

Abstract
NKAP and HDAC3 are critical for T cell maturation. NKAP and HDAC3 physically associate and a point mutation in NKAP, NKAP(Y352A), abrogates this interaction. To evaluate the significance of NKAP and HDAC3 association in T cell maturation, transgenic mice were engineered for cre-mediated endogenous NKAP gene deletion coupled to induction of NKAP(Y352A) or a wild type control transgene, NKAP(WT), in double positive (DP) thymocytes or Tregs. T cell maturation was normal in mice with endogenous NKAP deletion coupled to NKAP(WT) induction. However, severe defects occurred in T cell and Treg maturation, and in iNKT cell development when NKAP(Y352A) was induced, recapitulating NKAP deficiency. Conventional T cells expressing NKAP(Y352A) failed to enter the long-term T cell pool, did not produce cytokines and remained complement-susceptible while Tregs expressing NKAP(Y352A) were eliminated as recent thymic emigrants (RTEs) leading to lethal autoimmunity. Overall, these results demonstrate the significance of NKAP-HDAC3 association in T cells.
Introduction
Immunity and homeostasis depend on αβ T cells which can be broadly divided into conventional (CD4+ and CD8+ T cells), regulatory (Tregs) and invariant natural killer T (iNKT) cells (1). All three subtypes develop from αβ CD4+CD8+ double positive (DP) precursors in the thymus. After positive selection, most DP thymocytes become conventional CD4 or CD8 single positive (SP) cells (1). By contrast, thymic Tregs and iNKT cells are agonist selected at the CD4 SP and DP stages, respectively, via strong TCR interactions with cognate self-ligands (1). As positive selection is insufficient for conventional T cell and Treg functional competency, additional terminal maturation steps are required (2, 3).
T cell maturation begins in the thymus and continues in the periphery as recent thymic emigrants (RTEs) transition to mature naïve T cells (MNTs) (3). Maturation enables thymic egress and TCR/CD28 stimulation dependent proliferation and cytokine production (3). It also confers long-term survival by protection from death receptor signaling and resistance to complement proteins. In the case of Tregs, maturation facilitates the acquisition of an activated state critical for tissue-specific tolerance (4). The X-linked transcriptional regulator NKAP is indispensable for T cell maturation (5–7). In CD4-cre NKAP conditional knockout (cKO) mice, NKAP deletion at the DP stage impairs long-term persistence of peripheral T cells although SP thymocyte production and egress are intact (5). Peripheral NKAP-deficient naïve T cells are predominantly RTEs and fail to enter the long-lived naïve T cell pool. NKAP-deficient RTEs exhibit reduced cytokine production and increased complement deposition compared to WT RTEs. Consistently, expression of molecular markers associated with maturation, such as Qa2, CD45RB and CD55, are reduced.
Similarly, while Treg-specific NKAP-deletion (in Foxp3-YFP-cre NKAP cKO mice) does not impede thymic Treg development, it renders Tregs unable to persist and adopt a mature/activated state (7). Foxp3-YFP-cre NKAP cKO mice resemble Foxp3-mutant ‘scurfy’ mice that do not generate Tregs (7, 8). Both develop systemic autoimmunity with dermatitis, lymphocytic infiltration into vital organs, unchecked T cell proliferation, B cell tolerance breakdown and lethality by three weeks of age (7, 9–11). Foxp3-YFP-cre NKAP cKO females carry one XFoxp3-YFP-cre, NKAP-fl allele and an XNKAP-fl allele, and are healthy natural chimeras with a mix of NKAP-sufficient and NKAP-deficient Tregs due to random X-inactivation (7). Unlike NKAP-sufficient Tregs, that develop normally and persist, NKAP-deficient Tregs are rapidly eliminated at the RTE stage revealing a cell-intrinsic survival defect in Foxp3-YFP-cre NKAP cKO female chimeras.
NKAP is a regulator of gene expression but lacks a defined DNA-binding domain and likely operates within larger molecular complexes (12). NKAP’s C-terminal domain associates with Histone Deacetylase 3 (HDAC3), a class-I HDAC that modifies gene expression by removing acetyl groups from histone and non-histone proteins. Similar to NKAP-deficient RTEs, HDAC3-deficient RTEs in CD4-cre HDAC3 cKO mice have decreased persistence, impaired cytokine production, increased complement binding and decreased CD55 expression (13). In contrast to NKAP-deficient T cells, HDAC3-deficient RTEs express normal levels of Qa2 and CD45RB demonstrating that these markers associated with maturation may not accurately indicate functional maturity (13). Additionally, although Foxp3-YFP-cre HDAC3 cKO mice develop multi-organ autoimmunity, they survive longer than Foxp3-YFP-cre NKAP cKO mice, suggesting a less severe form of disease (7, 14). Lastly, while loss of either NKAP or HDAC3 in conventional T cells and Tregs causes extra-thymic maturation defects, intra-thymic development of iNKT cells is severely curtailed at the DP stage in either CD4-cre NKAP cKO or CD4-cre HDAC3 cKO mice (15).
Given the phenocopy between mouse models with cKO of NKAP or HDAC3, and their known interaction, the importance of NKAP association with HDAC3 was recently examined in hematopoietic stem cells (HSCs) (16). Truncation analysis coupled with alanine scanning identified a single point mutation (Y352A) sufficient to abrogate the association of NKAP with HDAC3. A conditional deletion/re-expression mouse model was used to couple deletion of native NKAP in HSCs with induction of either YFP-tagged wild type (WT) or Y352A mutant NKAP transgenes (designated YFP-NKAP(WT) or YFP-NKAP(Y352A)). Induction of YFP-NKAP(WT) but not YFP-NKAP(Y352A) rescued the defects in HSC maintenance and survival resulting from NKAP deficiency, showing that the Y352A mutation impairs the function of NKAP in vivo. Although HSCs require NKAP association with HDAC3, proliferation of mouse embryonic fibroblasts (MEFs) was restored when YFP-NKAP(Y352A) was expressed (16). Therefore, the necessity for NKAP to with HDAC3 is context-dependent. These observations prompted us to interrogate the requirement of NKAP to associate with HDAC3 in the context of T cells.
Here, the consequence of Y352A substitution on NKAP’s function was assessed in T cells. Mice expressing either CD4-cre or Foxp3-YFP-cre drove endogenous NKAP deletion and simultaneous induction of either YFP-NKAP(WT) or YFP-NKAP(Y352A). YFP-NKAP(Y352A) expression at the DP stage coupled to endogenous NKAP deletion failed to restore iNKT cell development, phenocopying CD4-cre NKAP cKO and CD4-cre HDAC3 cKO mice. Further, conventional T cells in which YFP-NKAP(Y352A) was substituted for endogenous NKAP showed decreased peripheral persistence, reduced induction of TNFα after in vitro TCR/CD28 stimulation and enhanced complement deposition. In addition to conventional T cells, the substitution of endogenous NKAP with YFP-NKAP(Y352A) in Tregs failed to reverse their disappearance at the RTE stage causing severe autoimmunity similar to Foxp3-YFP-cre NKAP cKO mice. As expected, substitution of YFP-NKAP(WT) reversed all effects of NKAP deficiency on iNKT development, and conventional as well as regulatory T cell maturation. Recent studies indicate that NKAP deletion causes increased lipid peroxidation in naïve CD4 T cells compared to WT naïve CD4 T cells (17). Lipid peroxidation is a hallmark of ferroptosis, a form of programmed cell involving iron dependent generation of reactive oxygen species (ROS) (18) and is likely the mechanism underlying NKAP-deficient T cell disappearance (17). Here, we found that HDAC3-deficient CD4 T cells also exhibit enhanced lipid peroxidation compared to WT naïve CD4 T cells indicating that NKAP and HDAC3 work together to prevent ferroptosis. In summary, these findings show that Y352 of NKAP, which is required for HDAC3 binding, is also critical for NKAP’s roles in T cell maturation and iNKT development and long-term T cell persistence.
Materials and methods
Mice:
YFP-NKAP(WT) and YFP-NKAP(Y352A) knock-in mice were previously described (16). These were crossed to mice with a lox-neo-lox tetracycline transactivator (lnl-tTA, The Jackson Laboratory, USA (19)) expression cassette to allow Cre-mediated induction of YFP-NKAP when subsequently crossed with CD4-cre NKAP cKO mice (5), CD4-cre mice (20), Foxp3-YFP-cre NKAP cKO mice (7) and Foxp3-YFP-cre mice (21). All mouse work was performed with approval from the Mayo Clinic Institutional Animal Care and Use Committee.
Flow Cytometry:
Single-cell suspensions were prepared from thymii, spleens and lymph nodes. Single cell suspensions from spleens were treated with ACK lysis buffer (Quality Biological, MD, USA). Prior to surface staining, cells were treated with rat and mouse serum in a 1:1 ratio (Invitrogen, CA, USA) or Fc-Block (BioLegend, CA, USA TruStain fcX, clone 93) to prevent non-specific antibody binding. All experiments included fixable viability dye (Tonbo, CA, USA, Ghost Dye Red 780) for exclusion of dead cells during analysis. Cell surface markers were detected with antibodies conjugated with fluorescent probes from BioLegend, Tonbo and eBioscience (CA, USA): CD4 (BV785, clones GK1.5 or RM4–5), CD8 (BV510, APC or PE, clones 53–6.7 or 2.43), CD44 (V450, clone IM7), CD62L (APC or BV510, clone MEL-14), CD55 (PE clone RIKO-3), GITR (PE or APC, clone DTA-1), NRP-1 (APC, clone 3E12) and CD25 (BV421, clone PC16), TCR-β (APC), H2-Kb (APC, clone AF6–88.5), CD69 (PE-Cy7, clone H57–597), CCR7 (PE, clone 4B12) and CD24 (Pacific Blue, clone M1/69). Complement deposition was detected using biotinylated antibody for complement C3 (Cedarlane, Canada, clone RmC11H9) followed by PE or APC conjugated streptavidin (eBioscience or Tonbo). For C3 detection, freshly harvested splenocytes were incubated in GVB++ buffer (Complement Technology, TX, USA) for 1h at room temperature and cells were washed with FACS buffer and stained with appropriate antibodies. Cytokine production was detected by overnight incubation of freshly harvested total splenocytes in 24 well plates coated with plate-bound 10 μg/ml anti-CD3 (clone 2C11, Bio-X-Cell, NH, USA) and soluble 1 μg/ml anti-CD28 (clone 37.51, Bio-X-Cell) antibodies in RPMI media (Corning, VA, USA) supplemented with 10% FBS, penicillin, streptomycin, HEPES, non-essential amino-acids, L-glutamate and beta-mercaptoethanol. Surface staining was performed followed by fixation and permeabilization with a cytofix/cytoperm kit (BD, NJ, USA). Cytokine presence was detected using PE conjugated anti-TNFα (BioLegend, clone MP6-XT22). PE or BV421 conjugated CD1d: PBS57 or CD1d: empty tetramers generously provided by the NIH Tetramer Facility were used to detect iNKT cells. Intracellular staining for Foxp3 (Tonbo PE or APC, clone 3G3) was done after fixing and permeabilizing cells (Tonbo Foxp3 staining kit), using antibodies from eBioscience and Tonbo. Data collection was done using the Attune NxT flow cytometer (Thermofisher, NA, USA). Data was analyzed using FlowJo (Tree Star) v9.8 or v10.
Detection of antibodies to double stranded DNA:
Antibodies to double stranded DNA were detected using an Autoimmune EIA Anti-dsDNA Test (BioRad, CA, USA). Serum was diluted 100-fold in phosphate buffered saline with 1% bovine serum albumin (PBS/BSA). The assay was completed according to the manufacturer’s instructions with the substitution of horseradish peroxidase coupled goat anti-mouse IgG for the detection antibody (Southern Biotechnology Associates, AL, USA) diluted 2000-fold in PBS/BSA. Absorbance at 450 nM was measured using a Molecular Devices Spectromax microplate reader.
Anti-nuclear antibody detection:
Anti-nuclear antibodies were detected by applying diluted sera (1:100) to slides coated with Kallestad HEp-2 cells (BioRad). The assay was completed according to the manufacturer’s instructions except for the substitution of AF488 coupled goat anti-mouse IgG for the detection antibody (InVitrogen, diluted to 4 μg/ml in PBS/BSA). Each well was examined at room temperature without immersion medium using a Leica, Germany, DMI3000B fluorescence microscope with a 20X lens (Plan Fluotar type, numerical aperture of 0.4) and a FITC/EGFP filter cube. Images were acquired using a Q-Imaging Q1-Click camera and Q Capture Pro 6 software and saved as TIFF files. Adobe Illustrator was used to prepare the representative field shown.
Histology:
Livers from WT, Foxp3-YFP-cre NKAP cKO, Foxp3 [WT→cKO] and Foxp3 [MUT→cKO] mice were isolated and kept in 10% formalin for fixation. Fixed samples were processed by paraffin embedding, sectioning and H&E stained as per standard procedures. Sections were viewed on a Leica DMI3000B microscope, at 20X magnification and captured using the Leica EC3 camera.
Detection of lipid peroxidation:
Freshly harvested splenocytes were treated with 1 μM BODIPY-C11 (Thermo Fisher) in RPMI 1640 media (Corning) supplemented with 10% FBS, penicillin and streptomycin at 37 °C for 1 hour. Cells were then washed and surface stained for CD4, CD44, CD62L and Fixable Viability Dye as described above. To examine the impact of ferroptosis inhibitors on lipid peroxidation, total splenocytes were incubated with 100 μM alpha-Tocopherol (Millipore Sigma) and/or 10 μM Ferrostatin-1 (Millipore Sigma) for 15 min at 37 °C followed by staining with 1μM BODIPY-C11 for 1 hour. Cells were then washed and surface stained as described above.
Statistical analysis:
All statistics were calculated using GraphPad Prism (GraphPad Software Inc., CA, USA) or Microsoft Excel. Unpaired student’s t-test, one-way ANOVA (Analysis of Variance) or two-way ANOVA were used as indicated in figure legends to examine significant differences across mice and experimental conditions. Values of p<0.05 were deemed significant and lack of significant differences (p>0.05) are indicated by ns (not significant).
Results
NKAP(Y352A) cannot substitute for endogenous NKAP during T cell maturation
Similar maturation defects in CD4-cre NKAP cKO and CD4-cre HDAC3 cKO T cells raise the possibility that NKAP and HDAC3 work together to regulate T cell maturation. A single point mutation, NKAP(Y352A), abrogates NKAP-HDAC3 binding (16). Here, the impact of this mutation on NKAP’s roles in T cell maturation was examined. We used a mouse model for cre-mediated deletion of endogenous NKAP coupled with cre-mediated expression of YFP-NKAP(WT) or YFP-NKAP(Y352A) (16). Mice were engineered to express YFP-NKAP(WT) or YFP-NKAP(Y352A) upon CD4-cre mediated endogenous NKAP gene deletion, i.e., CD4-cre NKAP cKO LNL-tTA YFP-NKAP(WT) and CD4-cre NKAP cKO LNL-tTA YFP-NKAP(Y352A) mice. For simplicity, these will be abbreviated CD4-cre [WT→cKO] and CD4-cre [MUT→cKO], respectively. In addition, CD4-cre LNL-tTA YFP-NKAP(WT) and CD4-cre NKAP cKO LNL-tTA YFP-NKAP(Y352A) mice that express either YFP-NKAP(WT) or YFP-NKAP(Y352A) in the presence of endogenous NKAP were generated to account for potential dominant negative effects. These are abbreviated CD4-cre [WT→WT] and CD4-cre [MUT→WT], respectively.
Splenocytes from CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice were analyzed and compared to WT and CD4-cre NKAP cKO mice (Figure 1). Appropriate T cell maturation leads to long-term persistence of conventional T cells in the periphery (3). However, CD4-cre NKAP cKO mice harbor very few peripheral CD8+ and CD4+ T cells as a result of a block in T cell maturation (5, 6). In CD4-cre [WT→cKO], the frequencies and absolute cell counts of peripheral CD8+ and CD4+ T cells were restored to WT levels, indicating normal T cell maturation (Figure1A, 1C and Supplement Figure 1). In contrast to WT mice, CD4-cre [MUT→cKO] mice exhibited reductions in CD8+ and CD4+ peripheral T cell frequencies and absolute cell counts and were very similar to CD4-cre NKAP cKO mice indicating impaired long-term persistence. In particular, there were few naïve (CD62L+CD44lo) CD8+ and CD4+ T cells in both CD4-cre [MUT→cKO] and CD4-cre NKAP cKO mice (Figure 1A, 1C). Thus, NKAP(Y352A) could not substitute for endogenous NKAP to support T cell maturation, leading to the disappearance of peripheral CD4 and CD8 T cells.
Figure 1: NKAP(Y352A) cannot substitute for wildtype NKAP for peripheral T cell persistence.
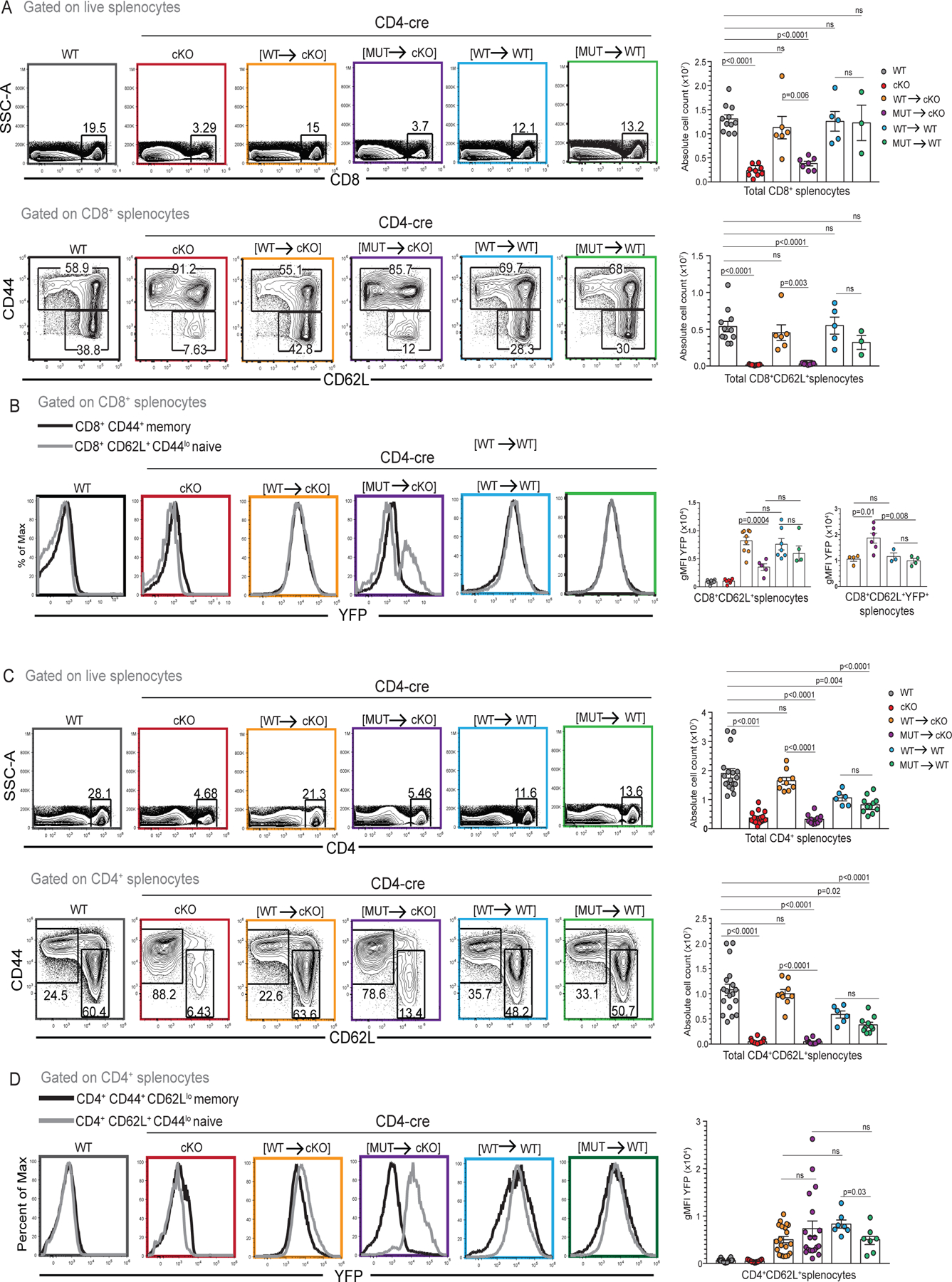
(A) Frequencies and absolute numbers of total CD8+ splenocytes and naïve CD8+CD62L+CD44lo splenocytes in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data is combined from at least 3 independent experiments with 11, 9, 6, 7, 5, and 3 mice in total for the genotypes listed, respectively. (B) YFP expression in naïve CD8+CD62L+CD44lo splenocytes in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data is combined from at least 3 experiments with 9, 7, 9, 5, 7, and 4 mice in total for the genotypes listed, respectively. YFP expression in naïve CD8+CD62L+YFP+ T cells. Data shown is combined from at least 3 experiments with 3, 4, 3, 4 mice in total for the genotypes listed, respectively. (C) Frequency and absolute cell numbers of total CD4+ splenocytes and naïve CD4+CD62L+CD44lo splenocytes in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data is combined from at least 6 independent experiments with 18, 20, 9, 12, 6, and 11 mice in total for the genotypes listed, respectively. (D) YFP expression in naïve CD4+CD62L+CD44lo splenocytes from WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data is combined from at least 4 independent experiments with 22, 17, 20, 19, 7, and 7 mice in total for the genotypes listed, respectively. (A-D) Flow-cytometry plots are representative of indicated cell populations. Bar graphs indicate mean and error bars indicate standard error of mean (SEM). All p-values were calculated using one-way ANOVA with multiple comparisons.
To determine whether the expression of YFP-NKAP(Y352A) exerted a dominant negative effect on peripheral T cell persistence, splenocytes from CD4-cre [WT→WT] and CD4-cre [MUT→WT] were analyzed. Absolute numbers of total CD8+ T cells and naïve CD8+ T cells in CD4-cre [WT→WT] and CD4-cre [MUT→WT] were similar to WT mice (Figure 1A). However, there was an approximately two-fold reduction in total and naïve CD4 T cells in CD4-cre [WT→WT] and CD4-cre [MUT→WT] compared to WT (Figure 1B). Therefore, YFP-NKAP(WT) or YFP-NKAP(Y352A) had no adverse effects on CD8+ frequencies and numbers, and had only a partial effect on CD4+ T cell numbers. However, as CD4+ and CD8+ T cell total and naïve numbers were very similar between CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice, the two-fold decrease observed in case of CD4+ T cells is likely an outcome of NKAP over-expression rather than mutation at Y352.
The failure of YFP-NKAP(Y352) to rescue peripheral T cell numbers could be a result of a reduction in its expression as compared to YFP-NKAP(WT). Next, YFP expression in CD8+ and CD4+ naïve and memory T cells was examined. YFP was expressed at similar levels between CD8+ naïve (CD62L+CD44lo) and CD8+ memory (CD44+) T cells (Figure 1B) and between CD4+ naïve (CD62L+CD44lo) and CD4+ memory (CD44+ CD62L−) T cells (Figure 1D) from CD4-cre [WT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. By contrast, no YFP expression was observed in memory CD4+ or memory CD8 T cells from CD4-cre [MUT→cKO]. Previously, memory T cells from CD4-cre NKAP cKO mice were shown to be NKAP gene non-deleters that underwent homeostatic expansion owing to a sparse T cell niche (5). YFP expression in CD4-cre [MUT→cKO] depends on cre activity and the lack of YFP expression in CD4+ and CD8+ memory cells is consistent with expansion of NKAP non-deleting cells that upregulate CD44 and downregulate CD62L. YFP expression across CD4+ naïve T cells from CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice was comparable. Interestingly, there was bi-modal expression of YFP-NKAP(Y352A) in naïve CD8+ cells with a YFP+ population and a YFP− population, indicating that a fraction of naïve T cells escaped cre-mediated NKAP deletion. Examination of only YFP+CD8+ naïve fraction shows that the average gMFI of YFP is not reduced in CD4-cre [MUT→cKO] compared to CD4-cre [WT→cKO]. In summary, the failure of YFP-NKAP(Y352A) to substitute for endogenous NKAP is not due to decreased expression as compared to YFP-NKAP(WT).
NKAP(Y352A) expression does not impair thymic T cell output.
WT and CD4-cre NKAP cKO mice do not exhibit significant differences in the production of DP and SP thymocytes, and the decrease in peripheral T cells in CD4-cre NKAP cKO mice is due to defects in long-term persistence (5, 6). Splenic T cell profiles of CD4-cre NKAP cKO and CD4-cre [MUT→cKO] mice were virtually indistinguishable, suggesting that YFP-NKAP(Y352A) expression failed to reverse defects in T cell persistence stemming from NKAP deficiency. However, another possibility was YFP-NKAP(Y352A) expression caused a reduction in thymic output. To examine this, frequency and absolute counts of CD4+ SP and CD8+ SP thymocytes were compared between WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice (Figure 2). In agreement with previous data, there was no significant difference in the numbers of CD4+ SP thymocytes in WT and CD4-cre NKAP cKO mice, and only a small difference in CD8+ SP thymocytes (Figure 2A). CD4-cre [WT→cKO] mice produced normal numbers of SP thymocytes (Figure 2A). In contrast, CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice had reduced CD4+ SP and CD8+ SP thymocytes numbers compared to WT. Importantly, SP thymocyte generation in CD4-cre [MUT→cKO] and CD4-cre [MUT→WT] mice were equivalent, although there was decreased peripheral T cell numbers in CD4-cre [MUT→cKO] mice compared to CD4-cre [MUT→WT] mice. Thus, peripheral T cell deficiency in CD4-cre [MUT→cKO] mice is not simply due to decreased thymic output but represents an inability of peripheral T cells to mature and accumulate in the long-term T cell pool.
Figure 2: Decreased thymic T cell output does not drive loss of peripheral T cells in CD4-cre [MUT→cKO] mice.
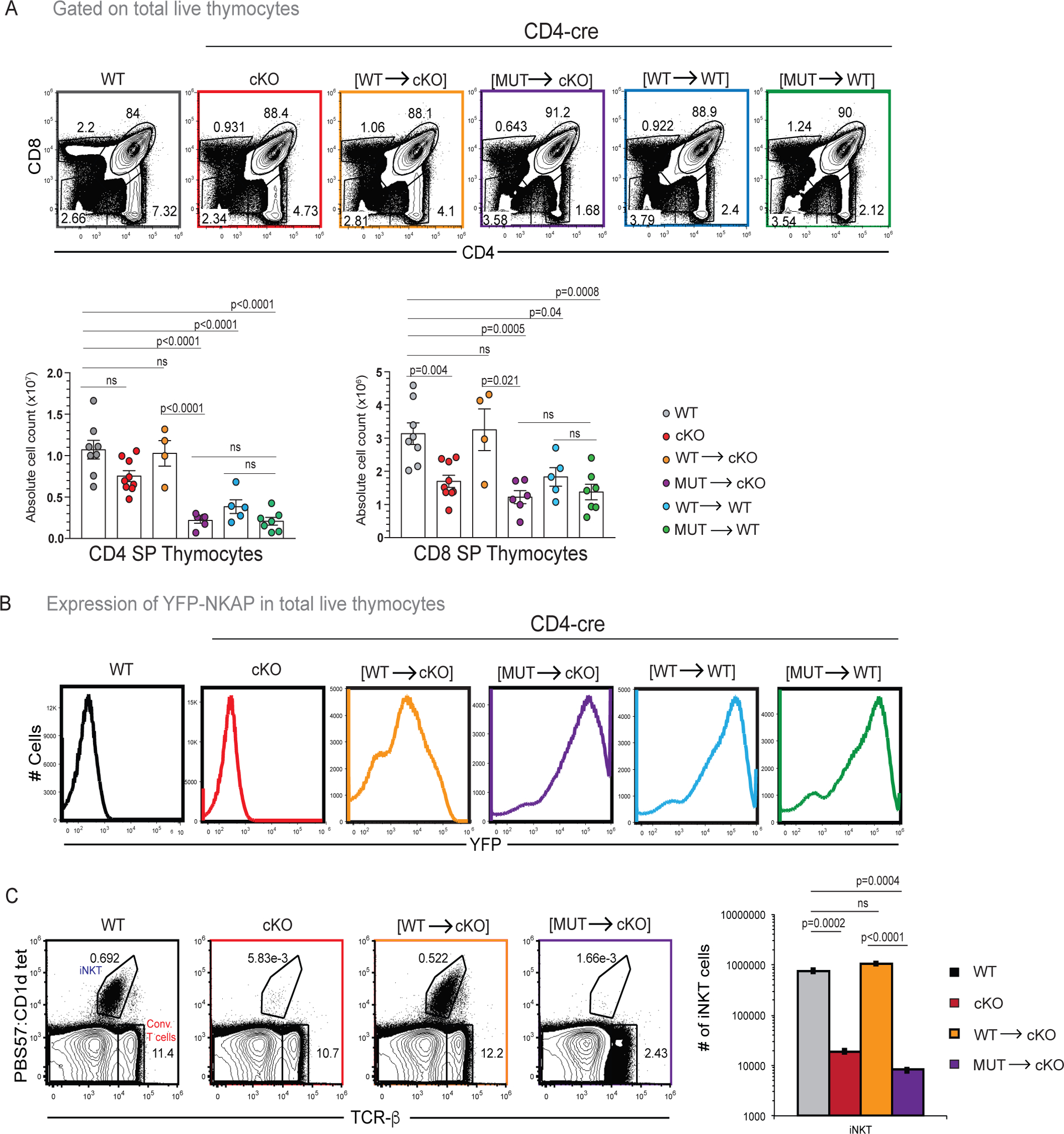
(A) CD8 SP and CD4 SP frequencies and absolute cell numbers in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice are shown. Data is combined from at least 4 experiments with 8, 9, 4, 6, 5, and 7 mice in total for the genotypes listed, respectively. (B) YFP expression in total thymocytes from WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data is representative of at least 4 experiments as described in (A). (C) Comparison of frequency and absolute cell counts of total CD1d-PBS57 tetramer+ TCRβ+ iNKT cells in thymus of WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO]. Data is combined from at least 3 experiments with 6, 4, 3 and 3 mice for the genotypes listed, respectively. (A-C) Bar graphs indicate mean absolute cell numbers and error bars indicate SEM. All p-values were calculated using one-way ANOVA with multiple comparisons.
Surprisingly, CD4-cre [MUT→cKO], CD4-cre [WT→WT] CD4-cre [MUT→WT] mice showed greater expression of YFP compared to CD4-cre [WT→cKO]. This was unexpected as the mice were identically engineered to express YFP-NKAP(WT) or YFP-NKAP(Y352A) (16). However, the reduction in SP thymocytes in CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice followed enhanced YFP expression suggesting that overexpression of YFP-NKAP in the thymus in these lines reduced SP generation compared to CD4-cre [WT→cKO] mice. To further examine whether NKAP overexpression reduces the size of SP population in the thymus, YFP-high and YFP-low CD4-cre [WT→WT] as well as CD4-cre [MUT→WT] thymocytes were compared with WT thymocytes expressing native NKAP only to assess the impact on SP thymocyte development and maturation (Supplement Figure 2). YFP-high and YFP-low CD4 and CD8 SP thymocytes were generated at similar levels in CD4-cre [WT→WT] and CD4-cre [MUT→WT] (Supplement Figure 2A). Maturation is a gradual process accompanied by an increase in the expression of the cell surface protein Qa2 and a decrease in the cell surface protein CD24. Using Qa2 and CD24, maturation was examined to assess whether NKAP-transgene expression level may alter maturation. In both CD4-cre [WT→WT] and CD4-cre [MUT→WT] thymocytes, YFP-low CD4+Qa2+CD24− mature cells were generated at frequencies equivalent to WT while YFP-high CD4+Qa2+CD24− mature cells were not produced. Thus, high YFP-NKAP expression was detrimental to the generation of mature CD4+Qa2+CD24− SP thymocytes and theY352A mutation did not alter this effect. Similarly, the generation of YFP-high CD8+Qa2+CD24− SP thymocytes was impeded while YFP-low CD8+Qa2+CD24− cells were generated at levels comparable to WT (data not shown). The process of intra-thymic maturation can be demarcated using additional maturation markers (3, 22). SP thymocytes can be divided into CCR7− CD24+ or H2-Kb- CD69+semi-mature (SM), CCR7+ CD24+ or H2-Kb+ CD69+ mature 1 (M1), and CCR7+ CD24− or H2-Kb+ CD69− mature 2 (M2) thymocytes (3, 22). CD4 and CD8 SM, M1, M2 populations were formed in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO] and CD4-cre [MUT→cKO] mice (Supplement Figure 2B, C, E, F). The examination of YFP expression in CD4 and CD8 SM, M1, M2 populations revealed that M1 and M2 SP thymocytes had lower YFP expression compared to SM SP thymocytes (Supplement 2D, G). This finding is consistent with the previous observation that high NKAP transgene expression is detrimental for mature SP generation and likely creates a bias for YFP-low cells to enter periphery. Further, YFP was expressed by all CD8 SP thymocytes indicating that bimodal expression of YFP starts in the periphery.
Although CD4-cre NKAP cKO and CD4-cre HDAC3 cKO mice do not present blocks in conventional T cell development, they suffer a severe block in iNKT development at the DP stage, leading to the development of very few thymic or peripheral iNKT cells (15). Unlike conventional T cells that have a diverse repertoire of TCRα chains, iNKT cells express a fixed TCRα chain and can be identified by CD1d tetramer loaded with the glycolipid PBS-57 (1, 15). iNKT cell development was examined in the thymus in WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO] and CD4-cre [MUT→cKO] mice using CD1d tetramer loaded with the glycolipid PBS-57 (Figure 2 C). Similar to CD4-cre NKAP cKO mice and CD4-cre HDAC3 cKO mice, very few iNKT cells were detected in the thymus of CD4-cre [MUT→cKO] mice, demonstrating the importance of Y352 of NKAP in iNKT development.
NKAP(Y352A) cannot substitute for endogenous NKAP for functional T cell maturation.
During T cell maturation, RTEs gain the ability to produce cytokines (such as TNFα) upon TCR/CD28 stimulation and acquire complement resistance (3). As the Y352A mutation impairs NKAP’s function in long-term T cell persistence, its effect on TNFα production was examined next. Total splenocytes from WT, CD4-cre NKAP cKO, CD4-cre [WT→WT], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice were stimulated by TCR/CD28 activation (Figure 3A, B). A smaller fraction of CD4-cre NKAP cKO CD4+CD44− T cells produced TNFα compared to WT T cells (Figure 3A). Compared to CD4+CD44−YFP+ cells from CD4-cre [WT→cKO], fewer CD4+CD44−YFP+ cells from CD4-cre [MUT→cKO] expressed TNFα, indicating a defect in functional T cell maturation (Figure 3B). The frequency of TNF production was similar in CD4+CD44−YFP+ cells from CD4-cre [WT→WT], CD4-cre [MUT→WT] and CD4-cre [WT→cKO] mice, ruling out the possibility of dominant negative effects of either YFP-NKAP(WT) or YFP-NKAP(Y352A) overexpression on TNFα production in peripheral T cells (Figure 3B).
Figure 3: NKAP(Y352A) fails to restore TNFα production and complement resistance.
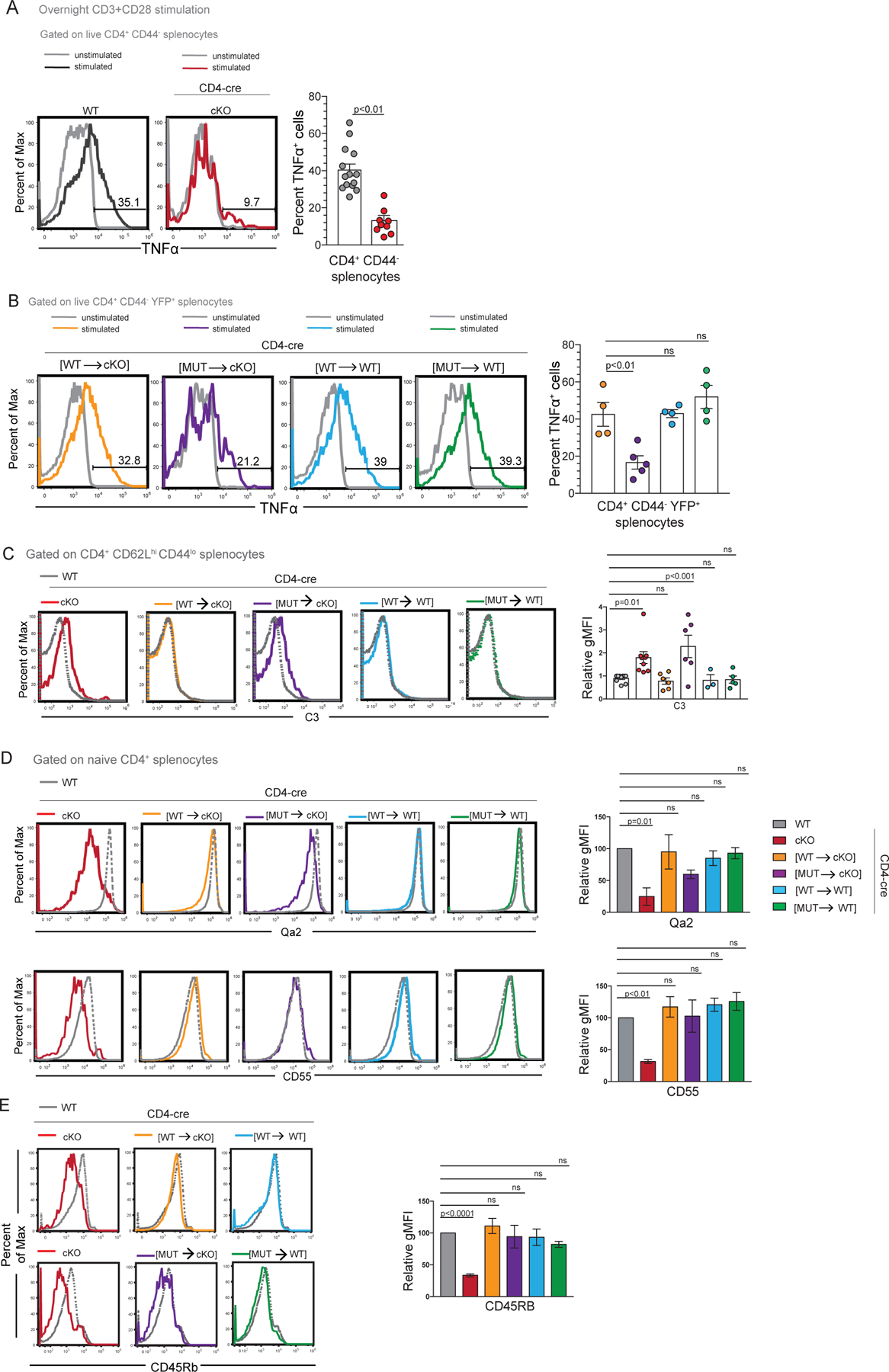
(A) Frequency of TNFα+ cells within CD4+CD44− gates from WT and CD4-cre NKAP cKO splenocytes following overnight stimulation with plate-bound anti-CD3 and soluble anti-CD28 antibodies. Flow-cytometry plots are representative of average TNFα+ mean frequencies and are overlaid on unstimulated cells. Bar graphs indicate TNFα+ average frequencies and error bars indicate SEM. All p-values were calculated using unpaired student’s T-test. Data shown is combined from at least 7 experiments with 14 and 9 mice in total per genotype, respectively. (B) Frequency of TNFα+ cells within CD4+CD44−YFP+ splenocytes from CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Flow-cytometry plots represent TNFα+ frequencies from all genotypes examined from a single experiment. Bar graphs indicate TNFα mean frequencies and error bars indicate SEM. Data shown is combined from 4 independent experiments with 4, 5, 4, and 4 mice in total for the genotypes, respectively. (C) Comparison of C3 binding between WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice. Data combined from at least 3 independent experiments with 10, 9, 6, 6, 5, 3 mice in total for the genotypes listed, respectively. (D, E) Expression of maturation markers by naïve CD4 T cells from WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] mice: Qa2 (combined data is from 3 independent experiments with 3 mice in total per group), CD55 (combined data is from at least 3 independent experiments with 5, 5, 4, 3, 4, 3 mice in total for the genotypes listed, respectively), CD45RB (combined data is from at least 3 independent experiments with 6, 8, 4, 3, 3, 4 mice in total for the genotypes listed, respectively). Bar graphs indicate mean geometric mean intensity relative to WT and error bars indicate SEM. (B-E) All p-values were calculated using one-way ANOVA with multiple comparisons.
Thymic maturation leads to gradual acquisition of complement resistance (3, 17). NKAP-deficient and HDAC3-deficient peripheral T cells exhibit elevated susceptibility to complement deposition (6, 13). To assess whether substitution of endogenous NKAP with NKAP(Y352A) could restore complement resistance, C3 binding to total CD4 naïve T cells from WT, CD4-cre NKAP cKO, CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] was examined (Figure 3C). C3 binding was enhanced specifically in naïve CD4+ T cells from CD4-cre [MUT→cKO], similar to that observed in naïve CD4+ T cells from CD4-cre NKAP cKO mice. Therefore, the Y352A mutant of NKAP cannot substitute for WT NKAP in protecting peripheral T cells from complement deposition.
Maturation is accompanied by increases in expression of cell surface proteins Qa2, CD45RB and CD55 (3, 5, 6, 13). Whether the NKAP(Y352A) mutation altered the ability of naive CD4 T cells to express these maturation markers was examined (Figure 3D, 3E). As previously shown, CD4+ naïve T cells from CD4-cre NKAP cKO mice expressed significantly lower levels of Qa2, CD55, and CD45RB as compared to WT (5, 6). On the other hand, CD55 and CD45RB were expressed at normal levels in CD4-cre [WT→cKO], CD4-cre [MUT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] cells. Qa2 was expressed at normal levels in CD4-cre [WT→cKO], CD4-cre [WT→WT] and CD4-cre [MUT→WT] but was only slightly reduced in CD4-cre [MUT→cKO] cells although this difference was not statistically significant. This is not surprising as HDAC3 deficiency does not lead to decreased Qa2 and CD45RB expression and causes a slight downregulation of CD55 (13). Thus, abrogation of HDAC3 association with NKAP by the Y352A mutation caused a phenotype resembling that of CD4-cre HDAC3 cKO mice, with defects in cytokine production and protection from complement but not reduction in Qa2, CD45RB and CD55.
NKAP(Y352A) cannot substitute for endogenous NKAP in regulatory T cells to prevent autoimmunity
Treg-specific NKAP deficiency (Foxp3-YFP-cre NKAP cKO) results in systemic autoimmunity and lethality by 3 weeks of age, similar to that observed in scurfy/Foxp3 mutant mice (7). Treg-specific HDAC3 deletion (Foxp3-YFP-cre HDAC3 cKO) also results in autoimmunity albeit less severe than Treg-specific NKAP deletion as Foxp3-YFP-cre HDAC3 cKO mice survive slightly longer (7, 14). Both exhibit extensive dermatitis, lymphocytic infiltration into various organs, T cell activation and proliferation as well as autoantibodies in sera indicative of Treg-mediated tolerance breakdown. To assess the impact of the NKAP(Y352A) mutation in Treg-mediated tolerance, a Treg-specific conditional deletion/re-expression system was used. Foxp3-YFP-cre NKAP cKO LNL-tTA YFP-NKAP(WT) mice express YFP-NKAP(WT) in Foxp3-YFP-cre NKAP cKO cells, and Foxp3-YFP-cre NKAP cKO LNL-tTA YFP-NKAP(Y352A) mice express YFP-NKAP(Y352A) in Foxp3-YFP-cre NKAP cKO cells. These are abbreviated Foxp3-YFP-cre [WT→cKO] and Foxp3-YFP-cre [MUT→cKO], respectively. To account for possible dominant negative effects, Foxp3-YFP-cre LNL-tTA YFP-NKAP(WT) and Foxp3-YFP-cre LNL-tTA YFP-NKAP(Y352A) that express YFP-NKAP-WT and YFP-NKAP(Y352A) in the presence of endogenous NKAP respectively were used. These are abbreviated as Foxp3-YFP-cre [WT→WT] and Foxp3-YFP-cre [MUT→WT], respectively.
Substitution of endogenous NKAP with YFP-NKAP(WT) in Foxp3-YFP-cre [WT→cKO] mice prevented autoimmunity (Figure 4A). However, substitution of endogenous NKAP with YFP-NKAP(Y352A) in Foxp3-YFP-cre [MUT→cKO] mice resulted in runted appearance, dermatitis, reduced weight gain and lethality by 3-weeks of age similar to Foxp3-YFP-cre NKAP cKO and scurfy mice. Total splenic absolute cell numbers were comparable between mice, however lymph node absolute cell numbers of in Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] mice were increased compared to WT or Foxp3-YFP-cre [WT→cKO] mice (Figure 4B). The thymus atrophies as DP thymocytes undergo increased apoptosis due to rampant inflammation (Figure 4B) (23). Examination of thymii from 3-week old mice showed severe reductions in thymic cellularity in Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] mice compared to WT or Foxp3-YFP-cre [WT→cKO] mice. Autoantibodies to nuclear antigens and double stranded DNA were detectable in the sera of all Foxp3-YFP-cre [MUT→cKO] mice similar to all Foxp3-YFP-cre NKAP cKO mice indicating a breach in B cell tolerance (Figure 4C, 4D). Further histological examination indicated lymphocytic infiltration into Foxp3-YFP-cre [MUT→cKO] livers similar to Foxp3-YFP-cre NKAP cKO livers while Foxp3-YFP-cre [WT→cKO] livers were similar to WT indicating peripheral tolerance breakdown (Figure 4E).
Figure 4. NKAP(Y352A) expression in Tregs fails to reverse systemic autoimmunity.
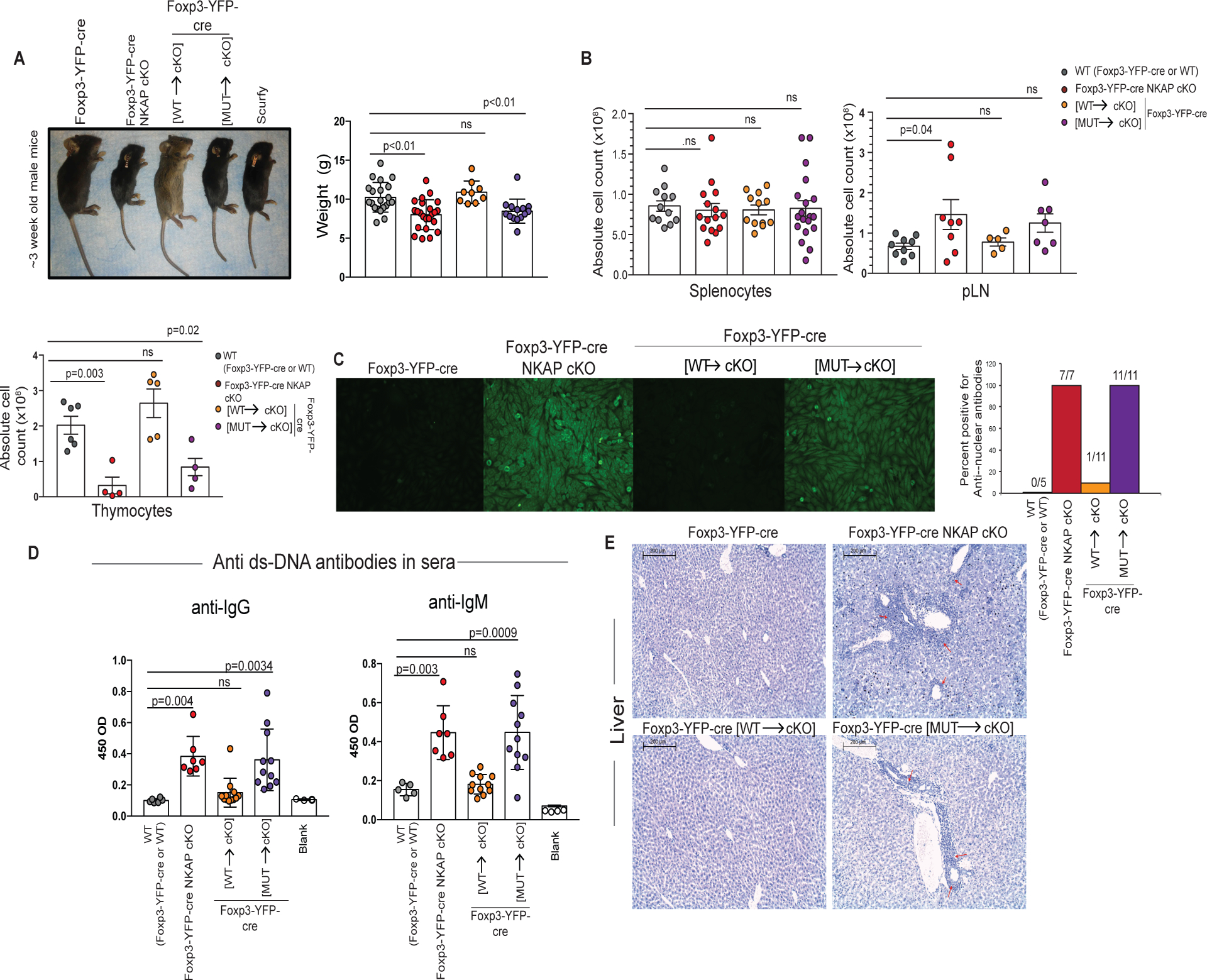
(A) Representative sizes of ~3-week old Foxp3-YFP-cre, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], Foxp3-YFP-cre [MUT→cKO], and scurfy mice. Weights of 3-week old WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice are shown. Data shown is combined from 22, 25, 10, and 14 mice for the genotypes are listed, respectively. Bar graphs indicate mean weight in grams and error bars indicate SEM. (B) Splenic, peripheral lymph node (pLN) and thymic total cell counts from WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice are shown. Splenic data from at least 10 independent experiments with 12, 15, 12, and 19 mice for the genotypes listed, respectively. pLN data is combined from at least 5 independent experiments with 9, 8, 5, and 7 mice for the genotypes listed, respectively. Thymic data is combined from 5 independent experiments with 6, 4, 5, 6 mice for the genotypes listed, respectively. Bar graphs indicate mean absolute cell counts and error bars indicate SEM. All p-values were calculated using one-way ANOVA with multiple comparisons. (C) The presence of Antinuclear antibodies (ANA) in sera collected from WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice was determined and summarized as bar graphs indicating percent mice positive for ANA. (D) Anti-double stranded DNA antibodies from WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice were measured. Bar graphs indicate mean absorbance at 450 OD and error bars indicate SEM. (C-D) Data was generated from a single experiment with serum from 5, 7, 11, and 11 mice for the genotypes listed respectively. All p-values were calculated using one-way ANOVA with multiple comparisons. (E) Lymphocytic infiltration was assessed using Hematoxylin and Eosin staining of liver sections from WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], or Foxp3-YFP-cre [MUT→cKO] mice. Images are representative of three independent experiments from at least 3 mice per group.
Unchecked T cell activation and proliferation is the driving cause of tolerance breakdown (10). In comparison to WT, Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] mice exhibited slightly increased CD4 and significantly increased CD8 absolute T cell counts (Supplement Figure 3A). In addition, memory/activated CD4+CD44+CD62L− and CD8+CD44+CD62L− absolute cell numbers were increased in Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] mice (Supplement Figure 3B). To exclude the possibility that NKAP(Y352A) exerted dominant negative effects leading to tolerance breakdown, Foxp3-YFP-cre [WT→WT] and Foxp3-YFP-cre [MUT→WT] mice were examined. No aberrant T cell expansion was found and mice survived to adulthood without any signs of autoimmunity (Supplement Figure 3C–E). Therefore, similar to Foxp3-YFP-cre NKAP cKO mice, Foxp3-YFP-cre [MUT→cKO] mice succumb to systemic autoimmunity characterized by B cell and T cell tolerance breakdown.
Substitution of NKAP with NKAP(Y352A) leads to Treg elimination at the RTE stage
Treg development is indispensable for ensuring tolerance in humans and mice (24–26). Mature thymic CD25+Foxp3+ Tregs develop from CD25+Foxp3− and CD25−Foxp3+ precursor populations (27). As rampant autoimmunity leads to thymic involution, 3-week old male Foxp3-YFP-cre NKAP cKO mice exhibit diminished overall thymic cellularity and Treg absolute numbers compared to WT mice (7). However, thymic Treg and Treg precursor frequencies in 3-week old Foxp3-YFP-cre NKAP cKO mice are similar to WT indicating normal thymic Treg production. Treg development was examined in male WT, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], Foxp3-YFP-cre [MUT→cKO], Foxp3-YFP-cre [WT→WT] and Foxp3-YFP-cre [MUT→WT] mice (Figure 5A). All mice had similar frequencies of CD25+Foxp3− and CD25−Foxp3+ precursors and CD25+Foxp3+ Tregs indicating that thymic Treg production occurs unhindered in Foxp3-YFP-cre [MUT→cKO] mice and the expression of YFP-NKAP(Y352A) in the presence of endogenous NKAP has no adverse effects on the generation of Tregs.
Figure 5. NKAP(Y352A) substitution does not alter thymic Treg development but leads to Treg elimination at the RTE stage.
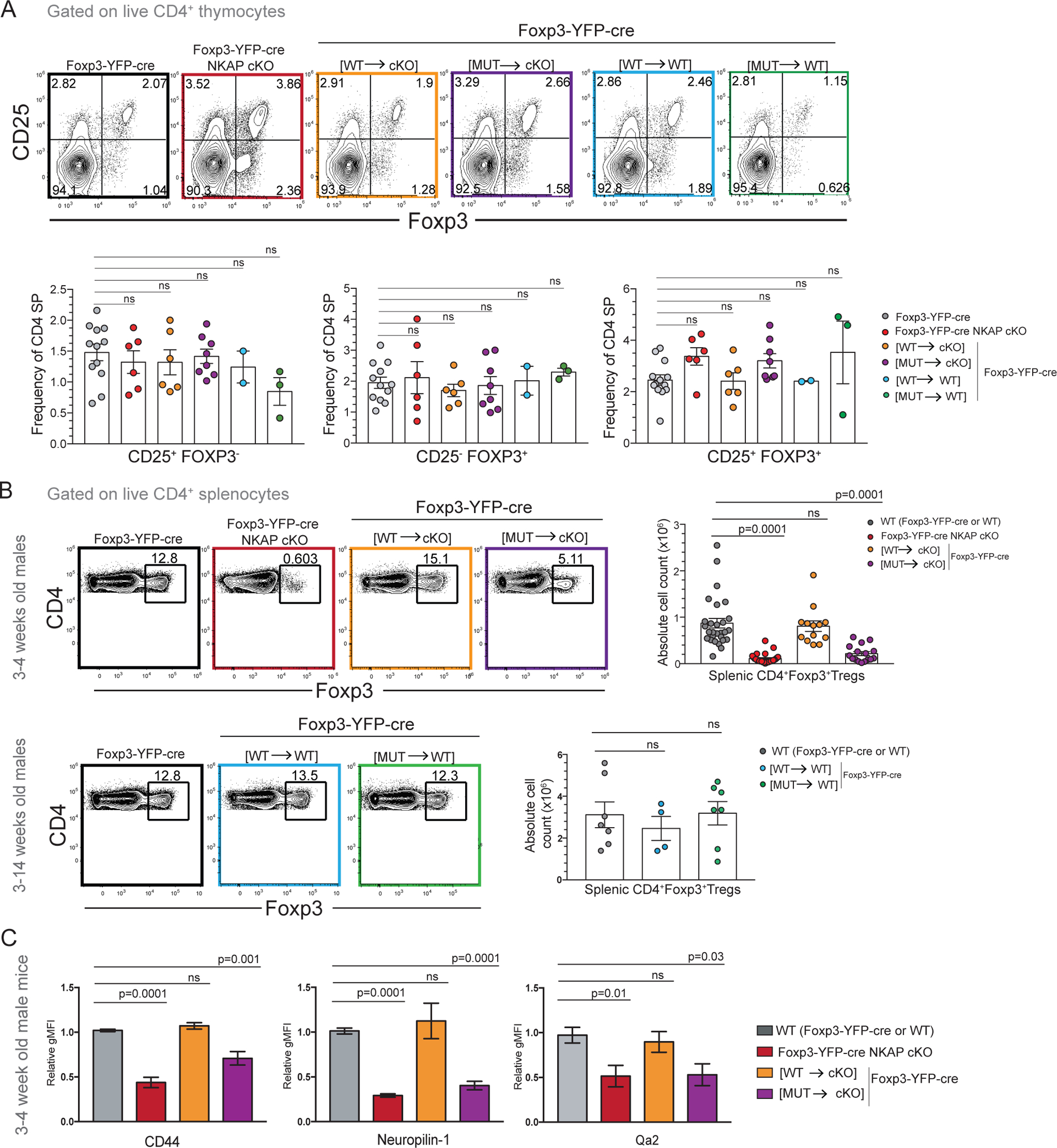
(A) Comparison of CD25+Foxp3− precursor, CD25−Foxp3+ precursor and CD25+Foxp3+ thymic Treg frequencies in Foxp3-YFP-cre, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], Foxp3-YFP-cre [MUT→cKO], Foxp3-YFP-cre [WT→WT] and Foxp3-YFP-cre [MUT→WT] mice. Data shown is combined from at least 2 independent experiments with 12, 6, 6, 8, 2, and 3 mice in total for the genotypes listed, respectively. Bar graphs indicate mean frequencies and error bars indicate SEM. (B) Examination of frequency and absolute cell counts of splenic Tregs in 3-week old WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice. Data shown is combined from 28, 27, 13, and 16 mice in total for the genotypes listed, respectively. 3–14-week old WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre [WT→WT] and Foxp3-YFP-cre [MUT→WT] mice were also examined. Data shown is combined from 7, 4, and 7 mice in total for the genotypes listed, respectively. Bar graphs indicate mean absolute cell numbers and error bars indicate SEM. (C) Examination of mean relative gMFI of Treg maturation markers CD44, Nrp-1 and Qa2 in WT (Foxp3-YFP-cre or WT), Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO] and Foxp3-YFP-cre [MUT→cKO] mice. Relative gMFI was calculated by dividing gMFI with WT gMFI of CD44, Nrp-1 and Qa2. Combined data from 3 independent experiments with at least 3 mice per genotype. Bar graphs indicate mean relative gMFI and error bars indicate SEM. (A-C) All p-values were calculated using one-way ANOVA with multiple comparisons.
The underlying cause of autoimmunity in Foxp3-YFP-cre NKAP cKO male mice is disappearance of peripheral Tregs (7). Foxp3 absolute cell numbers and frequencies were assessed in male WT, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] mice. Similar to Foxp3-YFP-cre NKAP cKO mice, Foxp3-YFP-cre [MUT→cKO] mice exhibited a severe decrease in absolute Treg counts in the spleen (Figure 5B). Examination of Foxp3-YFP-cre [MUT→WT] mice showed no defects in Treg persistence, ruling out dominant negative effects of YFP-NKAP(Y352A) expression. Previously, to bypass any indirect effects of inflammation, Foxp3-YFP-cre NKAP cKO female chimeras were compared to Foxp-3 YFP-cre WT female chimeras (7). Foxp3-YFP-cre WT chimeric females have XFoxp3-YFP-cre and a XNKAP fl alleles. Foxp3-YFP-cre NKAP cKO chimeric females have XFoxp3-YFP-cre NKAP fl and a XNKAP fl alleles. Because of random X-chromosome inactivation, both mice generate YFP+ (cre+) and YFP− (cre−) Tregs. However, YFP+ Tregs fail to persist in Foxp3-YFP-cre NKAP cKO female chimeras while similar fractions of YFP+ and YFP− Tregs are present in Foxp3-YFP-cre WT females (Supplement Figure 4A–C). Foxp3-YFP-cre [MUT→cKO] female chimeras had very few YFP+ Tregs in the periphery and showed a skewing toward YFP− WT population. Thus, Foxp3-YFP-cre [MUT→cKO] female chimeras are similar to Foxp3-YFP-cre NKAP cKO female chimeras. On the other hand, Foxp3-YFP-cre [WT→cKO] female chimeras have YFP+ Tregs at frequencies similar to Foxp3-YFP-cre female WT chimeras. In the presence of YFP− WT Tregs, YFP+ Foxp3-YFP-cre [WT→WT] Tregs or YFP+ Foxp3-YFP-cre [MUT→WT] Tregs did not exhibit any competitive survival disadvantages, demonstrating no adverse effects of YFP-NKAP(Y352A) in the presence of endogenous NKAP on Treg survival. There is considerable overlap between the YFP signal from Foxp3-YFP-cre (cre is fused to YFP) and YFP-NKAP precluding separation of these signals (Supplement Figure 4D, E). However, a slight increase in YFP gMFI occurs when both Foxp3-YFP-cre and YFP-NKAP transgene are expressed compared to expression of Foxp3-YFP-cre alone. This increase in YFP signal was used to confirm the expression of YFP-NKAP-(WT) and YFP-NKAP-(Y352A) transgenes in chimeric females (Supplement Figure 4 D, E).
Previously, NKAP-deficient YFP+ Tregs in Foxp3-YFP-cre NKAP cKO female chimeras were shown to be Nrp-1 low compared to NKAP-sufficient YFP+ Tregs from Foxp3-YFP-cre WT female chimeras (7). Nrp-1 low Tregs may represent peripherally derived Tregs or thymically derived RTE Tregs (28). Using RagGFP mice, RTE Tregs were shown to be CD44lo and CD62Lhi (29) . To distinguish between peripherally derived Tregs and RTE Tregs the expression of CD44, CD62L and Qa2 by NKAP-deficient YFP+ Tregs in Foxp3-YFP-cre NKAP cKO and YFP+ Tregs from Foxp3-YFP-cre WT female chimeras was previously examined (7). NKAP-deficient Tregs were found to be Nrp-1lo CD44lo Qa2lo while NKAP-sufficient Tregs from Foxp3-YFP-cre WT chimeric females were Nrp-1hi CD44hi Qa2hi (7). Further, NKAP-deficient Tregs had a greater abundance of TCR excision circles, which negatively correlates with the age of T cells(30). These results indicated that NKAP-deficient Tregs disappeared at the RTE stage. As Foxp3-YFP-cre [MUT→cKO] male mice develop autoimmunity at a rate similar to Foxp3-YFP-cre NKAP cKO mice, Foxp3-YFP-cre [MUT→cKO] Tregs likely disappear as RTEs as well. To determine whether abrogation of NKAP’s interaction with HDAC3 leads to Treg disappearance at the RTE stage, expression of Qa2, CD44 and Nrp-1 by Tregs from WT, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], and Foxp3-YFP-cre [MUT→cKO] male mice was determined (Figure 5C). Tregs from 3-week old male Foxp3-YFP-cre [MUT→cKO] mice are also Qa2loCD44loNrp-1lo compared to WT and Foxp3-YFP-cre [WT→cKO] indicating that they fail to transition to the more activated and mature state, and are eliminated at the RTE stage. To confirm the RTE phenotype of NKAP-deficient Tregs in a non-autoimmune environment, YFP+ and YFP− Tregs from Foxp3-YFP-cre, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], Foxp3-YFP-cre [MUT→cKO] female chimeras were examined for expression of CD44 and CD62L [Supplement Figure 4F, G]. Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] females harbored very few activated YFP+ CD44+ Tregs and compared to YFP− CD44+ Tregs. On the other hand, Foxp3-YFP-cre WT and Foxp3-YFP-cre [WT→cKO] females generated YFP+ CD44+ and YFP− CD44+ Tregs. Next, the expression of Qa2 by YFP+CD62L+ Tregs and YFP−CD62L+ Tregs from Foxp3-YFP-cre, Foxp3-YFP-cre NKAP cKO, Foxp3-YFP-cre [WT→cKO], Foxp3-YFP-cre [MUT→cKO] female chimeras was examined (Supplement Figure 4H). Qa2 was expressed at low and intermediate levels by YFP+CD62L+ Tregs from Foxp3-YFP-cre NKAP cKO and Foxp3-YFP-cre [MUT→cKO] chimeric females, respectively, compared to YFP−CD62L+ Tregs in the same mice. Qa2 expression by YFP−CD62L+ Tregs and YFP+CD62L+ Tregs from Foxp3-YFP-cre WT and Foxp3-YFP-cre [WT→cKO] females were comparable. Cumulatively, these results indicate that abrogation of NKAP’s interaction with HDAC3 leads to rapid loss of Tregs at the RTE stage replicating the phenotype of NKAP deficiency.
The absence of either NKAP or HDAC3 leads to enhanced lipid peroxidation, a hallmark of ferroptosis.
Maturing thymocytes gradually gain complement resistance prior to egress from the thymus (17). NKAP deletion results in the inability of peripheral naïve T cells to attain protection from complement proteins (6). NKAP-deficient T cells are rapidly opsonized by C3 as well as other complement molecules following thymic egress implicating complement activation as a possible mechanism driving their disappearance in secondary lymphoid organs (6). In a recent study (17), CD4-cre NKAP cKO mice were crossed with mice also deficient in C1q, MBL1 and MBL2 encoding genes to examine whether combined deletion of the classical (C1q) and lectin pathways (MBL1 and MBL2) could restore NKAP-deficient T cells to normal numbers. Although C3 deposition was prevented in these mice, NKAP-deficient T cells failed to persist in the absence the classical and lectin pathways indicating that another mechanism mediated T cell clearance. The classical and lectin pathways mediate C3 deposition (31). C3 deficiency did not curb the loss of peripheral T cells in CD4-cre NKAP cKO mice either (17). In addition, there was no evidence of mitochondrial dysfunction or apoptosis in NKAP-deficient T cells (6, 17).
Interestingly, CD4-cre NKAP cKO mice exhibited enhanced lipid peroxidation compared to WT mice (17). Accumulation of lipid peroxides is a defining feature of ferroptosis, an iron-dependent form of programmed cell-death. Increased lipid peroxidation in NKAP-deficient naïve T cells was detected using C-11 BODIPY 581/591, a lipophilic reactive oxygen species (ROS) sensor that gains fluorescence in the FITC channel following reaction with peroxyl radicals (17, 18, 32, 33). Lipid peroxidation stemming from NKAP-deficiency could be inhibited in vitro by treatment of cells with the lipophilic antioxidant alpha-tocopherol (vitamin E) and/or an inhibitor of ferroptosis, Ferrostatin-1, bolstering the idea that NKAP-deficiency leads to ferroptosis (17).
To determine whether loss of peripheral T cells in CD4-cre HDAC3 cKO mice occurred by a mechanism similar to CD4-cre NKAP cKO mice, the effect of C3 deficiency was examined (Figure 6A, B). The loss of HDAC3-deficient T cells was not mitigated in C3 KO CD4-cre HDAC3 cKO mice. Next, Ferroptosis was examined using BODIPY-C11 581/591 (Figure 6C). HDAC3-deficient naïve T cells exhibited enhanced lipid peroxidation in CD4-cre HDAC3 cKO as well as C3 KO CD4-cre HDAC3 cKO mice. In vitro, lipid peroxidation of HDAC3-deficient T cells was inhibited by alpha-tocopherol or Ferrostatin-1, or by a combination of both similar to NKAP-deficient T cells (Figure 6D). Further, compared to WT, C3 KO CD4-cre HDAC3 cKO mice also exhibit lipid peroxidation that can be reversed by alpha-Tocopherol and Ferrostatin-1 in combination (Figure 6E) or individually (data not shown). Overall, our data indicate that susceptibility to complement attack and ferroptosis are regulated by NKAP and HDAC3 in a similar manner, consistent with the evidence that their interaction is important for T cell maturation.
Figure 6. HDAC3 and NKAP are required to prevent lipid peroxidation, a hallmark feature of ferroptosis.
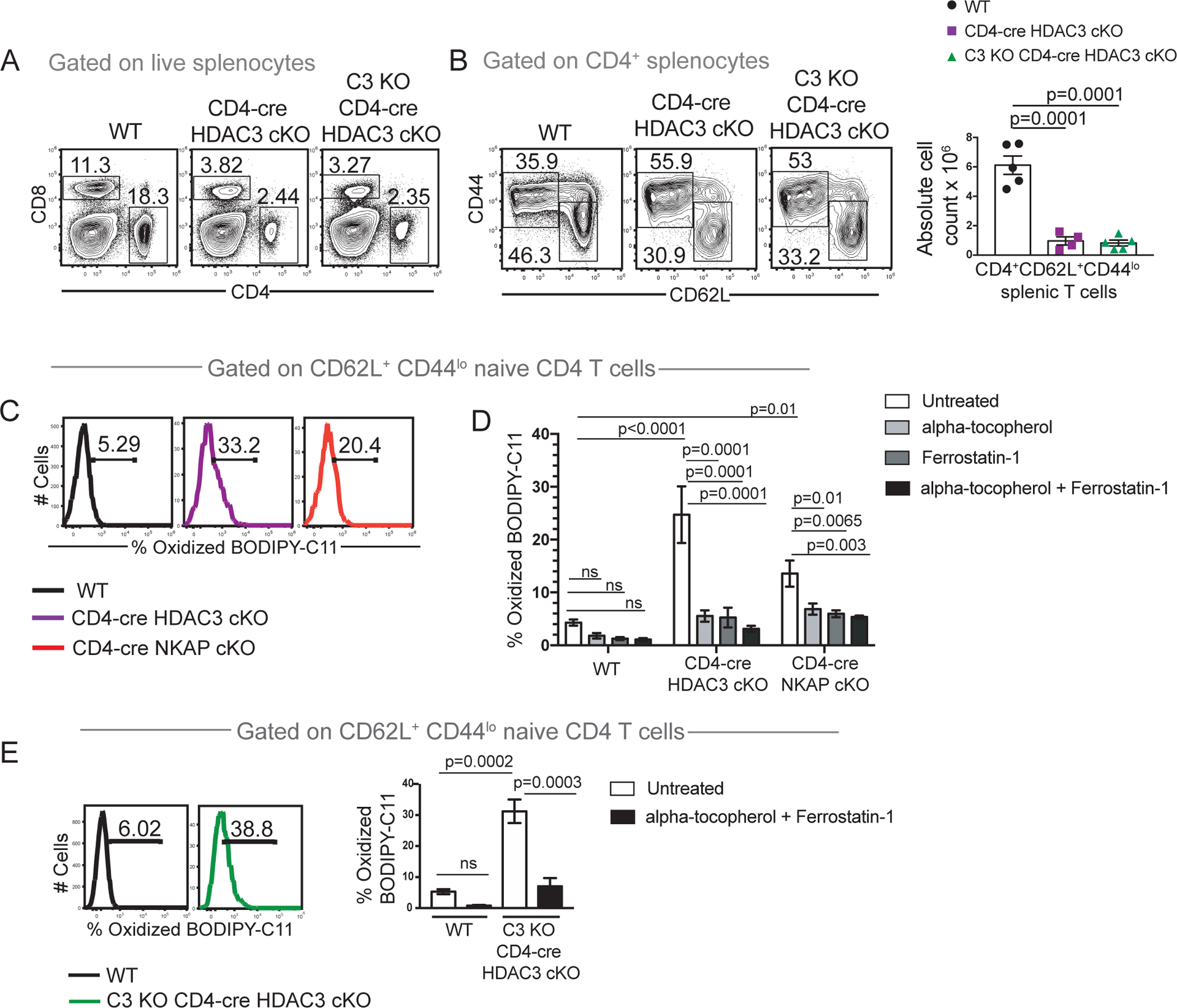
(A) Examination of frequencies of total CD4 and total CD8 T cells in WT, CD4-cre HDAC3 cKO and C3 KO CD4-cre HDAC3 cKO mice. (B) Examination of frequency and absolute cell numbers of naïve CD4 T cells in WT, CD4-cre HDAC3 cKO and C3 KO CD4-cre HDAC3 cKO mice. (C) Assessment of frequency of WT, CD4-cre HDAC3 cKO and CD4-cre NKAP cKO naïve CD4 T cells T cells undergoing lipid peroxidation using BODIPY-C11 581/591 which fluoresces in the FITC channel upon oxidation (oxidized BODIPY-C11). Cells were analyzed after incubation with BODIPY-C11 581/591 for 1 hour in culture. (D) Comparison of frequencies of WT, CD4-cre HDAC3 cKO and CD4-cre NKAP cKO naïve CD4 splenocytes positive) after 15-minute treatments with alpha-tocopherol (vitamin E), Ferrostatin-1 or a combination of both followed by incubation with BODIPY-C11 581/591 for 1 hour in culture. Bar graphs are mean frequency of BODIPY-C11+ naïve T cells from 4 independent experiments with at least one mouse per genotype per experiment. Error bars indicate SEM. p-values were calculated using two-way ANOVA with multiple comparisons. (E) Assessment of frequency of naïve CD4 T cells undergoing lipid peroxidation in WT and C3 KO CD4-cre HDAC3 cKO mice using BODIPY-C11 581/591. Assessment of mean frequency of oxidized BODIPY-C11 cells in WT, CD4-cre HDAC3 cKO and C3 KO CD4-cre HDAC3 cKO mice after treatment with alpha-tocopherol (vitamin E) and Ferrostatin-1 in combination. Bar graphs are mean frequency of BODIPY-C11+ naïve T cells from 3 independent experiments with one mouse per genotype per experiment. Error bars indicate SEM. All p-values were calculated using two-way ANOVA with multiple comparisons within genotypes (untreated versus treatment with each inhibitor or inhibitors in combination) and across genotypes (WT versus CD4-cre HDAC3 cKO or CD4-cre NKAP cKO or C3 KO CD4-cre HDAC3 cKO).
Discussion:
Maturation endows T cells with functional capacity and is an important determinant of immunity and immune system homeostasis. A greater understanding of maturation mechanisms may be harnessed to improve T cell dependent immune responses. While regulators of early T cell development are very well-characterized, only a few regulators of maturation have been identified (2, 3, 5, 13, 22, 34–40). NKAP and HDAC3 have been separately shown to be required for T cell maturation and iNKT development (5, 13, 15). Neither NKAP nor HDAC3 has a defined DNA binding domain, and likely associate with chromatin as parts of larger protein complexes. Interestingly, NKAP and HDAC3 physically associate (12), and this interaction can be disrupted by a single amino acid substitution in the NKAP(Y352A) mutant (16). Here, we report that this mutation in NKAP also impairs its function during T cell maturation and iNKT cell development, providing evidence that the association between NKAP and HDAC3 is critical for T cell maturation and iNKT cell development.
Upon CD4-cre driven substitution of endogenous NKAP with NKAP(Y352A), a block occurred in intra-thymic iNKT cell development, while no defects in thymic T cell or Treg development were observed. This phenotype was essentially identical to that presented by CD4-cre NKAP cKO mice. Similarly, CD4-cre or Foxp3-YFP-cre driven substitution of NKAP(Y352A) for WT NKAP resulted in defects in T cell and Treg maturation similar to those observed in CD4-cre or Foxp3-YFP-cre NKAP cKO mice, respectively. Specifically, in conventional T cells, these defects included a failure in long-term persistence, impaired cytokine production, and a lack of resistance to complement. In Tregs, their disappearance at the RTE stage caused systemic lethal autoimmunity. In control mice, induction of an NKAP(WT) transgene prevented all effects of deletion of the native NKAP gene in each T cell population. Together, these results show that NKAP(Y352A) cannot substitute for WT NKAP during iNKT development in the thymus or during maturation of conventional T cells or Tregs. These results suggest that the ability to associate with HDAC3 is critical for the function of NKAP.
Interestingly, NKAP(Y352A) was able to functionally substitute for WT NKAP during conventional T cell maturation in one respect. Significantly reduced expression of Qa2 and CD45RB, two phenotypic markers associated with maturation, occurs in CD4-cre NKAP cKO RTEs, yet expression of these proteins was restored to nearly normal levels in cells expressing NKAP(Y352A) instead of WT NKAP. Thus, NKAP must influence the expression of these proteins independent of HDAC3. This idea is further supported by the observation that Qa2 and CD45RB expression were not reduced in HDAC3 deficient RTEs despite a general defect in T cell maturation (13). The participation of NKAP in both HDAC3-dependent and independent processes was also demonstrated in a previous study in which NKAP was deleted in multiple hematopoietic population with Mx1-cre along with induction of NKAP(Y352A) (16). While the mutant was unable to substitute for WT NKAP in the maintenance of the HSC pool, it was able to completely substitute in the erythroid lineage. Further, the Y352A mutation did not cause any reduction in the ability of NKAP to promote proliferation in fibroblasts (16). These results also suggest that NKAP may be present in molecular complexes whose individual components vary depending on the biological context, which will be investigated in the future.
Similar to NKAP, HDAC3 is also critical to prevent lipid peroxidation in naïve T cells and ferroptosis is the likely the cause of disappearance of both NKAP and HDAC3 deficient T cells. Abrogating the association of NKAP and HDAC3 would likely result in ferroptosis as well. Unfortunately, this cannot be tested in CD4-cre [MUT→cKO] mice due to overlap of BODIPY-C11 581/591 and YFP fluorescence. In vivo, ferroptosis results from complex interactions between several factors including iron accumulation, increased synthesis of acid-phospholipids, depletion of enzymes that reverse lipid peroxidation and unavailability of antioxidant molecules (18). In T cells, two well characterized inhibitors of ferroptosis are GPX-4 and Glutathione (g-L-glutamyl-L-cysteinylglycine or GSH); GPX-4 uses GSH to reduce lipid hydroperoxides to less harmful lipid alcohols and this process is critical for T cell persistence (41). Both GPX-4 and GSH were previously shown to be present at normal levels in NKAP-deficient CD4 naïve T cells, suggesting a GPX-4 independent mechanism of lipid-peroxidation (17).
While the effects of the Y352A substitution on association of NKAP with partners other than HDAC3 cannot be excluded, the findings presented here, combined with previous observations of similar requirements for NKAP and HDAC3 during T cell development, strongly motivate future investigations on interplay between the two proteins. For example, the functions of both NKAP and HDAC3 in T cell development earlier than the DP stage have been investigated using cre-mediated deletion at the DN stage (12, 42, 43). Deficiency in either protein resulted in different blocks in T cell development. While the deletion of HDAC3 caused a block at the DP stage, the deletion of NKAP caused a DN3 stage block. Thus, the tools presented in this study hold great potential to further dissect functions of NKAP and HDAC3 in early T cell development and other aspects of T cell biology to identify functions that require their association and functions that do not.
Supplementary Material
Key points:
NKAP association with HDAC3 is required for conventional T cell and Treg maturation.
NKAP association with HDAC3 is required for iNKT cell development.
Both NKAP and HDAC3 prevent lipid peroxidation, a defining feature of ferroptosis.
Acknowledgements:
We thank the members of lab of Virginia Smith Shapiro for critical reading of the manuscript and Paul Belmonte for technical assistance with lipid peroxidation studies. We thank members of Kay Medina and Hu Zeng laboratories for helpful discussions. We also thank the NIH tetramer facility and the Mayo Knockout and Transgenic Core Facility.
Funding:
This work was supported by NIH R01 AI083279 to V.S.S.
Abbreviations:
- HDAC3
Histone Deacetylase 3
- HSC
Hematopoietic Stem Cells
- DP
Double positive
- SP
Single Positive
- RTE
Recent thymic emigrants
- MNT
Mature naïve T cells
- Tregs
Regulatory T cells
- iNKT
invariant Natural Killer T cells
- cKO
conditional knockout
- WT
Wildtype
Footnotes
Competing interests: The authors declare that they have no competing interests.
Bibliography
- 1.Hogquist KA, and Jameson SC. 2014. The self-obsession of T cells: how TCR signaling thresholds affect fate “decisions” and effector function. Nat. Immunol 15: 815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fink PJ, and Hendricks DW. 2011. Post-thymic maturation: young T cells assert their individuality. Nature 11: 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hogquist KA, Xing Y, Hsu F-C, and Shapiro VS. 2015. T Cell Adolescence: Maturation Events Beyond Positive Selection. J. Immunol 195: 1351–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan X, Moltedo B, Mendoza A, Davydov AN, Faire MB, Mazutis L, Sharma R, Pe’er D, Chudakov DM, and Rudensky AY. 2018. CD49b defines functionally mature Treg cells that survey skin and vascular tissues. J. Exp. Med 215: 2796–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu F-C, Pajerowski AG, Nelson-Holte M, Sundsbak R, and Shapiro VS. 2011. NKAP is required for T cell maturation and acquisition of functional competency. J. Exp. Med 208: 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu F, Shapiro MJ, Chen MW, McWilliams DC, Seaburg LM, Tangen SN, and Shapiro VS. 2014. Immature recent thymic emigrants are eliminated by complement. J. Immunol 193: 6005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash B, Shapiro MJ, Chung JY, Romero Arocha S, and Shapiro VS. 2018. Treg-specific deletion of NKAP results in severe, systemic autoimmunity due to peripheral loss of Tregs. J. Autoimmun 89: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyon MF, Peters J, Glenister PH, Ball S, and Wright E. 1990. The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc. Natl. Acad. Sci 87: 2433–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet 27: 68–73. [DOI] [PubMed] [Google Scholar]
- 10.Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, and Ramsdell F. 1999. Cellular and Molecular Characterization of the scurfy Mouse Mutant. J. Immunol 162: 2546–2554. [PubMed] [Google Scholar]
- 11.Aschermann S, Lehmann CHK, Mihai S, Schett G, Dudziak D, and Nimmerjahn F. 2013. B cells are critical for autoimmune pathology in Scurfy mice. Proc. Natl. Acad. Sci. U. S. A 110: 19042–19047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pajerowski AG, Nguyen C, Aghajanian H, Shapiro MJ, and Shapiro VS. 2009. NKAP Is a Transcriptional Repressor of Notch Signaling and Is Required for T Cell Development. Immunity 30: 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu F, Belmonte PJ, Constans MM, Chen MW, McWilliams DC, Hiebert SW, and Shapiro VS. 2015. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol 195: 1578–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Liu Y, Han R, Beier UH, Bhatti TR, Akimova T, Greene MI, Hiebert SW, and Hancock WW. 2015. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J. Clin. Invest 125: 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thapa P, Das J, McWilliams D, Shapiro M, Sundsbak R, Nelson-Holte M, Tangen S, Anderson J, Desiderio S, Hiebert S, Sant’Angelo DB, and Shapiro VS. 2013. The transcriptional repressor NKAP is required for the development of iNKT cells. Nat. Commun 4:1582. doi: 10.1038/ncomms2580 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shapiro MJ, Lehrke MJ, Chung JY, Arocha SR, and Shapiro VS. 2019. NKAP Must Associate with HDAC3 to Regulate Hematopoietic Stem Cell Maintenance and Survival. J. Immunol 202: 2287–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dash B, Belmonte PJ, Fine SR, Shapiro MJ, Chung JY, Schwab AD, McCue SA, Rajcula MJ, and Shapiro VS. 2019. Murine T Cell Maturation Entails Protection from MBL2, but Complement Proteins Do Not Drive Clearance of Cells That Fail Maturation in the Absence of NKAP. J. Immunol 203 DOI: 10.4049/jimmunol.1801443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Overholtzer M, Woerpel KA, Torti FM, Zhang DD, Ran Q, Salnikow K, Jiang X, Linkermann A, Fulda S, V Torti S, Hatzios SK, Rosenfeld CS, Tang D, Park J, Murphy ME, Kagan VE, Gascón S, Oyagi A, Toyokuni S, Dixon SJ, Noel K, and Pagnussat GC. 2017. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Sharma K, Deng HX, Siddique T, Grisotti G, Liu E, and Roos RP. 2007. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol. Dis 29: 400–408. [DOI] [PubMed] [Google Scholar]
- 20.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pé rez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, and Wilson CB. 2001. A Critical Role for Dnmt1 and DNA Methylation in T cell Development, Function, and Survival. Immunity 15: 763–774. [DOI] [PubMed] [Google Scholar]
- 21.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Muller W, and Rudensky AY. 2008. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity 28: 546–558. [DOI] [PubMed] [Google Scholar]
- 22.Xing Y, Wang X, Jameson SC, and Hogquist KA. 2016. Late stages of T cell maturation in the thymus involve NF-κB and tonic type I interferon signaling. Nat. Immunol 17: 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dooley J, and Liston A. 2012. Molecular control over thymic involution: From cytokines and microRNA to aging and adipose tissue. Eur. J. Immunol 42: 1073–1079. [DOI] [PubMed] [Google Scholar]
- 24.d’Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, Noya FJ, Jabado N, Mazer B, and Piccirillo CA. 2009. FOXP3 Forkhead Domain Mutation and Regulatory T Cells in the IPEX Syndrome. N. Engl. J. Med 361: 1710–1713. [DOI] [PubMed] [Google Scholar]
- 25.Yang S, Fujikado N, Kolodin D, Benoist C, and Mathis D. 2015. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. 348: 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao X 2010. Regulatory T cells and immune tolerance to tumors. Immunol. Res 46: 79–93. [DOI] [PubMed] [Google Scholar]
- 27.Lio CWJ, and Hsieh CS. 2008. A Two-Step Process for Thymic Regulatory T Cell Development. Immunity 28: 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Von Schack D, and Bluestone JA. 2012. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med 209: 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ. 2014. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med 211: 121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fink PJ 2013. The Biology of Recent Thymic Emigrants. Annu. Rev. Immunol 31: 31–50. [DOI] [PubMed] [Google Scholar]
- 31.Sarma JV, and Ward PA. 2011. The complement system. Cell Tissue Res. 343: 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, and Tang D. 2016. Ferroptosis: process and function. Cell Death Differ. 23: 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, and Stockwell BR. 2012. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinreich MA, and Hogquist KA. 2008. Thymic Emigration: When and How T Cells Leave Home. J Immunol Ref. J. Immunol 181: 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, Wani MA, Lingrel JB, Hogquist KA, and Jameson SC. 2006. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442: 299–302. [DOI] [PubMed] [Google Scholar]
- 36.Rothenberg EV, Zhang J, and Li L. 2010. Multilayered specification of the T-cell lineage fate. Immunol. Rev 238: 150–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothenberg EV 2014. Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol 32: 283–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu FC, Shapiro MJ, Dash B, Chen CC, Constans MM, Chung JY, Romero Arocha SR, Belmonte PJ, Chen MW, McWilliams DC, and Shapiro VS. 2016. An Essential Role for the Transcription Factor Runx1 in T Cell Maturation. Sci. Rep 6. doi: 10.1038/srep23533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham CA, Helm EY, and Fink PJ. 2018. Reinterpreting recent thymic emigrant function: defective or adaptive? Curr. Opin. Immunol 51: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boursalian TE, Golob J, Soper DM, Cooper CJ, and Fink PJ. 2004. Continued maturation of thymic emigrants in the periphery. Nat Immunol 5: 418–425. [DOI] [PubMed] [Google Scholar]
- 41.Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, and Kopf M. 2015. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med 212: 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Philips RL, Chen MW, McWilliams DC, Belmonte PJ, Constans MM, and Shapiro VS. 2016. HDAC3 Is Required for the Downregulation of RORγt during Thymocyte Positive Selection. J. Immunol 197: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stengel KR, Zhao Y, Klus NJ, Kaiser JF, Gordy LE, Joyce S, Hiebert SW, and Summers AR. 2015. Histone Deacetylase 3 Is Required for Efficient T Cell Development. Mol. Cell. Biol 35: 3854–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.