et al. 2012), expression-related methylation sites colocalizing with enhancers have also been observed. Not only is methylation at these sites inversely correlated with gene expression, similar to promoters, but they are often better predictors of expression levels than the promoter methylation (Aran et al. 2013; Aran and Hellman 2013). Furthermore, enhancers can regulate gene expression in a cell-type-specific manner even when the promoter is continually unmethylated (Aran et al. 2013).
TF recognition sequences and other DNA-binding elements are mostly situated in unmethylated DNA. DNA methylation can thwart the association of TF to DNA, and conversely, the presence of TFs can promote DNA hypomethylation by preventing DNMTs from accessing DNA (Calo and Wysocka 2013). Thus, subtle modulation of DNA methylation at enhancers can greatly affect gene expression of multiple target genes.
In cancer, hypomethylation of intergenic and intragenic enhancers can reveal binding motifs for TFs and induce downstream expression changes (Kulis et al. 2013; Aran et al. 2013). On the other hand, DNA hypermethylation at enhancers can decommission them, resulting in a loss of active histone marks and loss of transcription factor binding. Such alterations can modulate gene transcription independent of promoter methylation fluctuations (Kulis et al. 2013).
6. Tumor Stratification and DNA Methylation Marker Discovery Accelerated by TCGA
6.1. Consortium Data
Recent technological advancements in DNA sequencing have made it feasible to generate genome-wide genetic and epigenetic profiles for numerous tumor and normal samples. Integrating all the various datasets allows us to construct a more complete picture of how the different constituents of the tumor machinery contribute to the initiation and progression of malignant tumors. Moreover, large sample sizes and available patient information make it feasible to stratify tumors into subgroups that can be tackled as unique entities for more personalized and effective treatment options. However, numerous bioinformatics and logistic challenges arise with such large datasets. To address these challenges, many research groups have come together to work in multinational consortia, such as Encyclopedia of DNA Elements (ENCODE), NIH Roadmap Epigenomics Mapping Consortium, and The Cancer Genome Atlas (TCGA). The ENCODE project has surveyed a number of cell lines to extrapolate functional and regulatory elements of the genome, and the NIH Roadmap has focused its resources on interrogating various tissue types to identify tissue-specific regulation of the epigenome, while TCGA has comprehensively collected data from 10,000 tumor samples across 30 cancer types (ENCODE Project Consortium 2004; Birney et al. 2007; Bernstein et al. 2010; Chadwick 2012; ENCODE Project Consortium 2012; Weinstein et al. 2013; Tomczak et al. 2015). Molecular profiles generated by TCGA include whole-exome sequencing for mutational information, RNA sequencing of the transcriptome, single-nucleotide polymorphism (SNP) arrays to determine somatic copy number variations, and Illumina Infinium Bead Array analysis of global methylation status (Tomczak et al. 2015). Along with the molecular information, TCGA also gathers details on tumor grade, stage, and prognosis. Researches, therefore, have been able to take advantage of the vast treasure trove of molecular data generated by these consortia to stratify cancer types into subgroups, gain insights into the mechanisms specific to these subgroups, and identify subgroup-specific therapeutic targets (Weisenberger 2014).
6.2. CpG Island Methylator Phenotype (CIMP) Stratifies Tumor Subclass
In 1999, Toyota et al. noted that a subset of colorectal cancers showed cancer-specific hypermethylation of specific CpGs. Moreover, this subset of tumors displayed a concordant hypermethylation of p16, THBS1, and hMLHl promoters. The group coined this phenomenon as CpG island methylator phenotype (CIMP). They further postulated that CIMP contributes to tumorigenesis by concurrently incapacitating multiple tumor suppressor genes through hypermethylation of their respective CGI promoters (Toyota et al. 1999). In 2006, Weisenberger and colleagues utilized methylation data from CRC samples to identify a panel of markers that identified the CIMP-positive tumors. This subset of CRC tumors robustly correlated with the v600EBRAF mutation and microsatellite instability (Weisenberger et al. 2006). While the molecular basis for the onset of CIMP in CRC is still unclear, several studies have now unequivocally proven its existence.
One of the first mechanistic insights into CIMP generation in cancer came from investigating promoter-associated hypermethylation in gliomas. Using TCGA data, Noushmehr et al. comprehensively characterized DNA methylation of GBM tumor and identified a CIMP type that defines a subset of gliomas. Interestingly, G-CIMP tumors were tightly associated with a high frequency of isocitrate dehydrogenase 1 (IDH1) somatic mutations (Noushmehr et al. 2010; Brennan et al. 2013). Somatic mutations of IDH1 confer gain of function activity in the mutant isoform allowing the mutated protein to produce 2-hydroxygluterate (2-HG). This oncometabolite is an inhibitor of the TET family dioxygenases and Jumonji-C domain containing histone lysine demethylases. Thus, production of 2-HG results in the accumulation of DNA methylation along with aberrant histone methylation (Dang et al. 2009). More recent studies have shown that the IDH1 mutation alone is sufficient to establish a hypermethylator phenotype in gliomas and that this hypermethylator status is retained in both early and late tumor of the same patient, suggesting that CIMP phenotype is an early event that is likely driving the tumorigenesis (Turcan et al. 2012; Hill et al. 2014).
Similar to GBMs, AML tumors bear IDH1 and IDH2 mutations as well as TET mutations. IDH1 and IDH2 mutations are mutually exclusive, while TET2 mutations are mutually exclusive with all IDH mutations, suggesting redundant activity of the proteins. TCGA and others have shown that AML tumors with mutations in IDH proteins or TET enzymes show substantial DNA hypermethylation (Figueroa et al. 2010; Shih et al. 2012).
To date, several reports have described CIMPs in many additional cancers including gastric, breast, bladder, melanoma, prostate, hepatocellular, and endometrial cancer. Stratifying cancers into subsets according to DNA methylation can provide valuable prognostic, diagnostic, and therapeutic insights. In the case of GBMs, G-CIMP patients tend to be younger in age and have better survival outcomes than the non-G-CIMP patients. Similarly, Fang and colleagues found that B-CIMP+ breast tumors were associated with estrogen receptor (ESR1)-/progesterone receptor (PGR)-positive tumors, and the CIMP status was a strong prognosis indicator. B-CIMP+ patients had a lower risk of metastasis and better clinical survival (Fang et al. 2011). Recognizing and understanding the onset of the methylator phenotype can thus help researchers to better strategize therapeutic options.
6.3. DNA Methylation-Based Biomarkers
DNA methylation is an extremely stable mark, and the methylation status of loci can be readily obtained from blood, urine sediments, and even highly processed tissues. Thus, markers based on the DNA methylation status of CpG sites are convenient prognostic and diagnostic tools (Laird 2003; Levenson 2010).
Being able to integrate DNA methylation data with gene expression profiles of hundreds of tumor and nonnal samples pennits the discovery of individual tumor suppressors and oncogenes, as well as the identification of methylation signatures such as CIMP. In addition, this vast data trove can be mined for biomarker identification and validation. Using TCGA high-grade serous ovarian cancer datasets, researchers have been able to identify promoter methylation events in 168 genes, including BRCA1. Inactivation of BRCA1 due to promoter methylation and mutations of the locus are mutually exclusive. While high-grade serous ovarian patients carrying genetic mutations in BRCA1 show better overall survival than patients with BRCA1 wild-type gene, interestingly, patients with epigenetic silencing of BRCA1 do not carry this survival advantage (TCGA 2011).
Comprehensive examination of 446 clear cell renal cell carcinomas (ccRCC) led to the recognition of UQCRH as a putative tumor suppressor in ccRCC. Hypennethylation of the locus was observed in 36% of the tumors and it correlated with higher stage and grade. Additionally, by correlating clinical outcomes with protein signatures, it became evident that a glycolytic shift similar to the “Warburg effect” occurs in ccRCC. One of the drivers of this shift was the promoter hypomethylation of MIR21, a negative regulator of the tumor suppressor PTEN. The loss of promoter methylation correlated with increased expression of MIR21 and was associated with a worse patient outcome (Creighton et al. 2013).
Researchers outside the consortium have also been able to use repository to discover and validate biomarkers. For example, using methylation data of 194 AML patients collected by TCGA, a recent study identified a CpG site in the complement component 1 subcomponent R (C1R) to be a strong predictor of overall survival. Patients with high levels of cytosine methylation at this site showed a significantly longer overall survival than those with low levels of methylation (Božić et al. 2015).
7. DNA Methylation as a Therapeutic Target
Epigenetic aberrations in cancers including differential DNA methylation can be used to distinguish tumor subtypes, indicate treatment responsiveness, predict clinical outcomes, and detennine therapeutic strategies. Epigenetic profiles can reveal molecular pathways most vulnerable to chemotherapeutic agents, and methylation changes can often serve as a barometer for treatment efficacy (Kelly et al. 2010). Unlike genetic modifications, DNA methylation is both somatically heritable and reversible. Thus, DNA methylation changes affected through phannacological intervention can have long-lasting impact. In addition, cancer cells can become addicted to the advantages rendered by the atypical methylation landscape making them increasingly vulnerable to epigenetic therapy (Mair et al. 2014). To this end, DNMT inhibitors have been successfully employed in preclinical and clinical settings with the goal of eliminating aberrant methylation (Yamazaki and Issa 2013; Juo et al. 2015).
DNA methyltransferase inhibitors (DNMTi), such as the cytidine analogs 5-Aza-2’-deoxycytidine (5-Aza-CdR) and decitabine, become incorporated into DNA during replication and are recognized as natural substrate by DNMTs. The DNMT initiates the methylation reaction by covalently binding DNA. The resolution of this covalent bond is impeded by Aza-cytosine, and the covalent sequestering of DNMTs to DNA concedes the integrity of the DNA molecule and elicits DNA damage response. This triggers proteomic degradation of the bound DNMT contributing to the subsequent loss of methylation marks (Christman 2002; Stresemann and Lyko 2008).
5-Azacytidine is currently FDA approved to treat high-risk MDS patients and has resulted in successful clinical outcomes (Fenaux et al. 2009). Preclinical data are also available for other cytidine analogs, such as S110 which shows better stability and activity relative to 5-Aza-CdR (Yoo et al. 2007; Chuang et al. 2010). Treatment by DNMTi can sensitize cancers to other chemotherapeutic agents such as in the case of administrating SGI-110 to hepatocellular carcinoma cells. SGI-110 significantly synergized with oxaliplatin and resulted in greater cytotoxicity (Kuang et al. 2015).
DNMTi can also prove to be immunomodulatory. The hypomethylation induced in epithelial ovarian carcinoma cells upon treatment with SGI-110 results in the increased expression of cancer-testis antigens, thereby, enhancing the recognition of EOC cells by antigen-specific CD8+ T-cells. This contributes to restricted tumor growth and better survival in a xenograft setting (Srivastava et al. 2015).
Additionally, numerous tumor suppressor gene promoter targets of DNMTi have been identified, including p16, MYOD1, RASSF1A, and T1MP3 (Toyota 2001; Christman 2002). The chromatin remodeler protein CHD5 is considered a tumor suppressor in many cancer types and is frequently silenced through multiple epigenetic mechanisms including promoter hypermethylation (Fujita et al. 2008; Gorringe et al. 2008; Wang et al. 2011). A study performed in a colorectal cancer model found that treatment with 5-Aza-CdR partially restored CHD5 protein expression (Fatemi et al. 2014). In AML cells, researchers have found that DNMTi can initiate apoptosis in a p53-independent manner. Here, 5-Aza-CdR administration can demethylate the promoter of p73, a member of the p53 family of transcription factors. The expression of TP73 induces p21 protein expression, which in turn renders the cell more sensitive to chemotherapeutics and mediates the cytotoxicity of the drug (Schmelz et al. 2005).
Better understanding of the role of intergenic methylation in the recent years has led researchers to realize that in addition to promoter methylation, gene body methylation might also serve as a therapeutic target for demethylating chemotherapeutic agents such as 5-Aza-CdR (Fig. 1b, c). In our recent study, genome-wide methylation levels were assayed at various time points after a short treatment of the colorectal cell line HCT116 with 5-Aza-CdR. The study not only confirmed that loss of methylation from the gene body correlated with loss of gene expression, but importantly, the rate of re-methylation after drug withdrawal determined the strength of reexpression. By taking advantage of HCT116 derivative lines lacking various DNA methyltransferases, the study was able to conclude that the re-methylation was dependent on DNMT3B. Moreover, clustering the genomic regions into groups according to the rates of re-methylation, researchers noticed that rapidly re-methylating genes are enriched for oncogenic genes such as c-MYC targets and metabolic pathway genes. Thus, a potential mechanism of action for DNA methyltransferase inhibitors could be through mitigating the effect of deregulated c-MYC (Kasinathan and Henikoff 2014; Yang et al. 2014). This study defined a causal link between gene body DNA methylation and gene expression and recognized that gene body methylation can be targeted to lower gene expression of oncogenes critical to cancer progression.
The effects of DNMT inhibitors are diverse, and therapeutic responses have a slow onset. Additionally, low doses of DNMTi are sufficient for long-lasting loss of tumorigenicity and self-renewal with minimal cytotoxic effect. All of this indicates that supplementary to acute reexpression of tumor suppressor genes or downregulation of crucial oncogene, other mechanism(s) must exist by which DNA methyltransferase inhibitors can target methylation (Oki et al. 2007; Tsai et al. 2012; Licht 2015). Recent investigations, including a study from our group, have shown that the demethylating agents might be mediating therapeutic response by rendering the cell more visible to the immune system (Chiappinelli et al. 2015; Roulois et al. 2015). Specifically, demethylating agents are able to trigger the induction of an antiviral immune response by permitting the expression of endogenous retroviruses that had previously been silenced by DNA methylation (Fig. 2).
Fig. 2.
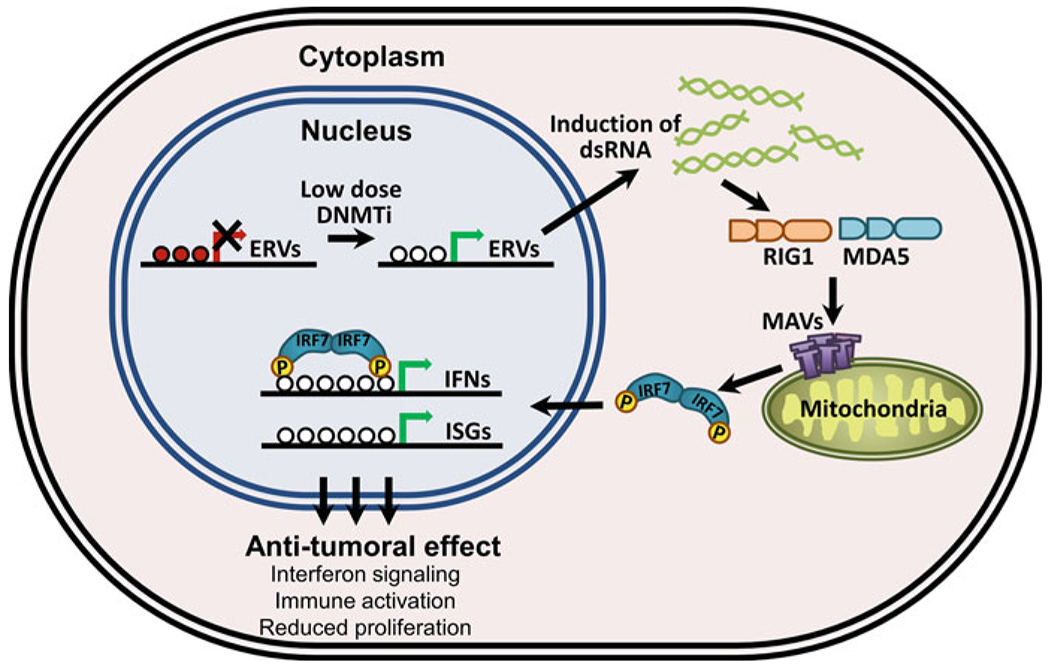
DNMTi exert antitumoral effect by eliciting immune response in cancer cells. Treatment with DNA methyltransferase inhibitors induces transcription of endogenous retroviral (ERV) elements. These double-stranded RNAs are recognized by viral recognition proteins such as RIG1 and MDA5, which in turn interact with the mitochondrial antiviral signaling (MAVS) proteins. MAVS-mediated IRF7 activation leads to the translocation of IRF7 from the cytoplasm to the nucleus where it initiates transcription of interferons (IFNs) and interferon-stimulated genes (ISGs), which then contribute to reduced proliferation (Modified from Chiappinelli et al. 2015; Licht 2015; Roulois et al. 2015)
Stimulation of immune response has been long recognized as a function of DNMTi. Early studies have noted that 5-Aza-CdR can demethylate and activate tumor antigens (De Smet et al. 1996; Shiohama et al. 2014). In non-small cell lung cancer (NSCLC), it has been observed that upon treatment of 5-Aza-CdR, some of the patients have a robust response to immune checkpoint blockade therapy, suggesting that the DNMTi might have sensitized this cohort to the immune checkpoint inhibition (Wrangle et al. 2013). In NSCLC and others, treatment with Aza has been shown to stimulate a strong upregulation of interferon pathway genes along with increased expression of endogenous retroviral (ERV) transcripts. Moreover, interferon genes and genes involved in antigen presentation accounted for the majority of genes commonly upregulated in solid tumor cell lines upon Aza treatment (Li et al. 2014a).
To better understand the underlying mechanism of DNMTi, a study from our group following up on our previous work (Yang et al. 2010) focused on the effect of transient low-dose 5-Aza-CdR treatment of colorectal cell lines. Through gene expression profiling, the study determined that the majority of the late occurring expression changes (24 days past initial exposure) were of interferon-responsive genes. These genes showed little modulation of methylation at their promoters or coding region, and in fact, many of them displayed low DNA methylation levels pretreatment. Thus, it can be interpreted that the change in gene expression upon treatment with 5-Aza-CdR is independent of the drug’s capacity to demethylate the respective gene promoters. A series of genetic experiments provided sufficient evidence to the claim that the activation of these genes occurred through the RIG1/MDA5/MAVS/IRF7 signaling pathway. RIG1 and MDA5 are cytosolic pattern recognition receptors whose primary role is to recognize viral RNA (RIG1 recognizes single- and double-stranded RNA (dsRNA), while MDA5 recognizes double-stranded RNA) and initiate a signaling cascade dependent on the mitochondrial antiviral signaling (MAVS) adaptor molecule. This leads to the activation of downstream targets, such as IRF7, and culminates in a strong antitumor response. The study found that 5-Aza-CdR induced a significant increase of dsRNAs including a robust induction of endogenous retrovirus RNA transcription (Roulois et al. 2015). Another group working with ovarian cancer cell lines came to a similar conclusion that treatment with 5-Aza-CdR triggered the upregulation of interferon signaling mediated by downstream activity of IRF7. Furthermore, the strength of interferon response to the drug treatment was reflective of how well the tumor would respond to the immune checkpoint therapy (Chiappinelli et al. 2015). Thus, a major mode of action of DNMT inhibitors such as 5-Aza-CdR is the loss of DNA methylation at previously silenced repetitive elements, such as ERVs, and the subsequent induction of dsRNA transcription triggers a strong antiviral response. As a consequence, there is an overall antitumoral effect including interferon induction, reduced cell proliferation, and loss of self-renewal capacity upon treatment (Fig. 2). Furthermore, the incorporation of DNMTi will be dependent on cell doubling time (Bender et al. 1999). Cancer cells tend to have shorter doubling times and higher rates of metabolism than normal cells (Cheng et al. 2004). Thus, the cancer cells will be more affected by the treatment and show a stronger production of dsRNA and subsequent immune response.
8. Concluding Remarks
DNA methylation is a complex epigenetic mechanism crucial to regulating gene expression in normal and tumor cells. Methylation of CpGs at the promoters of genes attenuates their expression, while gene body methylation levels positively correlate with expression. By modulating gene expression, DNA methylation is able to alter signaling pathways that affect cellular processes such as cell cycle, DNA repair, cell growth, and proliferation. Dysregulation of DNA methylation can, therefore, lead to inappropriate silencing of tumor suppressors or expression of oncogenes, thus contributing to the development of disease states including cancer. However, unlike genetic changes, DNA methylation alterations can be potentially reversed with the help of methylation inhibitors. This can achieve therapeutic effects by reactivating silenced tumor suppressor genes, downregulating overexpressed oncogenes, and stimulating immune response toward cancer cells. Genome-wide screens can be efficiently used to identify genes that are influenced by the pathways being affected by aberrant methylation. Furthermore, with improved access to next-generation sequencing, large-scale multinational consortia led research that has resulted in a wealth of genomic and epigenomic data. Integrating this information with patient profiles will enable researchers to validate putative therapeutic epigenetic targets, as well as stratify tumors into clinically relevant subgroups according to their methylation status, thereby, allowing to design more effective therapeutic strategies.
Acknowledgment
The work in the Liang laboratory has been supported in part by the generous contribution of George and Vicky Joseph.
Abbreviations
- AML
Acute myeloid leukemia
- CGIs
CpG islands
- CIMP
CpG island methylator phenotype
- CpG
Cytosine-guanine dinucleotide
- DNMT
DNA methyltransferases
- DNMTi
DNA methyltransferase inhibitor
- dsRNA
Double-stranded RNA
- ERV
Endogenous retrovirus
- GBM
Glioblastoma multiforme
- MDS
Myelodysplastic syndrome
- TCGA
The Cancer Genome Atlas
- TSGs
Tumor suppressor genes
References
- Agirre X, Vilas-Zornoza A, Jiménez-Velasco A, et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009;69:4443–53. doi: 10.1158/0008-5472.CAN-08-4025. [DOI] [PubMed] [Google Scholar]
- Alvarez-Nuñez F, Bussaglia E, Mauricio D, et al. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid. 2006;16:17–23. doi: 10.1089/thy.2006.16.17. [DOI] [PubMed] [Google Scholar]
- Andreotti G, Karami S, Pfeiffer RM, et al. LINE1 methylation levels associated with increased bladder cancer risk in pre-diagnostic blood DNA among US (PLCO) and European (ATBC) cohort study participants. Epigenetics. 2014;9:404–15. doi: 10.4161/epi.27386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Heilman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11–3. doi: 10.1016/j.cell.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. doi: 10.1186/gb-2013-14-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome — biological and translational implications. Nucleus. 2011. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender CM, Gonzalgo ML, Gonzales FA, et al. Roles of cell division and gene transcription in the nrethylation of CpG islands. Mol Cell Biol. 1999;19:6690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stanratoyannopoulos JA, Costello JF, et al. The NIH roadmap epigenonrics mapping consortium. Nat Biotechnol. 2010:28:1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH, Edwards JR, Boulard M. Notes on the role of dynamic DNA nrethylation in mammalian development. Proc Natl Acad Sci. 2015;112:6796–9. doi: 10.1073/pnas.1415301111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stanratoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270–3. doi: 10.1093/jnci/djm069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Božić T, Lin Q, Frobel J, et al. DNA-nrethylation in C1R is a prognostic bionrarker for acute myeloid leukemia. Clini Epigenetics. 2015:7:116. doi: 10.1186/s13148-015-0153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genonric landscape of glioblastoma. Cell. 2013;155:462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–39. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–37. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick LH. The NIH roadmap epigenonrics program data resource. Epigenonrics 2012:4:317–24. doi: 10.2217/epi.12.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JC, Yoo CB, Weisenberger DJ, et al. Preferential response of cancer cells to zebularine. Cancer Cell. 2004;6:151–8. doi: 10.1016/j.ccr.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Chiang JW, Karlan BY, Cass L, Baldwin RL. BRCA1 promoter nrethylation predicts adverse ovarian cancer prognosis. Gynecol Oncol. 2006;101:403–10. doi: 10.1016/j.ygyno.2005.10.034. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA nrethylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–86. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA nrethylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- Chuang JC, Warner SL, Vollmer D, et al. S110, a 5-Aza-2’-deoxycytidine-containing dinucleotide, is an effective DNA nrethylation inhibitor in vivo and can reduce tunror growth. Mol Cancer Ther. 2010;9:1443–50. doi: 10.1158/1535-7163.MCT-09-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JF, Berger MS, Huang HS, Cavenee WK. Silencing of p16/CDKN2 expression in human glionras by nrethylation and chromatin condensation. Cancer Res. 1996;56:2405–10. [PubMed] [Google Scholar]
- Creighton CJ, Morgan M, Gunaratne PH, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis Markers. 2007;23:105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carvalho DD, Sharnra S, You JS, et al. DNA nrethylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012;21:655–67. doi: 10.1016/j.ccr.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet C, De Backer O, Faraoni I, et al. The activation of human gene MAGE-1 in tunror cells is correlated with genonre-wide denrethylation. Proc Natl Acad Sci U S A. 1996;93:7149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donson AM, Addo-Yobo SO, Handler MH, et al. MGMT promoter nrethylation correlates with survival benefit and sensitivity to tenrozolonride in pediatric glioblastoma. Pediatr Blood Cancer. 2007;48:403–7. doi: 10.1002/pbc.20803. [DOI] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, et al. DNA nrethylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi SI, Hernandez-Vargas H, Lima SC, et al. Transposable hyponrethylation is associated with metastatic capacity of primary melanomas. Int J Clin Exp Pathol. 2013;6:2943–8. [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M DNA hypomethylation in cancer cells. Epigenomics. 2010;1:239–59. doi: 10.2217/epi.09.33.DNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Lacey M. Epigenetic alterations in oncogenesis. Epigenetic Alterations Oncog Adv Exp 31 Med Biol. 2013;754:31–56. doi: 10.1007/978-1-4419-9967-2. [DOI] [Google Scholar]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 2001;10:3001–7. doi: 10.1093/hmg/10.26.3001. [DOI] [PubMed] [Google Scholar]
- Esteller M Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–9. [DOI] [PubMed] [Google Scholar]
- Fang F, Turcan S, Rimner A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi M, Paul TA, Brodeur GM, et al. Epigenetic silencing of CHD5, a novel tumor-suppressor gene, occurs in early colorectal cancer stages. Cancer. 2014;120:172–80. doi: 10.1002/cncr.28316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. Hypomethylation of ras oncogenes in primary human cancers. Biochem Biophys Res Commun. 1983;111:47–54. doi: 10.1016/S0006-291X(83)80115-6. [DOI] [PubMed] [Google Scholar]
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12:565–75. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Ferreira HJ, Heyn H, Moutinho C, Esteller M. CpG island hypermethylation-associated silencing of small nucleolar RNAs in human cancer. RNA Biol. 2012;9:881–90. doi: 10.4161/rna.19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Igarashi J, Okawa ER, et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008;100:940–9. doi: 10.1093/jnci/djn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-sosa MA, Slagel VA, Trewyn RW. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. [DOI] [PubMed] [Google Scholar]
- Gorringe KL, Choong DY, Williams LH, et al. Mutation and methylation analysis of the chromodomain-helicase-DNA binding 5 gene in ovarian cancer. Neoplasia. 2008;10:1253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- Helbo A, Treppendahl M, Aslan D, et al. Hypermethylation of the VTRNA1-3 promoter is associated with poor outcome in lower risk myelodysplastic syndrome patients. Genes (Basel). 2015;6:977–90. doi: 10.3390/genes6040977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91:9700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill VK, Shinawi T, Ricketts CJ, et al. Stability of the CpG island methylator phenotype during glioma progression and identification of methylated loci in secondary glioblastomas. BMC Cancer. 2014;14:506. doi: 10.1186/1471-2407-14-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoue T, Weisenberger DJ, Lange CPE, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–82. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins MP, Wong JJL, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–11. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo Y- Y, Gong X- J, Mishra A, et al. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics. 2015;7:215–35. doi: 10.2217/epi.14.73. [DOI] [PubMed] [Google Scholar]
- Kaneda A, Feinberg AP. Loss of imprinting of IGF2: a common epigenetic modifier of intestinal tumor risk. Cancer Res. 2005;65:11236–40. doi: 10.1158/0008-5472.CAN-05-2959. [DOI] [PubMed] [Google Scholar]
- Karami S, Andreotti G, Liao LM, et al. LINE1 methylation levels in pre-diagnostic leukocyte DNA and future renal cell carcinoma risk. Epigenetics. 2015;10:282–92. doi: 10.1080/15592294.2015.1006505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Henikoff S. 5-Aza-CdR delivers a gene body blow. Cancer Cell. 2014;26:449–51. doi: 10.1016/j.ccell.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–78. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497–506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Y, El-Khoueiry A, Taverna P, et al. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol Oncol. 2015;9:1799–814. doi: 10.1016/j.molonc.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- Kulis M, Queirós AC, Beekman R, Martín-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta Gene Regul Mech. 2013;1829:1161–74. doi: 10.1016/j.bbagrm.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- Lay FD, Liu Y, Kelly TK, et al. The role of DNA methylation in directing the functional organization of the cancer epigenome. 2015:1–11. doi: 10.1101/gr.183368.114.Freely. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K- S, Park J- L, Lee K, et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget. 2014;5:3944–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson VV. DNA methylation as a universal biomarker. Expert Rev Mol Diagn. 2010;10:481–8. doi: 10.1586/erm.10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–5. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014a;5:587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Huang Q, Zeng F, et al. The prognostic value of global DNA hypomethylation in cancer: a meta-analysis. PLoS One. 2014b;9:e106290. doi: 10.1371/journal.pone.0106290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yao X, Wang Y, et al. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013;8:e59064. doi: 10.1371/journal.pone.0059064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Chan MF, Tomigahara Y, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–91. doi: 10.1128/MCB.22.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht JD. DNA Methylation Inhibitors in cancer therapy: the immunity dimension. Cell. 2015;162:938–9. doi: 10.1016/j.cell.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Lin JC, Jeong S, Liang G, et al. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–44. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou S, Lee H- M, Qin H, et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014;15:408. doi: 10.1186/s13059-014-0408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Song G, Tang Q, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF- k B signaling. J Clin Invest. 2015;125:1–18. doi: 10.1172/JCI78437.Iroquois. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair B, Kubicek S, Nijman SMB. Exploiting epigenetic vulnerabilities for cancer therapeutics. Trends Pharmacol Sci. 2014;35:136–45. doi: 10.1016/j.tips.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen EL, Stark AL, Zhang W, et al. The role of gene body cytosine modifications in MGMT expression and sensitivity to temozolomide. Mol Cancer Ther. 2014;13:1334–44. doi: 10.1158/1535-7163.MCT-13-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Zhang B, Bell RJA, et al. Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Res. 2014;24:761–74. doi: 10.1101/gr.164707.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenol-type that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Oki Y, Aoki E, Issa J-PJ. Decitabine – bedside to bench. Crit Rev Oncol Hematol. 2007;61:140–52. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, Makino T, Bailly-Bechet M, et al. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci U S A. 2012;109:5346–51. doi: 10.1073/pnas.1116763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res. 2000;450:155–66. doi: 10.1016/S0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- Pontier DB, Gribnau J. Xist regulation and function explored. Hum Genet. 2011;130:223–36. doi: 10.1007/s00439-011-1008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenel JD, Broman KW, Perlman EJ, et al. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of wilms tumor. JNCI J Natl Cancer Inst. 2001;93:1698–703. doi: 10.1093/jnci/93.22.1698. [DOI] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–6. [DOI] [PubMed] [Google Scholar]
- Rhee I, Jair KW, Yen RW, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003–7. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–73. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saied MH. Marzec J. Khalid S. et al. Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS One. 2012;7:e33213. doi: 10.1371/journal.pone.0033213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y Liang G. Egger G. et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006:9:435–43 doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Sakai T Toguchida J. Ohtani N. et al. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet. 1991;48:880–8. [PMC free article] [PubMed] [Google Scholar]
- Sandoval J Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012:22:50–5. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Schmelz K Wagner M. Dörken B. Tamm I. 5-Aza-2’-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. Int J Cancer. 2005;114:683–95. doi: 10.1002/ijc.20797. [DOI] [PubMed] [Google Scholar]
- Sharma S De Carvalho DD. Jeong S. et al. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet. 2011:7, e1001286. doi: 10.1371/journal.pgen.1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S Kelly TK. Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H Laird PW. Interplay between the cancer genome and Epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH. Abdel-Wahab O. Patel JP. Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- Shiohama Y Ohtake J. Ohkuri T. et al. Identification of a meiosis-specific protein. MEIOB, as a novel cancer/testis antigen and its augmented expression in demethylated cancer cells. Immunol Lett. 2014;158:175–82. doi: 10.1016/j.imlet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Silber JR. Bobola MS. Blank A. Chamberlain MC. O(6)-methylguanine-DNA methyltransferase in glioma therapy: promise and problems. Biochim Biophys Acta. 2012;1826:71–82. doi: 10.1016/j.bbcan.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD. Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Søes S Daugaard IL. Sprensen BS. et al. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience. 2014;1:367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P Paluch BE. Matsuzaki J. et al. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics. 2015;10:237–46. doi: 10.1080/15592294.2015.1017198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stresemann C Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- Su S-F, de Castro Abreu AL. Chihara Y. et al. A panel of three markers hyper- and hypomethylated in urine sediments accurately predicts bladder cancer recurrence. Clin Cancer Res. 2014;20:1978–89. doi: 10.1158/1078-0432.CCR-13-2637. [DOI] [PubMed] [Google Scholar]
- Taniguchi T Sullivan MJ. Ogawa O. Reeve AE. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci U S A. 1995;92:2159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczak K Czerwińska P. Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Poznań, Poland). 2015;19:A68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M Methylation profiling in acute myeloid leukemia. Blood. 2001;97:2823–9. doi: 10.1182/blood.V97.9.2823. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treppendahl MB, Qiu X, Søgaard A, et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood. 2012;119:206–16. doi: 10.1182/blood-2011-06-362541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H-C, Li H, Van Neste L, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–46. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–83. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrba L, Garbe JC, Stampfer MR, Futscher BW. A lincRNA connected to cell mortality and epigenetically-silenced in most common human cancers. Epigenetics. 2015;10:1074–83. doi: 10.1080/15592294.2015.1106673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen H, Fu S, et al. The involvement of CHD5 hypermethylation in laryngeal squamous cell carcinoma. Oral Oncol. 2011;47:601–8. doi: 10.1016/j.oraloncology.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Collisson EA, Mills GB, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ. Characterizing DNA methylation alterations from the cancer genome atlas. J Clin Invest. 2014;124:17–23. doi: 10.1172/JCI69740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Liang G. Contributions of DNA methylation aberrancies in shaping the cancer epigenome. Transl Cancer Res. 2015;4:219–34. doi: 10.3978/j.issn.2218-676X.2015.05.01. [DOI] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Wolff EM, Byun H- M, Han HF, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrangle J, Wang W, Koch A, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4:2067–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MS, Wang HP, Lin CC, et al. Loss of imprinting and overexpression of IGF2 gene in gastric adenocarcinoma. Cancer Lett. 1997;120:9–14. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Issa J-PJ. Epigenetic aspects of MDS and its molecular targeted therapy. Int J Hematol. 2013;97:175–82. doi: 10.1007/s12185-012-1197-4. [DOI] [PubMed] [Google Scholar]
- Yang X, Han H, De Carvalho DD, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–46. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegnasubramanian S, Haffner MC, Zhang Y, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68:8954–67. doi: 10.1158/0008-5472.CAN-07-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–40. [DOI] [PubMed] [Google Scholar]
- Yoo CB, Jeong S, Egger G, et al. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–8. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnett OJ, Sahgal A, Gosio J, et al. Treatment of elderly patients with glioblastoma: a systematic evidence-based analysis. JAMA Neurol. 2015;72:589–96. doi: 10.1001/jamaneurol.2014.3739. [DOI] [PubMed] [Google Scholar]