

Abstract
PURPOSE
Mutations in DNA mismatch repair genes and PTEN, diagnostic of Lynch and Cowden syndromes, respectively, represent the only established inherited predisposition genes in endometrial cancer to date. The prevalence of other cancer predisposition genes remains unclear. We determined the prevalence of pathogenic germline variants in unselected patients with endometrial cancer scheduled for surgical consultation.
PATIENTS AND METHODS
Patients prospectively consented (April 2016 to May 2017) to an institutional review board–approved protocol of tumor-normal sequencing via a custom next-generation sequencing panel—the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets—that yielded germline results for more than 75 cancer predisposition genes. Tumors were assessed for microsatellite instability. Per institutional standards, all tumors underwent Lynch syndrome screening via immunohistochemistry (IHC) for mismatch repair proteins.
RESULTS
Of 156 patients who consented to germline genetic testing, 118 (76%) had stage I disease. In 104 patients (67%), tumors were endometrioid, and 60 (58%) of those tumors were grade 1. Twenty-four pathogenic germline variants were identified in 22 patients (14%): seven (4.5%) had highly penetrant cancer syndromes and 15 (9.6%) had variants in low-penetrance, moderate-penetrance, or recessive genes. Of these, five (21%) were in Lynch syndrome genes (two MSH6, two PMS2, and one MLH1). All five tumors had concordant IHC staining; two (40%) were definitively microsatellite instability–high by next-generation sequencing. One patient had a known BRCA1 mutation, and one had an SMARCA4 deletion. The remaining 17 variants (71%) were incremental findings in low- and moderate-penetrance variants or genes associated with recessive disease.
CONCLUSION
In unselected patients with predominantly low-risk, early-stage endometrial cancer, germline multigene panel testing identified cancer predisposition gene variants in 14%. This finding may have implications for future cancer screening and risk-reduction recommendations. Universal IHC screening for Lynch syndrome successfully identifies the majority (71%) of high-penetrance germline mutations.
INTRODUCTION
Endometrial cancer (EC) is the most common gynecologic malignancy, with approximately 60,000 cases diagnosed in the United States annually.1 The majority of patients (70%) present with early-stage disease and are cured.1
CONTEXT
Key Objective
To determine the prevalence of pathogenic germline variants in unselected patients with endometrial cancer.
Knowledge Generated
A custom next-generation sequencing panel, the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets—MSK-IMPACT—identified 24 pathogenic germline variants in 22 patients (14%), including 4.5% who had pathogenic variants in high-penetrance genes. Mismatch repair protein immunohistochemistry (IHC) screening identified all patients with Lynch syndrome (LS; 3% of the overall cohort), representing the majority of high-penetrance variants.
Relevance
In patients with predominantly low-risk endometrial cancer, identification of pathogenic germline variants via a multigene panel test may have implications in altering cancer screening and risk-reduction recommendations. IHC for LS screening identifies the majority of high-penetrance variants.
Inherited mutations in mismatch repair (MMR) genes (MLH1, MSH2, PMS2, MLH6, and EPCAM) diagnostic for Lynch syndrome (LS) and PTEN diagnostic for Cowden syndrome are the only established EC predisposition genes to date. They are associated with a significant lifetime risk of EC, but they account for less than 5% of diagnoses.2 Data regarding inherited risk in EC have been generated from selected patient populations, often ascertained from cancer genetics clinics from patients who have been referred because of a personal or family cancer history. For women with LS, EC is often the sentinel event,3 and universal tumor screening of EC via MMR protein staining or microsatellite instability (MSI) assessment is recommended.4
Data regarding the breadth of inherited genetic mutations in an unselected EC patient population are limited. Only one prior study explored the role of germline multigene panel testing, which identified pathogenic variants in 9% of ECs using banked samples.5 We sought to determine the prevalence of germline cancer predisposition mutations in a prospectively collected unselected cohort of newly diagnosed EC patients scheduled for surgery.
PATIENTS AND METHODS
From April 2016 to May 2017, unselected newly diagnosed patients with EC scheduled for hysterectomy consented to an institutional review board–approved protocol (ClinicalTrials.gov identifier: NCT01775072) for tumor and germline DNA sequencing. This pilot project rolled out at various times across gynecologic oncology clinics. Patients with uterine sarcoma are excluded. Patients viewed a standard pretest educational video on germline genetic testing. All patients with pathogenic or likely pathogenic variants were offered genetic counseling. Variants of uncertain significance were not reported. Electronic medical records were reviewed for demographic and clinical variables, including family history.
Sequencing, Variant Calling, and Reporting of Results
Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a 468-gene targeted capture panel, was used for tumor sequencing. Germline analysis initially included a 76-gene hereditary predisposition panel which was later revised to an 88-gene panel (Appendix Table A1).6-8 For patients sequenced on the original panel, targeted review was performed for mutations in POLE and POLD. All variants with less than 1% population frequency in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/faq) were interpreted. A clinical molecular geneticist or molecular pathologist interpreted variants per American College of Medical Genetics criteria.9 Mutations were classified as high (relative risk [RR] > 4), moderate (RR of 2 to 4), or low (RR < 2) penetrance or recessive.10-12 If relevant, loss of heterozygosity (LOH) in the tumor at the locus was assessed using the Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) algorithm.13
Comparison of Germline Data With Public Databases
To assess associations of specific variants and tumor phenotype, population allele frequencies (AFs) in patients with cancer were compared with AFs in patients who did not have cancer obtained from the ExAC public database minus patients obtained from The Cancer Genome Atlas (TCGA).14 Comparisons of AFs in Ashkenazi Jewish patients were restricted to Ashkenazi Jewish individuals in the Genome Aggregation Database (gnomAD) version r2.02 (genome aggregation database; http://gnomad.broadinstitute.org). AFs were compared using Fisher’s exact test in R version 3.3.2 using RStudio Version 1.0.136 to compute the odds ratios (ORs), CIs, and P values. Clinical variables in subsets defined by mutation status were compared by analysis of variance using GraphPad Prism version 7.01 (GraphPad, San Diego, CA)
Tumor Analysis for MMR Deficiency and LS Screening
Per institutional standards, all ECs undergo universal screening for LS via MMR protein immunohistochemistry (IHC) staining for MLH1, MSH2, MSH6, and PMS2.15 If MLH1 or PMS2 are absent, reflex MLH1 promoter methylation analysis is performed. In this study, MSI status was also assessed via MSI sensor, a computational algorithm that analyzes sequencing reads at designated microsatellite regions in tumor-normal pairs and reports the percentage of unstable loci as a cumulative score.16 An MSI sensor score of 10 or greater indicates MSI-high status (MSI-H), a score of 3 to 9 indicates MSI-intermediate status (MSI-I), and a score of less than 3 indicates microsatellite stable status (MSS).
RESULTS
Patient Characteristics
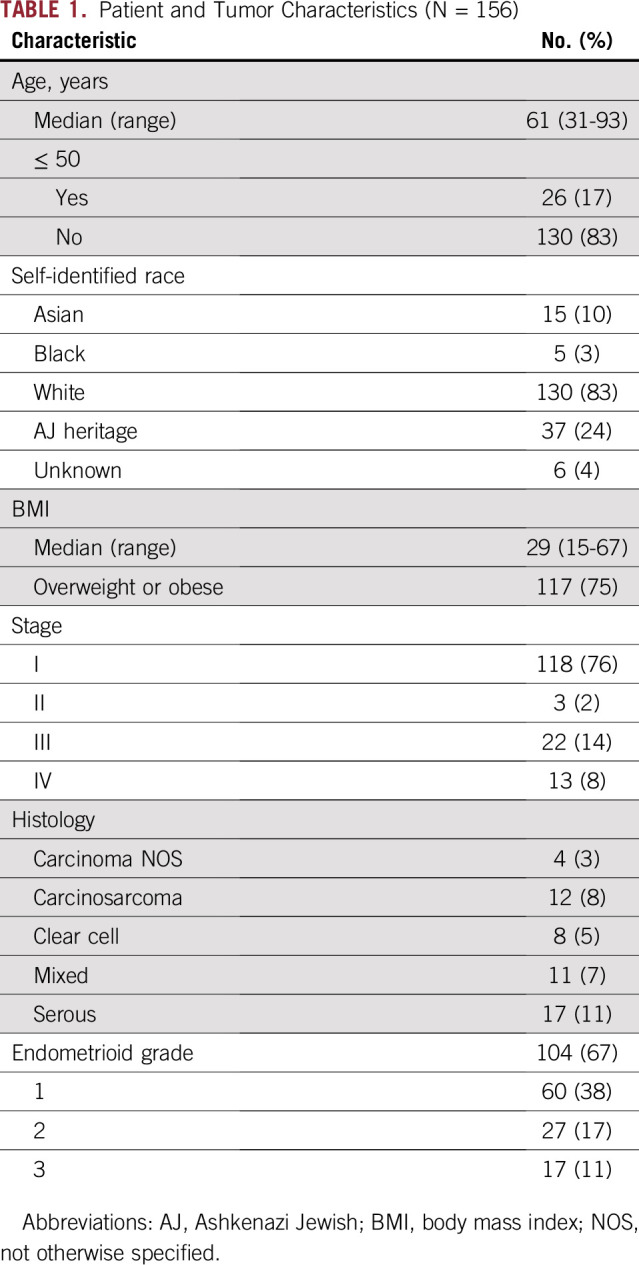
Germline genetic testing was performed in 156 women (Fig 1). Baseline patient and tumor characteristics are provided in Table 1. The median age was 61 years, 75% of patients were overweight or obese, and 76% had early-stage cancer. The majority self-identified as white (83%), and 24% were Ashkenazi Jews.
FIG 1.

Patient flow diagram.
TABLE 1.
Patient and Tumor Characteristics (N = 156)
Germline Analysis
Twenty-four germline mutations were identified in 22 patients (14%; 95% CI, 9.5% to 20.4%; Table 2). Five patients (3%; 95% CI, 1.4% to 7.3%) had LS (two MSH6, two PMS2, and one MLH1), representing 71% of high-penetrance variants. All had endometrioid or clear cell EC histology and absent MMR IHC staining. One patient met Amsterdam criteria and two had significant family histories, but had no cancer diagnoses reported at younger than age 50 years in the family. One patient had a sister with EC in her 30s and no other LS-associated cancers in her family, and one patient had limited family history available (Table 2).
TABLE 2.
Pathogenic Germline Mutations

Beyond the MMR genes, two other high-penetrance germline mutations were identified in BRCA1 and SMARCA4 (Table 2). The BRCA1 mutation carrier had previously tested positive for this known familial mutation and underwent risk-reducing salpingo-oophorectomy (RRSO) without hysterectomy. She had a stage III International Federation of Gynecology and Obstetrics grade 3 endometrioid EC, and her tumor showed LOH at BRCA1. She did not have prior breast cancer or exposure to tamoxifen. A 72-year-old woman who had clear cell EC lobular breast cancer when she was in her 40s and a family history of breast and prostate cancer had a deletion of SMARCA4 exon 17-18 identified with SMARCA4 IHC loss in her tumor.
Of these seven patients with high-penetrance mutations, six (86%) met criteria for testing for the implicated gene because of their personal or family cancer history. The SMARCA4 deletion carrier had no personal or family history of small cell ovarian tumors or rhabdoid tumor predisposition syndrome (Table 2).
Low- and Moderate-Penetrance or Recessive-Only Genes
The other 17 (71%) pathogenic germline mutations (in 15 patients) were in low- and moderate-penetrance or recessive-only genes (Table 2). Only one of these, an ATM (c.4741dupA [p.Ile1581Asnfs*5]) mutation, demonstrated tumor LOH. This 41-year-old patient had no additional history of cancer, although family history was significant for prostate cancer. Two additional women had ATM mutations without LOH, one with no personal or family history of cancer and one with a family history of postmenopausal breast cancer. Five patients (21%) harbored the polymorphism APC I1307K. The remaining mutations were in CHEK2 (missense variant I157T), MRE11A, MUTYH, and REQL4.
Seven (47%) of the 15 patients with variants in low- or moderate-penetrance or recessive genes met National Comprehensive Cancer Network (NCCN) criteria for BRCA testing on the basis of personal or family history. Of these, one (14%) had genetic testing before her EC diagnosis.
There was no association of pathogenic germline mutations in these low- or moderate-penetrance or recessive-only genes compared with population controls; however, the analysis is limited by the small size of the cohort. The excess of APC I130K mutations was no longer significant when corrected for the prevalence of Ashkenazi Jewish patients (Appendix Table A2). There was no difference in age at presentation or body mass index by mutation status (Table 3). Given the small sample size, tumor stage and histology per mutation status are described but not formally compared.
TABLE 3.
Description of Clinical and Tumor Variables Per Mutation Grouping

Concordance of Germline Genetic Testing for LS With Tumor Phenotype
MMR IHC staining was absent in 43 patients (28%); the majority (74%; n = 32) exhibited MLH1/PMS2 loss driven by MLH1 promoter methylation (Fig 2A). Five (45%) of the remaining 11 patients had a germline MMR gene mutation (Fig 2A). Four (67%) of the six remaining patients had dual somatic mutations accounting for the IHC loss; two of these were somatic POLE-associated cancers. The remaining two patients were discordant, with each having a single MMR gene somatic mutation in a gene that does not correspond to the IHC loss.
FIG 2.
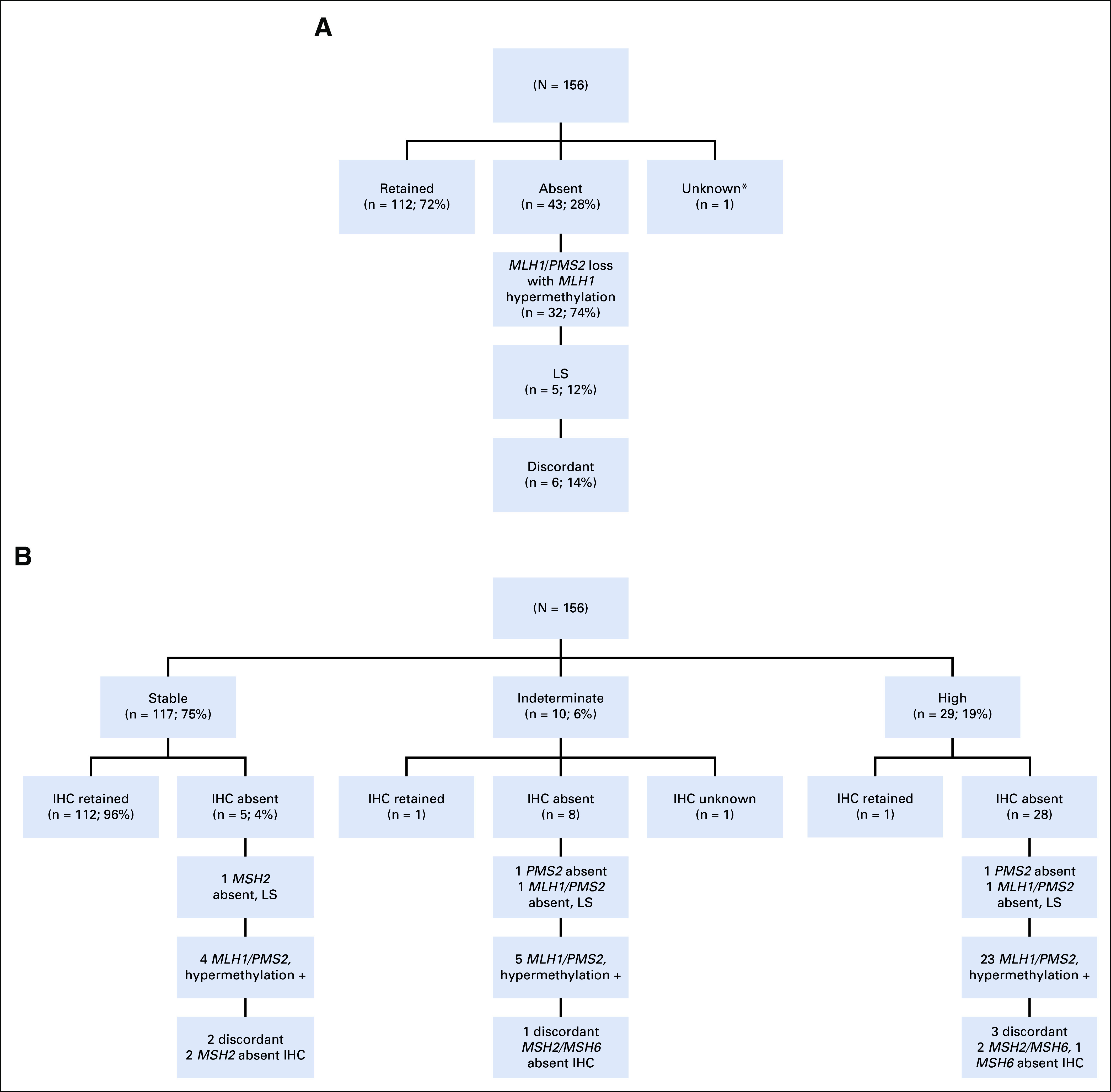
(A) Mismatch repair immunohistochemical status. (B) Microsatellite instability and correlation with immunohistochemistry (IHC) and germline findings. Hypermethylation +, MLH1 promoter hypermethylation identified; LS, Lynch syndrome. (*) Biopsy at outside hospital; no tumor remaining at hysterectomy.
MSI sensor score was concordant with IHC in 140 patients (90%; 112 MSS/IHC retained and 28 MSI-H/MMR-deficient; Fig 2B). Ten (6%) were MSI-I, warranting additional testing. Of these, one was IHC retained, eight were IHC absent, and one was IHC unknown. However, only two LS-associated tumors (40%) had an MSI-H tumor phenotype—two were MSI-I and one was MSS (Table 4). The MSS and one of the MSI-I tumors had less than 20% tumor purity, potentially affecting the result. The other LS-associated MSI-I tumor had a PMS2 mutation, 21 somatic mutations, and absence of MLH1/PMS2 on IHC. In addition, one patient had an MSI-H EC with retained IHC, no germline LS mutation, and no personal or family history suggestive of LS. Her tumor had 46 somatic mutations, including dual MLH1 mutations, possibly driving the MSI phenotype.
TABLE 4.
MSI and Correlation With IHC and Germline Findings in Lynch Syndrome Endometrial Cancer

DISCUSSION
Although next-generation sequencing has facilitated the widespread adoption of multigene panel germline testing, its clinical utility remains unclear.17,18 Studies of unselected patients with colorectal,19 ovarian,20 and breast cancers21 have helped define the range and prevalence of germline pathogenic cancer predisposition genes in these diseases. In addition, with broader genetic testing of patients with cancer, it has become evident that cancer stage may influence the prevalence of germline mutations. For example, DNA repair gene mutations are enriched in advanced-stage prostate cancer.8 Conversely, early-stage colon cancer is enriched for germline mutations in the MMR genes.22 Data regarding the spectrum of inherited risk in EC remains limited; only one prior study5 has explored the role of germline multigene panel testing in a clinical cohort of patients with EC, testing for 25 genes using banked samples. LS mutations were identified in 6% of patients with EC, and another 3% had mutations in other genes. This 6% LS prevalence is on the upper end of what might be expected, with 2% to 6% reported in the literature and a generally accepted rate of approximately 3%,23-27 raising the possibility of an ascertainment bias in the banked samples that were analyzed.5 Applying a broad gene panel, analyses of selected patients with EC from TCGA suggested that 16% of patients with EC harbor cancer predisposition mutations.28 We sought to determine the prevalence of cancer germline predisposition mutations in an unselected real-world clinical cohort of newly diagnosed patients with EC presenting for hysterectomy by using a large research multigene panel test. We have identified high-penetrance germline mutations in 4.5% of patients, and low- to moderate-penetrance or recessive germline mutations were identified in an additional 10%, confirming the overall rate of germline mutations identified in the heavily selected TCGA cohort.28
LS was identified in 3% of our patients, and all met the criteria for testing per current NCCN guidelines.29 Compared with the more permissive NCCN guidelines, the Amsterdam II/Revised Bethesda criteria do not perform as well in the setting of EC.30 Moreover, specific MMR genes have different phenotypic presentations, including variable cancer risk and age at cancer onset.31 As opposed to the predominance of MLH1 and MSH2 mutations in colorectal cancer, MSH6 mutations are more common and are associated with a median age of onset of ≥ 50 years in EC.31 In our series, two MSH6 mutations (40%), two PMS2 mutations (40%), and one MLH1 mutation (20%) were identified, similar to previous EC data.5,23 Only one patient met Revised Bethesda criteria, reinforcing the need for universal tumor screening in all ECs.
Similar to results from prior reports,23,32 28% of tumors in our study had absent MMR IHC staining, with the majority driven by MLH1 promoter hypermethylation.2 Of the remainder, approximately half were LS and half were discordant, whereby the loss of IHC is not associated with a corresponding germline mutation. These are now understood to be frequently driven by dual somatic mutations rather than occult LS germline mutations,33 as was the case in our cohort. MSI sensor performed reasonably well in the whole cohort, but IHC performed more consistently for identifying LS. This reflects the experience with traditional polymerase chain reaction–based MSI testing in which LS-associated ECs have a lower proportion of unstable markers per tumor compared with colon cancer.34 In addition, unlike next-generation sequencing–based MSI analysis, IHC testing is not vulnerable to low tumor purity.
Patients with BRCA mutations may be at increased risk of developing EC, although data are conflicting and confounded by tamoxifen use.28,35 Potential EC risk has important implications for counseling, because these women routinely undergo RRSO without hysterectomy. Similar to prior data, the patient with a BRCA mutation in our study had a high-grade EC.28,35 Her tumor demonstrated LOH at BRCA1, suggesting the contribution of the BRCA1 mutation to development of EC. Shu et al35 reported only seven ECs among approximately 1,000 women with BRCA mutations prospectively observed for a median of 5.1 years. Although a discussion of potential EC risk with BRCA mutation carriers undergoing RRSO is reasonable, routine hysterectomy in this population is not currently warranted.
SMARCA4 germline mutations result in rhabdoid tumor predisposition syndrome type 2, an autosomal dominant cancer syndrome associated with aggressive soft tissue rhabdoid tumors, commonly in the brain (atypical teratoid/rhabdoid tumors). For women, germline SMARCA4 mutations are associated with small cell carcinoma of the ovary, hypercalcemic type; however, association with EC is unknown.36 Median age at presentation is in the 20s; no patients older than age 60 years have been reported.36,37 Interestingly, IHC loss was demonstrated in our patient with EC and germline SMARCA4 mutation, raising the possibility that the mutation had a role in tumor development.
The majority (71%) of identified germline mutations were in low- or moderate-penetrance or autosomal recessive-only genes and most likely represent incidental findings. There was no significant increase in identified mutations compared with population controls. In addition, only one woman with a germline ATM mutation had LOH in her tumor. Given our relatively young population, of whom the majority had curable stage I cancer, the identification of incidental germline mutations may have significant clinical implications for future risk reduction and identification of at-risk family members.
Although a number of these patients met current NCCN criteria for BRCA testing,38 the majority did not have prior breast or ovarian cancer gene testing. Nationally reported rates of genetic testing are poor; less than a third of eligible patients with breast cancer and 15% of patients with ovarian cancer have discussions about genetic testing.39 We have shown that genetic testing is acceptable to newly diagnosed EC patients if they are approached and offered testing.
ATM is not known to be associated with EC. In a previous study of 381 patients with EC undergoing clinical multigene panel testing, one ATM mutation was found in a patient with breast cancer.5 Data from TCGA studies identified no germline ATM mutations in patients with EC (n = 528).28 Biallelic germline ATM mutations are associated with the autosomal recessive ataxia telangiectasia leading to progressive cerebellar ataxia in early childhood.40 Monoallelic ATM mutations are found in approximately 3% of white populations41 and are associated with a moderately increased risk of breast cancer (RR, 2.8; 90% CI, 2.2 to 3.7).11 Breast cancer screening with mammography beginning at age 40 years is recommended, with consideration for additional screening using magnetic resonance imaging.18 Recent data have shown that germline ATM mutations are associated with inherited pancreas42 and prostate cancer43,44 and potentially a small increased risk of ovarian cancer (OR, 1.7).45
The APC I1307K variant, present in approximately 7% of Ashkenazim, is associated with a modest (OR, 1.96; 95% CI, 1.37 to 2.79) increased risk of colorectal cancer but does not cause the highly penetrant familial adenomatous polyposis syndrome.46 Four of our patients (80%) with APC I1307K reported Ashkenazi Jewish heritage. Current guidelines support colonoscopy starting at age 45 years at 5-year intervals for APC I1307K carriers.46
The CHEK2 missense variant I157T has been associated with a small increased risk of breast and colon cancer.18 Although considered a low-penetrance variant by most commercial laboratories, one laboratory considers it to be a variant of uncertain clinical significance.47 In contrast to the CHEK2 1100delC truncating variant,48,49 in which the estimated RR for breast cancer is 3.0 (90% CI, 2.6 to 6.5),11 data are less clear for CHEK2 I157T, and risks are lower (RR, 1.58; 95% CI, 1.42 to 1.75).18 For women with CHEK2 1100delC, mammogram starting at age 40 years is recommended, with consideration of additional screening using magnetic resonance imaging.18 Given only a modest increase of lifetime risk of breast cancer with the CHEK2 I157T variant (18% compared with a population risk of 12% and 32% with CHEK2 1100delC),18 risk does not exceed population cumulative lifetime risk until age 65 to 69 years, and breast cancer screening per population guidelines is considered sufficient.18 CHEK2 I157T is associated with a modest increased risk of colon cancer (RR, 1.56; 95% CI, 1.32 to 1.84), and starting colonoscopy screening at age 45 years with 5-year intervals has been proposed.46
There were three REQL4, two MUTYH, and one MRE11A mutations identified. These genes are associated with the autosomal recessive Rothmund-Thompson syndrome, MUTYH-associated polyposis, and ataxia telangiectasia-like disorder, respectively. As opposed to biallelic MUTYH mutation carriers diagnostic of MUTYH-associated polyposis,50 monoallelic MUTYH carriers have only a modest increased risk of colorectal cancer (RR, 1.17; 95% CI, 1.01 to 1.34), and in the absence of a family history of colon cancer, enhanced screening over population-based guidelines is not recommended.46 Rothmund-Thompson syndrome and ataxia telangiectasia-like disorder are rare, serious childhood conditions, and carrier information may be used for reproductive planning.40,51
Given the rarity of Cowden syndrome and our cohort size, it is not surprising that we did not identify any PTEN mutations. Nor did we identify any germline mutations in POLE or POLD1 genes, which are associated with MMR proofreading and DNA replication fidelity. Although somatic POLE mutations are relatively common, it is not clear that germline mutations predispose to EC.52 Germline POLD1 mutations have been associated with EC, but they are rare.52
In conclusion, we report a pilot study of large multigene panel germline testing for patients with newly diagnosed EC presenting for surgery. Although our study is limited by a small sample size, predominantly white cohort, and overrepresentation of Ashkenazi Jewish heritage, it reflects a prospective unselected cohort of patients with early-stage EC.
We demonstrated the feasibility, acceptability, and yield of multigene germline testing in unselected patients with early-stage EC, and we found that the 3% incidence of LS in our study is comparable to that in the literature. Although not all patients met the classic criteria for LS testing, all met current NCCN guidelines and all were identified by our IHC screening protocol. Two additional patients with high-penetrance germline mutations were identified. BRCA1 was previously associated with a potential for EC risk, but its role in this setting remains unclear. For SMARCA4, there is no prior association with EC development, and data regarding this gene remain limited. The identification of pathogenic germline mutations in 14% of this largely cured cohort represents an opportunity of intervention for patients and family members.
Appendix
TABLE A1.
Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) List of Germline Genes
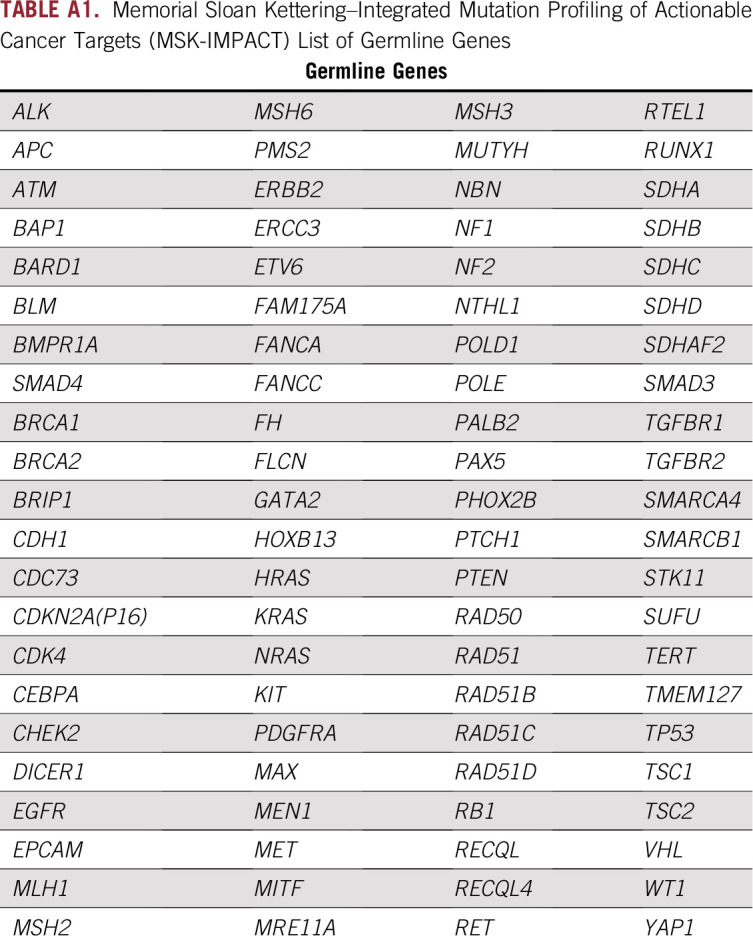
TABLE A2.
Comparison With Population Controls

Footnotes
Supported in part by the Romeo Milio Lynch Syndrome Foundation, the Robert and Kate Niehaus Center for Inherited Cancer Genomics, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, and Grant No. P30 CA008748 from the National Cancer Institute, National Institutes of Health.
AUTHOR CONTRIBUTIONS
Conception and design: Karen A. Cadoo, Mark E. Robson, Kenneth Offit, Carol Aghajanian, Zsofia Stadler
Financial support: Karen A. Cadoo, Kenneth Offit
Administrative support: Karen A. Cadoo, David M. Hyman, Kenneth Offit
Provision of study materials or patients: Karen A. Cadoo, Angela G. Arnold, Margaret Sheehan, Ginger J. Gardner, Elizabeth L. Jewell, Mario M. Leitao Jr, Yukio Sonoda, Oliver Zivanovic, David M. Hyman, Liying Zhang, Mark E. Robson, Kenneth Offit
Collection and assembly of data: Karen A. Cadoo, Carolyn Stewart, Daire Hurley, Yelena Kemel, Angela G. Arnold, Margaret Sheehan, Nisha Pradhan, Ginger J. Gardner, David M. Hyman, Liying Zhang, Kenneth Offit, Zsofia Stadler
Data analysis and interpretation: Karen A. Cadoo, Diana L. Mandelker, Semanti Mukherjee, Deborah DeLair, Vignesh Ravichandran, Preethi Srinivasan, Nisha Pradhan, Vijai Joseph, Dennis S. Chi, Ginger J. Gardner, Elizabeth L. Jewell, Mario M. Leitao Jr, Kara Long Roche, Jennifer J. Mueller, Yukio Sonoda, Oliver Zivanovic, Michael Walsh, Maria I. Carlo, Michael F. Berger, Liying Zhang, Mark E. Robson, Kenneth Offit, Nadeem R. Abu-Rustum, Zsofia Stadler
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Karen A. Cadoo
Research Funding: AstraZeneca (Inst), Syndax Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Semanti Mukherjee
Employment: Regeneron Pharmaceuticals
Stock and Other Ownership Interests: Regeneron Pharmaceuticals
Research Funding: Regeneron Pharmaceuticals
Dennis S. Chi
Leadership: CSurgeries
Stock and Other Ownership Interests: Bovie Medical, Verthermia, Intuitive Surgical, TransEnterix
Consulting or Advisory Role: Bovie Medical, Verthermia
Elizabeth L. Jewell
Consulting or Advisory Role: Covidien/Medtronic
Speakers' Bureau: Covidien/Medtronic
Mario M. Leitao Jr
Honoraria: Intuitive Surgical
Research Funding: KCI
Travel, Accommodations, Expenses: Intuitive Surgical
Kara Long Roche
Travel, Accommodations, Expenses: Intuitive Surgical
Yukio Sonoda
Patents, Royalties, Other Intellectual Property: Patent pending for a surgical instrument (uterine manipulator)
Maria I. Carlo
Consulting or Advisory Role: Pfizer
Michael F. Berger
Consulting or Advisory Role: Roche
Research Funding: Illumina
David Hyman
Consulting or Advisory Role: Atara Biotherapeutics, Chugai Pharmaceutical, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Genentech
Research Funding: AstraZeneca, Puma Biotechnology, Loxo Oncology
Travel, Accommodations, Expenses: Genentech, Chugai Pharmaceutical
Liying Zhang
Employment: Shanghai Genome Center (I)
Leadership: Shanghai Genome Center (I)
Honoraria: Future Technology Research
Travel, Accommodations, Expenses: Shanghai Genome Center (I), Roche Diagnostics Asia Pacific
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca, Merck, Pfizer
Research Funding: AstraZeneca (Inst), Myriad Genetics (Inst), InVitae (Inst), AbbVie (Inst), TESARO (Inst), Medivation (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Carol Aghajanian
Consulting or Advisory Role: Clovis Oncology, TESARO, Mateon Therapeutics, Immunogen
Research Funding: Genentech (Inst), AbbVie (Inst), Clovis Oncology (Inst), AstraZeneca (Inst)
Nadeem R. Abu-Rustum
Honoraria: prIME Oncology
Research Funding: Stryker/NOVADAQ (Inst), Olympus (Inst), GRAIL (Inst)
Travel, Accommodations, Expenses: prIME Oncology
Zsofia Stadler
Allergan (I), Genentech (I), Regeneron Pharmaceuticals (I), Optos (I), Adverum Biotechnologies (I), BioMarin Pharmaceuticals (I), Alimera Sciences (I), Novartis (I), Spark Therapeutics (I), Fortress Biotech (I), REGENXBIO (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Bruegl AS, Djordjevic B, Batte B, et al. Evaluation of clinical criteria for the identification of Lynch syndrome among unselected patients with endometrial cancer. Cancer Prev Res (Phila) 2014;7:686–697. doi: 10.1158/1940-6207.CAPR-13-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu KH, Dinh M, Kohlmann W, et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105:569–574. doi: 10.1097/01.AOG.0000154885.44002.ae. [DOI] [PubMed] [Google Scholar]
- 4.Bruegl AS, Kernberg A, Broaddus RR. Importance of PCR-based tumor testing in the evaluation of Lynch syndrome-associated endometrial cancer. Adv Anat Pathol. 2017;24:372–378. doi: 10.1097/PAP.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ring KL, Bruegl AS, Allen BA, et al. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol. 2016;29:1381–1389. doi: 10.1038/modpathol.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet Med. 2015;17:70–87. doi: 10.1038/gim.2014.147. [DOI] [PubMed] [Google Scholar]
- 11.Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weitzel JN, Blazer KR, MacDonald DJ, et al. Genetics, genomics, and cancer risk assessment: State of the art and future directions in the era of personalized medicine. CA Cancer J Clin. 2011;61:327–359. doi: 10.3322/caac.20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stone JG, Robertson D, Houlston RS. Immunohistochemistry for MSH2 and MHL1: A method for identifying mismatch repair deficient colorectal cancer. J Clin Pathol. 2001;54:484–487. doi: 10.1136/jcp.54.6.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middha S, Zhang L, Nafa K, et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis Oncol. doi: 10.1200/PO.17.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domchek SM, Bradbury A, Garber JE, et al. Multiplex genetic testing for cancer susceptibility: Out on the high wire without a net? J Clin Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- 18.Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13:581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. 2017;35:1086–1095. doi: 10.1200/JCO.2016.71.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tung N, Lin NU, Kidd J, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–471.23. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson SE, Aronson M, Pollett A, et al. Performance characteristics of screening strategies for Lynch syndrome in unselected women with newly diagnosed endometrial cancer who have undergone universal germline mutation testing. Cancer. 2014;120:3932–3939. doi: 10.1002/cncr.28933. [DOI] [PubMed] [Google Scholar]
- 26.Egoavil C, Alenda C, Castillejo A, et al. Prevalence of Lynch syndrome among patients with newly diagnosed endometrial cancers. PLoS One. 2013;8:e79737. doi: 10.1371/journal.pone.0079737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32:90–100. doi: 10.1200/JCO.2013.51.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C, Xie M, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015;6:10086. doi: 10.1038/ncomms10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S, Provenzale D, Regenbogen SE, et al. NCCN Guidelines insights: Genetic/familial high-risk assessment: Colorectal, version 3.2017. J Natl Compr Canc Netw. 2017;15:1465–1475. doi: 10.6004/jnccn.2017.0176. [DOI] [PubMed] [Google Scholar]
- 30.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan NAJ, Morris J, Green K, et al. Association of mismatch repair mutation with age at cancer onset in Lynch syndrome: Implications for stratified surveillance strategies. JAMA Oncol. 2017;3:1702–1706. doi: 10.1001/jamaoncol.2017.0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosgrove CM, Cohn DE, Hampel H, et al. Epigenetic silencing of MLH1 in endometrial cancers is associated with larger tumor volume, increased rate of lymph node positivity and reduced recurrence-free survival. Gynecol Oncol. 2017;146:588–595. doi: 10.1016/j.ygyno.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations . Gastroenterology. 2014;147:1308–1316.e1. doi: 10.1053/j.gastro.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuismanen SA, Moisio AL, Schweizer P, et al. Endometrial and colorectal tumors from patients with hereditary nonpolyposis colon cancer display different patterns of microsatellite instability. Am J Pathol. 2002;160:1953–1958. doi: 10.1016/S0002-9440(10)61144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu CA, Pike MC, Jotwani AR, et al. Uterine cancer after risk-reducing salpingo-oophorectomy without hysterectomy in women with BRCA mutations. JAMA Oncol. 2016;2:1434–1440. doi: 10.1001/jamaoncol.2016.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foulkes WD, Kamihara J, Evans DGR, et al. Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res. 2017;23:e62–e67. doi: 10.1158/1078-0432.CCR-17-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witkowski L, Goudie C, Ramos P, et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol. 2016;141:454–460. doi: 10.1016/j.ygyno.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 1.2018. https://www2.tri-kobe.org/nccn/guideline/gynecological/english/genetic_familial.pdf
- 39.Childers CP, Childers KK, Maggard-Gibbons M, et al. National estimates of genetic testing in women with a history of breast or ovarian cancer. J Clin Oncol. 2017;35:3800–3806. doi: 10.1200/JCO.2017.73.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh MF, Chang VY, Kohlmann WK, et al. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res. 2017;23:e23–e31. doi: 10.1158/1078-0432.CCR-17-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swift M, Morrell D, Cromartie E, et al. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39:573–583. [PMC free article] [PubMed] [Google Scholar]
- 42.Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35:3382–3390. doi: 10.1200/JCO.2017.72.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abida W, Armenia J, Gopalan A, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. doi: 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurian AW, Hughes E, Handorf EA, et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precis Oncol. doi: 10.1200/PO.16.00066. [DOI] [PubMed] [Google Scholar]
- 46.Katona BW, Yurgelun MB, Garber JE, et al. A counseling framework for moderate-penetrance colorectal cancer susceptibility genes. Genet Med. 2018;20:1324–1327. doi: 10.1038/gim.2018.12. [DOI] [PubMed] [Google Scholar]
- 47.Balmaña J, Digiovanni L, Gaddam P, et al. Conflicting interpretation of genetic variants and cancer risk by commercial laboratories as assessed by the prospective registry of multiplex testing. J Clin Oncol. 2016;34:4071–4078. doi: 10.1200/JCO.2016.68.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.CHEK2 Breast Cancer Case-Control Consortium CHEK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74:1175–1182. doi: 10.1086/421251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weischer M, Nordestgaard BG, Pharoah P, et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol. 2012;30:4308–4316. doi: 10.1200/JCO.2012.42.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the Familial Risk-Colorectal Cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33:209–217. doi: 10.1200/JCO.2014.58.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine: Points to consider—A joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125:653–662. doi: 10.1097/AOG.0000000000000666. [DOI] [PubMed] [Google Scholar]
- 52.Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18:325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]