

Abstract
We have previously used RNA interference to demonstrate that GRK2 regulates endogenously expressed H1 histamine receptor in HEK-293 cells. In this report, we investigate the regulation of endogenously expressed M3 muscarinic acetylcholine receptor (M3 mAchR). We show that knockdown of GRK2, GRK3 or GRK6, but not GRK5, significantly increased carbachol-mediated calcium mobilization. Stable-expression of wild-type GRK2 or a kinase-dead mutant (GRK2-K220R) reduced calcium mobilization following receptor activation, while GRK2 mutants defective in Gαq binding (GRK2-D110A, GRK2-R106A and GRK2-R106A/K220R) had no effect on calcium signaling, suggesting that GRK2 primarily regulates Gq following M3 mAchR activation. The knockdown of arrestin-2 or arrestin-3 also significantly increased carbachol-mediated calcium mobilization. Knockdown of GRK2 as well as the arrestins also significantly enhanced carbachol-mediated activation of ERK1/2 while prolonged ERK1/2 activation was only observed with GRK2 or arrestin-3 knockdown. We also investigated the role of casein kinase-1α (CK1α) and found that knockdown of CK1α increased calcium mobilization but not ERK activation. In summary, our data suggest that multiple proteins dynamically regulate M3 mAchR-mediated calcium signaling while GRK2 and arrestin-3 play the primary role in regulating ERK activation.
Activation of G protein-coupled receptors (GPCRs) by agonist occupancy leads to a conformational change in the receptor that promotes the activation of heterotrimeric G proteins, which in turn activate a variety of effectors leading to downstream signaling events (Pierce et al., 2002). Activated GPCRs are regulated by three principal mechanisms: desensitization, internalization, and down-regulation. Receptor desensitization is initiated by the phosphorylation of serine/threonine residues by GPCR kinases (GRKs) which promotes the high affinity binding of arrestins, uncoupling the receptor from G protein and terminating signaling (Krupnick and Benovic, 1998).
There are seven members of the GRK family that are grouped into three subfamilies based on sequence and functional similarity: GRK1 and GRK7; GRK2 and GRK3; and GRK4, GRK5, and GRK6. GRK2, GRK3, GRK5, and GRK6 are ubiquitously expressed, while GRK1, GRK4, and GRK7 have a restricted expression pattern. Much of the research determining specific GPCR-GRK interaction has relied on techniques such as heterologous overexpression, dominant-negative constructs, and more recently RNA interference (Krupnick and Benovic, 1998; Iwata et al., 2005; Kim et al., 2005).
The non-visual arrestins, arrestin-2 (β-arrestin1) and arrestin-3 (β-arrestin2) bind to activated, phosphorylated GPCRs subsequently terminating G protein activation and targeting the receptors to clathrin coated pits for internalization (Moore et al., 2007). Arrestins have also been shown to act as scaffolding proteins to promote downstream signaling events, such as activation of mitogen-activated protein kinases (Lefkowitz and Shenoy, 2005).
The muscarinic acetylcholine receptors (mAchRs) represent a subfamily of GPCRs with five subtypes, M1 – M5. The M3 mAchR couples to Gq resulting in phospholipase C-β (PLC-β) activation, and production of inositol trisphosphate (IP3) and diacylglycerol (DAG), which leads to calcium release from intracellular stores and protein kinase C (PKC) activation. Additionally, the M3 mAchR can activate extracellular signal-regulated kinase (ERK) in a calcium independent and PKC dependent manner (Kim JY et. al., 1999, Wylie PG et. al., 1999).
Upon activation, the M3 mAchR is rapidly phosphorylated on serine/threonine residues within the third intracellular loop (Tobin et al., 1997) and C terminal tail (Budd et al., 2000), although it is unclear which kinases mediate receptor phosphorylation and regulation. Wu et al. showed that GRK2 phosphorylates the M3 mAchR in a Gβγ dependent manner and mapped the phosphorylation sites to 331SSS333 and 348SASS351 in the third intracellular loop (Wu et al., 2000). GRK3 also has the ability to phosphorylate the receptor but receptor regulation appears to occur primarily through modulation of PLC-β activity (Willets et al., 2001; Willets et al., 2002; Willets et al., 2003). Willets and coworkers also showed that GRK6 regulates the M3 mAchR by phosphorylation while GRK2 and GRK5 were found to have no effect on the receptor in SH-SY5Y cells (Willets et al., 2001; Willets et al., 2002; Willets et al., 2003). In addition to GRK-mediated phosphorylation, casein kinase 1α (CK1α) has also been shown to phosphorylate the M3 mAchR in an agonist dependent manner although this alone was insufficient to mediate receptor desensitization (Budd et al., 2000). Finally, arrestins do not appear to be required for M3 mAchR internalization (Lee et al., 1998; Mundell and Benovic, 2000), but are involved in receptor desensitization with no discernable specificity between arrestin-2 and arrestin-3 (Mundell and Benovic, 2000).
One major unanswered question regarding the physiological regulation of GPCRs is to understand which GRKs and arrestins regulate a given receptor subtype. To date, a limited number of GRKs and arrestins have been identified, whereas more than 700 mammalian GPCRs have been cloned (Gainetdinov et al., 2004). Studies over the past decade have defined the ability of individual GRKs, second messenger dependent kinases (e.g., PKA or PKC), and arrestins to regulate GPCRs in model systems. However, the mechanisms by which GRKs target endogenous GPCRs are still unknown. Using either wild type GRK2, kinase dead GRK2, or mutants deficient in Gαq binding, we previously showed that the human H1 histamine receptor was specifically regulated by GRK2 mainly through regulation of activated Gq (Iwata et al., 2005). In this report, we used RNA interference to target proteins specifically involved in the agonist dependent regulation of the endogenous M3 mAchR in HEK-293 cells. We found that there was differential GRK-mediated regulation of this receptor as assessed by calcium signaling and ERK activation. In addition, knockdown of either arrestin-2 or arrestin-3 resulted in enhanced signaling from the receptor, with different temporal effects. Furthermore, we show that, in addition to GRKs, CK1α has a negative role in M3 mAchR mediated calcium mobilization. Taken together, our results show that multiple proteins mediate agonist-dependent regulation of M3 mAchR signaling.
Materials and Methods
Materials.
HEK-293 cells were from Microbix Biosystems, Inc (Toronto, Canada) while carbachol was from EMD Biosciences (San Diego, CA). Pirenzepine and p-fluorohexahydrosila-difenol (pFHHsiD) were from Sigma-Aldrich (St. Louis, MO) and Lipofectamine™ 2000 and Opti-MEM® were from Invitrogen (Carlsbad, CA). Phospho-specific p44/p42 polyclonal antibody was from Cell Signaling Technologies (Beverly, MA). Polyclonal ERK2, CK1α and GRK3 antibodies were from Santa Cruz (Santa Cruz, CA). Anti-β-arrestin monoclonal antibody was from BD Biosciences Pharmagen (San Diego, CA). Anti-GRK4–6 monoclonal antibody was from Upstate Cell Signaling Solutions (Waltham, MA) while the GRK2 monoclonal antibody was produced in our laboratory and anti-α-tubulin monoclonal antibody was from Sigma (St. Louis, MO).
Synthesis of small interfering RNAs (siRNAs).
All siRNAs were chemically synthesized by Dharmacon, Inc (Chicago, IL). The GRK2, GRK5 and CK1α siRNAs were reported previously (Iwata et al., 2005; Kim et al., 2005; Liu et al., 2002). The GRK3 siRNA sequence was 5´-GCAGAAGUCGACAAAUUUA-3′ while 5´-GCGCUUGGCCUACGCCUAU-3´ was used for GRK6. Arrestin-2 and −3 siRNAs were purchased as a SMARTpool®. Non-specific control siRNA VIII (5´-AAACUCUAUCUGCACGCUGAC-3´) was used as the control for all siRNA experiments.
Cell Culture and siRNA transfection:
HEK-293 cells were maintained in Dulbecco’s modified Eagles Media supplemented with 10% FBS, 25 mM HEPES, pH 7.2, and 0.1 mM non-essential amino acids in a 5% CO2 incubator at 37°C. For transfection of GRK and casein kinase siRNAs, HEK-293 cells grown to 85 to 90% confluence in 100-mm dishes were transfected with 600 pmol of siRNA using Lipofectamine 2000 in Opti-MEM. After 6 hr, cells were split 1:2 and a second transfection of 600 pmol was performed 24 hr after the initial transfection. Forty-eight hr after the second transfection, cells were split for assay the following day. For arrestin SMARTpool® siRNAs, cells ~70% confluent were transfected with 600 pmol of siRNA corresponding to either arrestin-2 or arrestin-3. Forty-eight hr later, cells were split for assay the following day. Control siRNA was transfected in a similar fashion as described above for each transfection condition.
Immunoblotting:
To analyze siRNA target proteins, siRNA transfected HEK-293 cells in a 6-well plate were washed twice with ice cold PBS and lysed with buffer (20 mM HEPES, pH 7.5, 10 mM EDTA, 150 mM NaCl, 1% Triton X-100 and one tablet of Complete Inhibitor (Roche) per 50 ml) at 4°C on a rocker for 30 min. The lysates were centrifuged at 4°C at 30,000 rpm in a TLA45 rotor for 30 min. The supernatants were electrophoresed on a 10% SDS polyacrylamide gel, transferred to nitrocellulose, and immunoblotted using monoclonal anti-GRK2 (1:1000), polyclonal anti-GRK3 (1:200), monoclonal anti-GRK4–6 (1:3000), monoclonal anti-β-arrestin-1 (1:1000) or polyclonal anti-CK1α (1:200), HRP-labeled secondary antibodies, and chemiluminescence. The blots were stripped and reprobed using an anti-tubulin (1:7500) monoclonal antibody.
Measurement of intracellular calcium mobilization.
Calcium mobilization was performed as previously described with slight modifications (Iwata et al., 2005). In brief, HEK-293 cells transfected with siRNAs were harvested with Cellstripper (Mediatech, Herndon, VA), washed twice with phosphate-buffered saline, and resuspended at 5 × 106 cells/ml in Hanks’ balanced salt solution (140 mM NaCl, 5 mM KCl, 10 mM HEPES, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, 1 mg/ml glucose) (Invitrogen) containing 0.025% bovine serum albumin. The cells were then loaded with 3 μM Fura-2 acetoxymethyl ester derivative (Fura-2/AM) (Molecular Probes, Eugene, OR) for 30 min at 37°C. The cells were washed once in Hanks’ solution, resuspended in Hanks’ solution containing 0.025% bovine serum albumin, incubated at room temperature for 15 min, washed twice in Hanks’ solution, and then resuspended in Hanks’ at a concentration of 3 × 107 cells/ml. A typical experiment contained 1.5 × 106 cells/1.6 ml in a quartz cuvette and stimulation with different concentrations of carbachol. Calcium mobilization was measured using excitation at 340 and 380 nm and emission at 510 nm in a fluorescence spectrometer (LS55, Perkin-Elmer Life Sciences). Calibration was performed using 0.1% Triton X-100 for total fluorophore release and 15 mM EGTA to chelate free calcium. When antagonists were used, cells were preincubated with the indicated antagonist for 30 sec prior to starting the fluorescent spectrometer and an additional 30 sec prior to stimulation with carbachol. Intracellular calcium concentrations were calculated using a fluorescence spectrometer measurement program.
ERK activation assays.
HEK-293 cells, ~90% confluent in 6-well plates, were serum starved for at least 6 hr. Following serum starvation, cells were stimulated with 100 μM carbachol as indicated and washed once with ice cold PBS. Lysis buffer (1% Triton X-100, 20 mM HEPES, pH 7.2, 150 mM NaCl, 10 mM EDTA, 1 μM sodium orthovanadate, 3 mM sodium pyrophosphate, 10 mM sodium fluoride, and 1 Complete Inhibitor tablet per 50 ml) was added and plates were stored at −80°C until harvesting. Cells were thawed and scraped into lysis buffer on ice, vortexed briefly, and debris was cleared by centrifugation at 14,000 rpm for 15 min. Equal amounts of whole cell lysate were separated by electrophoresis on a 10% SDS polyacrylamide gel, transferred to nitrocellulose, and proteins detected by immunoblotting. Nitrocellulose membranes were blocked for 1 hr at room temperature in a 1:3 dilution of ODYSSEY® blocking buffer (LI-Cor® Biosciences). A mixture of primary antibodies directed at ERK2 (monoclonal, Santa Cruz) and phospho-ERK1/2 (polyclonal, Cell Signaling Technologies) in 100% ODYSSEY® blocking buffer were incubated overnight at 4°C. Nitrocellulose membranes were washed with Tris Buffered Saline containing 0.1% Tween-20 (TBS-T) over 40 min. The membranes were then incubated for 1 hr at room temperature with a mixture of goat anti-rabbit Alexa® Fluorophore 680 conjugated (Molecular Probes) and goat anti-mouse IRDye 800 conjugated (Rockland Immunochemicals) antibodies. Following a 1 hr incubation, the membranes were washed with TBS-T for 60 min. Fluorescence was detected simultaneously using the ODYSSEY® infrared imaging system (LI-Cor® Biosciences). When antagonists were used, cells were incubated at 37°C with the indicated antagonist for 5 min prior to stimulation with carbachol. Fluorescence intensity of phosphorylated ERK2 was normalized to total ERK2 fluorescence, and data are represented as fold-increase over basal (+/− SEM).
Statistical Analysis.
Results were analyzed using a paired, two-tailed, students T-Test with significance at p≤0.05.
Results
Pharmacological characterization of the muscarinic acetylcholine receptor subtype endogenously expressed in HEK-293 cells.
Using RNAi, we have previously shown that GRK2 regulates the endogenously expressed H1 histamine receptor in HEK-293 cells (Iwata et al., 2005). We wanted to expand this approach to determine the regulation of other endogenous GPCRs. Previous work has shown that HEK-293 cells respond to stimulation with carbachol, a non-specific mAchR agonist, with robust IP3 production and calcium mobilization that had been attributed to the M1 mAchR subtype (Mundell and Benovic, 2000). However, a recent microarray analysis of commonly used cell lines suggested that the mAchR endogenously expressed in these cells is the M3 receptor subtype (Hakak et al., 2003). In light of this, we sought to pharmacologically determine which mAchR subtype is actually expressed in HEK-293 cells. Cells loaded with the ratiometric calcium indicator Fura-2/AM display a robust increase in calcium mobilization in response to carbachol stimulation (Figure 1A), with an EC50 of 20 μM (data not shown). Incubation with the antagonist p-FHHsiD, which has some selectivity for the M3 mAchR (pKi 7.7) as compared to the M1 mAchR (pKi 7.1) (de la Vega et al., 1997), completely inhibited calcium mobilization in response to carbachol while the selective M1 mAchR antagonist pirenzepine, only slightly inhibited calcium mobilization (Figure 1A). This result is in line with previous reports demonstrating that pirenzepine selectively inhibits the M1 mAchR (pKi 8.0), but at higher concentrations is able to inhibit the M3 subtype (pKi 6.7) (de la Vega et al., 1997). In addition, there was no calcium response when the cells were stimulated with the M1/M4 mAchR selective agonist McN-A-343 (data not shown).
Figure 1.
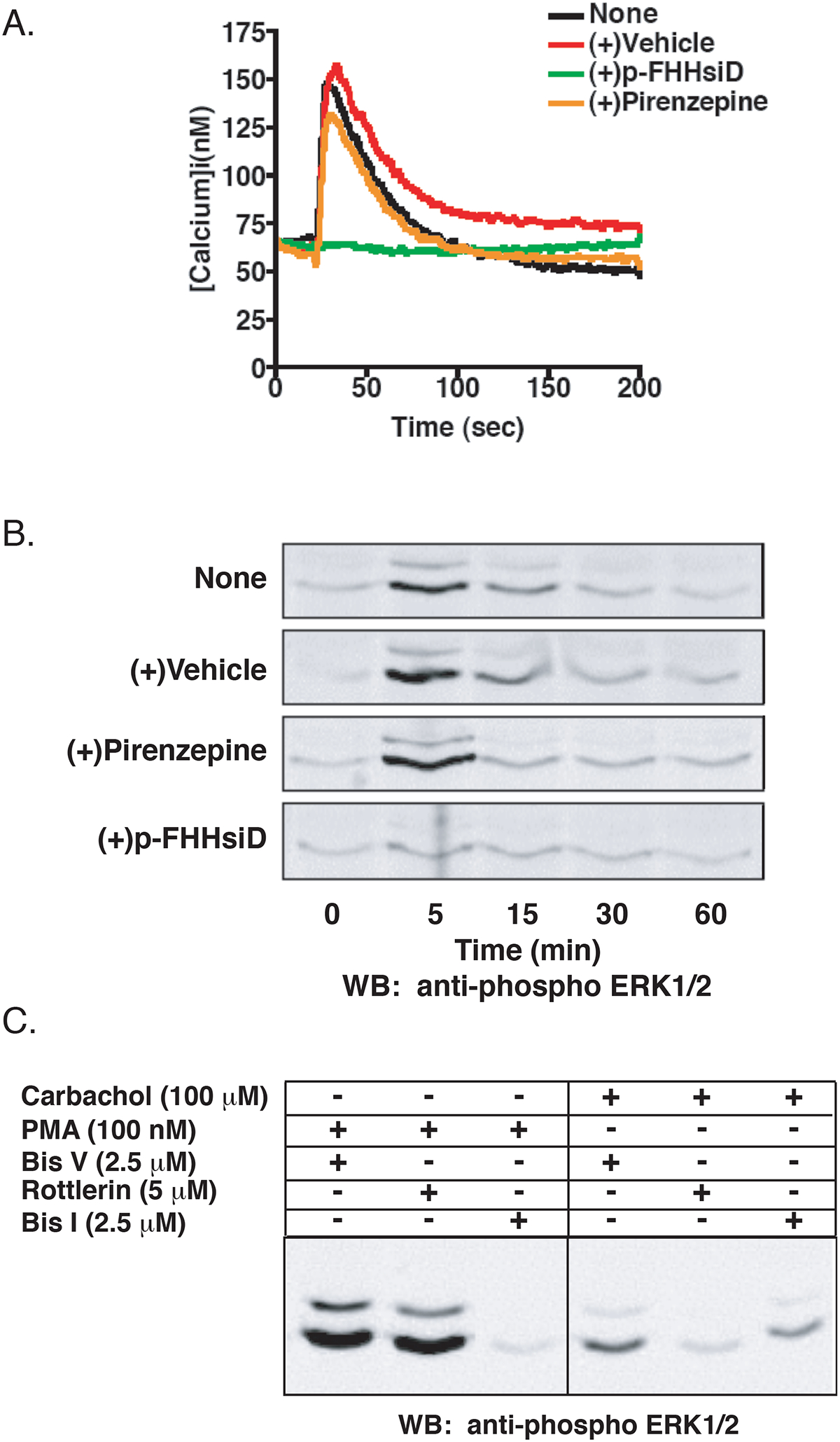
Characterization of the Muscarinic Acetylcholine Receptor Subtype Endogenously Expressed in HEK-293 Cells. A) HEK-293 cells loaded with the ratiometric calcium indicator Fura2/AM were incubated with 100 nM pirenzepine (orange), 1 μM p-FHHsiD (green), vehicle (red), or not pretreated (black) and stimulated with 100 μM carbachol. Changes in calcium mobilization were assayed by monitoring the change in Fura-2AM fluorescence. Shown is a representative tracing from three independent experiments. B) Following a 6 hr serum starve, HEK-293 cells were incubated with 100 nM pirenzepine, 1 μM p-FHHsiD, vehicle, or not pretreated and stimulated with 100 μM carbachol for the indicated times. Cells from a 6-well plate were harvested and equal amounts of total cellular lysate were separated by SDS-PAGE and probed for phospho-ERK1/2 as described in Materials and Methods. Shown is a representative immunoblot of three independent experiments. C) Cells were treated with Bis I (2.5 μM), Bis V (2.5 μM) or rottlerin (5 μM) for 30 min prior to stimulation with carbachol (100 μM) for 5 min or PMA (100 nM) for 15 min.
To further investigate the subtype of mAchR expressed, we also analyzed the effects of the M1 and M3 selective antagonists on carbachol-stimulated ERK activation. GPCRs activate ERK1/2 via a number of pathways (Werry et al., 2005) and both the M1 and M3 mAchRs have been shown to activate ERK1/2 in a number of cell types (Budd et al., 1999; Guo et al., 2001). Carbachol-mediated ERK1/2 activation in HEK-293 cells is dose dependent (EC50 ~8 μM), peaking at 5 min and returning to basal levels by 60 min (Figure 1B, top panel). The addition of p-FHHsiD completely blocked ERK1/2 activation in response to carbachol, whereas pirenzepine had no effect (Figure 1B). These results confirm that the primary mAchR subtype in HEK-293 cells is the M3.
We also wanted to determine whether PKC was responsible for ERK activation following M3 mAchR stimulation. Previous evidence suggests that the novel PKC isoforms are responsible for M3 mAchR-mediated ERK activation including PKCε in SK-N-BE2(C) cells (Kim JY et. al., 1999) and a calcium independent PKC in CHO cells (Wylie PG et. al., 1999). Furthermore, it has recently been shown that the M3 mAchR regulates the Kir 3.1/3.2 potassium channel through activation of PKC-δ in HEK-293 cells (Brown et al., 2005). To establish whether PKC-δ is involved in M3 mAchR-mediated ERK activation, we used Bisindolylmaleimide I (Bis I), a general PKC inhibitor, and rottlerin, which selectively inhibits PKC-δ (Gschwendt et al., 1994). Rottlerin significantly inhibited carbachol-mediated ERK activation while Bis I only partially inhibited ERK activation (Figure 1C). The specificity of these inhibitors was confirmed by the demonstration that rottlerin had minimal effects on PMA-induced ERK activation while Bis I completely inhibited PMA-promoted ERK activation (Figure 1C). Taken together, we conclude that HEK-293 cells endogenously express the M3 mAchR and that carbachol-mediated activation of the ERK1/2 cascade is dependent on PKC-δ.
Regulation of M3 mAchR-mediated calcium mobilization in HEK-293 cells.
We next evaluated the effect of knocking down various regulatory proteins on M3 mAchR signaling. Since the phosphorylation of activated GPCRs by GRKs is often an early step in signal termination, we initially determined the effect that GRK knockdown would have on calcium mobilization following carbachol treatment. As shown in Figure 2A and 2B, we were able to selectively and specifically knockdown each of the four individual GRKs expressed in HEK-293 cells. A modest increase in GRK3 expression was observed when other GRKs, in particular GRK2, were knocked down (Figure 2B). Knockdown of GRK2, GRK3, and GRK6 led to increases of 210% (p<0.001), 190% (p<0.001) and 230% (p<0.001), respectively, in the peak calcium transients while knockdown of GRK5 had no effect on calcium mobilization (Figure 3A and 3B). This effect was also observed when methacholine was used to activate the M3 mAchR (data not shown). These data suggest that multiple GRKs are involved in the desensitization of the M3 mAChR.
Figure 2.
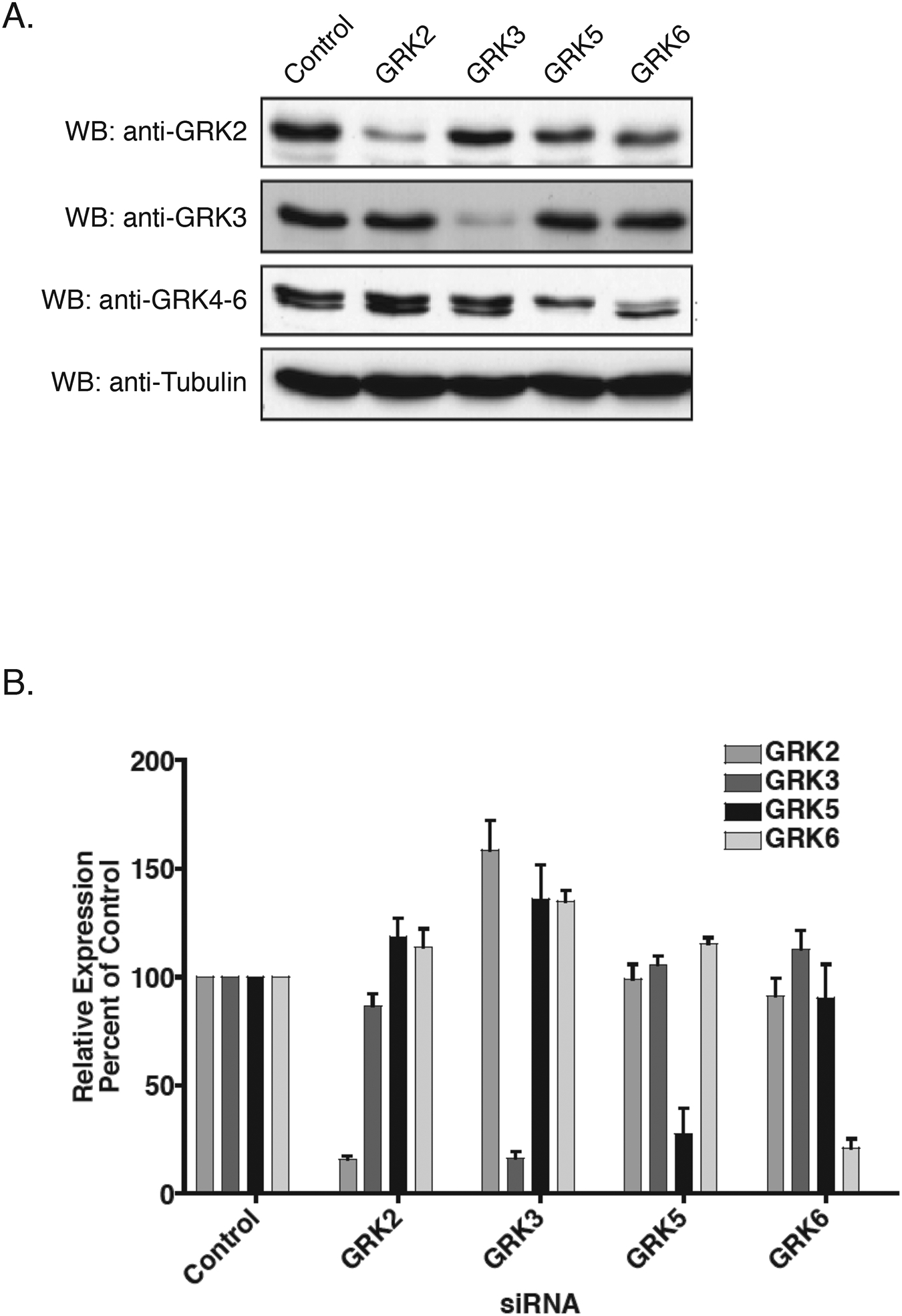
Knockdown of Endogenous GRK Isoforms in HEK-293 Cells. A) HEK-293 cells were transfected twice within a 24 hr interval with GRK-specific or non-specific control siRNA. 72 hr after the second transfection, cells were harvested and equal amounts of total cellular lysate was separated by 10% SDS-PAGE, transferred to nitrocellulose and incubated with the indicated antibodies. Blots were stripped and re-probed for α-tubulin to control for loading. Shown is a representative immunoblot. B) Mean relative level of GRK expression following siRNA quantified by densitometry from five separate experiments.
Figure 3.
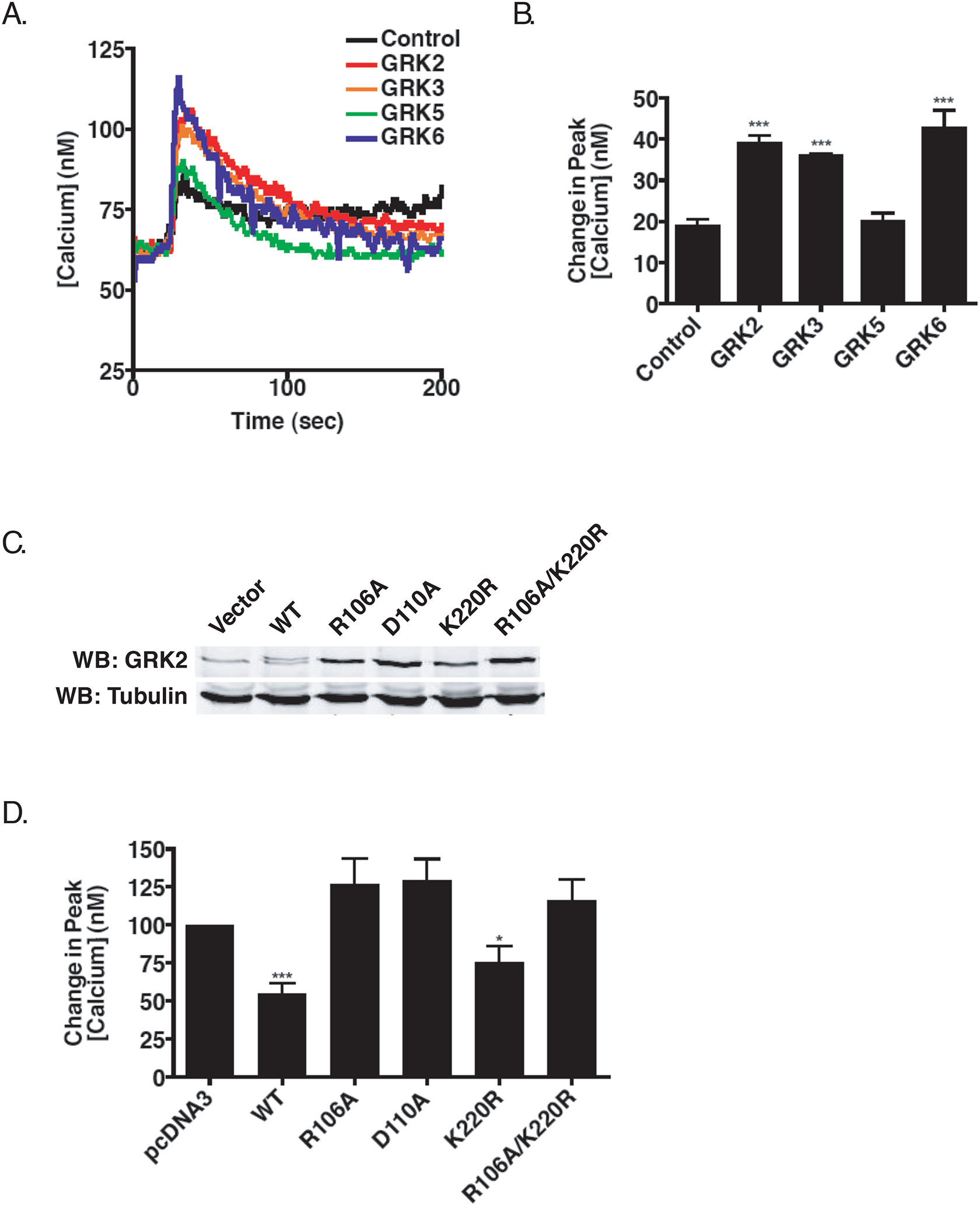
GRK-Mediated Regulation of Calcium Mobilization Following M3 mAchR Activation. A) Effect on calcium mobilization. 72 hr after the second siRNA transfection, HEK-293 cells were loaded with Fura2/AM and stimulated with 10 μM carbachol. B) Mean (+/− SEM) increase in the peak calcium transient following stimulation with 10 μM carbachol from five individual experiments (***p<0.001 using two-tailed T test). C) Representative immunoblot showing relative levels of GRK2 stably expressed in HEK-293 cells. D) Calcium mobilization in HEK-293 cells stably expressing bovine GRK2. Mean (+/− SEM) increase in peak calcium mobilization in cells expressing vector (pcDNA3), wild type, Gq-binding deficient (R106A; D110A), kinase-dead (K220R), or the Gq-binding deficient/kinase dead (R106A/K220R) bovine GRK2 (*p<0.05 for GRK2-K220R, ***p<0.001 for wild type GRK2).
GRK2 interaction with Gq is primarily responsible for increased calcium mobilization.
The enhanced mobilization of calcium seen following silencing of GRK2 may arise from phosphorylation-dependent and/or phosphorylation-independent mechanisms (Ribas et al., 2007). Therefore, we next sought to further delineate the underlying mechanism observed for calcium mobilization when GRK2 was knocked down. Since we previously showed that GRK2 interacts with Gαq through the RGS-homology domain of GRK2 (Carman et al., 1999), the increase in peak calcium mobilization could be a result of a loss of receptor phosphorylation, a loss of the ability of GRK2 to inhibit activated Gq, or both. To address this, we generated cell lines that stably express either wild-type bovine GRK2, kinase dead GRK2 (K220R), GRK2 point mutants defective in binding Gαq (R106A, D110A), or a GRK2 mutant that was both kinase-dead and Gq-deficient (R106A/K220R). Cloned cell lines expressing wild type or mutant bovine GRK2 at levels close to endogenous GRK2 levels (1–5-fold overexpression) were selected for study (Figure 3C). SDS-PAGE revealed that bovine GRK2 ran slightly slower than endogenous human GRK2 when expressed in HEK-293 cells (Figure 3C). Stable expression of either wild type or the kinase dead mutant reduced carbachol-stimulated calcium mobilization by ~50% (Figure 3D). In striking contrast, stable expression of the Gαq-binding deficient mutants (R106A and D110A) or the double mutant (R106A/K220R) had no effect on calcium mobilization (Figure 3D). This suggests that GRK2 primarily regulates the activity of the M3 mAchR through its ability to interact with the activated pool of Gαq.
The non-visual arrestins negatively regulate M3 mAchR-promoted calcium mobilization.
Our data suggest that GRK-mediated phosphorylation of the M3 mAchR may contribute to subsequent desensitization. Since GRK phosphorylation often promotes arrestin binding, we next determined the effect siRNA knockdown of arrestin-2 and arrestin-3 had on calcium mobilization. Pooled siRNAs targeting either arrestin-2 or arrestin-3 specifically reduced protein expression by ~90% (Figure 4A and 4B). As shown in Figure 4C and 4D, knockdown of either arrestin-2 or arrestin-3 resulted in a significant increase in the peak calcium transient upon stimulation with carbachol. The increase seen with arrestin-3 was slightly higher (74% increase) than that seen with arrestin-2 (65% increase), although silencing of arrestin-3 also led to an increase in the prolonged phase of the calcium transient (Figure 4C), suggesting prolonged IP3 production.
Figure 4.
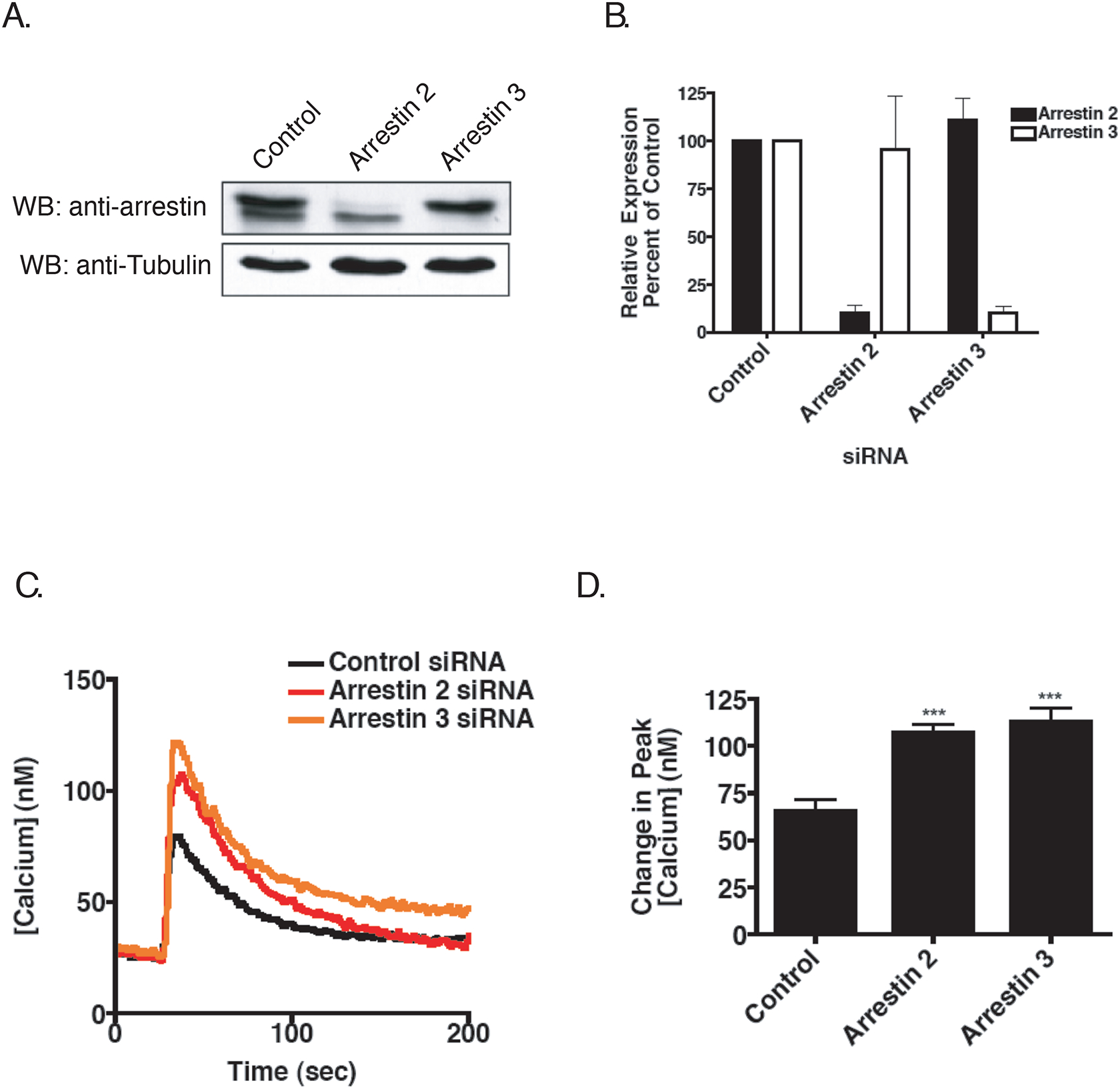
Effect of Arrestin Knockdown on Calcium Mobilization Following M3 mAchR Activation. A) Cells were transfected with SMARTpool siRNA and harvested 72 hr later. Blots were incubated with a monoclonal antibody for arrestin-2 that cross-reacts with arrestin-3. Blots were stripped and re-probed for α-tubulin to control for loading. Shown is a representative immunoblot. B) Mean relative level of arrestin expression following siRNA quantified by densitometry from five separate experiments. C) Effect on calcium mobilization. Cells were harvested 72 hr post-transfection and processed as described previously. Shown is a representative calcium trace from five independent experiments. D) Mean (+/− SEM) increase in the peak calcium transient following stimulation with 100 μM carbachol from five individual experiments (***p<0.001 using two-tailed T test).
Regulation of M3 muscarinic acetylcholine receptor-mediated activation of the ERK cascade.
We next focused on understanding the roles of GRKs and arrestins in regulating activation of ERK1/2 following M3 mAchR stimulation. The kinetics of ERK1/2 activation showed a consistent peak at 5 min that returned to basal levels by 60 min (Figure 1C). As shown in Figure 5A and 5B, knocking down GRK2 resulted in a 2.5-fold increase in the peak of ERK1/2 activation as well as prolonged ERK1/2 activation (Figure 5B). Silencing of GRK5 or GRK6 also enhanced ERK1/2 activation following a 5 min stimulation, although the effects were modest and not statistically significant (1.3- and 1.5-fold increase, respectively) (Figure 5A and 5B). GRK knockdown did not change basal phospho-ERK1/2 levels (data not shown). Interestingly, in contrast to calcium mobilization, knocking down GRK3 had no effect on ERK1/2 activation (Figure 5A and 5B). Collectively, these data demonstrate that signaling pathways downstream of M3 mAchR activation are regulated by multiple GRKs in HEK-293 cells, in a separate but coordinated fashion.
Figure 5.
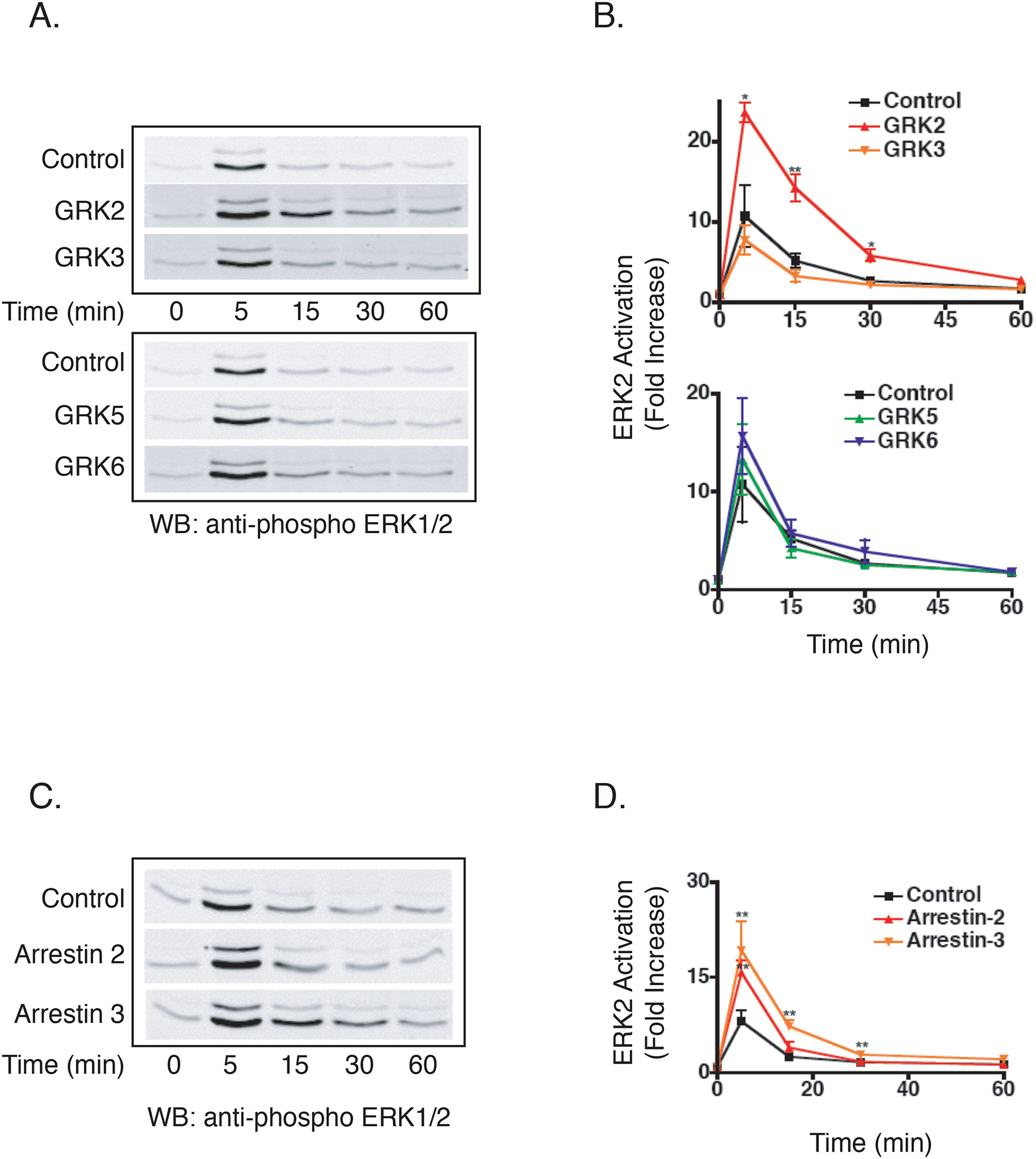
Effect of GRK and Arrestin Knockdown on M3 mAchR ERK Activation. A) Effect of GRK knockdown on ERK1/2 activation. Following a 6 hour serum starve, cells were treated with 100 μM carbachol for indicated times. Shown is a representative immunoblot from six independent experiments. B) Mean fold increase in ERK2 activation. Blots were incubated simultaneously with primary antibodies specific for phospho-ERK1/2 and total ERK2 overnight. Phospho-ERK1/2 fluorescence was normalized to total ERK2 fluorescence and data are presented as fold-increase in ERK2 activation over basal (n=6, +/− SEM; *p<0.05, **p<0.01). C) Effect of arrestin knockdown on ERK1/2 activation. Following a 6 hour serum starve, cells were treated with 100 μM carbachol for indicated times. Shown is a representative immunoblot from eight independent experiments. D) Mean fold increase in ERK2 activation. Blots were incubated simultaneously with primary antibodies specific for phospho-ERK1/2 and total ERK2 overnight. Phospho-ERK1/2 fluorescence was normalized to total ERK2 fluorescence and data are presented as fold-increase in ERK2 activation over basal (n=8, +/− SEM; **p<0.01).
In contrast to some GPCRs (Ahn et al., 2004; Lefkowitz and Shenoy, 2005), internalization is not required for M3 mAchR-mediated ERK activation (Budd et al., 1999). Thus, it was not surprising that knockdown of either arrestin-2 or arrestin-3 resulted in an ~2-fold increase in ERK activation, with differential temporal effects (Figure 5C and 5D). Silencing of arrestin-2 led to enhanced ERK1/2 activation at 5 min while silencing of arrestin-3 led to both enhanced and prolonged activation (Figure 5D). These data suggest that under normal physiological conditions, either arrestin-2 or arrestin-3 is sufficient to negatively regulate acute signaling events upon M3 mAchR activation, although arrestin-3 appears to play a larger role in terminating signaling in response to prolonged agonist exposure.
Regulation of the M3 muscarinic acetylcholine receptor by casein kinase 1α.
CK1α also phosphorylates the M3 receptor in an agonist dependent manner although it does not appear to be required for desensitization of the receptor (Budd et al., 2000; Budd et al., 2001; Tobin et al., 1997). CK1α has also been shown to phosphorylate the M1 mAchR and rhodopsin in vitro (Tobin et al., 1997; Waugh et al., 1999). To determine whether CK1α has a role in regulating the endogenous M3 mAchR, HEK-293 cells were transfected with CK1α siRNA that specifically reduced CK1α protein levels to ~40% of that seen in control cells (Figure 6A). Knockdown of CK1α resulted in a significant increase (62%, p<0.01, n=4) in the peak calcium transient as compared to cells treated with control siRNA (Figure 6B). To determine if this effect was specific to CK1α mediated regulation of the M3 mAchR and not to some other aspect of the Gq signaling pathway, we also tested the ability of CK1α to regulate the histamine H1 receptor which is regulated by GRK2 in HEK-293 cells (Iwata et al., 2005). Knockdown of CK1α had no effect on calcium mobilization upon stimulation with 100 μM histamine (data not shown), suggesting that the effect of CK1α knockdown was specific for M3 mAchR signaling. Interestingly, knockdown of CK1α had no effect on carbachol-mediated activation of ERK1/2 (Figures 6C and 6D). These data demonstrate that, in addition to the GRK family, the agonist activated M3 mAchR is also regulated by CK1α.
Figure 6.
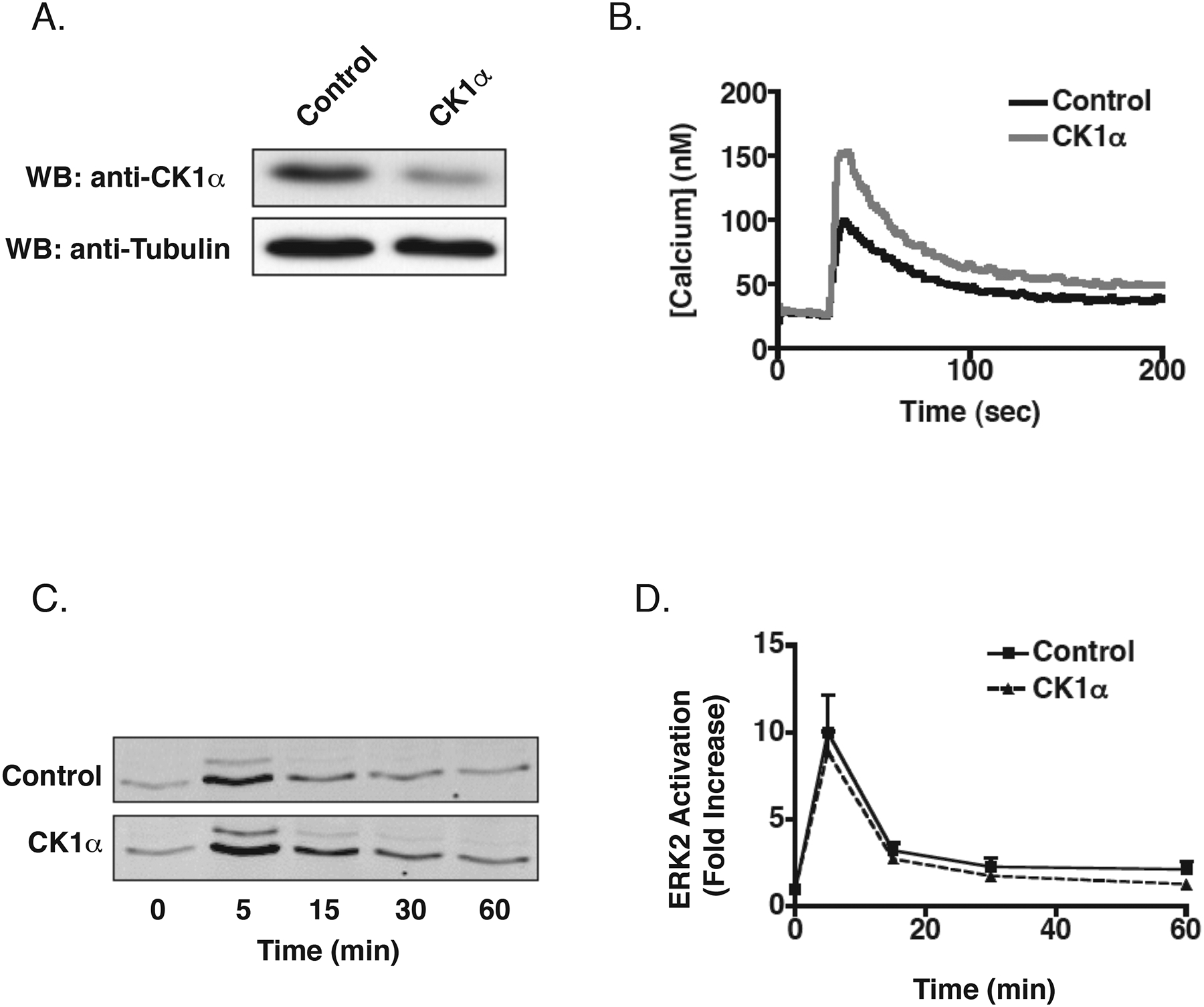
Effect of CK1α Knockdown on M3 mAchR Signaling. A) 72 hr after the second siRNA transfection, cells were harvested and equal amounts of total cellular lysate were separated by SDS-PAGE and immunoblotted for CK1α using a specific antibody. Blots were stripped and re-probed for α-tubulin to control for loading, Shown is a representative immunoblot. B) Effect on calcium mobilization. 72 hr after the second siRNA transfection, cells were loaded with Fura-2/AM and stimulated with 100 μM carbachol. Shown is a representative tracing from four independent experiments (control: 103 ± 10 nM, CK1α siRNA: 163 ± 15 nM, p<0.01). C) Effect on ERK1/2 activation. Following a 6 hr serum starve, cells were stimulated with 100 μM carbachol for indicated times. Shown is a representative immunoblot from eight independent experiments. D) Mean activation of ERK2. Blots were incubated simultaneously with primary antibodies specific for phospho-ERK1/2 and total ERK2 overnight. Phospho-ERK1/2 fluorescence was normalized to total ERK2 fluorescence and data are presented as fold-increase over basal (n=8, +/− SEM).
Discussion
GPCRs transduce extracellular stimuli into specific intracellular signals that regulate a variety of cellular functions. GPCR desensitization is classically mediated by members of the GRK family, which specifically phosphorylate the agonist-occupied receptor, promoting the subsequent high-affinity binding of arrestins. For most GPCRs, the specificity of GRKs and arrestins in cells remains poorly defined. In this report, we used a siRNA-based approach in HEK-293 cells to characterize the role of these proteins in M3 mAchR signaling. We found that the M3 mAchR displays a complex pattern of regulation, such that GRK2, GRK3, GRK6, arrestin-2, arrestin-3, and CK1α all participate to negatively regulate calcium signaling upon receptor activation.
Previously, it was shown that GRK2 can be recruited to and phosphorylate the M3 mAchR at two separate serine clusters within the third intracellular loop (Wu et al., 2000). In addition to receptor phosphorylation, GRK2 is able to bind both GTP-bound Gαq (Carman et al., 1999) and free Gβγ (Pitcher et al., 1992). The crystal structure of GRK2 (Tesmer et al., 2005) suggests that it may simultaneously sequester both active Gαq and free Gβγ, which in addition to receptor phosphorylation may increase the strength and effectiveness of GRK2-mediated receptor regulation. Previously, we and others demonstrated that GRK2 regulated GPCRs, such as the H1 histamine (Iwata et al., 2005), M1 mAchR (Willets et al., 2005), metabotropic glutamate (Dhami et al., 2005) and mouse cytomegalovirus GPCR M33 (Sherrill and Miller, 2006), involved the regulation of Gq. Studies analyzing GRK-mediated regulation of the M3 mAchR in SH-SY5Y cells have shown that GRK3 and GRK6 differentially regulate the receptor whereas GRK2 and GRK5 did not appear to be involved (Willets et al., 2001; Willets et al., 2002; Willets et al., 2003). Overexpressed GRK3 could phosphorylate the M3 mAchR, however, GRK3-mediated regulation appeared to be the result of altering the activity of PLC-β and not via receptor phosphorylation (Willets et al., 2001; Willets et al., 2002; Willets et al., 2003). In contrast, overexpressed GRK6 could phosphorylate the M3 mAchR leading to a decrease in signaling. This effect was reversed upon expression of a kinase dead GRK6 (Willets et al., 2003).
Using siRNA coupled with stable expression of low levels of various GRK2 mutants, we found that the enhanced calcium mobilization observed upon GRK2 knockdown is primarily due to a loss in regulation of activated Gq following M3 mAchR stimulation (Figure 3). Furthermore, we showed that loss of GRK2 leads to enhanced and prolonged activation of the ERK1/2 cascade (Figure 5). The observed effects of GRK2 knockdown are two-fold: the enhanced calcium mobilization appears to be primarily due to the loss of inhibition of activated Gq, while the enhanced and prolonged activation of ERK1/2 likely reflects enhanced DAG production/PKC-δ activation and a relief of inhibition of mitogen-activated protein kinase kinase 1 (MEK1) (Jimenez-Sainz MC et. al., 2006). However, we cannot completely rule out the possibility that GRK2 also mediates receptor phosphorylation since endogenous M3 mAchR levels are too low to evaluate phosphorylation (Tovey and Willars, 2004).
We have also found that GRK3 and GRK6 negatively regulate calcium mobilization following M3 mAchR stimulation. While knockdown of either kinase led to significant increases in calcium mobilization (Figure 3A and 3B), silencing of GRK3 had no effect on activation of ERK1/2 while loss of GRK6 had only a minor effect (Figure 5A and 5B). The possibility exists that there is overlap between these kinases and that regulation might involve a competition for receptor binding as has been suggested for the angiotensin receptor (Kim et al., 2005). These previous studies suggested that GRK2 and GRK3 negatively regulate while GRK5 and GRK6 positively regulate ERK1/2 activation and that differences in the phosphorylation pattern mediated by GRK2/3 or GRK5/6 could alternatively promote the binding of arrestin-2 or arrestin-3, respectively (Kim et al., 2005). However, our results suggest that the M3 mAchR is not subject to this type of overlapping regulation. Furthermore, the GRKs do not play a positive role in M3 mAchR signaling. There is a growing number of non-receptor substrates that have been identified for the GRKs (Ribas et al., 2007), and in line with previous findings, GRK3 could be primarily regulating PLC-β activity via binding to Gβγ or Gαq (Willets et al., 2001). This might allow for a very rapid and robust production of IP3 and subsequent calcium release that is not evident at later time points because other kinases (e.g., GRK6) may phosphorylate the receptor resulting in desensitization. Additionally, mechanisms regulating downstream signaling events (e.g., IP3 hydrolysis, calcium reuptake, etc) also shape both calcium mobilization and ERK1/2 activation responses following carbachol stimulation. As we have identified three GRKs that are involved in M3 mAchR regulation, multiple proteins may need to be knocked-down simultaneously in order to produce more prolonged signaling.
Previously, we reported that an ~50% reduction in arrestin levels using antisense strategies had no effect on calcium mobilization in HEK-293 cells (Mundell and Benovic, 2000). In the present study, we were able to reduce protein levels by ~90% and show that the loss of either arrestin-2 or arrestin-3 enhanced the peak calcium transient seen upon activation of the M3 mAchR (Figure 4C and 4D). Taking into consideration previous reports demonstrating that the M3 mAchR internalizes in an arrestin-independent manner (Lee et al., 1998), our results suggest that arrestins primarily mediate desensitization of the M3 mAchR following agonist activation. Consistent with this and with previous reports (Budd et al., 1999), knockdown of either arrestin-2 or arrestin-3 also enhanced ERK1/2 activation (Figure 5C and Figure 5D). This is in contrast to the emerging paradigm that has been proposed for a number of other GPCRs where arrestins promote G protein independent signaling pathways (reviewed in Lefkowitz and Shenoy, 2005) or even have opposing effects to one another as has been shown for the angiotensin II receptor (Ahn et al., 2004). In light of the fact that HEK-293 cells express similar levels of endogenous arrestin-2 and arrestin 3 (unpublished results), our data suggest an inherent specificity for the M3 mAchR by arrestin-3 as both calcium mobilization and ERK activation were enhanced and prolonged with arrestin-3 knockdown. This also suggests that the PLC-β/PKC arm of signaling is responsible for ERK activation, consistent with previous reports (Budd et al., 1999; Kim et al., 1999; Wylie et al., 1999). Interestingly, arrestins can also terminate muscarinic receptor signaling by recruiting diacylglycerol kinases and enhancing the degradation of the second messenger DAG, thereby coordinately terminating GPCR/G protein interaction and promoting second messenger degradation (Nelson et al., 2007). Taken together, the prolonged ERK activation observed following GRK2 and arrestin-3 knockdown can be attributed to enhanced Gq activity, sustained DAG production and subsequent PKC-δ activation (Figure 7).
Figure 7.
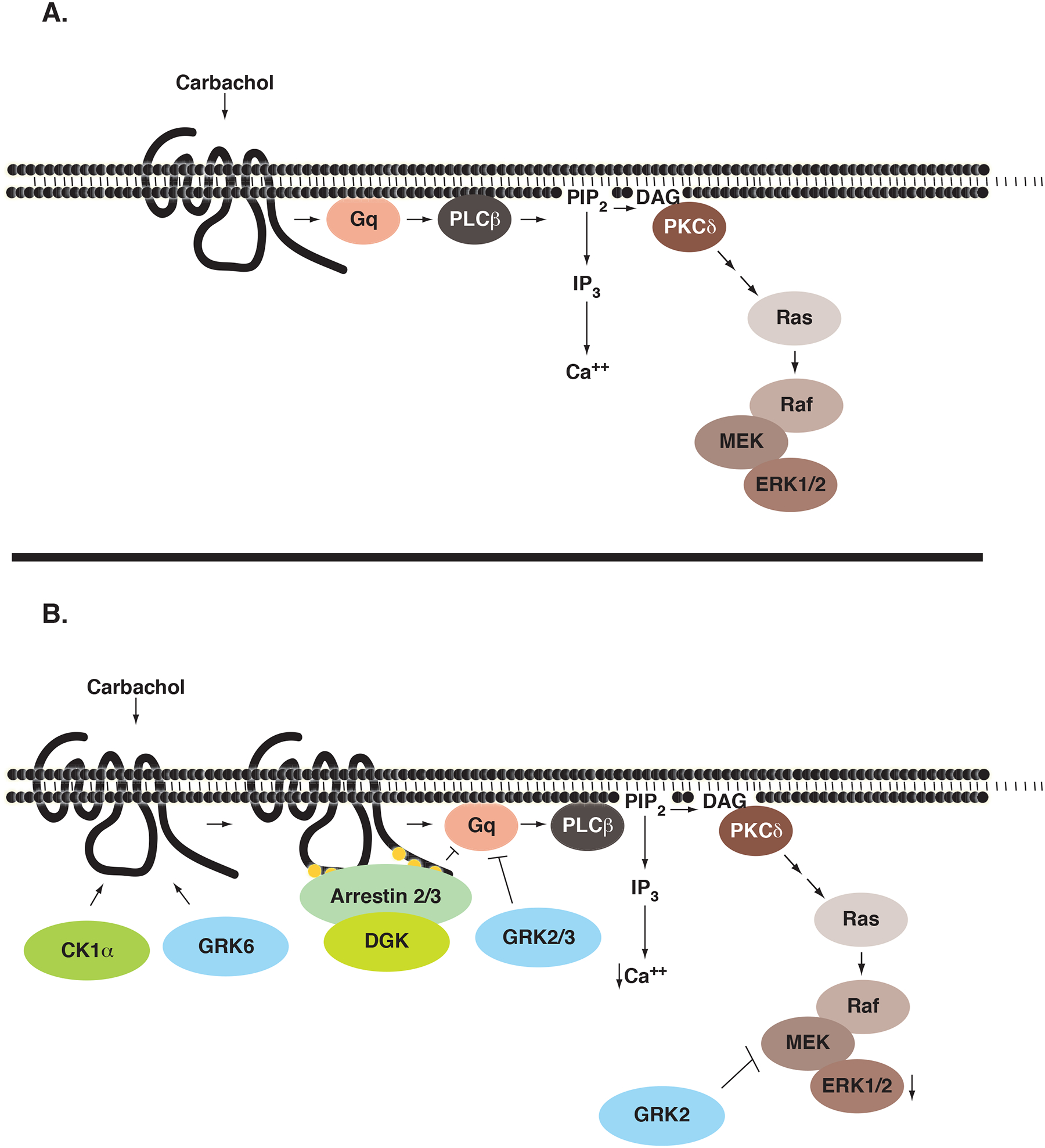
Regulation of the Endogenous M3 mAchR in HEK-293 Cells. A) Carbachol binding to the M3 mAchR results in activation of the Gq family of heterotrimeric G proteins leading to the dissociation of Gαq and Gβγ. Activated Gαq activates PLC-β resulting in the hydrolysis of PIP2 to form the second messengers IP3 and DAG. IP3 interacts with the IP3 receptor located at the endoplasmic reticulum, resulting in a robust but transient increase in cytosolic calcium. The formation of DAG recruits and activates the novel PKC-isoform, PKC-δ. Once activated, PKC-δ leads to the activation of a Ras-Raf-MEK-ERK1/2 cascade. B) Phosphorylation of the M3 mAchR by GRK6 and possibly CK1α recruits arrestin-2 and arrestin-3 to the receptor, preventing further G protein activation and terminating signaling. In addition, arrestins are able to recruit diacylglycerol kinases (DGK) to the membrane and terminate the PKC-dependent arm of the signaling cascade. GRK2 and GRK3, through a conserved RGS-domain, are able to interact with and sequester free Gαq and prevent activation of PLC-β. This results in the inhibition of both calcium mobilization and activation of the ERK1/2 cascade. GRK2 is also able to regulate activation of the ERK1/2 cascade by interacting with and negatively regulating the activity of MEK1.
CK1α has a variety of functions within the cell (Knippschild et al., 2005) and recently has been shown to regulate heterologously expressed M3 mAchR in HEK-293 and COS7 cells (Budd et al., 2000; Tobin et al., 1997). These studies showed that CK1α phosphorylated the receptor in an agonist dependent manner, and that deletion of a portion of the third intracellular loop or transient expression of a dominant-negative CK1α construct caused an increase in IP3 production upon receptor stimulation. Similarly, in the present study, we demonstrate that CK1α knockdown results in enhanced calcium mobilization upon M3 receptor activation, suggesting that CK1α is also involved in desensitization of endogenous M3 mAchR in HEK-293 cells. Knockdown of CK1α had no effect on calcium mobilization upon H1 histamine receptor activation, demonstrating that this effect was specific to the M3 mAchR. Previous studies have also shown that expression of a peptide corresponding to the CK1α binding region or overexpression of a mutated receptor lacking a portion of the third intracellular loop led to a decrease in ERK1/2 activation upon receptor stimulation, suggesting that CK1α-mediated phosphorylation was necessary for ERK activation (Budd et al., 2001). While we show that knockdown of CK1α has no effect on ERK1/2 activation (Figures 6C and 6D), indicating CK1α only plays a partial role in regulation of M3 mAchR similar to GRK3 and GRK6, this may be due to the fact that we only achieved ~60% knockdown of CK1α. It is interesting to note that the peptide expressed in previous studies to sequester CK1α also contained a portion of the Gβγ binding site of the third intracellular loop (Budd et al., 2001; Wu et al., 2000). While free Gβγ was preferred, the heterotrimeric G protein complex could also bind to this region of the receptor (Wu et al., 2000). Therefore, overexpression of this peptide could result in sequestration of the G protein, decreasing activation of downstream signaling. The third intracellular loop of the M3 mAchR contains 12 putative CK1α phosphorylation motifs (Tobin, 2002), two of which overlap with the proposed GRK2 phosphorylation sites (Wu et al., 2000). Thus, under physiological conditions, there could be competition between these kinases for receptor binding and phosphorylation.
In this study, we demonstrate that multiple proteins coordinately regulate the activity of the endogenous M3 mAchR in HEK-293 cells (Figure 7). Knockdown of GRK2, GRK3, GRK6, and CK1α, but not GRK5, enhanced receptor calcium signaling, suggesting that multiple kinases regulate downstream signaling following M3 mAchR activation. The effect of GRK2 on calcium flux could be enhanced by both wild type and a kinase-dead mutant but not by Gαq-binding defective mutants demonstrating that GRK2 primarily regulates activated Gq. Interestingly, only silencing of GRK2 led to both an enhanced and prolonged ERK activation. Consistent with our findings that GRK2 primarily regulated Gq activity, this is likely a result of enhanced activation of the Gq/PLC-β/PKC-δ signaling pathway (Figure 7). Finally, both arrestin-2 and arrestin-3 are involved in negatively regulating the M3 mAchR as knockdown of either protein enhanced calcium mobilization and ERK activation. Overall, our data suggest that multiple proteins dynamically regulate M3 mAchR-mediated signal transduction.
Acknowledgments
Work was supported by NIH grants GM44944 and GM47417 (J.L.B). JMB is supported by a predoctoral fellowship from the American Heart Association.
List of non-standard abbreviations:
- Bis I
Bisindolymaleimide I
- Bis V
Bisindolymaleimide V
- CK1α
casein kinase 1-alpha
- DAG
diacylglycerol
- ERK
extracellular signal-regulated kinase
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- IP3
inositol trisphosphate
- M3 mAchR
muscarinic acetycholine receptor subtype 3
- pFHHsiD
p-fluorohexahydro-sila-difenol
- PKC
protein kinase C
- PLC-β
phospholipase C-β
References
- Ahn S, Shenoy SK, Wei H and Lefkowitz RJ (2004) Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem 279:35518–35525. [DOI] [PubMed] [Google Scholar]
- Brown SG, Thomas A, Dekker LV, Tinker A and Leaney JL (2005) PKC-delta sensitizes Kir3.1/3.2 channels to changes in membrane phospholipid levels after M3 receptor activation in HEK-293 cells. Am J Physiol Cell Physiol 289:C543–556. [DOI] [PubMed] [Google Scholar]
- Budd DC, McDonald JE and Tobin AB (2000) Phosphorylation and regulation of a Gq/11-coupled receptor by casein kinase 1alpha. J Biol Chem 275:19667–19675. [DOI] [PubMed] [Google Scholar]
- Budd DC, Rae A and Tobin AB (1999) Activation of the mitogen-activated protein kinase pathway by a Gq/11-coupled muscarinic receptor is independent of receptor internalization. J Biol Chem 274:12355–12360. [DOI] [PubMed] [Google Scholar]
- Budd DC, Willars GB, McDonald JE and Tobin AB (2001) Phosphorylation of the Gq/11-coupled m3-muscarinic receptor is involved in receptor activation of the ERK-1/2 mitogen-activated protein kinase pathway. J Biol Chem 276:4581–4587. [DOI] [PubMed] [Google Scholar]
- Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL and Kozasa T (1999) Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem 274:34483–34492. [DOI] [PubMed] [Google Scholar]
- de la Vega MT, Nunez A and Arias-Montano JA (1997) Muscarinic M1 and M3 receptors in rat striatum: a binding study. Arch Med Res 28:493–497. [PubMed] [Google Scholar]
- Dhami GK, Babwah AV, Sterne-Marr R and Ferguson SS (2005) Phosphorylation-independent regulation of metabotropic glutamate receptor 1 signaling requires g protein-coupled receptor kinase 2 binding to the second intracellular loop. J Biol Chem 280:24420–24427. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ and Caron MG (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27:107–144. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Kittstein W and Marks F (1994) Elongation factor-2 kinase: effective inhibition by the novel protein kinase inhibitor rottlerin and relative insensitivity towards staurosporine. FEBS Lett 338:85–88. [DOI] [PubMed] [Google Scholar]
- Guo FF, Kumahara E and Saffen D (2001) A CalDAG-GEFI/Rap1/B-Raf cassette couples M(1) muscarinic acetylcholine receptors to the activation of ERK1/2. J Biol Chem 276:25568–25581. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Shrestha D, Goegel MC, Behan DP and Chalmers DT (2003) Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett 550:11–17. [DOI] [PubMed] [Google Scholar]
- Iwata K, Luo J, Penn RB and Benovic JL (2005) Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J Biol Chem 280:2197–2204. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sainz MC, Murga C, Kavelaars A, Jurado-Pueyo M, Krakstad BF, Heijnen CJ, Mayor F Jr., Aragay AM. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell 17:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H and Lefkowitz RJ (2005) Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A 102:1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Yang MS, Oh CD, Kim KT, Ha MJ, Kang SS and Chun JS (1999) Signalling pathway leading to an activation of mitogen-activated protein kinase by stimulating M3 muscarinic receptor. Biochem J 337 (Pt 2):275–280. [PMC free article] [PubMed] [Google Scholar]
- Knippschild U, Wolff S, Giamas G, Brockschmidt C, Wittau M, Wurl PU, Eismann T and Stoter M (2005) The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 28:508–514. [DOI] [PubMed] [Google Scholar]
- Krupnick JG and Benovic JL (1998) The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38:289–319. [DOI] [PubMed] [Google Scholar]
- Lee KB, Pals-Rylaarsdam R, Benovic JL and Hosey MM (1998) Arrestin-independent internalization of the m1, m3, and m4 subtypes of muscarinic cholinergic receptors. J Biol Chem 273:12967–12972. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ and Shenoy SK (2005) Transduction of receptor signals by beta-arrestins. Science 308:512–517. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X and He X (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847. [DOI] [PubMed] [Google Scholar]
- Moore CA, Milano SK and Benovic JL (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69:451–482. [DOI] [PubMed] [Google Scholar]
- Mundell SJ and Benovic JL (2000) Selective regulation of endogenous G protein-coupled receptors by arrestins in HEK293 cells. J Biol Chem 275:12900–12908. [DOI] [PubMed] [Google Scholar]
- Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK and Lefkowitz RJ (2007) Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science 315:663–666. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT and Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3:639–650. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG and Lefkowitz RJ (1992) Novel role for the βγ subunits of heterotrimeric G proteins: Targeting of beta-adrenergic receptor kinase to membrane bound receptors. Science 257:1264–1267. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, Aymerich I and Mayor F Jr. (2007) The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta 1768:913–922. [DOI] [PubMed] [Google Scholar]
- Sherrill JD and Miller WE (2006) G protein-coupled receptor (GPCR) kinase 2 regulates agonist-independent Gq/11 signaling from the mouse cytomegalovirus GPCR M33. J Biol Chem 281:39796–39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T and Tesmer JJ (2005) Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science 310:1686–1690. [DOI] [PubMed] [Google Scholar]
- Tobin AB (2002) Are we beta-ARKing up the wrong tree? Casein kinase 1 alpha provides an additional pathway for GPCR phosphorylation. Trends Pharmacol Sci 23:337–343. [DOI] [PubMed] [Google Scholar]
- Tobin AB, Totty NF, Sterlin AE and Nahorski SR (1997) Stimulus-dependent phosphorylation of G-protein-coupled receptors by casein kinase 1alpha. J Biol Chem 272:20844–20849. [DOI] [PubMed] [Google Scholar]
- Tovey SC and Willars GB (2004) Single cell imaging of intracellular Ca2+ and phospholipase activity revelas that RGS 2, 3, and 4 differentially regulate signaling via the Galphaq/11-linked muscarinic M3 receptor. Mol Pharm 66: 1453–1464. [DOI] [PubMed] [Google Scholar]
- Waugh MG, Challiss RA, Berstein G, Nahorski SR and Tobin AB (1999) Agonist-induced desensitization and phosphorylation of m1-muscarinic receptors. Biochem J 338 (Pt 1):175–183. [PMC free article] [PubMed] [Google Scholar]
- Werry TD, Sexton PM and Christopoulos A (2005) “Ins and outs” of seven-transmembrane receptor signalling to ERK. Trends Endocrinol Metab 16:26–33. [DOI] [PubMed] [Google Scholar]
- Willets JM, Challiss RA, Kelly E and Nahorski SR (2001) G protein-coupled receptor kinases 3 and 6 use different pathways to desensitize the endogenous M3 muscarinic acetylcholine receptor in human SH-SY5Y cells. Mol Pharmacol 60:321–330. [DOI] [PubMed] [Google Scholar]
- Willets JM, Challiss RA and Nahorski SR (2002) Endogenous G protein-coupled receptor kinase 6 Regulates M3 muscarinic acetylcholine receptor phosphorylation and desensitization in human SH-SY5Y neuroblastoma cells. J Biol Chem 277:15523–15529. [DOI] [PubMed] [Google Scholar]
- Willets JM, Mistry R, Nahorski SR and Challiss RA (2003) Specificity of g protein-coupled receptor kinase 6-mediated phosphorylation and regulation of single-cell m3 muscarinic acetylcholine receptor signaling. Mol Pharmacol 64:1059–1068. [DOI] [PubMed] [Google Scholar]
- Willets JM, Nahorski SR and Challiss RA (2005) Roles of phosphorylation-dependent and - independent mechanisms in the regulation of M1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 in hippocampal neurons. J Biol Chem 280:18950–18958. [DOI] [PubMed] [Google Scholar]
- Wu G, Bogatkevich GS, Mukhin YV, Benovic JL, Hildebrandt JD and Lanier SM (2000) Identification of Gbetagamma binding sites in the third intracellular loop of the M(3)-muscarinic receptor and their role in receptor regulation. J Biol Chem 275:9026–9034. [DOI] [PubMed] [Google Scholar]
- Wylie PG, Challiss RA and Blank JL (1999) Regulation of extracellular-signal regulated kinase and c-Jun N-terminal kinase by G-protein-linked muscarinic acetylcholine receptors. Biochem J 338 (Pt 3):619–628. [PMC free article] [PubMed] [Google Scholar]