

SUMMARY
SORL1/SORLA is a sorting receptor involved in retromer-related endosomal traffic and an Alzheimer’s disease (AD) risk gene. Using CRISPR-Cas9, we deplete SORL1 in hiPSCs to ask if loss of SORL1 contributes to AD pathogenesis by endosome dysfunction. SORL1-deficient hiPSC neurons show early endosome enlargement, a hallmark cytopathology of AD. There is no effect of SORL1 depletion on endosome size in hiPSC microglia, suggesting a selective effect on neuronal endosomal trafficking. We validate defects in neuronal endosomal traffic by showing altered localization of amyloid precursor protein (APP) in early endosomes, a site of APP cleavage by the β-secretase (BACE). Inhibition of BACE does not rescue endosome enlargement in SORL1-deficient neurons, suggesting that this phenotype is independent of amyloidogenic APP processing. Our data, together with recent findings, underscore how sporadic AD pathways regulating endosomal trafficking and autosomal-dominant AD pathways regulating APP cleavage independently converge on the defining cytopathology of AD.
Graphical Abstract
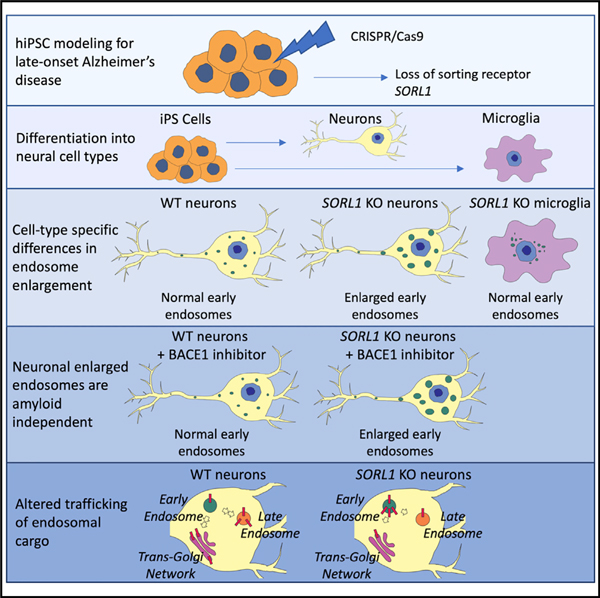
In Brief
Enlarged endosomes are an early cytopathology of Alzheimer’s disease (AD). Knupp et al. deplete the AD risk gene SORL1, a sorting receptor, in hiPSCs and report endosome enlargement in differentiated neurons but not microglia. Endosome enlargement is not dependent on amyloidogenic processing of APP but affects its trafficking within neurons.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the elderly, affecting nearly 40 million people worldwide (Alzheimer’s Association, 2019), and there is no treatment that alters disease progression. Recent therapeutic designs have focused on the main neuropathologic hallmarks of AD, accumulations of amyloid beta (Aβ) in senile plaques and abnormally phosphorylated tau protein in neurofibrillary tangles. The nearly universal failures of these trials to date, the vast majority of which have focused on removing or modulating Aβ, argues that other cellular pathways should be mechanistically studied for therapeutic development. Both genetics and pathology point to endosomal abnormalities and dysfunction as an early pathway in AD pathogenesis (Cataldo et al., 2000; Karch and Goate, 2015; Offe et al., 2006; Rogaeva et al., 2007). In particular, the SORL1 gene, which encodes the sorting protein SORLA, is highly relevant, being associated with both late-onset and early-onset forms of AD (Bettens et al., 2008; Holstege et al., 2017; Pottier et al., 2012; Reitz et al., 2011; Rogaeva et al., 2007). SORLA, first identified as a neuronal sorting receptor (Andersen et al., 2005; Hermans-Borgmeyer et al., 1998), is expressed in nearly all central nervous system (CNS) cell types (Zhang et al., 2014) and has multiple roles in endocytic sorting (Dumanis et al., 2015; Glerup et al., 2013; Herskowitz et al., 2012; Klinger et al., 2011; Nielsen et al., 2007), retromer-dependent retrograde trafficking (Fjorback et al., 2012), and amyloid precursor protein (APP) processing regulation (Andersen et al., 2005; Mehmedbasic et al., 2015; Rogaeva et al., 2007; Young et al., 2015). SORLA expression decreases in sporadic AD (SAD) (Dodson et al., 2006; Ma et al., 2009; Sager et al., 2007), and protein coding variants identified in early-onset AD families may lead to functional defects in the sorting of Aβ in cells (Caglayan et al., 2014). Rare loss-of-function truncation mutations have been found to be causal of late-onset AD (Holstege et al., 2017; Raghavan et al., 2018). We previously evaluated SORL1 activity in human induced pluripotent stem cell (hiPSC)-derived neurons from AD patients and controls and demonstrated that SORL1 expression induction with neurotrophic factors and its subsequent effect on neuronal Aβ peptides can be affected by the presence of AD-associated risk variants (Young et al., 2015).
Because of its role as a sorting receptor and because it may be decreased in AD, we hypothesized that SORL1 deficiency would affect endosome pathology and, by default, trafficking of cargo in the endo-lysosomal network. To evaluate this hypothesis, we generated SORL1-deficient hiPSC lines using CRISPR-Cas9 genome editing. We examined endosomal size in two cell types differentiated from these hiPSCs: neurons and microglia. We also tested whether inhibiting amyloidogenic processing of APP by inhibiting BACE modulated endosome enlargement in hiPSC-derived neurons. In this study, we report that loss of SORL1 by itself induces enlarged endosomes in hiPSC-derived neurons and that this phenotype is not altered by BACE inhibition. We also observe that SORL1 deficiency alters APP localization within the neuronal endosomal network. Interestingly, SORL1 loss does not induce endosome enlargement in hiPSC-derived microglial-like cells, suggesting cell type-specific differences in this early AD cytopathology. Taken together, our data suggest that loss of the known AD risk gene SORL1 induces early AD cytopathology in neurons and that, although it affects trafficking of APP, the endosomal pathology occurs in an amyloid-independent manner. We observe important endosome pathology differences in two CNS cell types, underscoring the complexity of this cellular pathway in the brain.
RESULTS
SORL1 Depletion in Human iPSC-Derived Neurons Leads to Enlarged Early Endosomes
We hypothesized that depletion of SORL1 in human neurons would allow us to investigate early features of AD that may involve endosomal network dysfunction. We established isogenic SORL1-knockout (KO) and wild-type (WT) hiPSC lines using CRISPR-Cas9 technology. We targeted exon 6 of the SORL1 gene, inducing indels that disrupted the reading frame, leading to complete loss of SORLA (SORL1 KO) protein in hiPSCs, differentiated neural progenitor cells (NPCs), and neurons (Figure 1A; Figure S1). In neurons differentiated from the SORL1-KO hiPSC lines, we quantified staining of endogenous EEA1 and Rab5 (markers of early endosome morphology) using confocal microscopy. We observed significantly increased fluorescence intensity from EEA1+ puncta (Figures 1B and 1C) and Rab5+ puncta (Figure S2A) in SORL1-KO neurons. Western blot analysis showed no change in total EEA1 protein between WT and SORL1-KO neurons (Figure S3A), suggesting that the increased fluorescence intensity is due to enlarged or fused early endosomes. We quantified endosome size and binned populations on the basis of ≥0.5 or <0.5 μm2 area distributions (Figure 1D). We observed significantly more endosomes ≥0.5 mm2 and a significant increase in the mean EEA1+ puncta area in SORL1-KO neurons (Figures 1E and 1F). Interestingly, we also noticed significantly enlarged endosomes in NPCs from the SORL1-KO lines (Figure S2B) suggesting that loss of SORL1 affects endosome morphology early in the neural lineage, after hiPSCs are driven to neuroectoderm. Finally, we also tested a short hairpin (shRNA) against SORL1 in WT neurons and again observed significantly enlarged endosomes (Figure S2C), suggesting that an acute reduction of SORL1 expression also leads to this phenotype.
Figure 1. Depletion of SORL1 Leads to Enlarged Early Endosomes in hiPSC-Derived Neurons.
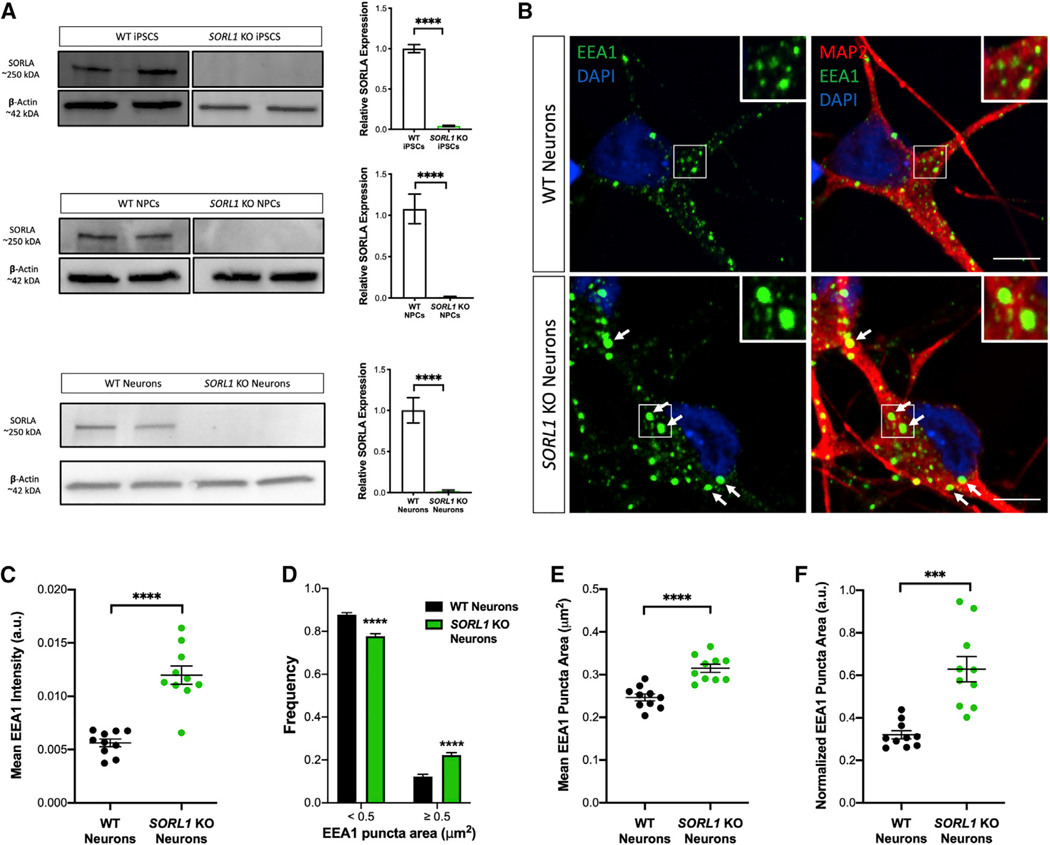
(A) Representative western blots and quantitation show reduction of SORLA protein levels to nearly zero in the KO hiPSCs, NPCs, and neurons. Quantification for all cell types includes two isogenic clones of each genotype (four total), n = 6 biological replicates. All values represent mean ± SD. All normally distributed data were analyzed using two-tailed unpaired t tests (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
(B) Representative immunofluorescence images of WT and SORL1-KO neurons stained with EEA1 (green), MAP2 (red), and DAPI (blue). White boxes indicate regions of interest (ROIs) magnified in insets. Arrows indicate enlarged EEA1+ puncta. Scale bars, 5 μm.
(C) Quantification of immunofluorescence images demonstrates increased EEA1+ puncta fluorescence intensity in SORL1-KO neurons compared with WT. n = 10 images (18–22 cells).
(D) Size distribution of EEA1+ puncta in SORL1-KO neurons compared with WT demonstrates increased frequency of EEA1+ puncta ≥0.5 μm2 in SORL1-KO neurons. n = 10 images (18–22 cells).
(E) Quantification of mean EEA1+ puncta area in WT compared with SORL1-KO neurons demonstrates increased mean EEA1+ puncta area in SORL1-KO neurons compared with WT. n = 10 images (18–22 cells).
(F) Normalization of mean EEA1+ puncta area by cell area in WT compared with SORL1-KO neurons. n = 10 images (18–22 cells).
For (B)–(F), all experiments represent two isogenic clones of each genotype (four total). All values represent mean ± SEM. Normally distributed data (C–F) were analyzed using parametric statistical tests. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by two-tailed unpaired t tests (C, E, and F) and two-way ANOVA (D).
Enlarged Early Endosomes in SORL1-Deficient Cells Are Present in hiPSC-Derived Neurons but Not Microglial-like Cells
AD-associated risk genes and their biological pathways may function differently between unique CNS cell types. Indeed, recent work using hiPSC-derived, gene-edited cells demonstrates that the strongest AD genetic risk factor, APOE ε4, affects different cellular AD phenotypes in a cell type-specific manner (Lin et al., 2018; Wang et al., 2018). Microglia, the innate immune cells of the CNS, also express SORL1 (Zhang et al., 2014), but the role of SORL1 in microglia is undefined, and functionality of the endosomal network is likely very different in these highly phagocytic cells compared with neurons, which are professional secretory cells. We differentiated SORL1-KO hiPSCs to microglial-like cells using a previously published protocol (McQuade et al., 2018) (Figure S4) and analyzed endogenous EEA1 staining using similar protocols as used with hiPSC-derived neurons and NPCs. Surprisingly, we did not observe differences in EEA1 fluorescence intensity (Figures 2A and 2B) or puncta size (Figures 2C and 2D) when we compared WT and SORL1-KO microglial-like cells. Microglia are derived from mesoderm/hematopoietic lineages, while NPCs and neurons are derived from neural ectoderm. Our findings suggest that the endosomal trafficking and sorting functions of SORL1 may depend on cell lineage and that SORL1-dependent early endosome pathology is specific to neuronal cells.
Figure 2. Depletion of SORL1 Does Not Lead to Enlarged Early Endosomes in hiPSC Microglial-like Cells.
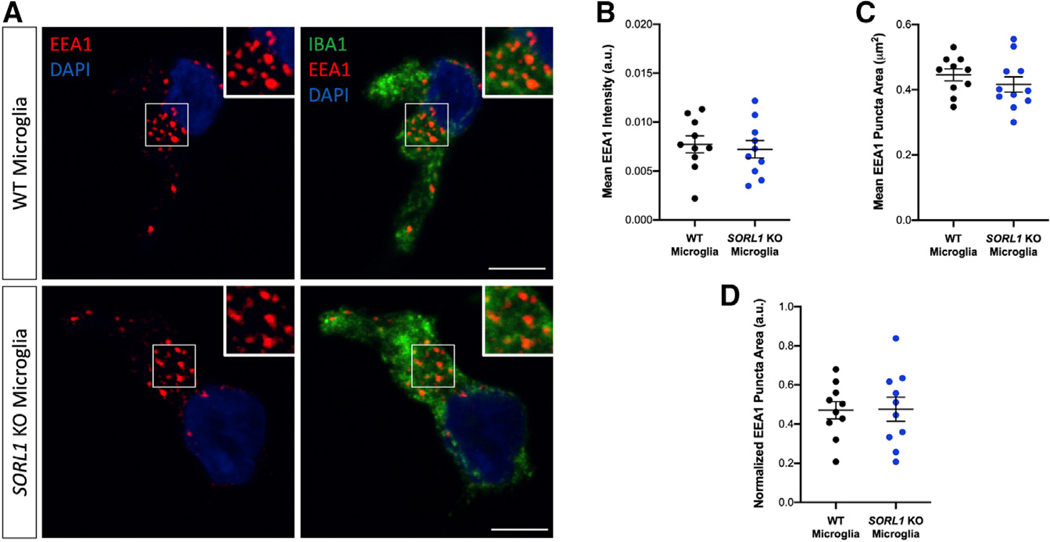
(A) Representative immunofluorescence images of WT and SORL1-KO microglial-like cells stained with EEA1 (red), Iba1 (green), and DAPI (blue). White boxes indicate regions of interest (ROIs) magnified in insets. Scale bars, 5 μm.
(B) Quantification of immunofluorescence images demonstrates no significant change in mean EEA1+ puncta intensity in SORL1-KO microglial-like cells compared with WT. n = 10 images (10–14 cells).
(C) Quantification of immunofluorescence images demonstrates no significant change in EEA1+ puncta area in SORL1-KO microglial-like cells compared with WT. n = 10 images (10–14 cells).
(D) Normalization of mean EEA1+ puncta area by cell area in WT compared with SORL1-KO microglial-like cells. n = 10 images (18–22 cells). All experiments represent two isogenic clones of each genotype (four total). Normally distributed data (B–D) were analyzed using two-tailed unpaired t tests and found non-significant (ns).
Loss of SORL1 in hiPSC-Derived Neurons Alters APP Trafficking and Processing in the Endosomal Network
Enlarged endosomes are indicative of endosomal traffic jams and may delay the proper maturation and progression of vesicles and cargo for processing and degradation (Kaur and Lakkaraju, 2018). One well-characterized cargo of SORL1-dependent trafficking is APP (Fjorback et al., 2012). Previous studies have shown that molecules that enhance retrograde trafficking away from the early endosome toward the Golgi or back to the plasma membrane reduce amyloidogenic cleavage and decrease colocalization of APP with early endosomal markers (Mecozzi et al., 2014; Young et al., 2018). We quantified the localization of APP in various compartments of the endo-lysosomal network in SORL1-deficient neurons using confocal microscopy. We observed an increase in colocalization of APP with EEA1 (Figures 3A and 3B) and a decrease in colocalization of APP with TGN38, a marker for the trans-Golgi network (TGN) (Figures 3C and 3D), confirming reduced retrograde transport of APP in the absence of SORL1. We also observed decreased colocalization of APP with Rab7, a marker of maturing endosomes (Figures 3E and 3F), suggesting either that in the context of SORL1 deficiency, vesicles with APP cargo are not maturing into late endosomes/lysosomes, or that the trafficking of APP itself to these compartments is impaired.
Figure 3. SORL1 Depletion in hiPSC-Derived Neuron Alters APP Localization in the Endosomal Network.
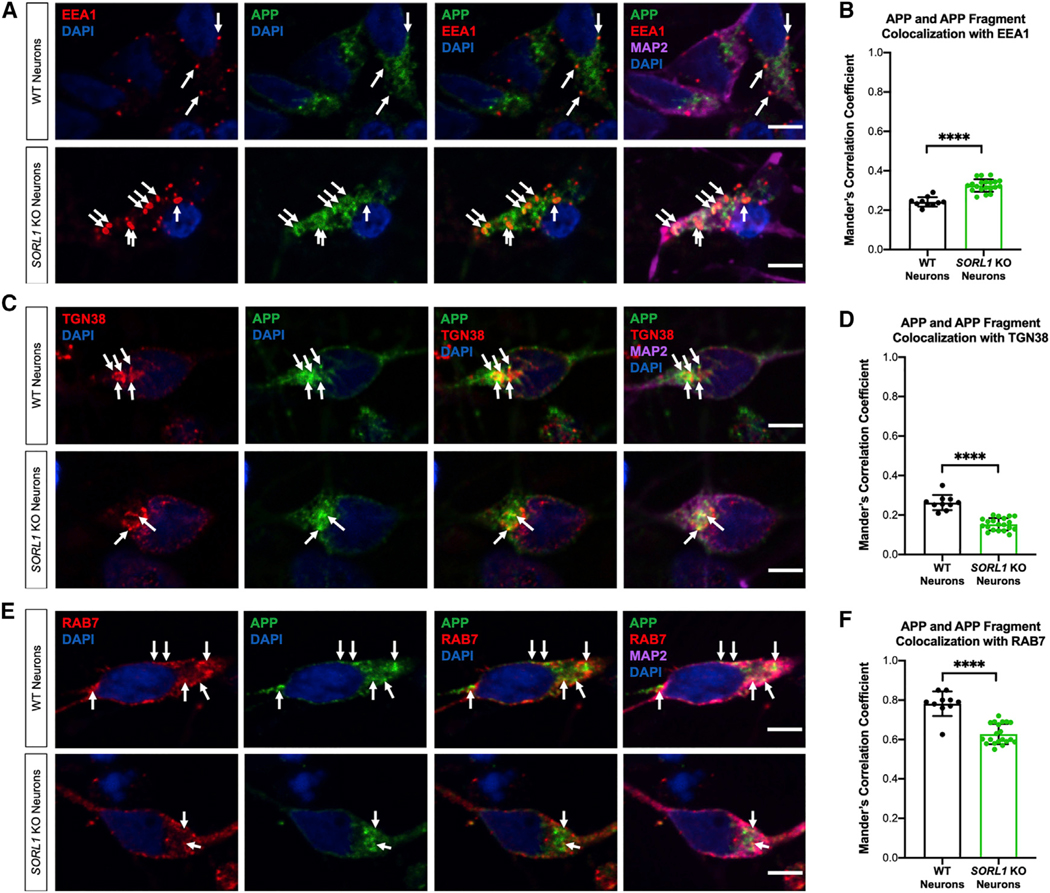
(A) SORL1 depletion results in APP accumulation in EEA1+ early endosomes. Representative immunofluorescence images of WT and SORL1-KO neurons stained with EEA1 (red), APP C-terminal antibody (green), MAP2 (far-red), and DAPI (blue). Arrows indicate colocalization of EEA1+ puncta with APP and APP fragments. Scale bars, 5 μm.
(B) Quantification of colocalization of EEA1 with APP. n = 10–20 images.
(C) SORL1-KO neurons show reduced colocalization of APP and APP fragments with TGN38+ puncta. Representative immunofluorescence images of WT and SORL1-KO neurons stained with TGN38 (red), APP C-terminal antibody (green), MAP2 (far-red), and DAPI (blue). Arrows indicate colocalization of TGN38+ puncta with APP and APP fragments. Scale bars, 5 μm.
(D) Quantification of colocalization of TGN38 with APP. n = 10–20 images.
(E) SORL1-KO neurons show reduced colocalization of APP and APP fragments with Rab7+ puncta. Representative immunofluorescence images of WT and SORL1-KO neurons stained with Rab7 (red), APP C-terminal antibody (green), MAP2 (far-red), and DAPI (blue). Arrows indicate colocalization of Rab7+ puncta with APP and APP fragments. Scale bars, 5 μm.
(F) Quantification of colocalization of Rab7 with APP. n = 10–20 images.
All experiments represent two isogenic clones of each genotype (four total). All values represent mean ± SD. All normally distributed data were analyzed by parametric statistical tests. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by two-tailed, unpaired t test (B, D, and F).
Cleavage of APP to amyloidogenic fragments occurs in acidic early endosomes (Small and Gandy, 2006). The impact of SORL1 expression on APP processing has been previously described by us and others in studies showing that knockdown of SORL1 increases Aβ peptides in non-neuronal cells (Rogaeva et al., 2007) and in hiPSC-derived neurons (Young et al., 2015). We confirmed that SORL1-KO neurons have increased Aβ peptides released into the culture media and that both Aβ1–40 and Aβ1–42 species were equally increased, without inducing a significant change in the Aβ 42:40 ratio (Figures 4E–4G). We did not observe a change in APP holoprotein expression in SORL1 KO neurons (Figure S3B).
Figure 4. Enlarged Early Endosomes in SORL1-Deficient Neurons Are Independent of Amyloidogenic APP Processing.
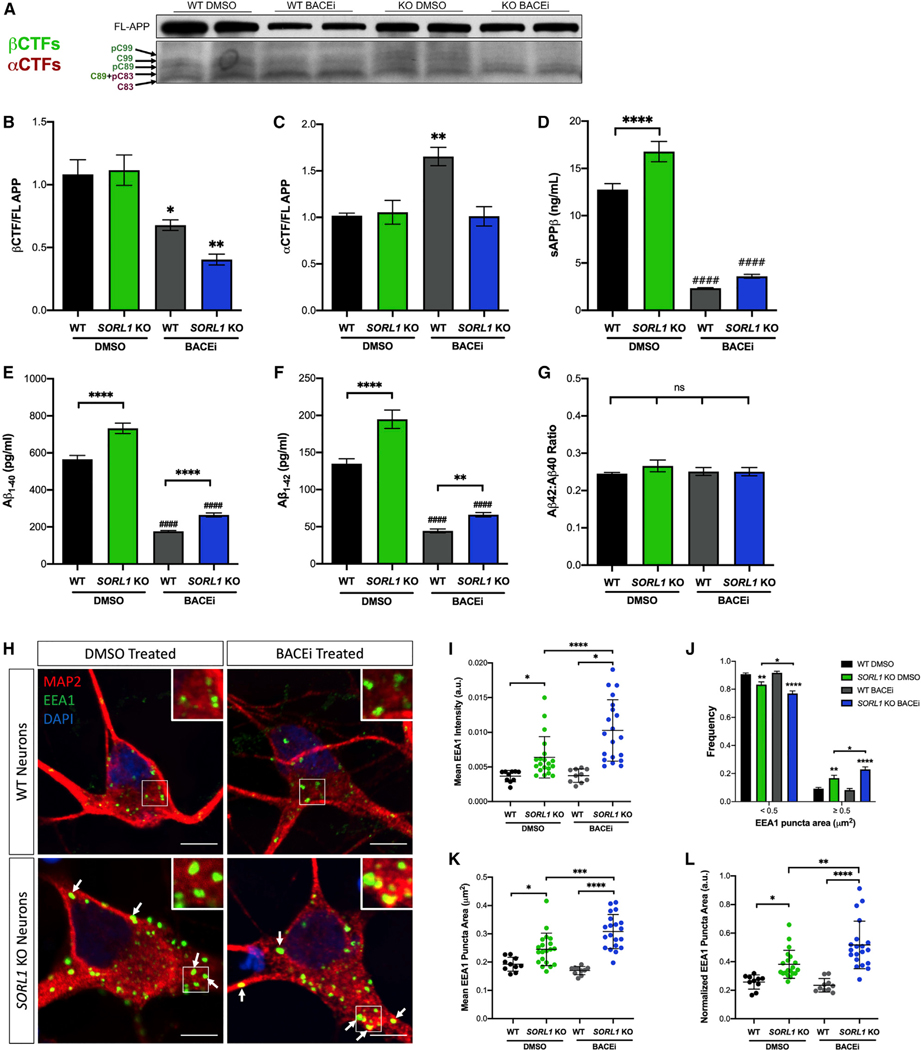
(A) Representative western blot of full-length (FL) APP and APP CTFs in WT and SORL1-KO cell lines treated with BACEi or DMSO control.
(B) βCTFs of APP are significantly reduced in BACEi-treated neurons as determined by western blot.
(C) αCTFs of APP are significantly higher in WT neurons treated with BACEi but unchanged in SORL1-KO neurons treated with BACEi, as determined by western blot.
(D) sAPPβ fragments are significantly reduced in WT and SORL1-KO neurons with BACEi treatment, as determined by ELISA. n = 3–6 technical replicates.
(E and F) SORL1-KO neurons secrete higher levels of (E) Aβ1–40 and (F) Aβ1–42 peptides than WT cells, indicated by asterisks. BACEi significantly reduces these peptides in both genotypes compared with DMSO controls, indicated by hashmarks. In the presence of BACEi, SORL1-KO neurons still secrete increased levels of (E) Aβ1–40 and (F) Aβ1–42 peptides, indicated by asterisks. n = 3–6 technical replicates.
(G) Neither SORL1-KO nor treatment with BACEi changes the ratio of Aβ1–42 to Aβ1–40 peptides. n = 3–6 technical replicates.
(H) Representative immunofluorescence images of WT and SORL1-KO neurons treated with either DMSO or BACEi for 72 h. Neurons were stained with EEA1 (green), MAP2 (red), and DAPI (blue). White boxes indicate regions of interest (ROIs) magnified in insets. Arrows indicate enlarged EEA1+ puncta. Scale bars, 5 μm.
(I) Quantification of immunofluorescence images demonstrates increased mean EEA1+ puncta intensity in BACEi-treated SORL1-KO neurons compared with DMSO-treated controls. n = 10–20 images (42–58 cells).
(J) Size distribution of EEA1+ puncta in SORL1-KO neurons compared with WT neurons with and without BACEi treatment. n = 10–20 images (42–58 cells).
(K) Quantification of immunofluorescence images demonstrates enlarged EEA1+ puncta area in BACEi-treated SORL1-KO neurons compared with DMSO-treated controls. n = 10–20 images (42–58 cells).
(L) Normalization of mean EEA1+ puncta area by cell area in BACEi-treated WT and SORL1-KO neurons. n = 10–20 images (42–58 cells).
All experiments represent two isogenic clones of each genotype (four total). All values represent mean ± SEM. Normally distributed data (B–G and J) were analyzed using parametric statistical tests. Non-normally distributed data (I–L) were analyzed using non-parametric statistical tests. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by one-way ANOVA (B–G), by two-way ANOVA (J), or by Kruskal-Wallis test (I–L).
Enlarged Early Endosomes in the Context of SORL1 Depletion Occur Independent of Amyloidogenic APP Processing
Recent work has shown that neurons with introduced familial AD (FAD) mutations also show enlarged endosome morphology that is FAD gene dose dependent (Kwart et al., 2019). In Kwart et al., (2019) study, endosome enlargement was dependent on β-secretase processing of APP. To test whether amyloidogenic processing of APP also contributed to early endosome enlargement in the context of SORL1 loss, we treated SORL1-KO and isogenic WT neurons with a BACE inhibitor (BACEi) or DMSO (vehicle control). This treatment reduced the β-C-terminal fragment (βCTF) and the soluble APPβ (sAPPβ) fragments of APP (Figures 4A, 4B, and 4D) and reduced Aβ peptide levels in our neurons (Figures 4E and 4F). Interestingly, in the SORL1-defifient cells we observed an even stronger increase in mean EEA1 intensity (Figures 4H and 4I) and no amelioration of early endosome enlargement compared with SORL1-KO cells treated with DMSO (Figures 4J–4L). There was no difference in endosome size or EEA1 intensity in WT cells treated with BACEi compared with DMSO (Figures 4H–4L). These data suggest that the mechanism by which loss of SORL1 expression induces endosome enlargement is independent of amyloidogenic APP processing and differs from how FAD mutations affect the endosomal network. Although BACEi significantly reduced Aβ levels in all cell lines, there were still significantly higher levels of Aβ peptides secreted into the culture media in the SORL1-KO neurons (Figures 4E and 4F). Together these data suggest that loss of SORL1 influences APP processing, but early endosome enlargement is independent of amyloidogenic cleavage of APP. The levels of αCTFs and βCTFs did not differ between SORL1-KO and WT neurons under DMSO conditions, although we observed an increase in αCTFs in WT neurons with BACEi treatment (Figures 4A–4C). This suggests that α cleavage may be exacerbated under these conditions, as α- and β-secretase have been shown to have competitive activity (Netzer et al., 2017; Skovronsky et al., 2000). We did not observe an increase in αCTFs in SORL1-KO neurons (Figures 4A–4C), which is consistent with the observation that loss of SORL1 retains APP in the early endosome, where it is unavailable for α cleavage.
DISCUSSION
Endosome enlargement is an early cytopathology in AD (Cataldo et al., 2000), and abnormalities in the endo/lysosomal network are prevalent throughout human neurodegenerative disorders (Vagnozzi and Praticò, 2019). Multiple genetic studies have also identified risk loci in or near genes involved in endosomal trafficking (Van Acker et al., 2019). This points to protein trafficking as an important, and potentially modifiable, pathway for AD and related disorders. As defects in the endosomal network are an early event in AD pathogenesis, this is an important target whose modulation may affect downstream pathologies. Indeed, our previous work has shown that molecules that enhance endosomal trafficking pathways affect Aβ and tau independently, supporting the premise that trafficking dysfunction is an early driver of AD (Young et al., 2018). In this study, we used hiPSC and CRISPR-Cas9 technology to ask whether loss of an established AD risk gene, SORL1, induces early endosome pathology in neurons and other cell types affected in AD. We document significantly enlarged endosomes in hiPSC-derived neurons lacking SORL1, demonstrating that loss of this sorting receptor is sufficient to induce this pathology in neurons (Figures 1A–1F).
Enlarged early endosomes were first reported in neurons in post-mortem brain (Cataldo et al., 2000), and models using hiPSCs with FAD mutations or from SAD patients have also shown endosome enlargement in neurons (Israel et al., 2012; Lin et al., 2018; Raja et al., 2016). In addition to neurons, microglia are also highly reliant on the endosomal network for the trafficking of internalized substrates (Solé-Domènech et al., 2016). We differentiated microglial-like cells from our SORL1-deficient hiPSCs and performed the same analysis as with hiPSC-derived neurons. Interestingly, we did not observe significant early endosome enlargement, as marked by EEA1+ puncta in SORL1-deficient microglial-like cells (Figures 2A–2D). These data demonstrate that loss of SORL1 affects microglial-like cells differently than neuronal cells, emphasizing important differences in cell type-specific responses to insults involving the endosomal network. For example, recent work reported that loss of the endosomal adaptor protein TOM1 in microglia led to reduced microglial branching and impaired phagocytosis, while in neurons it resulted in upregulation of inflammatory signaling genes (Martini et al., 2019). Because of the vastly different roles microglia and neurons play in the CNS, future work on analyzing the functionality of endosomal trafficking, in addition to endosome size, in SORL1-deficient microglial-like cells is warranted.
In neurons, one of the best characterized cargos of SORL1 is APP. The protein SORLA directly binds APP (Andersen et al., 2006) and serves as an adaptor molecule, via VPS26, in retromer-dependent retrograde trafficking of APP (Fjorback et al., 2012). We observed that loss of SORL1 alters APP trafficking in the endosomal network, leading to increased colocalization of APP in early endosomes and a decrease in its localization in the TGN (Figures 3A–3D). Thus, we confirm in human neurons that SORL1 functions as a retromer receptor that traffics APP out of endosomes. We also observe decreased APP localization in Rab7+ late endosomes/lysosomes (Figures 3E and 3F), which suggests a defect in either endosomal maturation or trafficking of APP toward degradative compartments.
Previous studies have established that the APP βCTFs are toxic and can themselves cause endosomal enlargement (Israel et al., 2012; Kim et al., 2016; Kwart et al., 2019; Lauritzen et al., 2016; Tamayev et al., 2012; Xu et al., 2016). Recently, in a large cohort of FAD hiPSC-derived neurons, endosome enlargement due to FAD mutations was rescued by BACEi treatment (Kwart et al., 2019). In our study, BACEi treatment reduced Aβ peptides (Figures 4E and 4F) but did not ameliorate, and in fact exacerbated, endosomal size increases induced by loss of SORL1 in hiPSC-derived neurons (Figures 4H–4L). This suggests that endosomal homeostasis may require a delicate balance of cargo and that endosome pathology is a global event in AD pathogenesis affected by many different factors, which likely differ between early- and late-onset forms of the disease. Interestingly, in SORL1-KO neurons the βCTF of APP is not significantly increased compared with WT (Figures 4A and 4B), and we document an even more significant reduction of βCTFs with BACEi in SORL1-KO versus WT cells (Figure 4B). Together, these data indicate that the enlarged endosome phenotype we observe is independent of amyloidogenic processing of APP and is directly tied to the loss of an endosomal sorting receptor. We do observe an increase in Aβ peptides in SORL1-deficient neurons, as has been seen in other models (Andersen et al., 2005; Rogaeva et al., 2007). Therefore, it is plausible that the increase in Aβ peptides in SORL1-KO neurons may be due in part to reduced trafficking of Aβ or APP to lysosomes. Indeed, previous studies have demonstrated that the VPS10 region of SORL1 is important for trafficking Aβ to lysosomes for degradation (Caglayan et al., 2014).
The genetic mutations causing FAD (in APP or PSEN genes) affect APP processing. In contrast, genetic studies of SAD have identified approximately two dozen associated candidate genes, a group of which converge around vesicular trafficking and endocytosis pathways. Among these, SORL1 is common, and truncation mutations causing loss-of-function of the SORLA protein are shown to be causal for late-onset AD (Holstege et al., 2017; Raghavan et al., 2018), making SORL1 depletion a reliable approach for studying this pathway. Taken together with other recent findings (Kwart et al., 2019), our study establishes that two causative pathways in AD, APP cleavage and endosomal trafficking, independently cause AD’s hallmark cytopathology.
Our work confirms the importance of SORL1 in modulating processing APP and suggests that it may have a broader role in regulating endosomal network function. Our data support the idea that endosome pathology is an upstream event from amyloidogenic APP cleavage and Aβ generation and suggest that further disruptions of endosomal cargo processing or homeostasis (such as BACE inhibition) can enhance traffic jams or delay maturation in the absence of SORL1. Finally, our work demonstrates that hiPSC-derived neuronal models are valuable tools for dissecting early pathogenic events in AD and may help clarify the molecular mechanisms that underlie therapeutic failures.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jessica E. Young (jeyoung@uw.edu).
Materials Availability
There are restrictions to the availability of the cell lines due to the lack of an external centralized repository for their distribution and our need to maintain the stock. We are glad to share these cell lines with reasonable compensation by requestor for its processing and shipping. All requests for cell lines should be directed to the Lead Contact.
Data and Code Availability
No datasets were generated during this study. CellProfiler software is available at https://cellprofiler.org/. The images and CellProfiler pipelines used in this study are available from the Lead Contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
CRISPR/Cas9 Genome Editing
All genome editing was performed in the previously published and characterized CV background human induced pluripotent stem cell line (Young et al., 2015). This cell line is male and has a APOE ε3/ε4 genotype (Levy et al., 2007). Genome edited lines were generated using published protocols (Young et al., 2018). Briefly, guide RNAs (gRNAs) to SORL1 were generated using the Zhang Lab CRISPR Design website at MIT (http://zlab.bio/guide-design-resources) and selected to minimize off-target effects. gRNAs were cloned into vector px458 that co-expresses the Cas9 nuclease and GFP, and hiPSCs were electroporated with the plasmid. Electroporated hiPSCs were FACS sorted for GFP, plated in 10 cm plates at a clonal density (~1×10∧4 cells/plate), and allowed to grow for roughly 2 weeks. Colonies were picked into 96 well plates and split into two identical sets. One set was analyzed for sequence information by Sanger sequencing and one set was expanded for cell line generation. Four clones were chosen for experiments reported in this publication. Two wild-type clones, designated clone A6 and clone A7, and two SORL1 KO clones, designated clone E1 and clone E4, were selected. Sequencing data for all cell lines was confirmed by measuring protein expression using western blot techniques (Figure 1A). All four clones were shown to have normal karyotypes. Authentication: Sequencing data confirms CV cell lines by presence of a SNP unique to this genetic background (Figure S1). All cell lines are routinely karyotyped by Diagnostic Cytogenetics, Inc. (Seattle, WA), and tested for mycoplasma (MycoAlert).
CRISPR/Cas9 gRNA, ssODN, and Primer Sequences
gRNA: ATTGAACGACATGAACCCTC
ssODN: GGGAATTGATCCCTATGACAAACCAAATACCATCTACATTGAACGACATGAACCCTCTGGCTACTCCACGTCTTCCGA AGTACAGATTTCTTCCAGTCCCGGGAAAACCAGGAAG
Forward primer: ctctatcctgagtcaaggagtaac
Reverse primer: ccttccaattcctgtgtatgc
PCR amplifies 458 bp sequence
METHOD DETAILS
Western Blotting
Cell lysates were run on 4%–20% Mini-PROTEAN TGX Precast Protein Gels (#4561096; Biorad) or 16.5% Criterion Tris-Tricine Gel (#3450063; BioRad) and transferred to PVDF membranes. Membranes were probed with antibodies to Sortilin-related receptor 1 (SORLA) at 1:1000 (BD 611860 and abcam ab190684), β-actin (ACTB) at 1:2000 (EMD Millipore Corp MAB1501), early endosome antigen 1 (EEA1) at 1:5000 (BD 610456), and amyloid precursor protein (APP) at 1:500 (Invitrogen 14–9749-80) and APPY188 (Ab32136 Rabbit Y188; Abcam) at 1:1000. Imaging was performed with a BioRad ChemiDoc system and quantification was performed using ImageJ software.
hiPSC Neuronal Differentiation
All cell lines were maintained at 37°C in a 5% CO2 incubator. hiPSCs were differentiated to neurons using previously described dual-SMAD inhibition techniques (Rose et al., 2018). Briefly, hiPSCs were plated on Matrigel (Growth factor reduced basement membrane matrix; # 356231; Corning) coated 6-well plates at a density of 3.5 million cells per well and fed with Basal Neural Maintenance Media (1:1 DMEM/F12 (#11039047 Life Technologies) + glutamine media/neurobasal media (#21103049, GIBCO), 0.5% N2 supplement (# 17502–048; Thermo Fisher Scientific,) 1% B27 supplement (# 17504–044; Thermo Fisher Scientific), 0.5% GlutaMax (# 35050061; Thermo Fisher Scientific), 0.5% insulin-transferrin-selenium (#41400045; Thermo Fisher Scientific), 0.5% NEAA (# 11140050; Thermo Fisher Scientific), 0.2% β-mercaptoethanol (#21985023, Life Technologies) + 10 μM SB-431542 + 0.5 μM LDN-193189 (#1062443, Biogems). Cells were fed daily for seven days. On day eight, cells were incubated with Versene (#15640066, GIBCO), gently dissociated using cell scrapers, and split 1:3. On day nine media was switched to Basal Neural Maintenance Media + 20 ng/mL FGF (R&D Systems, Minneapolis, MN) and fed daily. On day sixteen, cells were passaged 1:3. Cells were fed until approximately day twenty-three. At this time, cells were FACS sorted to obtain the CD184/CD24 (#557145/561646 BD PharMingen) positive, CD44/CD271 (#555479/557196 BD PharMingen) negative neural precursor cell (NPC) population (Yuan et al., 2011). Following sorting, NPCs were expanded for neural differentiation. For cortical neuronal differentiation, NPCs were plated out in 10 cm plates at a density of 6 million cells/plate. After 24 hours, cells were switched to Neural Differentiation media (DMEM-F12 + glutamine, 0.5% N2 supplement, 1% B27 supplement, 0.5% GlutaMax) + 0.2 μg/mL brain-derived neurotrophic factor (#450–02 PeproTech) + 0.2 μg/Ml glial-cell-derived neurotrophic factor (#450–10 PeproTech) + 0.5 M dbcAMP (#D0260 Sigma Aldrich). Media was refreshed twice a week for three weeks. After three weeks, neurons were selected for CD184/CD44 negative population by magnetic-activated cell sorting and plated for experiments.
Purification of Neurons
Following three weeks of differentiation, cells were dissociated with Accutase (#AT104–500 Innovative Cell Tech) and resuspended in IMAG solution (PBS + 0.5% bovine serum albumin [Sigma Aldrich] + 2 mM ethylenediaminetetraacetic acid [ThermoFisher]). Following a modification of Yuan et al., 2011(Yuan et al., 2011), cells were incubated with PE-conjugated mouse anti-Human CD44 and mouse anti-Human CD184 antibodies (BD PharMingen) at a concentration of 5 μL/10 million cells. Following antibody incubation, cells were washed with IMAG solution and incubated with anti-PE magnetic beads (BD PharMingen) at a concentration of 25 μL/10 million cells. Bead-antibody complexes were pulled down using a rare earth magnet, supernatants were selected, washed, and plated at an appropriate density.
hiPSCs Microglial-like Cells Differentiation
SORL1 KO and WT hiPSCs were differentiated into microglial-like cells as previously described (McQuade et al., 2018). Briefly, hiPSCs were plated in mTESR™ 1 medium (STEMCELL Technologies) supplemented with ROCK Inhibitor (Y-27632; Apex Bio) on Matrigel (Growth factor reduced basement membrane matrix; # 356231; Corning) coated 6 well plates (#657160; CELLSTAR) at a dilution of 1:30. To begin hematopoietic progenitor differentiation, these cells were passaged to get a density of ~100 colonies (~150 cells per colony of iPSCs) per well of a 6 well plate. On day 0, mTESR™ 1 medium was replaced with STEMdiff Hematopoietic Supplement A medium from the STEMdiff Hematopoietic kit (# 05310; STEMCELL technologies). On day 3, when colonies became flattened, medium was replaced with STEMdiff Hematopoietic Supplement B medium from the STEMdiff Hematopoietic kit (# 05310; STEMCELL technologies). Cells remained in this medium for 7 additional days. By day 10, non-adherent hematopoietic progenitor cells (HPCs) coming off from the flattened colonies were harvested by removing medium. Any remaining HPCs/floating cells were collected by gentle PBS washes. At this point, HPCs were either frozen using Bambanker cell freezing medium (#BBH01; Bulldog-Bio) or plated at a density of 0.2 M cells per well of a Matrigel coated (1:60 dilution) 6 well plate in microglia differentiation medium for 25 days. Microglia differentiation medium comprised of DMEM-F12 (#11039047; Thermo Fisher Scientific), Insulin-transferrin-selenite (#41400045; Thermo Fisher Scientific), B27 (# 17504–044; Thermo Fisher Scientific), N2 (# 17502–048; Thermo Fisher Scientific), glutamax (# 35050061; Thermo Fisher Scientific), non-essential amino acids (# 11140050; Thermo Fisher Scientific), monothioglycerol (# M1753; Sigma), Insulin (# I2643; Sigma) freshly supplemented with TGF-β (#130–108-969, Miltenyl), IL-34 (# 200–34; Peprotech) and M-CSF (#PHC9501; Thermo Fisher Scientific). On day 25, this medium was supplemented with CD200 (#C311; Novoprotein) and CX3CL1 (#300–31; Peprotech) for maturation of microglial-like cells Cells remained in this medium for 3 days. On day 28, microglial-like cell differentiation was complete, and these cells were plated in laminin (#L2020; Sigma) coated coverslips (12mm diameter, #1760–012; cglifesciences) in a 24 well plate for immunocytochemistry with appropriate antibodies.
Amyloid Beta and sAPP Measurements
Aβ peptides were measured as previously described (Young et al., 2015). Briefly, purified neurons were seeded at a density of 200,000 cells/well of a 96-well plate and maintained in culture for 5 days. Medium and lysates were harvested from triplicate wells. To measure secreted Aβ peptides, media was run on an Aβ Triplex ELISA plate (Meso Scale Discovery #151200E-2). To measure sAPPβ, media was run on a sAPPα/sAPPβ ELISA plate (Meso Scale Discovery #K15120E-1).
Immunocytochemistry
Purified neurons were seeded at a density of 500,000 cells per well of a 24-well plate on glass coverslips coated with Matrigel. After 5 days in culture, cells were fixed in 4% paraformaldehyde (PFA, Alfa Aesar, Reston, VA) for 15 minutes. Cells were incubated in blocking buffer containing 2.5% bovine serum albumin and 0.1% Triton X-100 (Sigma Aldrich, St Louis, MO) for 30 minutes at room temperature then incubated in a primary antibody dilution in blocking buffer for 2 hours at room temperature. Cells were washed 3× with PBS + 0.1% Triton X-100 and incubated with a secondary antibody dilution in blocking buffer for 1 hour at room temperature. Cells were washed 3× in PBS and mounted on glass slides with ProLong Gold Antifade mountant (ThermoFisher, Waltham, MA). The following primary antibodies were used: Ras-related protein Rab-5A (RAB5A) at 1:500 (Synaptic Systems 108 011); early endosome antigen 1 (EEA1) at 1:500 (BD 610456); amyloid precursor protein (APP) at 1:250 (Abcam ab32136); microtubule-associated protein 2 (MAP2) at 1:1000 (Abcam ab92434); Nestin (NES) at 1:1000 (Santa Cruz Biotechnology sc23927); Trans-Golgi network integral membrane protein (TGN38) at 1:250 (Santa Cruz sc-166594); Ras-related protein Rab-7a (Rab7) at 1:1000 (Abcam ab50533); DAPI at 1 μg/mL final (Alfa Aesar).
Confocal Microscopy and Image Processing
All microscopy and image processing were performed under blinded conditions. Confocal z stacks were obtained using a Nikon A1R confocal microscope with ×63 and ×100 plan apochromat oil immersion objectives. Image processing was performed using ImageJ software (Schindelin et al., 2012). For endosome analysis, 10–20 fields were analyzed for a total of 10–58 cells. Maximum intensity projections of confocal stacks were generated, and background was subtracted using the rolling ball algorithm. Endosome channels were enhanced using contrast limited adaptive histogram equalization algorithms (CLAHE) and masked using cell body stains. Size and intensity measurements were performed using CellProfiler software (McQuin et al., 2018). Individual puncta were identified using automated segmentation algorithms. Mean intensity of each puncta was measured and has been presented as a mean value over all puncta per field. Similarly, pixel area of each puncta was measured and has been presented as a mean area over all puncta per field. Finally, mean puncta area normalized by total cell area calculated from cell body stains is also presented. For colocalization analysis, a minimum of 10 fields of confocal z stacks were captured using the ×100 plan apochromat oil immersion objective. Median filtering was used to remove noise from images and manual thresholding was applied to all images. The colocalization of APP with endocytic markers was quantified using JACOP plugin (Bolte and Cordeliè res, 2006) in ImageJ and presented as Mander’s correlation coefficient.
BACE Inhibition
Purified neurons derived from SORL1 KO and WT hiPSCs were plated on a matrigel coated 96 well plate at a density of 2×105 cells per well. After 5 days, cells were treated with either 25 nM β-secretase inhibitor (BACEi; LY2886721; # HY-13240; MedChemExpress) or DMSO (as a vehicle control) for 72h. All experiments were performed after 72 hours of drug treatment. At this point, medium from DMSO or BACEi treated neurons was harvested for quantification of Aβ1–40, Aβ1–42, and sAPPβ peptides secreted by neurons by ELISA. Additionally, cell lysates were harvested for determining protein levels of β-C-terminal fragment (βCTF) of amyloid precursor protein (APP) by western blot.
SORL1 shRNA design and transduction
Sequences for SORL1 shRNA and a scrambled (SCR) shRNA were designed previously (Young et al., 2015) and cloned into the pSI-COR-GFP plasmid (Ventura et al., 2004). Plasmids were packaged into lentivirus using HEK293FT cells. Virus was purified using PEG-it. Following differentiation and purification of neurons, 5 μL of purified virus was added to cells for 72 hours. Knockdown of SORL1 was confirmed by western blot.
QUANTIFICATION AND STATISTICAL ANALYSIS
We used two independent clones of homozygous knockout cell lines and two independent clones of isogenic WT cell lines (cells that underwent the CRISPR/Cas9 transfection and sub-cloning process, but in which editing events did not occur). For all imaging experiments the data was analyzed in a blinded manner. Experimental data was tested for normal distributions using the Shapiro-Wilk normality test. Normally distributed data was analyzed using parametric two-tailed unpaired t tests, one-way ANOVA tests, or two-way ANOVA tests. Non-normally distributed data was analyzed by non-parametric Kruskal-Wallis tests. Significance was defined as a value of p > 0.05. All statistical analysis was completed using GraphPad Prism software. Statistical details of individual experiments, including biological and technical replicate information, can be found in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-SORLA | Abcam | Cat#ab190684 |
| Purified mouse monoclonal anti-LR11 (clone 48) | BD Biosciences | Cat#611860; RRID: AB_399340 |
| Mouse monoclonal anti-Actin (clone C4) | EMD Millipore Corp | Cat#MAB1501; RRID: AB_2223041 |
| Mouse monoclonal anti-EEA1 | BD Biosciences | Cat#610456; RRID: AB_397829 |
| Mouse monoclonal anti-APP (clone 22C11) | Thermo Fisher Scientific | Cat#14-9749-80; RRID: AB_2572977 |
| Rabbit monoclonal anti-APP (clone Y188) | Abcam | Cat#ab32136; RRID: AB_2289606 |
| Mouse monoclonal anti-Rab5 | Synaptic Systems | Cat#108011; RRID: AB_887773 |
| Chicken polyclonal anti-Map2 | Abcam | Cat#ab92434; RRID: AB_2138147 |
| Mouse monoclonal anti-Nestin (clone 10c2) | Santa Cruz Biotechnology | Cat#sc23927; RRID: AB_627994 |
| Mouse monoclonal anti-TGN38 (clone B-6) | Santa Cruz Biotechnology | Cat#sc166594; RRID: AB_2287347 |
| Mouse monoclonal anti-Rab7 (clone Rab7-117) | Abcam | Cat#ab50533; RRID: AB_882241 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LY2886721 BACE1 Inhibitor | MedChemExpress | Cat#HY13240 |
| Critical Commercial Assays | ||
| Abeta V-PLEX Panel ELISA | Meso Scale Discovery | Cat#K15200E-2 |
| sAPPα/sAPPβ ELISA | Meso Scale Discovery | Cat#K15120E-1 |
| Experimental Models: Cell Lines | ||
| Human: Wildtype clone A6 iPS cell line | This paper | N/A |
| Human: Wildtype clone A7 iPS cell line | This paper | N/A |
| Human: SORL1 Knockout clone E1 iPS cell line | This paper | N/A |
| Human: SORL1 Knockout clone E4 iPS cell line | This paper | N/A |
| Oligonucleotides | ||
| gRNA targeting sequence: ATTGAACGACATGAACCCTC | This paper | N/A |
| ssODN: GGGAATTGATCCCTATGACAAACCAAATA CCATCTACATTGAACGACATGAACCCTCTGGCTA CTCCACGTCTTCCGAAGTACAGATTTCTTCCAG TCCCGGGAAAACCAGGAAG | This paper | N/A |
| SORLA: Forward primer ctctatcctgagtcaaggagtaac | This paper | N/A |
| SORLA: Reverse primer ccttccaattcctgtgtatgc | This paper | N/A |
| Software and Algorithms | ||
| CellProfiler | McQuin et al., 2018 | https://cellprofiler.org/ |
| ImageJ/FIJI | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| JACoP plugin | Bolte and Cordelières., 2006 | https://imagejdocu.tudor.lu/_media/plugin/analysis/jacop_2.0/just_another_colocalization_plugin/jacop_.jar |
Highlights.
Depletion of SORL1, an AD risk gene, in hiPSCs to model late-onset AD
Loss of SORL1 leads to early endosome enlargement in neurons but not microglia
Endosome in enlargement is independent of amyloidogenic APP processing
Loss of SORL1 leads to altered trafficking of APP
ACKNOWLEDGMENTS
We would like to thank the members of the Young laboratory, Harald Frankowski, Shannon Rose, and Yoshito Kinoshita for crucial discussions and Dr. Gregory Petsko for critical feedback during the preparation of the manuscript. We would like to acknowledge the UW SLU Cell Analysis Facility and the Garvey Imaging Core at the UW Institute for Stem Cell and Regenerative Medicine. This work was supported by NIH grants (R01AG062148) and BrightFocus Foundation grant (A2018656S) to J.E.Y., a Biogen Sponsored Research Agreement to J.E.Y., an NIH training grant (T32AG052354) to A.K., and a generous gift from the Ellison Foundation (to UW).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107719.
DECLARATION OF INTERESTS
S.A.S. is on the Scientific Advisory Board of Meira GTX and holds Equity in Imij Technologies.
REFERENCES
- Alzheimer’s Association (2019). 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 15, 321–387. [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al. (2005). Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U S A 102, 13461–13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OM, Schmidt V, Spoelgen R, Gliemann J, Behlke J, Galatis D, McKinstry WJ, Parker MW, Masters CL, Hyman BT, et al. (2006). Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45, 2618–2628. [DOI] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Engelborghs S, De Deyn PP, Van Broeckhoven C, and Sleegers K (2008). SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Hum. Mutat. 29, 769–770. [DOI] [PubMed] [Google Scholar]
- Bolte S, and Cordelières FP (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Caglayan S, Takagi-Niidome S, Liao F, Carlo AS, Schmidt V, Burgert T, Kitago Y, Füchtbauer EM, Füchtbauer A, Holtzman DM, et al. (2014). Lysosomal sorting of amyloid-β by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci. Transl. Med. 6, 223ra20. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, and Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson SE, Gearing M, Lippa CF, Montine TJ, Levey AI, and Lah JJ (2006). LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, Burgert T, Caglayan S, Füchtbauer A, Füchtbauer EM, Schmidt V, and Willnow TE (2015). Distinct functions for anterograde and retrograde sorting of SORLA in amyloidogenic processes in the brain. J. Neurosci. 35, 12703–12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, et al. (2012). Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 32, 1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glerup S, Lume M, Olsen D, Nyengaard JR, Vaegter CB, Gustafsen C, Christensen EI, Kjolby M, Hay-Schmidt A, Bender D, et al. (2013). SorLA controls neurotrophic activity by sorting of GDNF and its receptors GFRα1 and RET. Cell Rep. 3, 186–199. [DOI] [PubMed] [Google Scholar]
- Hermans-Borgmeyer I, Hampe W, Schinke B, Methner A, Nykjaer A, Süsens U, Fenger U, Herbarth B, and Schaller HC (1998). Unique expression pattern of a novel mosaic receptor in the developing cerebral cortex. Mech. Dev. 70, 65–76. [DOI] [PubMed] [Google Scholar]
- Herskowitz JH, Offe K, Deshpande A, Kahn RA, Levey AI, and Lah JJ (2012). GGA1-mediated endocytic traffic of LR11/SorLA alters APP intracellular distribution and amyloid-β production. Mol. Biol. Cell 23, 2645–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin N, Uitterlinden AG, et al. (2017). Characterization of pathogenic SORL1 genetic variants for association with Alzheimer’s disease: a clinical interpretation strategy. Eur. J. Hum. Genet. 25, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, and Lakkaraju A (2018). Early endosome morphology in health and disease. Adv. Exp. Med. Biol. 1074, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, Jiang Y, and Nixon RA (2016). Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 21, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger SC, Glerup S, Raarup MK, Mari MC, Nyegaard M, Koster G, Prabakaran T, Nilsson SK, Kjaergaard MM, Bakke O, et al. (2011). SorLA regulates the activity of lipoprotein lipase by intracellular trafficking. J. Cell Sci. 124, 1095–1105. [DOI] [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, and Tessier-Lavigne M (2019). A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 104, 1022. [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, and Checler F (2016). Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 132, 257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. (2007). The diploid genome sequence of an individual human. PLoS Biol. 5, e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Galasko DR, Ringman JM, Vinters HV, Edland SD, Pomakian J, Ubeda OJ, Rosario ER, Teter B, Frautschy SA, and Cole GM (2009). Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch. Neurol. 66, 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini AC, Gomez-Arboledas A, Forner S, Rodriguez-Ortiz CJ, McQuade A, Danhash E, Phan J, Javonillo D, Ha JV, Tram M, et al. (2019). Amyloid-beta impairs TOM1-mediated IL-1R1 signaling. Proc. Natl. Acad. Sci. U S A 116, 21198–21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, and Blurton-Jones M (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). Cell-Profiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, and Small SA (2014). Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 10, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmedbasic A, Christensen SK, Nilsson J, Rüetschi U, Gustafsen C, Poulsen AS, Rasmussen RW, Fjorback AN, Larson G, and Andersen OM (2015). SorLA complement-type repeat domains protect the amyloid precursor protein against processing. J. Biol. Chem. 290, 3359–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer WJ, Bettayeb K, Sinha SC, Flajolet M, Greengard P, and Bustos V (2017). Gleevec shifts APP processing from a β-cleavage to a nonamyloidogenic cleavage. Proc. Natl. Acad. Sci. U S A 114, 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Gustafsen C, Madsen P, Nyengaard JR, Hermey G, Bakke O, Mari M, Schu P, Pohlmann R, Dennes A, and Petersen CM (2007). Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol. Cell. Biol. 27, 6842–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, and Lah JJ (2006). The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 26, 1596–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, et al. ; PHRC GMAJ Collaborators (2012). High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 17, 875–879. [DOI] [PubMed] [Google Scholar]
- Raghavan NS, Brickman AM, Andrews H, Manly JJ, Schupf N, Lantigua R, Wolock CJ, Kamalakaran S, Petrovski S, Tosto G, et al. ; Alzheimer’s Disease Sequencing Project (2018). Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 5, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, and Tsai LH (2016). Self-Organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS ONE 11, e0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Cheng R, Rogaeva E, Lee JH, Tokuhiro S, Zou F, Bettens K, Sleegers K, Tan EK, Kimura R, et al. ; Genetic and Environmental Risk in Alzheimer Disease 1 Consortium (2011). Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch. Neurol. 68, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose SE, Frankowski H, Knupp A, Berry BJ, Martinez R, Dinh SQ, Bruner LT, Willis SL, Crane PK, Larson EB, et al. (2018). Leptomeninges-derived induced pluripotent stem cells and directly converted neurons from autopsy cases with varying neuropathologic backgrounds. J. Neuropathol. Exp. Neurol. 77, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager KL, Wuu J, Leurgans SE, Rees HD, Gearing M, Mufson EJ, Levey AI, and Lah JJ (2007). Neuronal LR11/sorLA expression is reduced in mild cognitive impairment. Ann. Neurol. 62, 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, and Lee VM (2000). Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J. Biol. Chem. 275, 2568–2575. [DOI] [PubMed] [Google Scholar]
- Small SA, and Gandy S (2006). Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron 52, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé-Domènech S, Cruz DL, Capetillo-Zarate E, and Maxfield FR (2016). The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 32, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, and D’Adamio L (2012). β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol. Med. 4, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi AN, and Praticò D (2019). Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol. Psychiatry 24, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Acker ZP, Bretou M, and Annaert W (2019). Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol. Neurodegener. 14, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, and Jacks T (2004). Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. U S A 101, 10380–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Weissmiller AM, White JA 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, et al. (2016). Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 126, 1815–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Boulanger-Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, Herrera C, Israel MA, Yuan SH, Edland SD, and Goldstein LS (2015). Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, and Goldstein LSB (2018). Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Reports 10, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, et al. (2011). Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS ONE 6, e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated during this study. CellProfiler software is available at https://cellprofiler.org/. The images and CellProfiler pipelines used in this study are available from the Lead Contact on request.