

Visual Abstract

Keywords: chronic kidney disease, anemia, hemodialysis, Erythropoiesis, hypoxia-inducible factor prolyl hydroxylase inhibitor, daprodustat, HIF, clinical trial, double-blind, hemoglobin, hepcidin
Abstract
Background and objectives
Daprodustat is an oral hypoxia-inducible factor prolyl hydroxylase inhibitor that stimulates erythropoiesis and regulates genes related to iron metabolism. The efficacy (noninferiority) and safety of daprodustat compared with standard therapy (darbepoetin alfa) was evaluated.
Design, setting, participants, & measurements
This was a randomized, phase 3, double-blind, active-control study in Japanese patients receiving hemodialysis with anemia of CKD. Participants’ treatment was switched from current erythropoiesis-stimulating agents (ESAs) to daprodustat 4 mg once daily or darbepoetin alfa 10–60 μg once weekly (on the basis of the prestudy ESA dose). Dose was adjusted every 4 weeks for daprodustat or every 2 weeks for darbepoetin alfa, according to a protocol-specified algorithm. The primary end point was mean hemoglobin during weeks 40–52 in the intent-to-treat population.
Results
Of 332 participants screened, 271 participants were randomized (safety evaluation: 271 participants; efficacy evaluation: 267 intent-to-treat population). The mean hemoglobin during weeks 40–52 were maintained within the target range in both groups (10.9 g/dl [95% confidence interval (95% CI), 10.8 to 11.0] for daprodustat, and 10.8 g/dl [95% CI, 10.7 to 11.0] for darbepoetin alfa). Daprodustat was noninferior to darbepoetin alfa, as the lower bound of the confidence interval for the treatment difference (0.1 g/dl; 95% CI, −0.1 to 0.2 g/dl) was greater than the noninferiority criterion of −1.0 g/dl. For most participants, hemoglobin was maintained within the target range (10.0–12.0 g/dl) during weeks 40–52 (88% daprodustat; 90% darbepoetin alfa). Geometric mean hepcidin levels decreased more at week 52 with daprodustat (−37%; 95% CI, −49 to −23) than with darbepoetin alfa (−20%; 95% CI, −36 to −1), and an increase in total iron-binding capacity was observed in the daprodustat group. Frequency of adverse events were generally similar between daprodustat and darbepoetin alfa.
Conclusions
Oral daprodustat was noninferior to darbepoetin alfa as measured by mean hemoglobin over weeks 40–52 in Japanese patients receiving hemodialysis switched from ESAs.
Clinical Trial registry name and registration number
201754, Clinicaltrials.gov, NCT02969655.
Introduction
Anemia is a common complication of CKD, caused by multiple factors including erythropoietin deficiency and reduced iron availability (1,2). Erythropoiesis-stimulating agents (ESAs) are used to treat anemia as standard of care along with iron supplementation (intravenous [IV] or oral) (3,4). However, treatment with ESAs has been associated with higher risk of major cardiovascular events and exacerbation of hypertension when treating to higher hemoglobin targets than recommended (5–8). It remains unclear whether the risk is related to the hemoglobin levels achieved, the rapid hemoglobin rise, the dose of ESA used, or the inability to achieve target hemoglobin levels. Hypoxia-inducible factor-prolyl hydroxylase inhibitors (HIF-PHIs) are a class of therapeutic agents being developed for the treatment of anemia of CKD (9). In phase 2 studies, the HIF-PHI daprodustat (0.5–25 mg once daily) achieved and maintained target hemoglobin levels in patients receiving and not receiving dialysis, with anemia of CKD (10–12). A phase 3 trial in Chinese patients on dialysis reported that another HIF-PHI, roxadustat, administered three times a week for 27 weeks was noninferior to erythropoietin with regard to mean change in hemoglobin (13).
In this phase 3 study, we tested the hypothesis that daprodustat is noninferior to darbepoetin alfa in maintaining hemoglobin levels in Japanese patients on hemodialysis with anemia of CKD currently being treated with an ESA. The 1-year safety of daprodustat was also evaluated.
Materials and Methods
Study Design
This was a phase 3, double-blind, active-controlled, parallel-group study to evaluate the efficacy (noninferiority) and safety of daprodustat compared with darbepoetin alfa for 52 weeks in Japanese patients on hemodialysis with anemia of CKD currently treated with ESAs (Clinicaltrials.gov identifier NCT02969655, registered November 21, 2016). The study was conducted between November 21, 2016 and July 2, 2018, with recruitment from the beginning of the study to May 30, 2017, at 50 centers in Japan, and included a 2- to 4-week screening period, a 52-week treatment phase, and a 2-week follow-up period. The primary efficacy evaluation period was weeks 40–52. No interim analysis was planned.
The study was approved by the ethics committee at every participating institution and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki. All participants provided written informed consent.
Eligibility Criteria
Patients aged ≥20 years who were on three times weekly hemodialysis or hemodiafiltration for ≥12 weeks before screening were included. Additional eligibility criteria included use of the same ESA (darbepoetin alfa 10–60 µg per week, epoetin [including biosimilars] ≤9000 IU per week, or epoetin beta pegol ≤250 µg per 4 weeks) for ≥10 weeks before screening; hemoglobin levels ≥9.5 g/dl and ≤12.5 g/dl, as determined by a hemoglobin analyzer (HemoCue) at the study site; and ferritin >100 ng/ml or transferrin saturation (TSAT) >20% at screening. The complete inclusion, exclusion, and study medication stopping criteria are listed in Supplemental Appendix 1.
Randomization and Intervention
Patients who met all eligibility criteria at the start of treatment (day 1) were randomized at a 1:1 ratio to receive daprodustat or darbepoetin alfa. A biostatistician generated the randomization codes using a company-validated system. The system concealed the actual randomization codes until unblinding. A random permutation of treatment assignments within blocks (block size was set to 4) was used for the randomization sequence. The investigational products containers were also randomized independently of the treatment randomization by a third-party vendor with no clinical involvement in the study. At each dispensing visit, the investigators accessed the Interactive Web Response System to obtain a particular container in a blinded manner and assigned the patients to interventions.
Study Treatments
Participants randomized to daprodustat received orally administered daprodustat once daily and IV darbepoetin alfa matching placebo once weekly. On day 1, treatment with 4 mg daprodustat was started. From week 4 onward, the dose was adjusted every 4 weeks within the range of 1–24 mg according to a prespecified dose adjustment algorithm (Supplemental Table 1) to achieve and/or maintain hemoglobin within the target range (10.0–12.0 g/dl), using HemoCue hemoglobin values.
Participants randomized to darbepoetin alfa received oral daprodustat matching placebo once daily and IV darbepoetin alfa once weekly. On day 1, darbepoetin alfa treatment was started at a corresponding dose to the prior ESA. From week 2 onward, the dose was adjusted every 2 weeks (on the basis of the Japanese label for darbepoetin alfa) within the range of 10–60 µg, according to a prespecified dose adjustment algorithm using HemoCue hemoglobin values (Supplemental Table 2).
Daprodustat could be administered without regard to food or hemodialysis. Gemfibrozil and rifampin were excluded medications from screening to 7 days after treatment completion. In both groups, IV iron or dose change for oral iron were not allowed from screening to week 4. From week 4 onward, supplemental iron therapy including IV and oral could be administered if ferritin was ≤100 ng/ml and TSAT was ≤20%, according to the Japanese Society for Dialysis Therapy guidelines for anemia in CKD (14).
Study End Points
The primary end point was mean hemoglobin during the primary efficacy evaluation period (weeks 40–52). The principal secondary end point was the percentage of participants with mean hemoglobin within target range during the primary efficacy evaluation period. Additional secondary end points included change from baseline and mean hemoglobin at each assessment visit. Exploratory end points included iron use during the treatment period and change from baseline in iron parameters (ferritin, TSAT, hepcidin, total iron-binding capacity [TIBC], and serum iron).
Safety assessments included adverse events (AEs), serious adverse events (SAEs), AEs of special interest, laboratory evaluations, and vital signs. AEs of special interest were defined a priori on the basis of nonclinical studies of daprodustat, theoretical or potential risks related to the mechanism of action of daprodustat, and the known safety profile of ESAs. An internal Safety Review Team conducted periodic case reviews in a blinded manner to evaluate which events constituted a potential “AE of special interest.” Comprehensive ophthalmologic exams (best corrected visual acuity, intraocular pressure, anterior segment examination, and funduscopic examination) were conducted in a blinded manner by a study-designated ophthalmology specialist at baseline, week 12, and week 48 to support assessment of ocular AEs of special interest.
Statistical Analyses
Assuming a noninferiority margin of −1.0 g/dl, a true treatment difference of 0.0 g/dl, and an SD of 1.5 g/dl for the primary end point, noninferiority test has at least 99% power at a one-sided significance level of 2.5%, with a sample size of 100 participants per group. Assuming a dropout rate of 25%, 135 participants per group were targeted to be randomized. This study was also designed to ensure long-term safety data from 100 patients on hemodialysis in the daprodustat group.
The primary end point, noninferiority to darbepoetin alfa, was evaluated in the intent-to-treat (ITT) population (all randomized participants with a baseline and one or more postbaseline hemoglobin assessment). Mixed model for repeated measures was used to estimate treatment difference in mean hemoglobin and 95% confidence intervals (95% CIs) during the primary efficacy evaluation period. This model included treatment groups, baseline hemoglobin, visits, interaction terms between treatment groups and visits, and interaction terms between baseline hemoglobin and visits.
The principal secondary end point was evaluated in the modified ITT population (all ITT participants with one or more hemoglobin measurement during the evaluation period). A logistic regression model, including treatment group and baseline hemoglobin as covariates, was used to estimate the odds ratio (daprodustat/darbepoetin alfa).
Treatment difference in changes from baseline in iron parameters were evaluated in the ITT population by the same mixed model for repeated measures model used for the primary end point. Other secondary end points and exploratory end points were evaluated in the ITT population and are summarized descriptively. The safety population included all participants who received one or more dose of study treatment. Imputation for missing data were not performed in any analyses.
Data Sharing
Within 6 months of this publication, anonymized individual participant data, the annotated case report form, protocol, reporting and analysis plan, data set specifications, raw data set, analysis-ready data set, and clinical study report will be available for research proposals approved by an independent review committee. Proposals should be submitted to www.clinicalstudydatarequest.com. A data access agreement will be required.
Results
Participant Disposition and Baseline Characteristics
Of 332 participants screened, 271 participants were randomized (136 daprodustat, 135 darbepoetin alfa) (Figure 1). A total of 115 participants (85%) in the daprodustat group and 120 (89%) in the darbepoetin alfa group completed the study. The most common reasons for study withdrawal in both treatment groups were AEs (ten daprodustat, eight darbepoetin alfa). All 271 participants were included in the safety population, 267 participants (133 daprodustat, 134 darbepoetin alfa) were included in the ITT population, and 245 participants (120 daprodustat, 125 darbepoetin alfa) were included in the modified ITT population.
Figure 1.
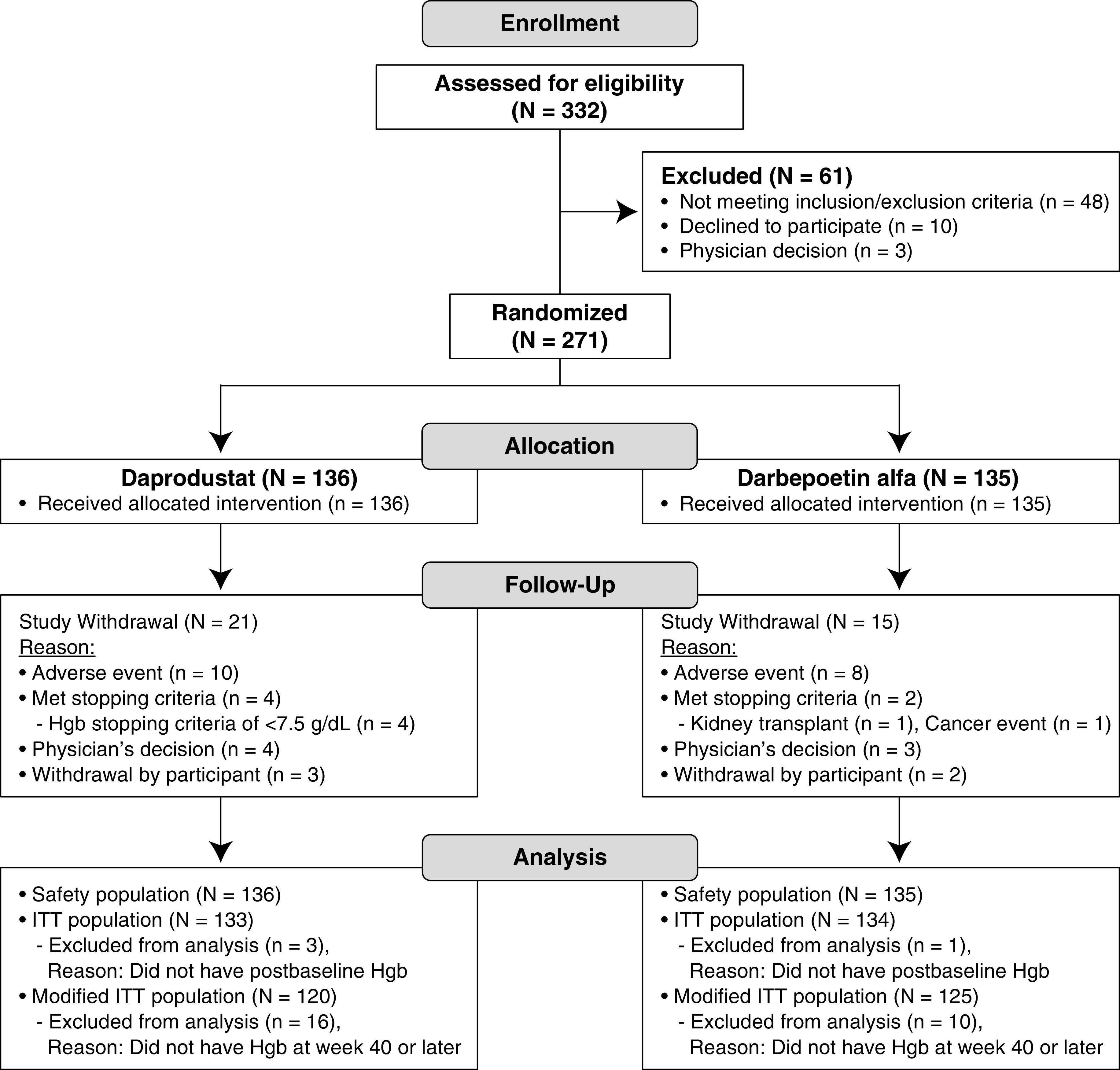
Two hundred seventy-one participants were randomized (136 participants in the daprodustat group, 135 participants in the darbepoetin alfa group) in this study. Hgb, hemoglobin; ITT, intent-to-treat.
Participant characteristics were generally balanced between treatment groups (Table 1). Overall, 66% of participants were men, and mean age was 64 years. Darbepoetin alfa was the most commonly used prior ESA for both treatment groups. Approximately two thirds of participants were undergoing hemodialysis and one third were undergoing hemodiafiltration, with an average time on dialysis of 8 years in both groups.
Table 1.
Baseline characteristics of participants in a randomized, clinical trial evaluating the efficacy (noninferiority) and safety of daprodustat compared with darbepoetin alfa in Japanese patients on hemodialysis (safety population)
| Characteristic | Daprodustat, n=136 | Darbepoetin Alfa, n=135 |
|---|---|---|
| Sex, n (%) | ||
| Male | 91 (67) | 89 (66) |
| Age, yr, mean±SD | 64±10 | 64±11 |
| BMI, kg/m2, mean±SD | 22±3.8 | 23±4.2 |
| Prior ESA type, n (%) | ||
| Epoetin | 53 (39) | 58 (43) |
| Darbepoetin alfa | 70 (51) | 64 (47) |
| Epoetin beta pegol | 13 (10) | 13 (10) |
| Prior ESA dose, IU/wk, mean±SDa | 4504±2318 | 4501±2474 |
| ERI, IU/kg per wk per g/dl, n (%)b,c | ||
| Tertile 1: <4.79 | 41/133 (31) | 48/134 (36) |
| Tertile 2: ≥4.79 and <8 | 45/133 (34) | 44/134 (33) |
| Tertile 3: ≥8.0 | 47/133 (35) | 42/134 (31) |
| Time on dialysis, yr, mean±SD | 7.9±6.9 | 7.9±7.1 |
| Mode of dialysis, n (%) | ||
| Hemodialysis | 86 (63) | 88 (65) |
| Hemodiafiltration | 50 (37) | 47 (35) |
| Hgb, g/dl, mean±SDc | 10.9±0.8 | 10.8±0.7 |
| Iron parametersc | ||
| Serum iron, µg/dl, mean (SD) | 68 (25) | 65 (23) |
| TIBC, µg/dl, mean (SD) | 254 (39) | 249 (38) |
| TSAT, %, mean (SD) | 27.0 (10.1) | 26.3 (8.9) |
| Ferritin, µg/L, geometric mean (CV %) | 86.5 (108.9) | 96.6 (124.5) |
| Hepcidin, ng/ml, geometric mean (CV %) | 54.1 (107.9) | 60.1 (105.0) |
| Hypertension, n (%) | 127 (93) | 125 (93) |
| Hyperlipidemia, n (%) | 57 (42) | 60 (44) |
| Diabetes mellitus, n (%) | 56 (41) | 52 (39) |
| Angina, n (%) | 23 (17) | 39 (29) |
| Eye disorders, n (%) | 52 (38) | 42 (31) |
| Diabetic retinopathy | 39 (29) | 39 (29) |
| Macular edema | 8 (6) | 5 (4) |
| Age-related macular degeneration | 5 (4) | 8 (6) |
BMI, body mass index; ESA, erythropoiesis-stimulating agent; ERI, erythropoietin resistance index; Hgb, hemoglobin; TIBC, total iron-building capacity; TSAT, transferrin saturation; CV, coefficient of variation; IV, intravenous; SC, subcutaneous; ITT, intent-to-treat.
Standardized to epoetin IV dose (IU/wk) as follows: epoetin SC dose (IU/wk) multiplied by 116/113; darbepoetin alfa dose (μg/wk) multiplied by 250; epoetin beta pegol dose (μg/wk) multiplied by 208.
ERI (IU/kg per week per g/dl)=standardized prior ESA dose (IU/wk)/baseline weight (kg)/baseline Hgb (g/dl).
ITT population.
Hemoglobin Levels
The point estimates of mean hemoglobin in the ITT population during the primary efficacy evaluation period were 10.9 g/dl (daprodustat) and 10.8 g/dl (darbepoetin alfa), with a between-group difference of 0.1 g/dl (95% CI, −0.1 to 0.2 g/dl). The lower limit of 95% CI for the treatment difference was greater than the prespecified noninferiority margin of −1.0 g/dl, demonstrating noninferiority of daprodustat to darbepoetin alfa. Mean change from baseline to week 52 is shown in Table 2.
Table 2.
Summary of hemoglobin efficacy data (intent-to-treat population)
| Hemoglobin | Daprodustat, n=133 | Darbepoetin Alfa, n=134 |
|---|---|---|
| Baseline Hgb, g/dl | ||
| n | 133 | 134 |
| Mean (SD) | 10.9 (0.8) | 10.8 (0.7) |
| Mean Hgb during wk 40–52, g/dl | ||
| n | 120 | 125 |
| Mean (SD) | 10.9 (0.7) | 10.8 (0.6) |
| Change from baseline (SD) | 0.0 (1.0) | 0.0 (0.8) |
| Adjusted mean Hgb during wk 40–52, g/dl | ||
| Adjusted mean (95% CI) | 10.9 (10.8 to 11.0) | 10.8 (10.7 to 11.0) |
| Adjusted treatment difference (95% CI) for mean Hgb during wk 40–52 | 0.1 (−0.1 to 0.2) | |
| No. (%) of participants with mean Hgb within target during wk 40–52a | ||
| n | 120 | 125 |
| No. of participants (%) | 105 (88) | 113 (90) |
Change from baseline in mean Hgb during weeks 40–52 was performed in a post hoc analysis. Adjusted values were estimated by a mixed model for repeated measures as specified in Statistical Analyses. ITT, intent-to-treat; Hgb, hemoglobin; 95% CI, 95% confidence interval.
Modified ITT population.
The percentage of participants with mean hemoglobin within the target range during the primary efficacy evaluation period was 88% (105 out of 120) in the daprodustat group and 90% (113 out of 125) in the darbepoetin alfa group (modified ITT population). This translates to the daprodustat group having unadjusted odds of being in the target range equal to 7 (responder/non-responder; 105/15) and the darbepoetin group having an unadjusted odds of 9.42 (responder/non-responder; 113/12). The odds ratio point estimate (daprodustat/darbepoetin alfa) was 0.76 (95% CI, 0.34 to 1.71), on the basis of a logistic regression analysis.
The mean (95% CI) hemoglobin levels were maintained within the target range for both groups at all time points during the treatment period (Figure 2). After switching to daprodustat, mean hemoglobin levels were lower relative to baseline from week 4 to week 16; however, the mean changes from baseline were <0.5 g/dl and the mean hemoglobin levels remained within target range. With subsequent dose adjustment, mean hemoglobin returned to baseline by week 20. The proportion of participants with a rapid hemoglobin increase (>2 g/dl for any 4 weeks) (Supplemental Table 3) and hemoglobin level >13 g/dl was similar in both groups (Supplemental Table 4).
Figure 2.

The mean hemoglobin levels were maintained within the target range for the daprodustat and the darbepoetin alfa groups during the treatment period. Area between dashed lines indicates hemoglobin target range. Error bars represent 95% confidence intervals. Data shown are nonadjusted values.
In a subgroup analysis according to erythropoietin resistance index (ERI), daprodustat maintained mean hemoglobin within the target range during weeks 40–52 in all three subgroups. However, initially after switching to daprodustat, decreases in mean hemoglobin were observed in tertile 2 (≥4.79 and <8.0 IU/kg per week per g/dl) and tertile 3 (≥8.0 IU/kg per week per g/dl) ERI subgroups, but not in tertile 1 ERI subgroup (<4.79 IU/kg per week per g/dl). With subsequent dose adjustment, mean hemoglobin returned to baseline in the tertile 2 and tertile 3 ERI subgroups. Correspondingly, median daprodustat dose was increased to 6 mg (from 4 mg starting dose) by week 12 in those participants. Median (25th percentile, 75th percentile) daprodustat doses during weeks 40–52 were 4.0 (2.7–6.0) mg, 6.0 (4.0–8.7) mg, and 6.0 (4.0–8.0) mg for the tertile 1, 2, and 3 ERI subgroups, respectively (Figure 3). Mean hemoglobin levels generally remained constant in all three ERI subgroups for the darbepoetin alfa group. In a post hoc analysis, median (25th percentile, 75th percentile) darbepoetin alfa doses during weeks 40–52 were 10.4 (4.2–15.0) μg, 15.0 (13.3–22.9) μg, and 25.8 (12.5–46.7) μg for the tertile 1, 2, and 3 subgroups, respectively, suggesting that a broader range of darbepoetin alfa dose was used during the study.
Figure 3.
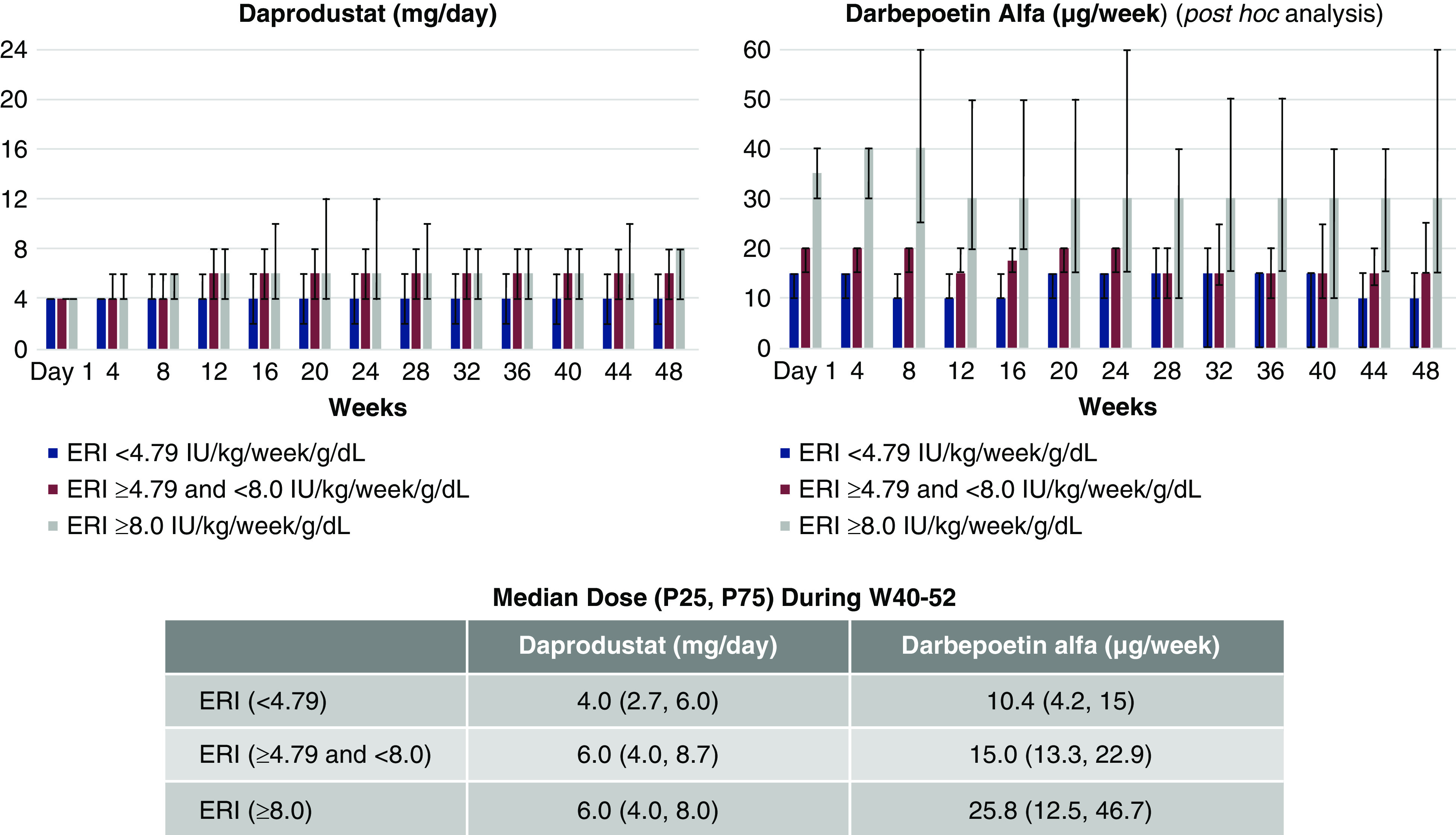
Daprodustat dose was increased by week 12 in the tertile 2 and 3 ERI subgroups. Daprodustat median dose during weeks 40–52 were 4.0 mg, 6.0 mg, and 6.0 mg for the tertile 1, 2, and 3 ERI subgroups, respectively. Error bars represent 25th percentile, 75th percentile for median dose. Median dose by ERI subgroup of darbepoetin alfa was a post hoc analysis. ERI, erythropoietin resistance index; W, week.
Iron Use
Fewer participants used IV iron in the daprodustat than in the darbepoetin alfa group (32% daprodustat, 43% darbepoetin alfa throughout the treatment period; 18% daprodustat, 27% darbepoetin alfa during weeks 40–52). The mean monthly dose of IV iron for all study participants (ITT population) was 14 mg (daprodustat) versus 17 mg (darbepoetin alfa) throughout the treatment period and 14 mg (daprodustat) versus 25 mg (darbepoetin alfa) during weeks 40–52. The percentage of participants using oral iron during the study was similar in both groups (32% daprodustat, 34% darbepoetin alfa).
Iron Parameters
Baseline iron parameters are shown in Table 1. Mean serum iron and TIBC (Figure 4, Table 3) increased with daprodustat treatment and remained near baseline with darbepoetin alfa throughout the treatment period. Mean TSAT remained near baseline with daprodustat treatment (after a brief increase at week 4) and decreased slightly with darbepoetin alfa.
Figure 4.
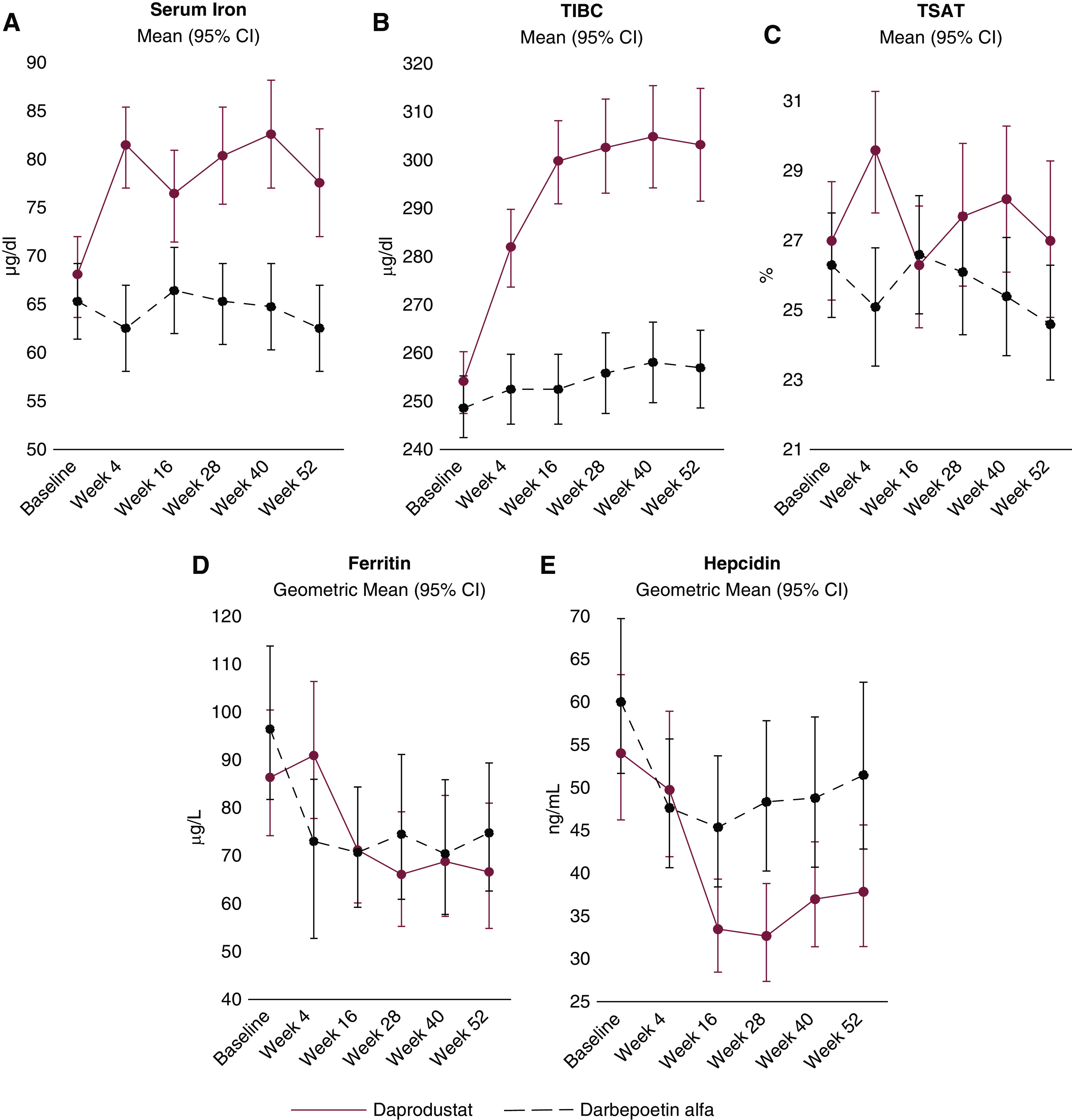
Iron parameters over time. In the daprodustat group, serum iron and TIBC increased, ferritin and hepcidin decreased, and TSAT remained near baseline. Error bars represent 95% CIs. 95% CI, 95% confidence interval; TIBC, total iron-binding capacity; TSAT, transferrin saturation.
Table 3.
Iron parameters at end of treatment (intent-to-treat population)
| Iron Parameter | Daprodustat, n=133 | Darbepoetin Alfa, n=134 |
|---|---|---|
| n | 115 | 120 |
| TSAT at wk 52, %a | ||
| Mean (SD) | 27.0 (12.3) | 24.6 (9.1) |
| Change from baseline (SD) | 0.3 (13.2) | −2.1 (11.4) |
| Adjusted treatment difference (95% CI) | 2.8 (0.1 to 5.4) | |
| TIBC at wk 52, µg/dl | ||
| Mean (SD) | 303 (64) | 257 (44) |
| Change from baseline (SD) | 49 (56) | 6 (34) |
| Treatment difference (95% CI) | 41 (30 to 53) | |
| Serum iron at wk 52, µg/dl | ||
| Mean (SD) | 78 (29) | 63 (24) |
| Change from baseline (SD) | 11 (34) | −4 (28) |
| Treatment difference (95% CI) | 16 (10 to 22) | |
| Ferritin, µg/Lb | ||
| Geometric mean (CV %) | 66.7 (143.5) | 74.9 (127.8) |
| Percent change from baseline (CV %) | −22 (132.2) | −20 (124.1) |
| Adjusted ratio for percent change from baseline (95% CI) | 0.95 (0.75 to 1.19) | |
| Hepcidin, ng/mlb | ||
| Geometric mean (CV %) | 37.9 (103.8) | 51.5 (111.5) |
| Percent change from baseline (CV %) | −37 (116.1) | −20 (133.6) |
| Adjusted ratio for percent change from baseline (95% CI) | 0.74 (0.57 to 0.95) | |
TSAT, transferrin saturation; 95% CI, 95% confidence interval; TIBC, total iron-binding capacity; CV %, coefficient of variation.
The distribution in TSAT had been assumed to be skewed but was found not to be skewed after the data review; therefore, the analysis of TSAT on the basis of a nonlog transformation was done as a post hoc analysis.
The distributions were assumed to be skewed and required a log transformation for the analysis. The distribution in ferritin had been assumed not to be skewed but was found to be skewed after the data review; therefore, the analysis of ferritin on the basis of a log transformation was done as a post hoc analysis.
Geometric mean ferritin decreased from baseline in both treatment groups. Geometric mean hepcidin decreased in both groups, but decreased more in the daprodustat group (percent change from baseline at week 52 was −37% for daprodustat and −20% for darbepoetin alfa; adjusted ratio, 0.74; 95% CI, 0.57 to 0.95).
Baseline mean transferrin levels (measured as a safety parameter) were 2.0 g/L in both treatment groups. Transferrin levels increased in the daprodustat group (mean change from baseline at week 52: 0.3 g/L [SD 0.5 g/L] for daprodustat and 0.0 g/L [SD 0.3 g/L] for darbepoetin alfa), which was aligned with changes in TIBC.
Safety and AEs
Most participants in both treatment groups experienced one or more AEs (93% daprodustat, 97% darbepoetin alfa). Of the AEs occurring in ≥5% of participants in any group (Table 4), contusion and diarrhea were more frequent (≥5% difference) in the daprodustat group, whereas nasopharyngitis and extremity pain was more frequent (≥5% difference) in the darbepoetin alfa group. Hyperkalemia was reported in 3% of participants in the daprodustat group versus 1% in the darbepoetin alfa group. SAEs occurred in 15% (daprodustat) and 27% (darbepoetin alfa) of participants. SAEs reported in two or more participants (≥1%) in any treatment group were shunt stenosis (3% daprodustat, 4% darbepoetin alfa), shunt occlusion (<1% daprodustat, 2% darbepoetin alfa), shunt malfunction (0% daprodustat, 1% darbepoetin alfa), pneumonia (<1% daprodustat, 1% darbepoetin alfa), sepsis (0% daprodustat, 1% darbepoetin alfa), and cardiac failure congestive (0% daprodustat, 1% darbepoetin alfa). No deaths were reported in the daprodustat group. One fatal event (sepsis) was reported during the follow-up period in the darbepoetin alfa group. AEs of special interest were generally reported in a similar proportion of participants in both groups for all categories (Supplemental Table 5).
Table 4.
Adverse events reported in ≥5% of participants in any group (safety population)
| Adverse Event | Daprodustat, n=136, n (%) | Darbepoetin Alfa, n=135, n (%) |
|---|---|---|
| Nasopharyngitis | 57 (42) | 73 (54) |
| Pharyngitis | 10 (7) | 6 (4) |
| Gastroenteritis | 7 (5) | 2 (1) |
| Shunt stenosis | 19 (14) | 20 (15) |
| Contusion | 17 (13) | 11 (8) |
| Skin abrasion | 10 (7) | 7 (5) |
| Procedural hypotension | 11 (8) | 5 (4) |
| Diarrhea | 20 (15) | 12 (9) |
| Vomiting | 15 (11) | 11 (8) |
| Nausea | 9 (7) | 12 (9) |
| Constipation | 8 (6) | 6 (4) |
| Abdominal discomfort | 3 (2) | 7 (5) |
| Back pain | 6 (4) | 10 (7) |
| Arthralgia | 5 (4) | 10 (7) |
| Muscle spasms | 7 (5) | 4 (3) |
| Pain in extremity | 1 (<1) | 10 (7) |
| Headache | 8 (6) | 6 (4) |
| Pyrexia | 7 (5) | 3 (2) |
| Hypertension | 5 (4) | 8 (6) |
There was little or no change from baseline in postdialysis systolic and diastolic BP in both treatment groups during the treatment period. The percentage of participants who had any change in antihypertensive medications because of increased BP was 38% (daprodustat) and 49% (darbepoetin alfa) (Supplemental Table 6). No clinically relevant change from baseline was seen in potassium in either group (Supplemental Table 7). The proportion of participants with low, within range, or high postbaseline potassium was similar in the daprodustat and the darbepoetin alfa groups (Supplemental Table 8).
Discussion
This was the first phase 3 study to evaluate the efficacy and safety of daprodustat compared with an ESA over 1 year of treatment, although a phase 3 trial of patients treated with roxadustat for 27 weeks has been reported (13). Noninferiority of daprodustat to darbepoetin alfa was demonstrated, with a high proportion of participants achieving and maintaining hemoglobin levels within the target range. The daprodustat starting dose (4 mg) and subsequent potential for monthly titration according to the predefined algorithm (with doses ranging from 0 to 24 mg) provided an appropriate balance between achieving target hemoglobin levels without rapid rises (>2.0 g/dl over 4 weeks) in hemoglobin or exceeding hemoglobin levels >13.0 g/dl.
Differences in hemoglobin variability in the two treatment groups in this study could potentially be because almost half (64 out of 135) of the darbepoetin alfa group were on the same drug and dosage before study commencement. Furthermore, the difference in the time interval for dose adjustment (once every 4 weeks for daprodustat versus once every 2 weeks for darbepoetin alfa) may have contributed to hemoglobin variability.
Interestingly, hemoglobin response varied between participants, as evidenced by the observation that some participants could maintain hemoglobin levels with 4 mg of daprodustat even in the tertile 3 (≥8.0 IU/kg per week per g/dl) ERI subgroup. Our results suggest that monitoring hemoglobin response upon treatment initiation with daprodustat is highly recommended. In addition, the use of lower daprodustat doses may reflect resolution of ESA resistance as ESA hyporesponsiveness can resolve over time.
Fewer participants in the daprodustat than the darbepoetin alfa group used IV iron throughout the double-blind treatment period, despite achieving comparable hemoglobin levels.
The observed decrease in ferritin with daprodustat treatment may be secondary to increased iron utilization and depletion of iron stores owing to erythropoiesis. The greater decrease in hepcidin, accompanied by decreases in ferritin and less IV iron use, may be indicative of better iron mobilization and utilization in participants treated with daprodustat compared with darbepoetin alfa. Observed increases in TIBC were likely determined by transferrin, which is increased by hypoxia-inducible factor (HIF) (15). HIF has been reported to upregulate divalent metal transporter 1 and duodenal cytochrome B to increase intestinal iron absorption (16). Increases in serum iron observed in the daprodustat group may be a consequence of upregulation of divalent metal transporter 1 and duodenal cytochrome B by HIF stabilization; however, further investigation is needed.
Daprodustat was generally well tolerated, with an AE profile comparable to the comparator darbepoetin alfa, including a similar incidence of AEs of special interest. Vascular access complication occurred in a similar proportion of participants in both groups. Contusion and diarrhea were reported more often in the daprodustat group; however, all events reported in the daprodustat group were mild or moderate and did not lead to discontinuation of study treatment (13).
Ocular AEs of special interest (proliferative retinopathy, macular edema, and choroidal neovascularization) were evaluated because of theoretical concerns relating to HIF stabilization resulting in increased vascular endothelial growth factor (VEGF) protein expression with potential secondary effects on retinal neovascularization. Prior phase 2 studies with daprodustat have shown moderate increases in endogenous erythropoietin with no change in circulating VEGF levels (10–12). In our study, 29% of participants had diabetic retinopathy and approximately 5% had macular edema at baseline in both groups. Considering that the incidence of ocular AEs of special interest was similar across treatment groups, daprodustat treatment did not appear to exacerbate these conditions. These findings, together with previous clinical data showing minimal VEGF elevation with daprodustat, suggest that daprodustat treatment for 52 weeks does not induce and/or worsen ocular AEs.
As mentioned above, treatment of Chinese patients on dialysis with roxadustat was evaluated in a phase 3 study. Effects of daprodustat on hemoglobin levels and iron parameters in our study were similar to the results observed with roxadustat; however, our study suggests the possibility of controlling hemoglobin with less IV iron use in patients treated with daprodustat, as physicians were allowed to select the route of iron administration as opposed to IV iron being limited to rescue use in the roxadustat study. Furthermore, our study provides 1-year safety data for daprodustat compared with ESAs. AEs reported more frequently with roxadustat, such as hyperkalemia, were not reported as a common AE (≥5%) in our study, although differences in study design and treatment period should be noted.
Study limitations included insufficient study duration (1 year) and sample size to conclusively evaluate cardiovascular and cancer events. In addition, patients with active malignancies or ESA hyporesponsiveness were excluded from the study. Two ongoing, large outcome studies (Anemia Studies in CKD: Erythropoiesis via a Novel PHI Daprodustat-Dialysis [ASCEND-D] and Anemia Studies in CKD: Erythropoiesis via a Novel PHI Daprodustat-Non-Dialysis [ASCEND-ND]) examining the effect of daprodustat on cardiovascular risk will help provide a definitive conclusion in this regard.
In conclusion, oral daprodustat is noninferior to darbepoetin alfa in the maintenance of hemoglobin concentration in Japanese patients undergoing hemodialysis who are switched from their current ESA treatment. Daprodustat was generally well tolerated, with an AE profile comparable to darbepoetin alfa.
Disclosures
T. Akizawa is a consultant for Astellas Pharma, Bayer Yakuhin, GlaxoSmithKline, Japan Tobacco, Kyowa Kirin, Nipro Corporation, Otsuka Pharmaceuticals, Sanwa Chemical, and Torii Pharmaceutical. Lecture fees were received from Bayer Yakuhin, Chugai Pharmaceutical, Fuso Pharmaceutical Industries, Kissei Pharmaceutical, Kyowa Kirin, Ono Pharmaceutical, and Torii Pharmaceutical. M. Nangaku has received grants and personal fees from Astellas Pharma, Chugai Pharmaceutical, Daiichi Sankyo, GlaxoSmithKline, Kyowa Kirin, Mitsubishi Tanabe Pharma, and Torii Pharmaceutical; grants from Bayer Yakuhin, Ono Pharmaceutical, and Takeda Pharmaceutical Company; and personal fees from AstraZeneca and JT Pharmaceuticals. T. Yonekawa, S. Kawamatsu, T. Onoue, and K. Hara are employees of GlaxoSmithKline. N. Okuda, Y. Endo, and A. Cobitz are employees of and hold equity in GlaxoSmithKline.
Funding
Funding for this study was provided by GlaxoSmithKline.
Supplementary Material
Acknowledgments
The authors wish to thank the participants, the principal investigators, and the clinical research coordinators for their participation and support in this study. The authors are grateful for the editorial support (Sarah Hummasti: assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services that were provided by AOIC, LLC, and were funded by GlaxoSmithKline.
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Dr. Tadao Akizawa, Dr. Masaomi Nangaku, Ms. Taeko Yonekawa, Mr. Yukihiro Endo, Mr. Katsutoshi Hara, and Dr. Alexander R. Cobitz contributed to the concept and design of the study, Dr. Tadao Akizawa, Dr. Masaomi Nangaku, Ms. Taeko Yonekawa, Mr. Nobuhiko Okuda, Mr. Shinya Kawamatsu, Mr. Tomohiro Onoue, Mr. Yukihiro Endo, Mr. Katsutoshi Hara, and Dr. Alexander R. Cobitz contributed to the data analysis and interpretation. All authors provided critical review and final approval of the publication and agree to take responsibility for its content.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.16011219/-/DCSupplemental.
Supplemental Appendix 1. Full inclusion and exclusion criteria.
Supplemental Table 1. Dose adjustment algorithm (daprodustat).
Supplemental Table 2. Dose adjustment algorithm (darbepoetin alfa).
Supplemental Table 3. Number (%) of participants with a hemoglobin increase of >2.0 g/dl over any 4 weeks (intent-to-treat population).
Supplemental Table 4. Number (%) of participants with a hemoglobin level >13.0 g/dl and number of episodes (intent-to-treat population).
Supplemental Table 5. Adverse events of special interest (safety population).
Supplemental Table 6. Systolic and diastolic BP at week 52 (safety population).
Supplemental Table 7. Change from baseline in potassium (mmol/L) (safety population).
Supplemental Table 8. Potassium results: postbaseline relative to baseline (safety population).
Supplemental Table 9. Institutional review board.
References
- 1.Babitt JL, Lin HY: Mechanisms of anemia in CKD. J Am Soc Nephrol 23: 1631–1634, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zumbrennen-Bullough K, Babitt JL: The iron cycle in chronic kidney disease (CKD): From genetics and experimental models to CKD patients. Nephrol Dial Transplant 29: 263–273, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group : KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl 2: 279–335, 2012 [Google Scholar]
- 4.Fishbane S, Spinowitz B: Update on anemia in ESRD and earlier stages of CKD: Core curriculum 2018. Am J Kidney Dis 71: 423–435, 2018. [DOI] [PubMed] [Google Scholar]
- 5.Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, Feyzi JM, Ivanovich P, Kewalramani R, Levey AS, Lewis EF, McGill JB, McMurray JJ, Parfrey P, Parving HH, Remuzzi G, Singh AK, Solomon SD, Toto R; TREAT Investigators : A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 361: 2019–2032, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, Reddan D; CHOIR Investigators : Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 355: 2085–2098, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Solomon SD, Uno H, Lewis EF, Eckardt KU, Lin J, Burdmann EA, de Zeeuw D, Ivanovich P, Levey AS, Parfrey P, Remuzzi G, Singh AK, Toto R, Huang F, Rossert J, McMurray JJ, Pfeffer MA; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators : Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 363: 1146–1155, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Szczech LA, Barnhart HX, Inrig JK, Reddan DN, Sapp S, Califf RM, Patel UD, Singh AK: Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 74: 791–798, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta N, Wish JB: Hypoxia-inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am J Kidney Dis 69: 815–826, 2017. [DOI] [PubMed] [Google Scholar]
- 10.Akizawa T, Tsubakihara Y, Nangaku M, Endo Y, Nakajima H, Kohno T, Imai Y, Kawase N, Hara K, Lepore J, Cobitz A: Effects of daprodustat, a novel hypoxia-inducible factor prolyl hydroxylase inhibitor on anemia management in Japanese hemodialysis subjects. Am J Nephrol 45: 127–135, 2017. [DOI] [PubMed] [Google Scholar]
- 11.Holdstock L, Cizman B, Meadowcroft AM, Biswas N, Johnson BM, Jones D, Kim SG, Zeig S, Lepore JJ, Cobitz AR: Daprodustat for anemia: A 24-week, open-label, randomized controlled trial in participants with chronic kidney disease. Clin Kidney J 12: 129–138, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meadowcroft AM, Cizman B, Holdstock L, Biswas N, Johnson BM, Jones D, Nossuli AK, Lepore JJ, Aarup M, Cobitz AR: Daprodustat for anemia: A 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clin Kidney J 12: 139–148, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, Liang X, Jiang G, Liu Z, Li X, Zuo L, Luo L, Wang J, Zhao MH, Liu Z, Cai GY, Hao L, Leong R, Wang C, Liu C, Neff T, Szczech L, Yu KP: Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med 381: 1011–1022, 2019. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto H, Nishi S, Tomo T, Masakane I, Saito K, Nangaku M, Hattori M, Suzuki T, Morita S, Ashida A, Ito Y, Kuragano T, Komatsu Y, Sakai K, Tsubakihara Y, Tsuruya K, Hayashi T, Hirakata H, Honda H: 2015 Japanese society for dialysis therapy: Guidelines for renal anemia in chronic kidney disease. Renal Replacement Therapy 3: 36, 2017 [Google Scholar]
- 15.Rolfs A, Kvietikova I, Gassmann M, Wenger RH: Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem 272: 20055–20062, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ: Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab 9: 152–164, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.