

Abstract
Liquid-liquid phase separation (LLPS) underlies the physiological assembly of many membrane-less organelles throughout the cell. However, dysregulation of LLPS may mediate the formation of pathological aggregates associated with neurodegenerative diseases. Here, we present complementary experimental approaches to study protein aggregation within and outside the context of LLPS in order to ascertain the impact of LLPS on aggregation kinetics. Techniques described include imaging-based approaches [fluorescence microscopy, atomic force microscopy (AFM), fluorescence recovery after photobleaching (FRAP)] as well as plate reader assays [Thioflavin-T (ThT) fluorescence intensity and turbidity]. Data and conclusions utilizing these approaches were recently reported for the low complexity domain (LCD) of the transactive response DNA binding protein of 43 kDa (TDP-43).
Keywords: Liquid-liquid phase separation, Amyloid, Protein aggregation, TDP-43, Thioflavin-T, Turbidity, Fluorescence recovery after photobleaching, Atomic force microscopy, Fluorescence microscopy
Background
Many proteins that aggregate in neurodegenerative disease also undergo LLPS to form dynamic, reversible liquid-like droplets ( Molliex et al., 2015 ; Conicella et al., 2016 ; Hofweber et al., 2018 ). LLPS is commonly mediated by the presence of intrinsically disordered regions (IDRs) that enable transient, weak, multivalent interactions ( Lin et al., 2017 ). These IDRs are often composed of only a few amino acid types and may be arranged in short, repetitive motifs. The low complexity domain (LCD) of TDP-43 forms pathologically-associated amyloid aggregates and was recently shown to phase separate ( Conicella et al., 2016 ; Lim et al., 2016 ). However, any relationship between these two processes remained unclear. We recently demonstrated that amyloid formation by the LCD can occur within the context of LLPS ( Babinchak et al., 2019 ). In this work, we implemented a combinatorial experimental approach to assess the kinetic role of LLPS in the formation of amyloids that can be applied to other aggregation-prone proteins that phase separate (Figure 1). This approach includes four major components: (1) imaging of amyloid and droplet-like species using fluorescence microscopy and atomic force microscopy (AFM); (2) assessment of LLPS propensity using turbidity measurements; (3) monitoring amyloid aggregation via Thioflavin-T (ThT) fluorescence intensity measurements; and (4) assessment of maturation of liquid-like droplets using fluorescence recovery after photobleaching (FRAP). Analysis of these results can provide quantitative insights for comparing aggregation under LLPS conditions and in the absence of LLPS.
Figure 1. Methods for studying the formation of TDP-43 LCD amyloid aggregates from liquid-like droplets.
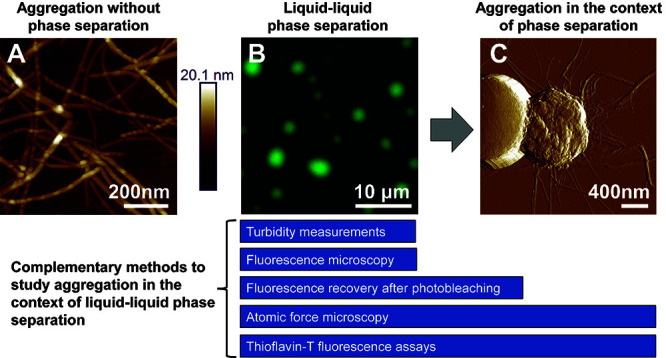
A. Atomic force microscopy (AFM) imaging of amyloid aggregates formed by the TDP-43 LCD in the absence of LLPS, which often appear as individual, non-clumped fibers. B. Fluorescence microscopy of liquid-like droplets formed by the TDP-43 LCD. C. With time, fibrils emanating from within mature droplets can be observed on AFM imaging and fibrils often appear clumped or intertwined. Complementary methods for studying aggregation within the context of liquid-liquid phase separation are presented. Turbidity measurements and fluorescence microscopy are optimal methods for studying liquid-like droplets, while ThT assays can be used to study amyloid formation. The transition from liquid-like droplets to amyloids can be captured using a combination of FRAP, AFM, and ThT assays. All images were originally published in J. Biol. Chem. ( Babinchak et al., 2019 ).
Materials and Reagents
Pipette tips
FluoroDish Cell Culture Dish (35 mm dish, 23 mm well, glass thickness: 0.17 mm) (World Precision Instruments, catalog number: FD35-100)
Amicon® Ultra–0.5 ml Centrifugal Filters (Ultracel–100,000 nominal molecular weight limit, NMWL) (Millipore, catalog number: UFC510096)
ScotchTM Tape
Adhesive tabs (Ted Pella, Inc., catalog number: 16079)
AFM specimen discs, 15 mm diameter (Ted Pella, Inc., catalog number: 16218)
Mica discs, 9.9 mm diameter (Ted Pella, Inc., catalog number: 50)
Corning® Assay Plate, 96-well half area (non-treated, no lid; black with clear flat bottom; polystyrene) (Corning, catalog number: 3880)
Sealing Tape, Advanced Polyolefin (Certified DNase-, RNase-, and Nucleic Acid-free) (Thermo ScientificTM, catalog number: 235307)
Millex®–GV filter unit, low protein binding Durapore® (PVDF) membrane, 0.22 μm (Millipore, catalog number: SLGV033RS)
Zeba spin desalting column (2 ml; 7 kDa molecular weight cut off) (Thermo ScientificTM, catalog number: 89890)
Microscope Cover Glass 12CIR-1 (Thermo ScientificTM, catalog number: 1254580)
Alexa Fluor 488TM C5 Maleimide (InvitrogenTM, catalog number: A10254)
Potassium phosphate monobasic, KH2PO4 (Fisher Scientific, catalog number: 7778-77-0)
Potassium phosphate dibasic, K2HPO4 (Fisher Scientific, catalog number: 7758-11-4)
Sodium acetate trihydrate, CH3COONa 3H2O (Fisher Scientific, catalog number: 127-09-3)
Acetic acid (glacial), CH3COOH (Fisher Scientific, catalog number: A38-212)
Urea (Fisher Scientific, catalog number: BP 169-500)
NaCl, 5 M solution (Sigma, catalog number: S5150-1L)
Imidazole (Fisher Scientific, catalog number: O3196)
Thioflavin-T (Sigma-Aldrich, catalog number: T-3516)
Tris (2-carboxyethyl) phosphine hydrochloride, TCEP (GoldBio, catalog number: TCEP25)
Trizma Base, Tris (Sigma-Aldrich, catalog number: T1503)
Hydrochloric acid, HCl (Fisher Scientific, catalog number: A144)
Type F immersion liquid (Leica Microsystems, catalog number: 11513859)
Silicon tip on nitride lever probe (Bruker, SCANASYST-AIR)
Milli-Q H2O (from Milli-Q Reference Water Purification System, MilliPore Sigma)
100 mM Phosphate and Acetate Stock Buffers (10x) (see Recipes)
Thioflavin-T stock buffer (see Recipes)
Labeling buffer (see Recipes)
Equilibration buffer (see Recipes)
Equipment
Plate Reader (Tecan Life Sciences, model: Infinite M1000)
Pipette
EppendorfTM Microcentrifuge (Fisher Scientific, model: 5424)
Computer
Vortex-Genie 2 Lab Mixer (Scientific Industries, Inc., model: G-560)
pH/mV meter (Fisher Scientific, model: Accumet AB150)
NanoDropTM Spectrophotometer (Thermo ScientificTM, model: 2000)
Leica TCS SP8 confocal microscope (Leica Microsystems)
All-in-one fluorescence microscope (Keyence, model: BZ-X710)
100x PlanApoλ objective (NA 1.45, oil immersion) (Nikon, catalog number: MRD31905)
GFP filter cube (Keyence, catalog number: OP-87763)
Multimode Atomic Force Microscope with Nanoscope V (Bruker)
Incubator (Fisher Scientific, model: 516D)
Software
i-controlTM 1.10 (for infinite reader) software (Tecan Life Sciences)
Leica Application Suite X (LAS X, Leica Microsystems)
Nanoscope 9.1 (Bruker)
Nanoscope Analysis 1.5 (Bruker)
BZ-X Viewer (Keyence)
BZ-X Analyzer (Keyence)
Procedure
Note (Introduction to Procedure A): Wild type and single-cysteine mutant proteins were expressed and purified as described in detail previously (Babinchak et al., 2019). In brief, this involves a two-step purification using nickel nitrilotriacetic acid affinity chromatography under denaturing conditions followed by C4 reverse phase HPLC. Purified protein is flash-frozen and lyophilized. Procedure A is designed to pick up after lyophilization and to remove any potential aggregates that may have formed. Procedure A is crucial to ensuring that experiments are formed with highly soluble, non-aggregated protein. In addition to the TDP-43 LCD, this protocol may be used to study other proteins that may phase separate and aggregate; however, users may need to amend Procedures A and D based on the specific purification protocol for their protein of interest.
-
A
Protein preparation
Suspend lyophilized protein in ~200-250 μl of Milli-Q H2O. Briefly vortex.
Pass soluble protein solution through 0.5 ml 100 kDa NMWL Amicon® centrifugal filter via centrifugation at > 21,000 × g for 7 min.
Collect “flow-through solution” that passes through the filter.
Measure “flow-through solution” absorbance at 280 nm (A280) to determine stock molar concentration using appropriate extinction coefficient [17990 M-1·cm-1 for LCD protein used in ( Babinchak et al., 2019 )] and the Beer-Lambert law.
Note (Introduction to Procedure B): Because induction of phase separation results in the scattering of light, the process can be quantified using turbidity measurements. These types of measurements provide a method for comparing a variety of conditions that promote varying degrees of phase separation. For the TDP-43 LCD, LLPS is regulated by both NaCl concentration and pH. Therefore, recipes to prepare solutions with different buffer conditions are provided (see Recipes).
-
B
Induction of phase separation and turbidity measurements
Calculations and dilutions from stocks in order to study phase separation by altering pH and NaCl concentration: Using a known protein stock concentration (see Procedure A), calculate the volume of 100 mM buffer (Recipe 1), 5 M NaCl, Milli-Q H2O, and protein stock solution necessary to create a final solution containing 10 mM buffer, the desired salt concentration (0-300 mM NaCl), and the desired protein concentration. To allow for technical replicates, we recommend a total volume of 300 μl that can be evenly distributed into 3 wells of the 96-well plate.
Experimental Solutions: Mix by pipetting reagents in the following order, mixing thoroughly at each step: (1) Milli-Q H2O, (2) buffer, (3) additional reagents (e.g., NaCl, polyethylene glycol, 1,6-hexanediol). Protein will be added last and just before taking measurements.
Blank Solutions: As in Steps B1-B2, calculate and prepare “blank” solutions using the same conditions but in the absence of protein (which will be replaced by water). Three technical replicates may also be used.
Add protein to experimental solutions. Mix thoroughly. Immediately add solutions to 96-well plate and measure absorbance at 600 nm (A600).
For experiments at 37 °C, prepare solutions as described in Steps B1-B3. Pre-heat plate reader to 37 °C. Pre-heat all solutions, remaining stocks, and materials (i.e., 96-well plate, pipette tips) using a 37 °C incubator for 15 min to minimize cooling during sample preparation. Once warmed, add protein to reaction buffer and measure A600. To minimize additional cooling effects during sample preparation, incubate the plate (with samples) for 5-10 min in the pre-heated plate reader before measuring A600.
Note (Introduction to Procedure C): Thioflavin-T has been used as a fluorescent probe to monitor amyloid formation for a number of proteins as well as to detect amyloids in histopathology samples from patient brains (Nielsen et al., 2001). Upon binding to amyloids, the intensity of fluorescence emission increases substantially, allowing for Thioflavin-T to be used to monitor the in vitro polymerization reaction. We found that the molecule itself does not appear to alter conditions under which phase separation is observed and its fluorescence intensity is not greatly altered by the presence of droplets, allowing us to assess amyloid formation within the context of phase separation. We typically monitor polymerization for 48-72 h, though the time to reaction completion is likely to be different for each protein.
-
C
Thioflavin-T (ThT) measurements to monitor aggregation
Pre-set plate reader temperature to 25 °C or 37 °C, depending on the temperature at which the user would like to study aggregation. This will be kept constant throughout the entirety of the experiment. Set up experimental parameters for Thioflavin-T aggregation kinetics in Tecan i-control software (Table 1).
Prepare solutions as described in Steps B1-B3 with the addition of 15 μM ThT (from 1.5 mM ThT stock; Recipe 2) that is added just before protein. Mix thoroughly.
Immediately add solutions to a 96-well plate and cover with advanced polyolefin sealing tape to prevent evaporation during time course of experiment.
Initiate protocol to measure fluorescence intensity every 10 min at a constant temperature. Tecan i-control 1.10 (for infinite reader) software will populate in real-time a Microsoft Excel file containing raw fluorescence intensity data as measurements are taken.
Table 1. Experimental parameters for Thioflavin-T aggregation kinetics.
| Measurement Parameter | Instrument Setting |
| Kinetic Interval | 10 min |
| Intermittent Agitation | None |
| Excitation Wavelength (Bandwidth) | 440 nm (5 nm) |
| Emission Wavelength (Bandwidth) | 485 nm (5 nm) |
| Gain | 100 |
| Number of Flashes | 50 |
| Integration time | 20 μs |
| Lag Time | 0 μs |
| Settle time | 0 ms |
| Z-Position | 23000 |
Note (Introduction to Procedure D): The use of a fluorescently-labeled molecule is critical to performing fluorescence microscopy. The exact choice of dye (i.e., Alexa FluorTM 488, Alexa FluorTM 594, etc.) is up to the user.
-
D
Fluorescent labeling of single-cysteine mutant protein
Perform Alexa FluorTM 488 (AF488) labeling using Alexa Fluor 488TM C5 maleimide according to manufacturer’s protocol in the presence of Labeling Buffer (Recipe 3).
Remove excess fluorescent label by passing protein through 7 kDa molecular weight cut-off (MWCO) Zeba spin desalting column (2 ml) equilibrated with Equilibration Buffer (Recipe 4). Follow manufacturer’s protocol guidelines for centrifugation and volumetric parameters.
Determine the concentration of AF488-labeled protein by measuring A280 and correcting for contribution of AF488 to absorbance (instructions provided in the manufacturer’s online manuals).
Note (Introduction to Procedure E): When performing microscopy imaging of droplets, we recommend the technique of “doping” unlabeled protein with a small amount of fluorescently-labeled protein, which can help to minimize costs by saving labeled protein. Which microscope is used depends on user preference and access provided by the laboratory; however, it is crucial to use a microscope with the capability of exciting and detecting emission of the user’s choice fluorophore (i.e., a GFP filter cube for AF488-labeled protein).
-
E
Imaging technique: Fluorescence microscopy of droplets
Prepare sample utilizing unlabeled protein from Procedure A and labeled protein from Procedure D with a ratio of 1:200-1:500 AF488-labeled to unlabeled protein. Steps in Procedure B should be used to design conditions. Pre-mix labeled and unlabeled protein before adding to remaining solution components. Mix thoroughly.
Add ~15 μl of protein solution to the surface of FluoroDish and cover with coverslip.
Image sample on BZ-X710 via bright field and with GFP filter cube. Use of 100x oil immersion objective can provide optimal resolution of droplets with diameters ranging from 1-5 μm on the BZ-X710, though other microscopes may provide adequate imaging at lower magnification. Imaging can be performed at the surface of the dish where droplets will sediment or in the solution above the dish surface. Imaging should not be performed near the edges of the coverslip, as evaporation of solution may cause artifacts. Droplets may be imaged immediately or over a long duration of time (for example, through use of time-lapse imaging). Typically, droplets will undergo growth, sedimentation, and fusion to eventually reach an equilibrium. Therefore, the timing of imaging should be consistent across conditions if a comparison is being performed.
Note (Introduction to Procedure F): Atomic force microscopy can provide more detailed insight into morphological differences between phase-separated and aggregated species. This is most clearly evident when fibrillar species can be identified within or surrounding droplets–a level of detail that is difficult to observe using fluorescence microscopy but is more easily seen with AFM. Importantly, AFM can therefore distinguish between droplets with and without fibrils; however, an inherent shortcoming should be noted wherein the provided protocol images species that are dried on mica. Whether artifacts may be introduced during this drying process is unclear. Therefore, it is helpful to perform both complementary atomic force microscopy and fluorescent microscopy imaging. Additionally, liquid-like character of droplets cannot be assessed by AFM in this manner (see Procedure G for more information on how to do this).
-
F
Imaging technique: Atomic force microscopy of aggregates and droplets
Prepare fibrils or droplets as described in Procedures A-C.
Attach a single mica disc to the surface of an AFM specimen disc using an adhesive tab. Use ScotchTM tape to strip mica surface (adhere tape to the surface of mica and press down firmly. Smoothly remove the tape from the surface of mica). Strip mica until the surface is smooth.
Add ~10 μl of sample to mica surface. Cover sample to prevent dust particles from entering. Incubate sample at ambient temperature for 5 min.
Rinse mica surface with 100 μl of Milli-Q H2O 4-5 times.
Dry mica surface for ~1 min using a gentle stream of nitrogen gas.
-
Image sample on multimode atomic force microscope using ScanAsyst in Air mode (with SCANASYST-AIR silicon tip on nitride lever).
Recommended parameters for imaging: 0.977-1.95 Hz scan rate; 512 samples/line; feedback gain: 5.289; 0.1 V peak force set point; ScanAsyst autocontrol: ON.
Note (Introduction to Procedure G): While fluorescence microscopy and AFM imaging can help to distinguish fibrillar from droplet-like species, maturation or aggregation within liquid droplets might occur before fibrils can be observed via imaging. Therefore, fluorescence recovery after photobleaching (FRAP) is ideal for assessing the dynamicity of phase-separated protein during this intermediary time period. This procedure provides a general approach to photobleaching liquid droplets to ascertain their dynamic nature. A number of works have utilized this approach but include slight variations in methodology and analysis. We recommend the following reference for users who might be interested in learning more (Taylor et al., 2019).
-
G
Fluorescence recovery after photobleaching (FRAP) to monitor droplet maturation
Turn on Leica HyVolution SP8 confocal microscope according to manufacturer’s guidelines. Allow 488 nm argon laser to warm up ~30 min.
Prepare a sample in a solution condition under which phase separation is observed. Liquid droplets are necessary to perform a FRAP experiment. Add sample to FluoroDish as described in Steps E1-E2.
Initial sample imaging and post-bleach frames should be collected using a laser intensity that does not bleach the sample. The absence of bleaching can be ascertained by recording 3-5 pre-bleach frames at the beginning of each FRAP experiment.
Identify droplets that sediment on the bottom of the microscope dish as these will move minimally and allow for ease in data analysis.
Using a consistent region of interest (ROI) diameter, bleach sample (2.4 mW or similar laser intensity) for 10 consecutive frames. Minimize time in between each bleach frame. Changes in ROI size may influence observed recovery and therefore should be kept consistent across samples.
Monitor recovery over 300 s (1 frame per 5 s) or over sufficient time to observe recovery.
Data analysis
Note (Introduction to Data Analysis): Data analysis is designed to emphasize identification of amyloids primarily through use of atomic force microscopy and of liquid droplets using fluorescence microscopy. FRAP experiments help to ascertain whether droplets are more dynamic (i.e., liquid-like) or less dynamic (i.e., potential aggregates). Quantitative assessment of amyloid formation and the extent of phase separation is performed using data from Thioflavin-T and turbidity assays, respectively.
-
Imaging Analysis:
Atomic Force Microscopy
Open AFM image in Nanoscope Analysis 1.5. A ‘height’ and ‘peak force error’ image should be present.
For the ‘height’ image, apply image flattening filter (Filter → Flatten) using the following recommended parameters: Flatten order: 1st; Flatten Z thresholding direction: no thresholding; Use histogram: OFF. Example imaging can be seen in Figure 1A.
In some cases, the height of dried droplets may be so great that a flattened ‘height’ image may not be best for visualization. In this case, ‘peak force error’ images can also provide information on sample morphology (Figure 1C).
Export image using Analysis → Journal Quality Export (recommended parameters: export type: TIFF; dots per inch: 300 or higher)
Fluorescence Microscopy
Open images in BZ-X analyzer software. All imaging analysis will be performed in this software.
For bright field images, a haze reduction filter may be used to facilitate better visualization of droplets. Recommended haze reduction settings: Blur size, 8; brightness, 3.9; reduction rate, 0.75
For fluorescence images, a black background correction may be used to allow greater clarity of droplets (Figure 1B).
-
Turbidity and Thioflavin-T data analysis using Microsoft Excel
General Analysis (Turbidity measurements and ThT traces)
In Microsoft Excel, average each set of technical replicates separately. For a specific condition, subtract the average value of blanks from the average value of the respective experimental traces to calculate a corrected value (A600 for turbidity measurements and fluorescence intensity values for ThT traces).
Calculate standard deviation from raw values.
To assess the effect of NaCl concentration on LLPS propensity, plot the average turbidity as a function of NaCl concentration (this can be applied to other experimental parameters, such as temperature).
To evaluate aggregation from Thioflavin-T measurements, for each condition plot the average fluorescence intensity as a function of time. Discrete lag and elongation phases should be readily apparent if aggregation has occurred (Figure 2).
Lag Time Analysis (Thioflavin-T Measurements)
Lag times should be calculated using non-corrected and non-averaged fluorescence intensity traces (i.e., individual raw ThT traces). Plot each trace as a function of time.
Using Microsoft Excel, insert a trend line through the lag phase data points and a second trend line through the elongation phase data points for a single trace. This can be performed in Excel by separating each individual trace into two datasets, one of which contains lag phase data points and the second of which contains elongation phase data points. We recommend minimizing the number of data points from the transition between lag and elongation phase, if possible (Figure 2). Do not include plateau phase data points.
Use Excel to calculate the slope and y-intercept of the trend line drawn through each set of data points. Calculate the x-value (time point) at which both lines intersect. This is the lag time for a single trace ( Nielsen et al., 2001 ).
After calculating for multiple traces, average lag times within a single condition and calculate the standard deviation.
-
FRAP analysis using Leica LAS X and Microsoft Excel
After FRAP experiment completion, LAS X built-in software procures a fluorescence intensity trace for the specific ROI used. Within the software, draw an additional ROI on an unbleached (control) droplet to monitor fluctuations in intensity as a result of z-axis drift. Intensity traces can be exported as ASCII or Excel files to be analyzed using Microsoft Excel.
In Excel, plot each individual bleached and control trace as a function of time (Figure 3A). If significant drift is present, subtraction of the control droplet intensity trace from the bleached droplet intensity trace can be used to reduce the effects of drift (Figure 3B).
Normalize each individual droplet intensity trace across pre-bleach and post-bleach frames.
Calculate the average and standard deviation from recovery traces (Figure 3C).
Figure 2. Representative fluorescence intensity trace of amyloid formation.

Amyloid formation can be described as having three distinct phases: (1) nucleation/lag phase, (2) elongation/exponential growth phase, and (3) plateau phase. Kinetic differences can be quantified by measuring the lag time. This is performed by generating a trend line through the lag phase data points (blue) and a second line through the elongation phase data points (red). The time point at which these two lines intersect is defined as the lag time ( Nielsen et al., 2001 ).
Figure 3. Analysis of FRAP Experiments.
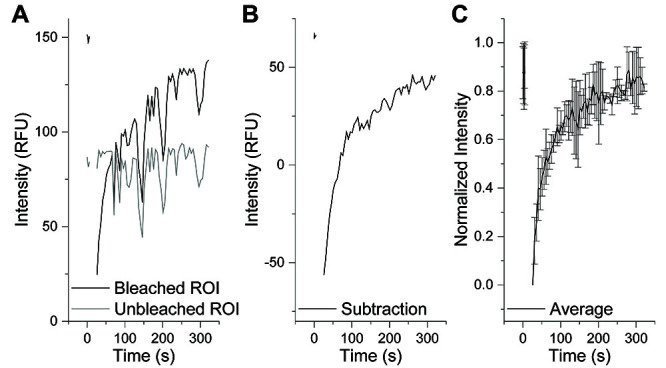
A. Individual traces commonly contain fluctuations in fluorescence intensity due to z-axis drift (black). This can be parsed out by drawing an ROI on a non-bleached droplet (grey). B. Subtraction of the unbleached ROI fluorescence trace from the bleached ROI fluorescence trace can correct for z-axis drift. C. Normalization and averaging of multiple FRAP experiments should be performed to assess the dynamicity of droplets under a specific condition.
Notes
-
Imaging
The number and observed size of droplets on the FluoroDish surface may be greatly influenced by incubation time. For more insight, please see McGuire et al., 1995 . We therefore recommend using a constant time window for imaging across sample conditions.
-
Turbidity and Thioflavin-T Fluorescence Assays
For Thioflavin-T aggregation traces, there was a tendency for the number of wells measured to have an impact on the lag phase length, possibly due to varying degrees of agitation involved with taking a higher number of fluorescence intensity readings by the plate reader at each time point. To work around this, we recommend standardizing all plate reader experiments to the same number of wells measured (i.e., 96 wells, even if sample is not present in all wells) and the same time interval between reads (10 min).
Introduction of bubbles or an air pocket into a well of a 96-well plate can cause artifact. We recommend tapping the side of the plate in order to remove bubbles. Measurements should not be considered valid if bubbles are present.
-
Fluorescence recovery after photobleaching
If many droplets are present within a single frame, multiple droplets can be bleached at one time to increase the number of technical repeats within an experiment.
When evaluating the effect of time on fluorescence recovery, we recommend preparing a reaction solution of sufficient volume in order to add a sample multiple times to a new dish for each time point of interest. As a result, the effect of droplet maturation on FRAP can be observed for droplets aging in the test tube and this minimizes potential aging effects resulting from droplet stasis on the bottom of the dish or interaction with the glass surface.
Recipes
-
100 mM Phosphate and Acetate Stock Buffers (10x)
Prepare 1 M stocks of KH2PO4, K2HPO4, CH3COONa, and CH3COOH in Milli-Q H2O. Prepare secondary stocks by diluting 10-fold in Milli-Q to reach a final concentration of 100 mM for each
To prepare phosphate buffers (pH 6, 7, or 7.3), mix 1:1 100 mM KH2PO4 and 100 mM K2HPO4 in the presence of a pH meter while stirring. Adjust to the desired pH by adding additional 100 mM KH2PO4 (to make more acidic) or 100 mM K2HPO4 (to make more basic). Once the desired pH is reached, ensure that pH remains constant for more than 15 min
To prepare acetate buffer (pH 4), mix 1:1 100 mM CH3COONa and 100 mM CH3COOH in the presence of a pH meter while stirring. Add additional 100 mM CH3COOH (to make more acidic) or 100 mM CH3COONa (to make more basic) until the desired pH is reached
Filter freshly made buffers before use with a Millex®-GV filter unit (0.22 μm). Store at ambient temperature
-
Thioflavin-T stock buffer
Prepare a fresh 1.5 mM Thioflavin-T stock solution in Milli-Q H2O by adding an appropriate volume of Milli-Q H2O to a known mass of Thioflavin-T powder
Filter before use with a Millex®–GV filter unit (0.22 μm). Store at 4 °C
-
Labeling Buffer
Please note that this labeling buffer is derived from the elution buffer used during 1-step nickel nitrilotriacetic acid affinity purification as described previously ( Babinchak et al., 2019 ).
Prepare 1 M stock of Tris. Prepare 500 mM stock of TCEP
To prepare 1 L of labeling buffer (20 mM Tris buffer at pH 8 containing 8 M urea, 200 mM NaCl, 250 mM imidazole, and 1 mM TCEP), combine 20 ml of 1M Tris, 480.48 g urea, 17.02 g imidazole, 2 ml of 500 mM TCEP, and 40 ml of 5 M NaCl. Fill with Milli-Q H2O to 1 L and adjust pH using HCl
-
Equilibration Buffer
Prepare 1 M stock of Tris
To prepare 50 ml of equilibration buffer (20 mM Tris buffer at pH 8, containing 8 M urea and 150 mM NaCl), combine 1 ml of 1 M Tris, 24.02 g urea, and 1.5 ml of 5 M NaCl. Fill with Milli-Q H2O to 50 ml and adjust pH using HCl
Acknowledgments
This research was originally published in the Journal of Biological Chemistry: Babinchak, WM; Haider, R; Dumm, BK; Sarkar, P; Surewicz, K; Choi, JK; Surewicz, WK. J. Biol. Chem. 2019; 294 (16): 6306-6317. This work was supported by NIH grants F30 AG059350 (W.M.B.), T32 NS077888, T32 GM 007250, P01 AI106705 (W.K.S.), R01 NS103848 (W.K.S.), and RF1 AG061797 (W.K.S.). Confocal microscopy work was supported by NIH ORIP grant S10 OD024996. We thank Benjamin Dumm and Raza Haider for critical evaluation and proofreading of this work.
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Babinchak W. M., Haider R., Dumm B. K., Sarkar P., Surewicz K., Choi J. K. and Surewicz W. K.(2019). The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J Biol Chem 294(16): 6306-6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Conicella A. E., Zerze G. H., Mittal J. and Fawzi N. L.(2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24(9): 1537-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M. D., Simons M., Niessing D., Madl T. and Dormann D.(2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173(3): 706-719 e713. [DOI] [PubMed] [Google Scholar]
- 4. Lim L., Wei Y., Lu Y. and Song J.(2016). ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol 14(1): e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin Y., Currie S. L. and Rosen M. K.(2017). Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J Biol Chem 292(46): 19110-19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGuire K. S., Laxminarayan A. and Lloyd D. R.(1995). Kinetics of droplet growth in liquid-liquid phase separation of polymer-diluent systems: experimental results. Polymer 36(26): 4951-4960. [Google Scholar]
- 7. Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., Mittag T. and Taylor J. P.(2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163(1): 123-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N. and Fink A. L.(2001). Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40(20): 6036-6046. [DOI] [PubMed] [Google Scholar]
- 9. Taylor N., Wei M., Stone H., Brangwynne C.(2019). Quantifying dynamics in phase-separated condensates using fluorescence recovery after photobleaching. Biophysical Journal 117(7): 1285-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]