

Summary
Nearly 90% of structural models in the Protein Data Bank (PDB), the central resource worldwide for three-dimensional structural information, are currently derived from macromolecular crystallography (MX). A major bottleneck in determining MX structures is finding conditions in which a biomolecule will crystallize. Here, we present a searchable database of the chemicals associated with successful crystallization experiments from the PDB. We use these data to examine the relationship between protein secondary structure and average molecular weight of polyethylene glycol and to investigate patterns in crystallization conditions. Our analyses reveal striking patterns of both redundancy of chemical compositions in crystallization experiments and extreme sparsity of specific chemical combinations, underscoring the challenges faced in generating predictive models for de novo optimal crystallization experiments.
Keywords: structural biology, macromolecular crystallography, protein crystallization, crystallization conditions, crystallization information, PDB
Graphical Abstract
Highlights
-
•
Provides an updatable Python script to extract details from PDB free text fields
-
•
Gives a standardized and searchable dataset of crystallization chemical conditions
-
•
Analyzes relationship between PEG MW and protein secondary structure profiles
-
•
Sparsity coupled with redundancy in chemical details make data mining challenging
The Bigger Picture
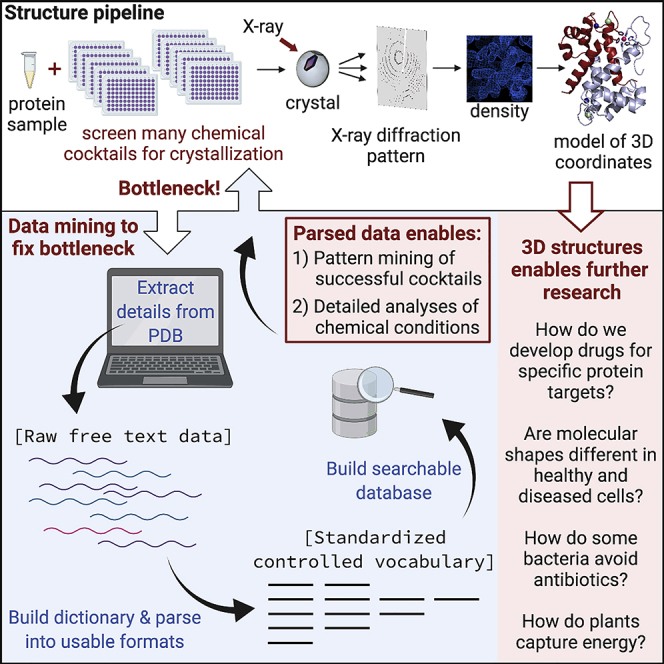
Determining structures of biological macromolecules is critical to advancing drug discovery and medical research. The majority (∼90%) of structures in the Protein Data Bank (PDB) derive from X-ray crystallography. To obtain a crystal structure, the first thing you need is a crystal. A key bottleneck to crystallographic methods is finding conditions in which a sample will crystallize. In addition to three-dimensional structural files, the PDB contains abundant metadata on crystallization details. Mining these data could unlock the bottleneck and facilitate structure acquisition. Crucial metadata on crystallization conditions are in free text fields in the PDB; parsing these data on a large scale is challenging. We have developed a tool to facilitate extraction and standardization. We provide the extraction tool, a curated dataset, and analyses of these metadata. This study enables PDB data mining by providing a customizable tool capable of imposing a controlled vocabulary on free text PDB metadata.
Free text formatted metadata from public databases are difficult to extract and leverage. We present a curated dataset of experimental details from the PDB, the primary repository of macromolecular structures. We contribute a software tool for parsing PDB free text fields for users to generate updated or customized datasets. Our parsing function handles irregular free text information to produce usable datasets with a controlled vocabulary. We illustrate extracted metadata use via analyses of relationships between chemicals and protein structure features.
Introduction
Structural biology is the study of the architecture of biological macromolecules; these structures sit at the base of a wide range of further scientific endeavors, from investigating enzymatic mechanisms that drive our understanding of energy production to the design of drugs capable of inhibiting disease progression. The worldwide repository for structural biology information is the Protein Data Bank (PDB), in which close to 160,000 structural models have been deposited since it was developed in 1971.1,2 Data from the PDB have a profound impact on an array of scientific discovery and innovation. Indeed, in 2017, over 679 million downloads of data from the PDB were reported, which averages to over 1.8 million structure data files downloaded per day.3,4 Scientists from all manner of disciplines rely on the wealth of information in the PDB to further their research programs. A recent analysis of the PDB found that 88% of the 210 new drugs that have been FDA approved between 2010 and 2016 depended on structural information from close to 6,000 different structures deposited in the PDB,5 illuminating how structural knowledge from the PDB empowers development of therapeutics. Nearly 90% of the structures available in the PDB are derived from experimental techniques requiring the sample to be in a crystalline form (the most common is macromolecular X-ray crystallography [MX], although electron crystallography and neutron diffraction are techniques that also require crystals). In these structural methods, a biomolecular crystal is exposed to an excitation source and diffraction patterns from the crystal are used to determine its structure. A critical step in this process is generating crystals of the biomolecules, and determining which conditions will drive crystal formation remains a central research area in structural biology.6, 7, 8, 9 The conditions that affect crystallization include the identity and amount of the chemical components in the crystallization condition (cocktail), the sample and/or cocktail pH, and the incubation temperature, among others. Experiments on the crystallization process have even been performed in space to investigate the role played by gravity.10 The crystallization parameter space is quite broad and is often approached experimentally with trial-and-error screening of different crystallization cocktail components. Once one or more cocktail hits (evidence of a nascent biomolecular crystal) are found in the initial crystallization screening, the conditions are typically optimized to increase diffraction quality by varying concentration and pH of component chemicals, as well as modulating other parameters such as temperature. Despite the extensive history and use of MX as a structural approach, the process of crystallization for macromolecular structure determination is nontrivial, as crystallization remains mysterious, even 100 years after the discovery that crystals will diffract X-rays.11 Formation of a crystal, however, is driven by fundamental underlying physical principles. A key factor in unearthing those principles is gathering enough information to tease out the complicated interactions between crystallization parameter space and crystal formation.
The PDB is an incredibly rich source of data about successful crystallization parameters, as it contains information on crystallization conditions in a free text field, “REMARK 280” in PDB format or “exptl_crystal_grow.pdbx_details” in the more recently developed mmCIF format. Mining this free text field for information about crystallization cocktails can provide insights about conditions in which macromolecular diffraction-quality crystals have formed. Despite the presence of the crystallization details in many of the structures deposited in the PDB, however, it is difficult to analyze these details across the entire PDB dataset. One of the difficulties in making extensive use of the crystallization condition data is the lack of standardized reporting within the free text field. Although the field has been available for depositing experimental details since 2000,12 not all crystal-based structures deposited in the PDB contain experimental crystallization information. Additionally, there are inconsistencies in naming conventions for compound identity (including typos and misspellings), in punctuation and spacing, and in exactly what information is listed (see specific examples in Tables S1 and S2). Efforts have been made to parse the details in this free text field, which have enabled assessment of which chemicals occur most frequently,12 estimation of impact of sample isoelectric point and calculated value for cocktail pH on successful crystallization,13 and, most recently, examination of correlations between protein sequence and crystallization conditions.14 Analyses have been performed to assess success rates (and number of screening conditions required)15 as well as data mining to better predict which cocktails will lead to positive crystallization outcomes.13,14,16,17 There has been long-standing interest in developing ways to better understand which parameters in the crystallization space enable biomolecular crystallization, but it remains a fundamental challenge to predict crystallization conditions that will be successful given details of the biomolecular target.
Here, we report the development of a searchable and updatable database of crystallization conditions extracted from the free text field containing crystallization details found in PDB entries. Recently the PDB file format has shifted to mmCIF format (mandatory submission of this type began in July 2019); both formats contain the same information regarding crystal growth conditions in a free text field. The crystallization database presented here (obtained December 9, 2019) contains 99,229 PDB IDs with crystallization details from the total 158,367 total structures deposited in the PDB as of that date. A focus of this work is investigation of the chemical patterns that occur within the data about crystallization conditions that are available from the PDB. A major goal is to investigate the parameter space of chemical components that are most prevalent in successful crystallization and which components are combined most frequently with one another. Ultimately this information could be used to develop a model for predicting, testing, and screening successful crystallization conditions. We summarize details regarding the chemical compounds and incubation temperature of successful crystallization experiments. Additionally, we have used these data to specifically probe how interactions between polyethylene glycol (PEG) and macromolecules enable crystallization by examining the relationship between average PEG molecular weight (MW) and the secondary structure composition for PDB entries in our database. We examine redundancy, providing examples of redundant proteins in the PDB with different crystallization conditions, to further illustrate the potential utility of the database. The wealth of information about crystallization conditions in the PDB is obfuscated by the difficulties in accessing the information in standardized form. We have extracted and standardized information from the PDB to enable analyses of relationships that we believe contribute to understanding the process of crystallization, and we make these data available for the research community. This database represents a snapshot of the parseable chemical conditions from the available PDB IDs as of December 2019 and our analyses of these data. We anticipate that additional research inquiries and analysis pipelines will be enabled by this data snapshot and will be expanded upon by making the data-extraction package easily updatable.
Results
Generation of Searchable and Updatable Database of Crystallization Conditions
Here, we present summaries and analyses of data downloaded from the PDB. As of December 9, 2019, the PDB repository contained 158,367 structures; details of how these data were parsed and standardized are provided in Experimental Procedures. The database we generate and analyze here contains 99,229 PDB IDs with crystallization details (Crystallization Details Dataset [CDD]). The CDD identifies 312 distinct chemical compounds; when chemical components are found at very low frequency within the PDB, they are less likely to appear in the final parsing. As one goal of this investigation is assessment of patterns among the chemical compounds and their combinations, the compound dictionary is more limited in cases of chemicals that appear with less frequency in the PDB. The PDB IDs that are not included in the CDD have been excluded for a number of reasons, including infrequent occurrence of chemicals and inability of the detail parsing function to identify specific compounds due to unexpected syntax, spacing, or punctuation. While it is possible to generate a report directly from the PDB that contains the crystallization condition details, in the CDD database presented here the details have been standardized and made more “searchable.” Examples of both successful and unsuccessful parsing for PDB entries show what works and reveal some of the difficulties encountered (Tables S1 and S2). The detail parsing function and manual parsing used to generate the CDD address a multitude of errors and inconsistencies, but some PDB IDs could not be parsed and some chemicals may have been too “incorrect” to even identify.
Previous research has focused on generating an extensive and full list of compounds that are used to crystallize macromolecules,12,16 including the chemical identity and concentration. Although we have extracted the concentration information for these data, not all of the PDB IDs within the CDD contain detailed concentration information. For the purposes of the analyses here, we do not make use of concentration information. As the analytical goal of this work is to identify broad patterns of behavior within the crystallization space, we have chosen to focus on the most commonly appearing chemicals within the PDB that are used in crystallization cocktails. These data have been mapped onto a matrix of PDB ID and presence/absence of each chemical compound (Table 1 shows a subset of the CDD). CDD data are provided in Data S1.
Table 1.
Subset of CDD Data Matrix
| PDB ID | Acetic Acid | PEG 4000 | Zinc(II) Sulfate | Reference | ||
|---|---|---|---|---|---|---|
| 1KHK | 0 | 0 | 1 | Le Du et al.18 | ||
| 2G0X | 0 | 0 | 0 | Aranda et al.19 | ||
| 3QG7 | 0 | 1 | 0 | Kirchdoerfer et al.20 | ||
| 5WL7 | 1 | 0 | 0 | Kaltenbach et al.21 |
The CDD is composed of 99,229 PDB IDs (row dimension) and 312 unique chemical compounds (column dimension), with a 0/1 indicator of absence/presence of a given compound in that PDB ID cocktail. Represented here is a subset of four PDB IDs (with literature citations) as examples of data that appear in the CDD.
We have also extracted secondary structure information from the ss.txt file available from the PDB for a subset of 97,782 PDB IDs with both crystallization details in the CDD and secondary structure information (Secondary Structure Dataset [SSD]). Table 2 shows an example subset of the SSD. We use these secondary structure data to examine the relationship between the composition of secondary structural elements and PEGs.
Table 2.
Subset of SSD Data Matrix
| PDB ID | Helix Count | Sheet Count | Total Count | Helix % | Sheet % | Other % |
|---|---|---|---|---|---|---|
| 1KHK | 276 | 187 | 898 | 30.73 | 20.82 | 48.44 |
| 2G0X | 121 | 0 | 154 | 78.57 | 0 | 21.42 |
| 3QG7 | 22 | 225 | 429 | 5.13 | 52.43 | 42.42 |
| 5WL7 | 84 | 62 | 226 | 37.17 | 27.43 | 35.40 |
The SSD is composed of 97,782 PDB IDs with columns representing counts of residues and percentage of each secondary structure component. Represented here is the same subset of four PDB IDs as those shown in Table 1.
Commonalities and Sparsity Observed in Crystallization Conditions
Of the 312 chemical compounds in the CDD, the most commonly appearing individual components include buffers, salts, and PEGs (Table 3 shows the ten most frequently occurring individual chemicals). Comparatively few of the crystallization cocktails are composed of only a single chemical compound (5.7%). The vast majority of cocktails (75.1%) contain either two or three components (Figure 1). The largest number of different chemical compounds (found in two PDB IDs) has 12 components, and only 5.9% of cocktails contain more than four different components.
Table 3.
Most Commonly Occurring Chemical Cocktail Components in the CDD
| Chemical Component | No. of PDB IDs | Chemical Component | No. of PDB IDs |
|---|---|---|---|
| PEG 3350 | 22,472 | Sodium chloride | 10,405 |
| Ammonium sulfate | 18,552 | PEG 8000 | 9,813 |
| PEG 4000 | 13,230 | Sodium citrate | 9,694 |
| Sodium acetate | 13,003 | MES | 9,590 |
| HEPES | 12,921 | Tris | 9,185 |
The ten most frequently identified chemical compounds in the CDD from the crystal growth detail free text field in the PDB and the number of the 99,229 PDB IDs in which these chemicals are found.
Figure 1.

Bar Plot of Number of Chemical Compounds in Crystallization Cocktails in the CDD
The count of PDB IDs in the CDD associated with a specific number of compounds in the crystallization cocktail. Most cocktails contain only either two or three components.
As temperature is another potential parameter of interest in the crystallization parameter space, we have extracted experimental incubation temperature for the CDD dataset. The majority (89.2%) of the CDD had temperature data reported for the crystallization conditions, ranging from 100 K to 328 K. Most crystallization experiments reported 277 K, 298 K, or 293 K (13.4%, 18.7%, and 29.8%, respectively, of those reporting a temperature). These data are provided in Data S2.
Redundancy Is Prevalent in Crystallization Conditions
From a chemical component point of view, there is enormous redundancy in the compositions of the different crystallization cocktails used for generating crystals that are used to generate structures in the PDB. Many crystallization components appear in multiple cocktails; in addition, many cocktails are associated with multiple PDB IDs. The top five chemical combinations are shown in Table 4. The 99,229 PDB IDs in the CDD are associated with 16,626 unique cocktails, in terms of chemical compound identity (not concentration). Of these 16,626 combinations of chemicals, 10,095 are found in only one PDB file, meaning that just over 10% of MX structures in the CDD have a unique set of chemical compounds that make up the crystallization cocktail. The remaining 89,134 PDB IDs in the CDD share the composition of crystallization cocktails with at least one other PDB ID in the CDD and make use of only 6,531 unique cocktails. Of these 6,531 unique chemical sets, 2,281 are associated with only one other PDB ID and a further 966 are associated with only two other PDB IDs. The median number of PDB IDs for a crystallization cocktail that is replicated elsewhere in the CDD is four. Note that these do not consider the concentration of the chemical components, only the chemical identity. Omitting concentration information is a limitation of the analyses with regard to examination of the entire crystallization parameter space. We have chosen to restrict our analyses to the domain of chemical component, as we found this to be the most efficient way to extract patterns given the extreme sparsity induced in the data when concentration is also included. Although some chemical sets are frequently found, the majority of cocktails appear quite sparsely within the PDB. In other words, more than 80% of the unique cocktails are replicated three times or less in the entire database and less than 20% of the unique cocktails account for crystallization components in the remaining 81,674 PDB IDs.
Table 4.
Most Commonly Occurring Chemical Combinations in the CDD
| Chemical Components | No. of PDB IDs |
|---|---|
| Ammonium acetate, sodium acetate, PEG 4000 | 814 |
| Ammonium sulfate | 808 |
| Bis-Tris, PEG 3350 | 726 |
| Ammonium sulfate, sodium acetate | 713 |
| Ammonium sulfate, sodium citrate | 599 |
The five most frequent combinations of the chemical compounds found in the crystallization cocktails in the CDD.
These are striking numbers. These results reflect the bias toward the available commercial crystallization cocktail screens,16 which is not surprising given that the commercial screens have been generated in large part in response to what has worked in the past.22,23 These results also highlight the extensive difficulties in generating predictive algorithms for crystallization conditions. It is a long-standing goal in the community to be able to predict potential crystallization conditions for macromolecules, given certain parameters of the macromolecule such as sequence and isoelectric point. The results presented here reveal the difficulties in developing these predictive models, as typically these types of predictive models require an extensive amount of well-annotated data. With such a combination of redundancy (of chemical compounds and cocktail combinations) and sparsity of the data, it is no wonder that the field has encountered problems in generating a predictive algorithm and generalizable guidelines for crystallization. Generating these types of predictions would benefit from the development of new machine-learning tools that can handle this type of sparsity.
Compositional Data Analyses Reveal the Relationship between PEGs and Secondary Structure Components
Since the first report of the successful use of PEGs in protein crystallization,24 PEGs have become one of the most common components of crystallization cocktails. In our dataset, 68,498 of the 99,229 PDBs contain at least one form of PEG (69.0% of all PDB files in the CDD), with 1,244 of those PDB IDs associated with more than one form of PEG (to a maximum of five different PEGs in the cocktail for two PDB IDs). The success of PEGs as precipitants in crystallization screening trials has led to widespread use of PEGs when screening for crystallization conditions as well as the development of PEG smears,25,26 which make use of mixtures of PEG polymers. A number of mixtures are commercially available; they are composed of different combinations of PEGs and are designed to efficiently screen the PEG chemical space for crystallization conditions. Often the PEG smears are optimized after the initial crystallization conditions are identified, although a variety of “PEG smears” occur in 86 PDB IDs that are not parsed (and therefore are not part of the CDD; see Table S2 for an example of a PEG smear that has not been parsed). Additionally, we note that although the use of multiple PEGs is not widespread in the final reported crystallization conditions, 1,244 of those PDB IDs in the CDD that have been parsed contain multiple PEG components, representing ∼1.2% of the total CDD data being examined here. Accounting for multiple PEGs in this analysis is challenging, as it makes pinpointing the contribution of the chemical components difficult, and for the purposes of the compositional data analyses we have not included PDB IDs containing multiple PEGs.
Adding PEGs to protein solutions affects the solubility of macromolecules through a variety of mechanisms.27,28 Although the underlying mechanisms by which PEGs promote crystallization are still not fully understood,13 it appears likely that when PEGs (inert, non-ionic, synthetic polymers) are included in solutions of crystallization cocktails, the volume of solvent accessible to the macromolecule is reduced, effectively increasing the net osmotic pressure and the interactions between macromolecules (Figure 2). One effect of PEG is therefore to decrease macromolecular solubility and increase the potential for crystallization. Solubility, including the effect of PEGs, has been modeled with the osmotic second virial coefficient B22; a slightly negative B22 has been correlated with crystallization success in these solubility trials, although B22 is affected by a number of complex interactions and factors.29,30 Here we investigate the question of whether the solubility effects of PEGs, which vary relative to average PEG molecular length and weight, affect the results on crystallization in proteins with different secondary structure content. In a comparison of nuclear magnetic resonance and crystal structure studies of the same macromolecules, Srivastava et al.31 observed that crystallization cocktail components influence protein secondary structure composition and hypothesized that PEGs are a driving factor for inducing secondary structure formation. Although the biophysical properties of proteins in solution have been explored with regard to impact of PEG MW,28 to the best of our knowledge no investigation has been performed with regard to protein secondary structure content. We are interested in determining whether secondary structure profiles for each PDB ID are differentially related to PEG MW for those cocktails containing one PEG compound that appears in the SSD (PEG-SSD). We performed regression analyses specific to modeling proportional outcomes to explore this question. We have as outcome data for each PDB ID multinomial counts of residues falling into each class of secondary structure (helix, sheet, and other) in the SSD. The counts of residues in each secondary structure class, bounded by total sequence length, give rise to compositional profiles of secondary structure for each PDB ID. A ternary plot illustrating a subset of the outcome data is presented in Figure 3. Each point in a ternary plot represents a three-way compositional proportion of helix, sheet, and other, summing to 100%, and is plotted at the barycentric coordinates on the ternary simplex.
Figure 2.

Schematic of PEG Impact on Crystallization Space
(A) PEGs are long-chain polymers where the number of ethylene oxide units typically varies but yields an average molecular weight for a specific PEG (for instance, PEG 4000 is a mixture of PEGs with an average MW of 4,000 g/mol). PEGs have many degrees of freedom of rotation.
(B) The effect of PEGs (represented by red ovoids) on two macromolecules (represented by two purple spheres) is to enhance the interaction between the molecules through an excluded volume effect. PEGs affect macromolecular solubility, increasing the potential for crystallization to occur.
Figure 3.

Ternary Plot of Secondary Structure Profiles for a Subset of PDB IDs
Each mark on the ternary plot represents the secondary structure composition of a single PDB ID in the subset of data. For illustration purposes, 400 points were randomly sampled and plotted from each group of PDB IDs with PEG 400 (black plus sign) and PEG 20000 (red cross). The plot shows 800 points of the 61,753 PDB IDs in the PEG-SSD dataset. In structures with a higher composition of helices (upper triangle, above the 60% mark for the H-ness axis on the left of the simplex), the points located in that sector are more likely to be black rather than red (associated with the low MW PEG instead of the high MW PEG). For the other dominated structures (triangle on the bottom left corner, above 60% mark on the bottom O-ness axis), the points are more likely to be red rather than black (associated with the high MW PEG instead of the low MW PEG). This gives a visual representation of the nature of the data used for modeling and underscores the model results.
We select a log-linear regression model of the Dirichlet multinomial (DMN) count profiles on PEG MW as continuous, non-zero predictor.32,33 The DMN distribution has been proposed as an extension to the multinomial for estimating proportions arising from count data in multiple categories, providing better handling of overdispersion by modeling the multinomial probability parameter as arising from a Dirichlet distribution.34 PEGs varied in MW from 200 to 35,000, with PEG 3350 and PEG 4000 appearing most often in cocktails (Table 3). The DMN regression model fit to the full PEG-SSD dataset of 61,753 PDB IDs (data and fitted values are shown in Figure 4) demonstrates a highly significant association (p ≪ 0.00001) of the secondary structure profiles with PEG MW. In our analysis, both β-sheet content and amount of other secondary structure have positive model coefficients and thus show increasing relative proportions with increasing PEG MW used in the cocktail. Most of this increase in relative proportion occurs in the other component. Helical content, however, has a negative coefficient; thus, higher PEG MW is associated with decreasing amounts of helical secondary structure content (see Figure S1 for a simplified plot of these trends). Regression on massive datasets can show high significance even for small effects. The sheer size of the dataset of PDB IDs being modeled has the potential to affect numerical stability of the algorithms as well as abetting significance determination for very small effects, so we present results from the analyses on the full data, as well as from models run on subsampled data, to understand the nature of the relation of PEG MW with the secondary structure profiles. Subsampling is a common strategy to provide inference, computational speed, and stability in modeling massive datasets.35,36 To assess whether the extreme significance we observe for the association of PEG MW with compositional profile of secondary structure is an artifact of the large sample size of our dataset, we also carried out subsampling. These results are stable under the subsampling analyses, with a high proportion of subsampled smaller datasets retaining significance. More than 84% of randomly sampled datasets of size n = 6,000 subsampled PDB IDs (less than 10% of the full PEG-SSD dataset) show significance at an alpha level of 0.05. Even when reducing the data size to n = 3,000 (less than 5% of the full PEG-SSD dataset), significance is retained in more than 57% of subsampled models. These results highlight that although the overall effects are subtle, they are consistently and strongly present in the data.
Figure 4.

Scatterplot of Secondary Structure Profiles by PEG MW and Model Fitted Values
Each PDB ID has a secondary structure profile, which contains the relative percentage of helix (pink), sheet (green), and other (blue). Each profile is plotted at the PEG MW in that PDB ID cocktail, which gives a scatterplot of the data (data points jittered). These data illustrate the non-uniform distribution of PEG MW and reveal that the number of PDB IDs associated with each PEG MW is highly variable. Overlaid on the scatterplot are the DMN regression model fitted values computed using model coefficients. These results show the relationship between each secondary structure class and PEG MW. For instance, the helix fitted regression line (pink) shows a clear decrease as PEG MW increases.
Investigating Protein Redundancy in the Chemical Condition Dataset
An investigation of PDB metadata examining the relationship between successful crystallization conditions and other parameters requires us to also consider the potential redundancy in the biomolecules that have been deposited in the PDB. There are a number of structures in the PDB from the same protein that have been solved by different groups, with different techniques, in different space groups, at different resolutions, and with different potential constructs, mutations, and small-molecule compounds. One consideration with regard to redundancy in the PDB is to define what it means to be a distinct deposited structure of a biomolecule.37 Mappings between those structures deposited in the PDB and protein amino acid sequences and annotations deposited in the UniProt Knowledgebase (UniProtKB) have been occurring since the Structure Integration with Function, Taxonomy and Sequences (SIFTS) resource was started in 2002.38 SIFTS enables annotations to be transferred between the protein sequence database (UniProtKB) and the PDB. We have used these mappings to query how prevalent protein redundancy is within the PDB from the reference point of UniProtKB accession numbers (ANs) and to examine whether protein redundancy biases result from investigation of crystallization conditions. For the 49,160 UniProtKB ANs with mappings to PDB IDs, 23,859 map to one distinct PDB ID (with a median of two distinct PDB IDs and an average of four distinct PDB IDs for every unique UniProtKB AN). Even within these mappings, however, it is difficult to define redundancy. In one example of a protein with multiple mappings between both databases, for instance, methyl-coenzyme M reductase from Methanotorris formicicus Mc-S-70 has three distinct UniProt AN subunits (HIKXL5, HIKXL9, and HIKXL6) and was crystallized in two different crystal forms, leading to two PDB IDs (5N28 and 5N2A).39 The protein sequences of the two PDB IDs are identical, but the crystallization conditions are different (and, indeed, share no components with one another except the presence of the protein itself). For the purposes of investigating crystallization chemical conditions, we would want to handle these two PDB IDs of identical biological molecules as two distinct entities.
To further investigate questions of redundancy with regard to the crystallization conditions within a subset of structures for the same macromolecule, we have examined hen egg white lysozyme (HEWL), which is a commonly used protein standard for crystallization studies.40 A search of the PDB for structures of HEWL deposited (UniProtKB AN P00698) yields 851 PDB entries (the maximum number of PDB IDs mapping to a single UniProtKB AN). Of these 851 HEWL structures, 846 (99.4%) are from studies using crystals or powder precipitate (821 MX structures, 10 electron crystallography structures, 2 neutron diffraction structures, and 13 powder diffraction structures). For the purposes of probing the subset of the chemical conditions successful for crystallizing lysozyme, we consider HEWL structures for which details have been successfully parsed, resulting in 471 PDB IDs in the CDD out of the 851 structures. For these 471 HEWL structures, there are 113 different crystallization cocktails. Seventy of the HEWL PDB IDs have distinct cocktails (found in only one PDB ID). Forty-three of the 113 different cocktails are duplicated at least once, with the most frequently occurring cocktail in this subset associated with 145 different HEWL PDB IDs. The HEWL subset of data is the most numerous of the UniProtKB ANs that have multiple PDB IDs, and even within this relatively large redundant set the most frequently occurring cocktail is only in 145 PDB IDs. This represents less than 0.15% of the entire CDD dataset. Notably, this summary of the subset of HEWL PDB IDs does not consider differences due to mutations or from different ligands bound (180 ligands are found in the 851 PDB IDs). Therefore, due to the extremely small amount of protein redundancy with regard to distinct chemical cocktails, we made the decision to not remove the small proportion of redundant proteins with the same crystallization conditions in our analyses. We note these details to alleviate any potential concern regarding bias introduced by redundancies in our analyses of the entire CDD and SSD dataset.
Discussion
In this work, we have developed and made available a searchable and updatable database of chemical components in crystallization cocktails mined from the PDB. As more structures are deposited in the PDB every year, more details about conditions that promote macromolecular crystal growth become available. Here, we present a snapshot of the data for crystallization conditions from December 9, 2019. The dataset presented contains 99,229 PDB IDs; the software for generating these data from the PDB is publicly available. As data science approaches expand and we step more firmly into the big data era, it is becoming more necessary to make data available in parseable formats; the dataset we provide here serves as a bridge to a usable version of the crystal growth metadata available in the PDB. Much of the PDB is extractable with queries of interest, but the challenge with regard to crystal growth conditions lies in the nature of it being a free text field in the PDB database (and all of the attendant difficulties that go with text mining). As we believe these data will serve as the basis for much further research, we plan to maintain this software and to update it as needed, as well as updating the parsed dataset links on a regular basis, further enabling analyses of the protein crystal growth metadata from the PDB. In this work, we have performed a formal analysis answering a complicated question using high-level modeling, but these data would be exploitable for many types of questions. These results can therefore be a significant resource for the protein structure community and will enable further analyses of the conditions that give rise to successful crystallization experiments.
Our analyses provide a window onto the simultaneous redundancy and extreme sparsity of crystallization cocktail information. It would be an encouraging step for depositions to the PDB to more fully incorporate naming conventions, controlled vocabularies, and standardized method descriptions in the crystal growth detail data field.41 Furthermore, we have specifically analyzed this dataset to examine the relationship of PEG MW with the secondary structure profiles of crystallized proteins with structures in the PDB. Our analyses demonstrate a strong association between PEG MW and secondary structure composition, with higher PEG MW associated with decreasing proportions of helical content for a given PDB ID. We speculate that the biophysical mechanism of this association might be related to a reduction in solvent-accessible surface area, which might drive stabilization of secondary structure; these correlations and potential mechanisms need to be more fully explored and serve as an intriguing direction for future research work. Given the bottleneck to the crystallization process of finding conditions to achieve macromolecular crystal growth, it is encouraging to observe relationships between features of the crystallization space and the structural outcomes.
We make these data and scripting available to enable further investigations. For instance, researchers interested in examining the previously successful crystallization conditions for a given set of PDB IDs (perhaps of the same protein from a different species, or a different construct of the same protein from the same species) could use the CDD data directly to probe the successful crystallization space. Similarly, if one were interested in how many times lysozyme crystallized when calcium is part of the crystallization cocktail, the CDD could be used to investigate that question very easily. Other parameters of interest to researchers can be extracted with the scripting provided. For example, if one were interested in examining the relationships between chemical crystallization space and MW of structures or the asymmetric unit cell, these additional parameters could be extracted from the PDB with the scripting provided. Finally, these data could be used as an example dataset for computational research in developing machine-learning methods and tools for extremely sparse data; we hope that the unique features of these data prompt computational tool building.
Finally, we note that our conclusions rely on the successful crystallization experiments that are reported in the PDB. What is missing from these analyses is an investigation of the chemical space that was explored: which cocktails generated crystals but were not further pursued, which chemical combinations were attempted, and which ones did not produce crystals? We believe that these data would contribute necessary information to more fully search the crystallization space for patterns. We hypothesize that to generate a predictive algorithm for successful crystallization conditions, information regarding the entire chemical search space and the outcomes (positive and negative) will need to be included. We hope, however, that the results presented here highlight the richness of information present in the PDB and emphasize the specific unique features that make it such a valuable resource.
Experimental Procedures
Resource Availability
Lead Contact
Sarah E.J. Bowman, PhD. sbowman@hwi.buffalo.edu.
Materials Availability
This study did not generate any new unique reagents or materials.
Data and Code Availability
Code and script annotations generated for extracting the data from the PDB are available at https://github.com/Hauptman-Woodward/crystallizationDatabase. The code on the github site will enable anyone to generate an updated database (as the PDB is updated on a weekly basis, the ability to update the data for future study is important). The github repository Structures directory also contains links to github large file storage to directly download the December 9, 2019 data scrape of the PDB (https://github.com/Hauptman-Woodward/crystallizationDatabase/tree/master/Structures). Additionally, we have provided flat file dataset versions of the CDD and the CDD-Kelvin data used in the analyses here (Data S1 and S2).
Generating the Crystallization Details Dataset
Data about crystallization conditions, sequences, and PDB IDs were downloaded with Python42 using the PDB API server for every structure that had information in that field. As of December 9, 2019, there were 158,367 PDB structure files in the PDB. Of these, 133,737 PDB IDs contain some crystallization condition information in the free text field. These data were standardized into a list of structures with associated information and stored as a list of Python objects serialized to binary file.
The detail parsing function extracted the raw plain English details in the free text field containing crystal growth details into a consistently formatted list of chemical compounds and concentrations, as well as temperature, when provided. A compound dictionary was manually generated to map the most common chemical compound names to a set of 312 unique standardized names, followed by manual checking of each compound name for potential synonyms. There is an application available to dynamically build and improve the dictionary to update the data with less frequently occurring chemical compounds as well as enabling updates to the database as the PDB is updated.
The source code, a thorough explanation of the data extraction, and a full description of all decisions made about the data from the PDB is publicly available at https://github.com/Hauptman-Woodward/crystallizationDatabase. Using the Python source code took less than 24 h to download these data as of December, 2019. The code can be used to update the database or extract a full set of new data from the PDB, and includes descriptions of how to add additional parameters of interest in the scripting. The data extracted December 9, 2019 are available in the Structures directory on the github site for large file storage (the raw data file is available as a.pkl file for download: structures.pkl, n = 133,737) as well as the parsed file in.pkl, .xml, and .csv formats (sensible_structures.pkl/.xml/.csv, n = 99,229); links are provided in Data Code and Availability. As the data files are updated, we will update the links to these files. Data in the sensible_structures.xml file were also extracted and placed into a flat data file (Data S1), with a matrix containing PDB IDs (row dimension) and 312 unique chemical compound names (column dimension) with a 0/1 indicator of absence/presence of a given compound in each PDB ID row.
Generating the Secondary Structure Dataset
Secondary structure data were downloaded from the PDB (December 9, 2019) as a ss.txt file, a FASTA-formatted file with amino acid sequences and secondary structure information generated using DSSP.43 A separate dataset was generated from this file with the total length of the coordinates of each PDB ID sequence, the total number of residues defined as “H,” “G,” or “I” (helix), the total count of residues defined as “B” or “E” (sheet), and added across chains (if multiple chains were in the PDB file). All residues not defined as helix or sheet are considered other for the purposes of these analyses. When residues with no secondary structure prediction from DSSP occur within the structural sequence, we have assigned those residues to other. Secondary structure data were checked and then merged with the 99,229 PDB IDs in the CDD for which we have chemical information from the free text crystal growth detail field, yielding an SSD of 97,782 for further analyses of the secondary structure information.
Statistical and Computational Methods
We extracted basic summary statistics regarding temperature, chemical component frequency, and chemical combinations in the CDD. For more detailed investigation, we probed the relationship between secondary structure composition and PEGs, as one of the primary components of crystallization cocktails, using Dirichlet Multinomial regression models with PEG MW as predictor variable. The data in the SSD provide a prediction of secondary structural identity on a per-residue basis. We used the number of residues in each secondary structure class (helix, sheet, and other), bounded by the total sequence length for each PDB ID, as multinomial outcome variable in the models. Parameter estimation for regression models was carried out using the MGLM software package version 0.2.0,44 implemented in the statistical computing environment R version 3.5.1.45 MGLM provides an efficient computational algorithm that increases stability of estimates and provides inference via likelihood ratio and Wald tests of model parameters. For all regression analyses, the constraints of the Dirichlet distribution required removing any observations that contained zero residue counts in any secondary structure class, resulting in 89,653 PDB IDs. Of these, 61,753 had cocktails containing one PEG compound available for use in the regression analysis (PEG-SSD). To examine model stability, we also performed regression on subsets of the full 61,753 PEG-SSD data. Subsampling was carried out by randomly sampling the available data with replacement, in batches of 1,000 samples of sizes n = 3,000 each (approximately 5% of the total PEG-SSD data) or n = 6,000 each (approximately 10% of the total PEG-SSD data), then carrying out model fits for each sampled dataset in the batch. Model parameter estimates from each model run on a subsampled dataset were extracted and stored, as well as likelihood values and significance levels. Ternary plots of the secondary structure profiles were created using the Ternary package version 1.0.2 in R.46
Acknowledgments
We thank Dr. William Bauer for insightful conversations about crystallization conditions for HEWL. We acknowledge support from the U.S. National Institutes of Health R24-GM124135 (M.L.L. and S.E.J.B.), the Seymour H. Knox Foundation (S.E.J.B.), and the Hauptman-Woodward Medical Research Institute Summer Internship Program (M.F.D.) for financial support.
Author Contributions
Conceptualization, M.L.L. and S.E.J.B.; Data Curation, M.L.L., M.F.D., and S.E.J.B.; Software, M.L.L. and M.F.D.; Formal Analysis, M.L.L.; Writing – Original Draft, M.L.L. and S.E.J.B.; Writing – Review & Editing, M.L.L. and S.E.J.B.
Declaration of Interests
The authors declare no competing interests.
Published: April 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.patter.2020.100024.
Supplemental Information
File is formatted with a header line and a column of row names (PDB IDs). Dimension of the flat datafile is 99,229 rows by 312 columns (without header or row names).
File is formatted with a header line and a column of row names (PDB IDs). Dimension of the flat datafile is 99,229 rows by 1 column (without header or row names).
References
- 1.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.wwPDB Consortium Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019;47:D520–D528. doi: 10.1093/nar/gky949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.wwPDB Foundation Download statistics. https://www.wwpdb.org/stats/download Accessed 12/9/2019.
- 4.Berman H.M., Bourne P.E., Westbrook J., Zardecki C. The Protein Data Bank. In: Chasman D.I., editor. Protein Structure. CRC Press; 2003. pp. 394–410. [Google Scholar]
- 5.Westbrook J.D., Burley S.K. How structural biologists and the protein data bank contributed to recent FDA new drug approvals. Structure. 2018;27:211–217. doi: 10.1016/j.str.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McPherson A. Vol. 586. Cold Spring Harbor Laboratory Press; 1999. (Crystallization of Biological Macromolecules). [Google Scholar]
- 7.Luft J.R., Collins R.J., Fehrman N.A., Lauricella A.M., Veatch C.K., DeTitta G.T. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J. Struct. Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 8.Chayen N.E., Saridakis E. Protein crystallization: from purified protein to diffraction-quality crystal. Nat. Methods. 2008;5:147. doi: 10.1038/nmeth.f.203. [DOI] [PubMed] [Google Scholar]
- 9.Bruno A.E., Charbonneau P., Newman J., Snell E.H., So D.R., Vanhoucke V., Watkins C.J., Williams S., Wilson J. Classification of crystallization outcomes using deep convolutional neural networks. PLoS One. 2018;13:e0198883. doi: 10.1371/journal.pone.0198883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McPherson A., DeLucas L.J. Microgravity protein crystallization. NPJ Micrograv. 2015;1:15010. doi: 10.1038/npjmgrav.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garman E.F. Developments in x-ray crystallographic structure determination of biological macromolecules. Science. 2014;343:1102–1108. doi: 10.1126/science.1247829. [DOI] [PubMed] [Google Scholar]
- 12.Peat T.S., Christopher J.A., Newman J. Tapping the Protein Data Bank for crystallization information. Acta Crystallogr. D. Biol. Crystallogr. 2005;61:1662–1669. doi: 10.1107/S0907444905033202. [DOI] [PubMed] [Google Scholar]
- 13.Kirkwood J., Hargreaves D., O'Keefe S., Wilson J. Analysis of crystallization data in the Protein Data Bank. Acta Crystallogr. F Struct. Biol. Commun. 2015;71:1228–1234. doi: 10.1107/S2053230X15014892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abrahams G.J., Newman J. BLASTing away preconceptions in crystallization trials. Acta Crystallogr. F Struct. Biol. Commun. 2019;75:184–192. doi: 10.1107/S2053230X19000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rupp B., Wang J. Predictive models for protein crystallization. Methods. 2004;34:390–407. doi: 10.1016/j.ymeth.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 16.Fazio V.J., Peat T.S., Newman J. A drunken search in crystallization space. Acta Crystallogr. F Struct. Biol. Commun. 2014;70:1303–1311. doi: 10.1107/S2053230X1401841X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newstead S., Ferrandon S., Iwata S. Rationalizing α-helical membrane protein crystallization. Protein Sci. 2008;17:466–472. doi: 10.1110/ps.073263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Du M., Lamoure C., Muller B., Bulgakov O., Lajeunesse E., Menez A., Boulain J.-C. Artificial evolution of an enzyme active site: structural studies of three highly active mutants of Escherichia coli alkaline phosphatase. J. Mol. Biol. 2002;316:941–953. doi: 10.1006/jmbi.2001.5384. [DOI] [PubMed] [Google Scholar]
- 19.Aranda R., Levin E.J., Schotte F., Anfinrud P.A., Phillips G.N. Time-dependent atomic coordinates for the dissociation of carbon monoxide from myoglobin. Acta Crystallogr. D Biol. Crystallogr. 2006;62:776–783. doi: 10.1107/S0907444906017318. [DOI] [PubMed] [Google Scholar]
- 20.Kirchdoerfer R.N., Garner A.L., Flack C.E., Mee J.M., Horswill A.R., Janda K.D., Kaufmann G.F., Wilson I.A. Structural basis for ligand recognition and discrimination of a quorum-quenching antibody. J. Biol. Chem. 2011;286:17351–17358. doi: 10.1074/jbc.M111.231258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaltenbach M., Burke J.R., Dindo M., Pabis A., Munsberg F.S., Rabin A., Kamerlin S.C., Noel J.P., Tawfik D.S. Evolution of chalcone isomerase from a noncatalytic ancestor. Nat. Chem. Biol. 2018;14:548. doi: 10.1038/s41589-018-0042-3. [DOI] [PubMed] [Google Scholar]
- 22.Jancarik J., Kim S.-H. Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl. Crystallogr. 1991;24:409–411. [Google Scholar]
- 23.Luft J.R., Newman J., Snell E.H. Crystallization screening: the influence of history on current practice. Acta Crystallogr. F Struct. Biol. Commun. 2014;70:835–853. doi: 10.1107/S2053230X1401262X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPherson A. Crystallization of proteins from polyethylene glycol. J. Biol. Chem. 1976;251:6300–6303. [PubMed] [Google Scholar]
- 25.Newman J., Egan D., Walter T.S., Meged R., Berry I., Ben Jelloul M., Sussman J.L., Stuart D.I., Perrakis A. Towards rationalization of crystallization screening for small-to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1426–1431. doi: 10.1107/S0907444905024984. [DOI] [PubMed] [Google Scholar]
- 26.Chaikuad A., Knapp S., von Delft F. Defined PEG smears as an alternative approach to enhance the search for crystallization conditions and crystal-quality improvement in reduced screens. Acta Crystallogr. D Biol. Crystallogr. 2015;71:1627–1639. doi: 10.1107/S1399004715007968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finet S., Vivarès D., Bonneté F., Tardieu A. Controlling biomolecular crystallization by understanding the distinct effects of PEGs and salts on solubility. Methods Enzymol. 2003;368:105–129. doi: 10.1016/S0076-6879(03)68007-9. [DOI] [PubMed] [Google Scholar]
- 28.Atha D.H., Ingham K.C. Mechanism of precipitation of proteins by polyethylene glycols. Analysis in terms of excluded volume. J. Biol. Chem. 1981;256:12108–12117. [PubMed] [Google Scholar]
- 29.Neal B., Asthagiri D., Velev O., Lenhoff A., Kaler E. Why is the osmotic second virial coefficient related to protein crystallization? J. Cryst. Growth. 1999;196:377–387. [Google Scholar]
- 30.Liu J., Yin D.-C., Guo Y.-Z., Wang X.-K., Xie S.-X., Lu Q.-Q., Liu Y.-M. Selecting temperature for protein crystallization screens using the temperature dependence of the second virial coefficient. PLoS One. 2011;6:e17950. doi: 10.1371/journal.pone.0017950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava S.K., Gayathri S., Manjasetty B.A., Gopal B. Analysis of conformational variation in macromolecular structural models. PLoS One. 2012;7:e39993. doi: 10.1371/journal.pone.0039993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Zhou H., Zhou J., Sun W. Regression models for multivariate count data. J. Comput. Graph. Stat. 2017;26:1–13. doi: 10.1080/10618600.2016.1154063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J., Li H. Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis. Ann. Appl. Stat. 2013;7:418–442. doi: 10.1214/12-AOAS592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosimann J.E. On the compound multinomial distribution, the multivariate β-distribution, and correlations among proportions. Biometrika. 1962;49:65–82. [Google Scholar]
- 35.Li R., Lin D.K., Li B. Statistical inference in massive data sets. Appl. Stoch. Model. Bus. Ind. 2013;29:399–409. [Google Scholar]
- 36.Wang H., Zhu R., Ma P. Optimal subsampling for large sample logistic regression. J. Am. Stat. Assoc. 2018;113:829–844. doi: 10.1080/01621459.2017.1292914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burra P.V., Zhang Y., Godzik A., Stec B. Global distribution of conformational states derived from redundant models in the PDB points to non-uniqueness of the protein structure. Proc. Natl. Acad. Sci. U S A. 2009;106:10505–10510. doi: 10.1073/pnas.0812152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dana J.M., Gutmanas A., Tyagi N., Qi G., O’Donovan C., Martin M., Velankar S. SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2018;47:D482–D489. doi: 10.1093/nar/gky1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner T., Wegner C.-E., Kahnt J., Ermler U., Shima S. Phylogenetic and structural comparisons of the three types of methyl coenzyme M reductase from Methanococcales and Methanobacteriales. J. Bacteriol. 2017;199 doi: 10.1128/JB.00197-17. e00197–00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strynadka N., James M. Lysozyme: a model enzyme in protein crystallography. EXS. 1996;75:185–222. doi: 10.1007/978-3-0348-9225-4_11. [DOI] [PubMed] [Google Scholar]
- 41.Newman J., Peat T.S., Savage G.P. What’s in a name? Moving towards a limited vocabulary for macromolecular crystallisation. Aust. J. Chem. 2014;67:1813–1817. [Google Scholar]
- 42.Python Core Team . Python Software Foundation; 2019. Python: A Dynamic, Open Source Programming Language.https://www.python.org [Google Scholar]
- 43.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 44.Kim J., Zhang Y., Day J., Zhou H. MGLM: an R package for multivariate categorical data analysis. R. J. 2018;10:73–90. doi: 10.32614/rj-2018-015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R Core Team . R Foundation for Statistical Computing; 2018. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 46.Smith M. 2017. Ternary: An R Package for Creating Ternary Plots. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File is formatted with a header line and a column of row names (PDB IDs). Dimension of the flat datafile is 99,229 rows by 312 columns (without header or row names).
File is formatted with a header line and a column of row names (PDB IDs). Dimension of the flat datafile is 99,229 rows by 1 column (without header or row names).
Data Availability Statement
Code and script annotations generated for extracting the data from the PDB are available at https://github.com/Hauptman-Woodward/crystallizationDatabase. The code on the github site will enable anyone to generate an updated database (as the PDB is updated on a weekly basis, the ability to update the data for future study is important). The github repository Structures directory also contains links to github large file storage to directly download the December 9, 2019 data scrape of the PDB (https://github.com/Hauptman-Woodward/crystallizationDatabase/tree/master/Structures). Additionally, we have provided flat file dataset versions of the CDD and the CDD-Kelvin data used in the analyses here (Data S1 and S2).