

Abstract
Understanding tumor resistance to T cell immunotherapies is critical to improve patient outcomes. Our study revealed a role for transcriptional suppression of the tumor-intrinsic HLA class I (HLA-I) antigen processing and presentation machinery (APM) in therapy resistance. Low HLA-I APM mRNA levels in melanoma metastases before immune checkpoint blockade (ICB) correlated with nonresponsiveness to therapy and poor clinical outcome. Patient-derived melanoma cells with silenced HLA-I APM escaped recognition by autologous CD8+ T cells. However, targeted activation of the innate immunoreceptor RIG-I initiated de novo HLA-I APM transcription, thereby overcoming T cell resistance. Antigen presentation was restored in interferon-sensitive (IFN-sensitive) but also immunoedited IFN-resistant melanoma models through RIG-I–dependent stimulation of an IFN-independent salvage pathway involving IRF1 and IRF3. Likewise, enhanced HLA-I APM expression was detected in RIG-Ihi (DDX58hi) melanoma biopsies, correlating with improved patient survival. Induction of HLA-I APM by RIG-I synergized with antibodies blocking PD-1 and TIGIT inhibitory checkpoints in boosting the antitumor T cell activity of ICB nonresponders. Overall, the herein-identified IFN-independent effect of RIG-I on tumor antigen presentation and T cell recognition proposes innate immunoreceptor targeting as a strategy to overcome intrinsic T cell resistance of IFN-sensitive and IFN-resistant melanomas and improve clinical outcomes in immunotherapy.
Keywords: Immunology, Oncology
Keywords: Antigen presentation, Melanoma

Introduction
Cytotoxic CD8+ T cells are critical mediators of clinical responses in tumor immunotherapy, killing cancer cells upon recognition of HLA class I (HLA-I) tumor antigen peptide complexes. Antigen processing and presentation requires the interplay of a complex cellular machinery involving components for antigen peptide generation (proteasome subunits LMP2, LMP7), transport (TAP1, TAP2), and loading (TAPBP) onto presenting HLA-I complexes (B2M-associated HLA-A, -B, and -C heavy chains). Genetic alterations in distinct components of the HLA-I antigen processing and presentation machinery (APM) of cancer cells can impair T cell recognition (1–10) and have recently been linked to primary and acquired resistance in immune checkpoint blockade (ICB) and adoptive T cell therapy (ACT) (4–7, 9). In contrast to genomic alterations, nongenetic mechanisms impairing HLA-I APM expression and their role in therapy resistance are still poorly understood.
Previous studies applying immunohistochemistry or proteomics revealed low HLA-I tissue expression in melanoma and other cancer types (11–13) and found it associated with poor clinical outcome (12, 13). This strongly suggests that patients would benefit from strategies enhancing tumor cell–intrinsic HLA-I antigen processing and presentation (14). Type I IFN (IFN-I, IFN-α/β) and type II IFN (IFN-II, IFN-γ) vigorously promote HLA-I APM gene expression via the JAK/STAT signaling pathway (15–17). Viral infections stimulate de novo IFN-I expression. This response is initiated by different innate pattern recognition receptors including the ubiquitous cytosolic RIG-I–like helicases (RLHs) RIG-I and MDA-5, both of them sensing discrete types of virus-derived double-stranded RNA (18–21). Upon ligand binding, RLHs undergo conformational changes and multimerization allowing them to associate with the adaptor protein MAVS. The latter in turn stimulates different pathways which finally activate several transcription factors including NF-κB and IRF3 (22). As a result, expression and release of IFN-β and other proinflammatory cytokines as well as chemokines are initiated, which orchestrate innate and adaptive immune responses. In tumor cells, RLHs can be activated by transfection with mimetics of viral RNA, polyI:C, and 3pRNA as ligands of MDA-5 and RIG-I, respectively (22). Intratumoral injection of synthetic RLH ligands enhances antitumor immune responses by IFN-I release in different preclinical models (23, 24). Thus, RLH activation could be a strategy to induce HLA-I APM expression in tumor cells by autocrine and paracrine IFN-I signaling. However, growing evidence suggests that melanoma as well as other tumor types acquire IFN resistance by downregulation or mutational inactivation of genes encoding crucial components of the JAK/STAT pathway, thereby avoiding antiproliferative and proapoptotic cytokine effects (4, 25–29). Recently, inactivation of IFN signaling turned out as a key mechanism in primary and acquired resistance to immune checkpoint blocking tumor therapies (4, 27–29). Moreover, it was demonstrated that JAK-STAT signaling–defective tumor cells acquire a stable T cell–resistant phenotype through HLA-I APM gene silencing (29), calling for IFN-independent strategies restoring HLA-I antigen presentation.
Here, we demonstrate that coordinated transcriptional suppression of HLA-I APM genes in melanoma is associated with primary resistance to ICB and poor clinical outcome. However, targeted activation of the immunoreceptor RIG-I overcomes HLA-I APM silencing and resensitizes melanoma cells toward autologous CD8+ T cells. RIG-I triggers an IFN-independent salvage pathway allowing for de novo HLA-I APM expression even in IFN-resistant JAK-STAT signaling–defective melanoma cells. Therefore, synthetic RIG-I agonists represent a valuable therapeutic tool to enhance the susceptibility of IFN-sensitive and IFN-resistant tumors to T cell–based immunotherapies.
Results
Nonresponsiveness to anti–CTLA-4 therapy and poor clinical outcome correlate with low HLA-I APM expression in melanoma.
The role of genetic defects in HLA-I APM components in resistance to immunotherapy is well accepted, while that of nongenetic abnormalities remains to be defined. Analysis of the TCGA melanoma data set (n = 462) revealed an association of shortened overall survival (OS) with low expression of HLA-I antigen processing (LMP2, LMP7, TAP1, TAP2, TAPBP) and presentation (B2M, HLA-A, HLA-B, HLA-C) components (Figure 1, A and B). Based on this observation, we hypothesized that melanoma cells evade T cell surveillance by coordinated transcriptional suppression of the different HLA-I APM genes, which in turn accelerates disease progression and impairs immunotherapy efficacy. To address the role of HLA-I APM downregulation in resistance to immunotherapy we performed an integrative analysis on published transcriptomic data from melanoma biopsies (n = 42) taken before anti–CTLA-4 treatment and related clinical data (30). The study cohort included 14 responders and 23 nonresponders (30). As shown in Figure 1C, tumors from ICB responders expressed higher levels of HLA-I APM components compared with nonresponders. Significant differences were observed for B2M, TAP1, and LMP2 (PSMB9). None of the individual genes passed multiple hypothesis correction (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI131572DS1), however, all HLA-I APM genes clearly trended toward higher expression in clinical responders compared with nonresponders (2-tailed binomial P value = 0.0039). Moreover, progression-free survival (PFS) and OS were significantly prolonged in the HLA-I APMhi melanoma group (Figure 1D). Overall, these data argue in favor of a functional role for transcriptional HLA-I APM suppression in ICB nonresponders, suggesting patient outcome could be improved by strategies enhancing tumor cell–intrinsic HLA-I APM expression.
Figure 1. Low HLA-I APM expression correlates with nonresponsiveness to anti–CTLA-4 therapy and poor clinical outcome.

(A) Schematic representation of HLA-I APM components. (B) Overall survival (OS) in the TCGA SKCM cohort (n = 462) stratified by high and low HLA-I APM (HLA-A, HLA-B, HLA-C, B2M, LMP2, LMP7, TAP1, TAP2, TAPBP) expression relative to the median, log-rank test. (C and D) Clinical relevance of altered HLA-I APM expression in an anti–CTLA-4–treated (αCTLA-4–treated) patient cohort (30). (C) Volcano plot showing overall upregulation of HLA-I APM genes in clinical responders (n = 14) versus nonresponders (n = 23) in the αCTLA-4–treated cohort. The x axis is the negative log10 value of the Mann-Whitney U P value; the y axis is the difference in mean rank between response groups. Red vertical dashed line, unadjusted P value of 0.05. (D) Kaplan-Meier survival curves of OS and PFS of high (n = 21) and low (n = 21) HLA-I APM expression groups, log-rank test. High and low expression groups were classified relative to the median HLA-I APM expression level in the entire cohort. (E) Clinical history of melanoma patient UKE-Mel-105 (ICB nonresponder). Horizontal line, time axis; above: diagnosis, therapeutic regimens, death; below: metastases development; arrows indicate cell lines established from metastases UKE-Mel-105b and UKE-Mel-105c. (F and G) Melanoma cells were transfected with 3pRNA, control (ctrl) RNA, or treated with IFNα-2a (IFNα) and subjected to further analysis following an incubation of 20 to 24 hours. HLA-I surface expression was measured by flow cytometry. (F) Representative histograms for UKE-Mel-105b and UKE-Mel-105c cells from 3 independent experiments. (G) HLA-I expression on Colo857 and Ma-Mel-54a melanoma cells. Relative MFI given as mean plus SEM, 2 independent experiments.
Seeking such strategies, we took advantage of short-term–cultured melanoma cell lines established from consecutive biopsies of the anti–CTLA-4 nonresponder UKE-Mel-105 (Figure 1E). Tumor cells (UKE-Mel-105b, UKE-Mel-105c) were treated either with clinically applied type I IFN (IFNα-2b) or transfected with a synthetic ligand (3pRNA) of the pattern recognition receptor RIG-I. We assumed that RLH activation, as elicited in the course of a viral infection, could boost HLA-I antigen presentation. As shown in Figure 1F, IFNα-2b modestly increased HLA-I expression on UKE-Mel-105b and UKE-Mel-105c cells whereas RIG-I activation strongly enhanced HLA-I levels. Superiority of RIG-I signaling in HLA-I upregulation compared with IFN-I signaling was confirmed using different melanoma cell lines (Figure 1G).
Tumor cell–intrinsic RIG-I activation enhances HLA-I–dependent CD8+ T cell recognition.
To mechanistically address the effect of RIG-I signaling on HLA-I APM component expression and determine its functional significance, we applied the patient model Ma-Mel-86, consisting of Ma-Mel-86c melanoma cells, expressing the tyrosinase antigen, and autologous tyrosinase–specific CD8+ T cells (3). We detected elevated levels of HLA-I and the adhesion molecule ICAM-1 (CD54) on 3pRNA-transfected Ma-Mel-86c cells in comparison to control cells treated with nonstimulatory control RNA (Figure 2, A and B). Similar results were obtained upon RIG-I activation in melanoma cells from distinct patient metastases (Supplemental Figure 1, A–C), suggesting a broader applicability of our findings.
Figure 2. Targeted RIG-I activation enhances HLA-I APM expression and CD8+ T cell recognition of melanoma cells.

(A–G, I and J) Melanoma Ma-Mel-86c cells were transfected with 3pRNA or control (ctrl) RNA and subjected to further analyses following an incubation of 20 to 24 hours. (A and B) HLA-I and ICAM-1 surface expression measured by flow cytometry. (A) Representative histograms, (B) relative MFI given as mean plus SEM from 3 independent experiments. (C) HLA-I APM component expression determined by qPCR. Relative expression given as mean plus SEM from 3 independent experiments. (D) Ma-Mel-86c cells were transfected with RIG-I (siRIG-I) or control (siCtrl) siRNA 24 hours before 3pRNA or ctrl RNA transfection and subsequently analyzed for APM component expression by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (E and F) 3pRNA- and ctrl RNA–transfected Ma-Mel-86c cells, preincubated with blocking anti–HLA-I mAb W6/32 or control IgG, were cocultured with an autologous tyrosinase–specific CD8+ T cell clone (Tyr-CD8+ Tc). T cell activation by autologous Ma-Mel-86c and allogenic HLA-I–mismatched Ma-Mel-62 cells was determined by IFN ELISpot assay. (E) Representative ELISpot results and (F) mean IFN-γ spots (+ SEM) from 3 independent experiments. Without, T cells without tumor cells. (G) Representative immunocytochemical staining of Ma-Mel-86c cells for HLA-I heavy chains from 3 independent experiments. (H) Ma-Mel-86c tumors grown subcutaneously on NOD/SCID mice were injected once with ctrl RNA (n = 4) or 3pRNA (n = 4). After 24 hours, tumors were excised and analyzed by immunohistochemistry for expression of HLA-I heavy chains and melanoma marker Melan-A. Representative staining, original magnification ×20. (I) OAS3 expression analyzed by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (J) IFNβ mRNA expression determined by qPCR. Relative expression given as mean plus SEM from 3 independent experiments. Significantly different experimental groups: **P < 0.01, ***P < 0.005 by 2-tailed paired t test.
Analysis of HLA-I APM component expression in 3pRNA-treated Ma-Mel-86c cells revealed a strong increase in B2M, HLA-A, LMP2, TAP1, TAP2, and TAPBP mRNA (Figure 2C). Upregulation of selected HLA-I APM proteins such as HLA-I heavy chains, TAP1, and LMP2 was confirmed by immunoblot. Knockdown of RIG-I before 3pRNA transfection abrogated this effect (Figure 2D). To demonstrate the functional relevance of HLA-I APM upregulation, we exposed 3pRNA- and control RNA–transfected Ma-Mel-86c cells to autologous tyrosinase–specific CD8+ T cells. Cytokine release by T cells was strongly increased in the presence of 3pRNA-transfected melanoma cells (Figure 2, E and F). Studies in an independent autologous tumor T cell model confirmed this result (Supplemental Figure 2A). Enhanced T cell stimulation was HLA-I– and T cell receptor–dependent, as it was abrogated in the presence of anti–HLA-I blocking antibodies and, accordingly, could not be induced by allogenic HLA-I–mismatched 3pRNA-transfected Ma-Mel-62 melanoma cells (Figure 2, E and F; Supplemental Figure 2B).
By immunocytochemistry we confirmed the pronounced upregulation of HLA-I heavy chains in 3pRNA-transfected Ma-Mel-86c cells (Figure 2G). To determine the impact of RIG-I activation on HLA-I heavy chain expression in vivo, we transplanted Ma-Mel-86c cells subcutaneously onto immunodeficient NOD/SCID mice. On day 9, palpable melanomas were injected with 3pRNA complexed with a polyethylenimine-based carrier system already used in clinical trials for intratumoral 3pRNA administration (NCT03065023, NCT03739138). Melanomas were explanted 24 hours after treatment and analyzed for HLA-I heavy chain expression by immunohistochemistry. As shown in Figure 2H, 3pRNA-treated melanomas strongly expressed HLA-I heavy chains in contrast to tumors subjected to control RNA. Both 3pRNA and control RNA–treated Ma-Mel-86c tumors expressed comparable levels of the melanoma marker Melan-A (Figure 2H), indicating a specific effect of RIG-I activation on HLA-I heavy chains.
Aside from HLA-I APM components, RIG-I stimulation triggered its own expression (Figure 2D) and upregulated OAS3, a member of the interferon-stimulated gene (ISG) family (Figure 2I). Consistent with the observed ISG induction, de novo IFNB expression was detected in 3pRNA-transfected Ma-Mel-86c cells (Figure 2J), raising the question of what extent IFN-β release and subsequent autocrine IFN-I signaling contributed to the upregulation of ICAM-1 and HLA-I APM components. Indeed, treatment of Ma-Mel-86c cells with recombinant IFNα-2b increased HLA-I antigen surface expression but only slightly affected ICAM-1 levels (Supplemental Figure 2C).
RIG-I activation upregulates HLA-I APM, even in IFN-I signaling–defective tumor cells.
To determine whether immunogenicity enhancement critically relies on IFN signaling, we examined the effects of RIG-I activation in IFN-I–resistant, JAK-STAT signaling–defective tumor cells, playing a key role also in primary and acquired melanoma resistance to ICB as recently demonstrated by us and others (4, 27–29).
In normal cells, IFN-I binds to the heterodimeric IFNAR1/IFNAR2 receptor complex, leading to the activation of the receptor-associated kinases TYK2 and JAK1 that in turn phosphorylate STAT1 and STAT2 proteins. Phosphorylated STAT1/STAT2 heterodimers associate with IRF9, forming the DNA-binding ISGF3 complex that recruits the transcriptional machinery to initiate expression of numerous ISGs (16). To decipher the role of the JAK-STAT signaling pathway in 3pRNA-induced enhancement of HLA-I antigen presentation, we took advantage of the STAT1-deficient U3A and the IFNAR2-deficient U5A fibrosarcoma cells (31, 32). As expected, U3A (STAT1–/–) cells did not respond to IFN-I (IFNα-2b) or to IFN-II (IFN-γ) treatment in terms of enhanced HLA-I and ICAM-1 expression (Figure 3A), whereas U5A (IFNAR2c–/–) cells responded to IFN-II but not IFN-I (Figure 3B). Upon transfection with 3pRNA, both U3A (Figure 3C) and U5A (Figure 3D) cells upregulated HLA-I and ICAM-1 surface expression. The increase in HLA-I heavy chain and TAP1/2 protein expression was confirmed by immunoblot (Figure 3, E and F). Similarly, protein levels of OAS3 and RIG-I were enhanced by 3pRNA transfection (Figure 3G). Taken together, these data demonstrate that 3pRNA is capable of stimulating the expression of HLA-I APM and ISGs despite defective interferon signaling.
Figure 3. RIG-I upregulates HLA-I APM expression in IFN-I–resistant tumor cells.

(A and B) Human fibrosarcoma cells U3A (STAT1–/–) (A) and U5A (IFNAR2c–/–) (B) were treated with IFNα or IFN-γ for 20 to 24 hours. Controls were left untreated. HLA-I and ICAM-1 surface expression was determined by flow cytometry. Relative MFI given as mean plus SEM of 2 (A) and 3 (B) independent experiments. (C–G) U3A and U5A cells were transfected with 3pRNA or control (ctrl) RNA and subjected to further analyses following an incubation of 20 to 24 hours. (C and D) HLA-I and ICAM-1 surface expression of U3A (C) and U5A (D) cells measured by flow cytometry. Left, representative histogram; right, relative MFI given as mean plus SEM from 3 independent experiments. (E and F) HLA-I APM component expression in U3A (E) and U5A (F) cells analyzed by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (G) RIG-I and OAS3 expression in U3A and U5A cells determined by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. Significantly different experimental groups: *P < 0.05, ***P < 0.005 by 2-tailed paired t test.
RIG-I signaling triggers HLA-I APM expression by IRF1 and IRF3.
To gain insight into the molecular mechanism(s) underlying HLA-I APM upregulation, we focused our analysis on transcription factors known to be activated in response to RIG-I signaling, including IRF1, IRF3 (22, 33), and the activator of HLA-I genes, NLRC5 (34, 35). RIG-I activation led to an upregulation of IRF1, phospho-IRF3 (pIRF3), and NLRC5 in IFN-sensitive Ma-Mel-86c as well as IFN-resistant STAT1- and IFNAR-defective U3A and U5A cells, respectively, indicating the existence of a STAT1-independent RIG-I–induced signaling pathway (Figure 4, A and B).
Figure 4. IRF1 and IRF3 mediate IFN-independent HLA-I APM upregulation upon RIG-I activation.

(A–D) Melanoma cells Ma-Mel-86c and fibrosarcoma cells U3A (STAT1–/–) and U5A (IFNAR2c–/–) were transfected with 3pRNA (+) or control RNA (–) and subjected to further analysis following an incubation of 20 to 24 hours. (A and B) Representative (p)STAT1 (A), IRF1 (A), (p)IRF3 (B), and NLRC5 (A) immunoblots from 3 independent experiments. GAPDH, loading control. (C and D) Ma-Mel-86c and U3A cells were transfected with siRNA targeting IRF3 (siIRF3) (C), IRF1 (siIRF1) (C), NLRC5 (siNLRC5) (D), or control siRNA (siCtrl) (C and D) 24 hours before 3pRNA (+) or control RNA (–) transfection. Protein expression was analyzed by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments.
To define the role of IRF transcription factors in HLA-I APM induction, we transfected Ma-Mel-86c cells with IRF1- or IRF3-specific siRNA before treatment with 3pRNA. IRF1 and IRF3 knockdown abrogated HLA-I APM upregulation as shown by immunoblot (Figure 4C). Furthermore, silencing of IRF1 or IRF3 blocked NLRC5 induction, suggesting a direct involvement of IRF1 and IRF3 upstream of NLRC5 transcription. Interestingly, NLRC5 downregulation by specific siRNA did not significantly affect 3pRNA-mediated HLA-I APM upregulation in IFN-sensitive Ma-Mel-86c and IFN-resistant U3A cells, arguing against NLRC5 as a critical HLA-I APM regulator under these conditions (Figure 4D). Taken together, our data indicate an essential role for IRF1 and IRF3 in the RIG-I signaling cascade, upregulating HLA-I APM components in IFN-sensitive and IFN-resistant tumor cells.
Targeted RIG-I activation induces de novo HLA-I APM expression in IFN-resistant melanoma cells and overcomes T cell resistance.
As mentioned, mutations abrogating IFN signaling in melanoma cells mediate primary and acquired resistance to T cell immunotherapy (4, 27–29). To determine the effect of RIG-I activation on HLA-I APM expression in IFN-resistant melanoma cells, we took advantage of patient model Ma-Mel-61 consisting of a set of short-term cultured melanoma cell lines (Ma-Mel-61b, Ma-Mel-61g, Ma-Mel-61h) established from consecutive metastases collected over a period of 2 years, with the latest ones, Ma-Mel-61g and Ma-Mel-61h, being JAK1 deficient (Figure 5A) (29). Accordingly, IFN signaling was intact only in early Ma-Mel-61b but not in immunoedited JAK1-G600W mutant Ma-Mel-61g and Ma-Mel-61h cells (Figure 5B, Supplemental Figure 3A). Aside from defective IFN signaling, Ma-Mel-61h cells lacked HLA-I surface expression (Figure 5A) due to the coordinated silencing of HLA-I APM genes (29). Previous analyses of corresponding tumor tissue by immunohistochemistry found HLA-I–negative melanoma cells to be present in both metastasis Ma-Mel-61h and Ma-Mel-61g (29), and tissue staining for B2M confirmed in vivo silencing of distinct HLA-I APM genes in IFN-resistant melanoma cells from patient Ma-Mel-61 (Figure 5C).
Figure 5. Targeted RIG-I activation overcomes HLA-I APM silencing in IFN-I–resistant melanoma cells and restores T cell sensitivity.

(A) Clinical history of melanoma patient Ma-Mel-61. Horizontal line, time axis; above: diagnosis, therapeutic regimens, death; below: metastases development; arrows indicate cell lines established from metastases Ma-Mel-61b (JAK1-wildtype, JAK1-WT), Ma-Mel-61g (JAK1-mutant, JAK1-G600W) and Ma-Mel-61h (JAK1-mutant, JAK1-G600W). HLA-I surface expression on cell lines established from corresponding lesions was determined by flow cytometry. Representative histograms from 3 independent experiments. (B) Cell lines treated with IFNα-2b or IFN-γ for 48 hours were analyzed for (p)STAT1 and IRF1 expression by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (C) Immunohistochemical staining of serial cryostat tissue sections from metastasis Ma-Mel-61g for melanoma marker GP100, HLA-I, B2M, and CD3. Top, tumor margin indicated by the dotted line; bottom, higher magnification of boxed regions; original magnifications: ×2.5 (top), ×10 (bottom). (D–I) Ma-Mel-61h cells were transfected with 3pRNA or control (ctrl) RNA and subjected to further analysis following an incubation of 20 to 24 hours. (D and E) HLA-I and ICAM-1 surface expression measured by flow cytometry. (D) Representative HLA-I dot plot and (E) relative MFI given as mean plus SEM from 3 independent experiments. (F) mRNA expression of APM components analyzed by qPCR. Relative expression given as mean plus SEM from 3 independent experiments. (G) Expression of indicated proteins analyzed by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (H and I) Activation of autologous CD8+ T cells by Ma-Mel-61h cells determined by IFN-γ ELISpot assay. (H) Representative ELISpot, (I) mean IFN-γ spots (+ SEM) from 3 independent experiments. without, incubation of T cells without tumor cells. Significantly different experimental groups: *P < 0.05, **P < 0.01, ***P < 0.005 by 2-tailed paired t test.
We concluded that loss-of-function mutation of JAK1 enabled Ma-Mel-61h cells to preserve their immune-evasive HLA-I–negative phenotype in the presence of interferons (Supplemental Figure 3B) (29). Strikingly, targeted activation of RIG-I restored HLA-I expression in IFN-resistant Ma-Mel-61h cells (Figure 5, D and E). Quantification of HLA-I APM component expression by qPCR revealed de novo transcription of genes including HLA-B, B2M, LMP2, TAP1, TAP2, and TAPBP (Figure 5F). De novo expression of HLA heavy chains and TAP1 was further confirmed at the protein level (Figure 5G). Restoration of HLA-I antigen presentation in response to 3pRNA transfection resensitized Ma-Mel-61h cells toward autologous CD8+ T cells (Figure 5, H and I), indicating tumor cell–intrinsic RIG-I signaling bypassed defective IFN signaling to overcome HLA-I APM silencing and T cell resistance.
RIG-I activation enhances CD8+ T cell recruitment.
Previous studies demonstrated that HLA-I–negative melanoma lesions lack T cell infiltrates (36). Accordingly, CD3+ T cells were primarily located within HLA-I–positive regions of metastasis Ma-Mel-61g, while HLA-I–negative areas were largely devoid of T cells (Figure 5C). Since RLH signaling induces expression of T cell-recruiting chemokines (37, 38), we asked to which extent chemokine release was retained in IFN-resistant Ma-Mel-61g cells. Besides enhanced HLA-I APM expression (Figure 6A, Supplemental Figure 4A), 3pRNA-transfected Ma-Mel-61g cells induced de novo transcription of IFNβ (Supplemental Figure 4B) and specific chemokine genes (CCL2, CCL4, CCL5, CXCL10) (Figure 6B). The strongest enhancement was observed for CCL5 and CXCL10, and their release was confirmed by ELISA (Figure 6C). Accordingly, conditioned media (CM) of 3pRNA-transfected Ma-Mel-61g cells, in contrast to CM of control RNA-transfected cells, led to enhanced migration of autologous CD8+ T cells in a Boyden chamber assay (Supplemental Figure 4, C and D), indicating that functionally relevant chemokine release could be induced by 3pRNA even if melanoma cells lost their IFN signaling competence.
Figure 6. IFN-I–independent chemokine release and CD8+ T cell recruitment in response to RIG-I signaling.

(A–C) Ma-Mel-61g cells were transfected with 3pRNA or control (ctrl) RNA and subjected to further analyses following an incubation of 20 to 24 hours. (A) Ma-Mel-61g cells were transfected with RIG-I siRNA (siRIG-I) or control siRNA (siCtrl) 24 hours before 3pRNA or ctrl RNA transfection and subsequently analyzed for protein expression by immunoblot. GAPDH, loading control. Representative data from 3 independent experiments. (B) Chemokine mRNA expression determined by qPCR. Relative expression given as mean plus SEM from 2 independent experiments. (C) Cell culture supernatants were analyzed for CCL5 and CXCL10 content by ELISA. Chemokine levels given as mean plus SEM from 3 independent experiments. Significantly different experimental groups: *P < 0.05, **P < 0.01 by 2-tailed paired t test. (D) Schematic representation of the chicken CAM model. Ma-Mel-86c cells transplanted onto 2 distant sites of each CAM, autologous tumor–reactive CD8+ T cells injected into accessible vein. (E) Human Ma-Mel-86c tumors grown on chicken CAM were analyzed by immunohistochemistry. The 2 tumors on each CAM were treated with either 3pRNA (n = 4) or ctrl RNA (n = 4) on 2 consecutive days. At 24 hours after RNA application, autologous T cells were injected into the blood vessels of the embryo. Tumors were harvested 20 hours after T cell injection. Representative CD8 and HLA-I heavy chain staining shown for each group; original magnifications: ×10 (top), ×40 (bottom).
To study the recruitment of autologous T cells in an in vivo model, we transplanted Ma-Mel-86c cells onto the chorioallantoic membrane (CAM) of fertilized chicken eggs (Figure 6D). Melanoma cells were applied on 2 distant sites of each CAM. Tumors formed within 4 days were treated with jetPEI-complexed 3pRNA or control RNA. Autologous CD8+ T cells were injected into the blood vessels of the embryo on the following day (Figure 6D). As shown in Figure 6E, 3pRNA-treated Ma-Mel-86c tumors showed marked HLA-I upregulation and enhanced CD8+ T cell infiltration in contrast to tumors treated with control RNA.
Taken together, the significant and biologically relevant upregulation and even de novo induction of HLA-I APM components by 3pRNA in an IFNAR2-, JAK1-, and STAT1-independent but RIG-I–dependent manner indicates the existence of an IFN-independent salvage pathway capable of restoring melanoma immunogenicity and inducing T cell recruitment.
Correlation of RIG-I expression with improved antigen presentation and T cell activation in melanoma tissues.
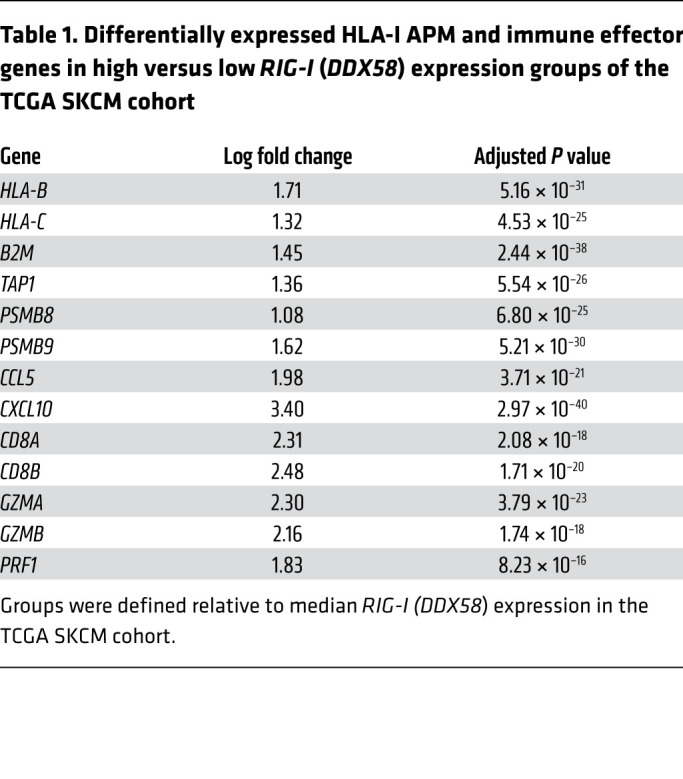
The striking effect of RIG-I activation on antigen presentation, T cell recognition, and T cell recruitment in different IFN-sensitive and IFN-resistant patient-derived melanoma models prompted us to extend our investigations to melanoma tissue samples. Analyzing transcriptomic data of the TCGA SKCM cohort (n = 462), we found HLA-I APM genes (HLA-B, HLA-C, TAP1, TAP2, TAPBP, B2M, PSMB8, PSMB9) to be differentially expressed in RIG-Ihi (DDX85hi) and RIG-Ilo (DDX58lo) tumors (Figure 7A). In addition to HLA-I APM components, RIG-I (DDX58) levels strongly correlated with T cell–recruiting chemokines (CCL5, CXCL10), markers of T cell infiltration (CD8A), and T cell cytotoxicity (GZMA, GZMB, PRF1) (Table 1). Integrative analyses of transcriptomic and annotated clinical data revealed a significant improvement in overall survival for patients with RIG-Ihi (DDX58hi) tumors compared with RIG-Ilo (DDX58lo) tumors (Figure 7B). The impact on survival was even more pronounced when tumors were segregated based on high and low expression of RIG-I (DDX58) pathway genes (DDX58, IRF1, IRF3) (Figure 7B). Overall, these data demonstrate a linkage between HLA-I APM expression and RIG-I (DDX58) expression in melanoma patient tumors, confirming our in vitro findings and favoring RIG-I as a valuable druggable therapeutic target to restore melanoma immunogenicity.
Figure 7. RIG-I (DDX58) expression in melanoma correlates with HLA-I APM expression and patient survival.

(A) HLA-I APM expression in high and low RIG-I (DDX58) expression groups relative to the median RIG-I (DDX58) expression level in the TCGA SKCM cohort. Mann-Whitney U P value is shown. (B) Overall survival in the TCGA SKCM cohort (n = 462) stratified by high and low expression of RIG-I (DDX58) (top) and RIG-I (DDX58) pathway genes (bottom) relative to the median. Log-rank P value shown.
Table 1. Differentially expressed HLA-I APM and immune effector genes in high versus low RIG-I (DDX58) expression groups of the TCGA SKCM cohort.
Targeted RIG-I activation synergizes with ICB in T cell stimulation.
Finally, we addressed the effect of combined ICB and RIG-I–driven HLA-I APM upregulation on T cell reactivity toward autologous melanoma cells. Here, we took advantage of patient model UKE-Mel-154, a nonresponder to anti–PD-1 therapy (Figure 8A). Tumor cells (UKE-Mel-154c) from a lymph node metastasis of this patient, excised before anti–PD-1 treatment, showed a HLA-Ilo phenotype (Figure 8B), suggesting impaired antigen presentation contributed to primary ICB resistance. Consistent with this finding, we observed an association between transcriptional HLA-I APM suppression and therapy resistance in a cohort of melanoma patients receiving anti–PD-1 ICB (n = 121) (39). Analyses of the recently published transcriptomic data from pre–anti-PD-1 melanoma biopsies revealed a trend toward lower HLA-I APM component expression in anti–PD-1 nonresponders (n = 62) compared with responders (n = 57) (2-tailed binomial P value = 0.039) (Figure 8C; Supplemental Table 2), similar to anti–CTLA-4 ICB (Figure 1C). In biopsies of both the anti–PD-1 and anti–CTLA-4 patient cohorts, we detected a strong correlation between expression of RIG-I (DDX58) pathway genes and HLA-I APM genes (rho = 0.81 for anti–CTLA-4, rho = 0.815 for anti–PD-1; Figure 8, D and E), nominating 3pRNA as a drug to overcome HLA-I APM downregulation. In line with this, tumor cells from ICB nonresponder UKE-Mel-154 acquired a HLA-Ihi/ICAM-1hi phenotype upon 3pRNA transfection (Figure 8B; Supplemental Figure 5A). Messenger RNA levels of the distinct HLA-I APM components and IFNβ followed the same response pattern (Supplemental Figure 5, B and C). In addition to enhanced HLA-I antigen presentation, we detected a strong upregulation of PD-L1 (Figure 8, F and G). Interestingly, phenotype analyses of corresponding autologous CD8+ tumor infiltrating lymphocytes (TILs) revealed positivity not only for PD-1 but also for TIGIT (Figure 8, A and H), another inhibitory checkpoint of tumor antigen–specific T cells currently targeted with blocking antibodies in different clinical trials (40). The TIGIT ligand CD155 (PVR) was highly expressed on UKE-Mel-154c cells and remained stable upon 3pRNA transfection (Figure 8, F and G).
Figure 8. Combination of 3pRNA and ICB improves TIL reactivity toward autologous melanoma cells.

(A) Clinical history of melanoma patient UKE-Mel-154. Horizontal line, time axis; above: diagnosis, therapeutic regimens, death; below: UKE-Mel-154c melanoma cell and CD8+ TILs obtained from lymph node (LN) metastasis. (B) HLA-I and ICAM-1 surface expression on 3pRNA- and ctrl RNA–transfected UKE-Mel-154c cells. Representative histograms from 3 independent experiments. (C) Volcano plot showing overall upregulation of HLA-I APM genes in clinical responders (n = 57) versus nonresponders (n = 62) in the αPD-1–treated cohort (39). The x axis is the negative log10 value of the Mann-Whitney U P value; the y axis is the difference in mean rank between response groups. Red vertical dashed line, unadjusted P value of 0.05. (D and E) Scatterplot of RIG-I (DDX58) pathway expression against HLA-I APM expression across all samples in the αPD-1 (n = 121) and αCTLA-4 (n = 42) cohort. (F and G) PD-L1 and CD155 surface expression on 3pRNA- or ctrl RNA–transfected UKE-Mel-154c cells. (F) Representative histograms, (G) relative MFI given as mean plus SEM from 3 independent experiments. (H) Representative PD-1 and TIGIT surface expression on TILs from 3 independent experiments. (I and J) Combination of targeted RIG-I activation and ICB enhances TIL reactivity toward autologous melanoma cells. (I) Concurrent treatment. TIL activation after 4-hour coincubation with 3pRNA- or ctrl RNA–treated UKE-Mel-154c cells in the presence of PD-1 or αTIGIT antibodies, by intracellular cytokines staining (ICS). (J) Sequential treatment. After 7-day preincubation with irradiated (irr.) UKE-Mel-154c cells in the absence or presence of αPD-1 or αTIGIT antibodies, TILs were harvested and activation was measured after 4-hour coincubation with 3pRNA- or ctrl RNA–treated UKE-Mel-154c cells. (I and J) Mean plus SEM fold change in frequency of IFN-γ+CD8+ T cells stimulated by ctrl RNA–transfected UKE-Mel-154c cells from 3 independent experiments. (G, I, and J) Significantly different experimental groups: *P < 0.05, **P < 0.01, ***P < 0.005 by 2-tailed paired t test.
Based on these data, we examined the influence of combined 3pRNA/anti–PD-1 ICB and 3pRNA/anti-TIGIT ICB on CD8+ TIL reactivity toward UKE-Mel-154c cells. To this end, TILs were coincubated with 3pRNA- and control RNA–treated melanoma cells in the absence or presence of anti–PD-1 and anti-TIGIT blocking antibodies. After 4 hours of incubation, intracellular cytokine staining (ICS) showed a strong increase in CD8+ T cell activation in the presence of 3pRNA-transfected UKE-Mel-154c cells compared with control RNA–treated tumor cells (Figure 8I). TIL responses toward 3pRNA-treated UKE-Mel-154c cells could be significantly enhanced when blocking anti–PD-1 and anti-TIGIT antibodies were added, indicating a synergistic effect of targeted RIG-I activation and ICB (Figure 8I). Improvement of T cell activation by anti–PD-1 ICB was achieved only in the presence of 3pRNA- but not control RNA–transfected tumor cells (Figure 8I), most likely attributable to the strong upregulation of PD-L1 upon RIG-I activation (Figure 8, F and G). In contrast, anti-TIGIT ICB significantly improved CD8+ T cell activation under both conditions (Figure 8I), due to constant high CD155 expression on UKE-Mel-154c cells (Figure 8, F and G).
In addition to concurrent treatment, we tested a sequential protocol of ICB and targeted RIG-I activation (Figure 8J). Initially, TILs were cocultured with irradiated UKE-Mel-154c cells in the absence or presence of anti–PD-1 and anti-TIGIT blocking antibodies. After an incubation period of 7 days, T cells were harvested and analyzed for their reactivity toward 3pRNA- and control RNA–transfected melanoma cells. Again, we observed a strong T cell activation only in the presence of 3pRNA- but not control RNA–transfected UKE-Mel-154c cells. Preincubation with anti–PD-1 and anti-TIGIT blocking antibodies clearly increased the fraction of TILs responding to 3pRNA-treated tumor cells, though significance was achieved only in the 3pRNA/anti-TIGIT setting (Figure 8J).
In summary, studies in the anti–PD-1 nonresponder melanoma model clearly demonstrated synergistic effects of HLA-I APM restoration by targeted RIG-I activation and ICB on antitumor T cell responses, suggesting combination therapy could improve clinical outcomes.
Discussion
Loss-of-function genetic alterations affecting tumor antigen presentation (4, 6, 7, 9) and IFN-γ signaling (4, 27–29) have recently been identified as key resistance mechanisms to ICB in different cancers. Studies applying CRISPR/Cas9 for tumor genome editing recapitulated those findings in mouse melanoma models, demonstrating that the efficacy of T cell–based therapy critically depends on intact antigen presentation and IFN-γ signaling (41, 42). Here, we provide evidence that, aside from genetic alterations, resistance of melanoma to ICB and T cells can be attributed to the coordinated transcriptional suppression of a gene set involved in antigen peptide generation (LMP2, LMP7), transport (TAP1, TAP2), and loading (TAPBP) onto presenting HLA-I complexes (HLA-A, HLA-B, HLA-C, B2M). We introduce tumor cell–intrinsic activation of the pattern recognition receptor RIG-I as a strategy to restore antigen processing and presentation and overcome T cell resistance independent of IFN signaling.
The association of low HLA-I APM mRNA levels and poor patient survival in melanoma prompted us to investigate the role of HLA-I APM downregulation in the outcome of immunotherapy. Analyzing a cohort of anti–CTLA-4–treated melanoma patients, we found low HLA-I APM levels associated with primary therapy resistance and shortened PFS and OS, indicating initial and durable clinical responses to this therapy require intact antigen processing and presentation. In line with our observation, a prior immunohistochemistry study correlated loss of melanoma membrane HLA-I expression with resistance in anti–CTLA-4 therapy (43). Moreover, we found the HLA-I APMlo tumor cell phenotype involved in primary resistance to anti–PD-1 therapy. Analysis of recently published transcriptomic data from pretreatment biopsies of a patient cohort receiving anti–PD-1 ICB revealed lower expression of HLA-I APM components in nonresponders compared with responders.
Aside from primary resistance, acquired resistance in adoptive T cell therapy (ACT) of melanoma (44) and anti–PD-1 ICB/ACT of Merkel cell carcinoma (45, 46) has recently been linked to the downregulation of single or multiple HLA-I APM components. Though the latter studies comprised only single patients, the total data suggest a cancer-overarching relevance of nongenetic HLA-I APM alterations in resistance to different types of T cell–based therapy.
Taking advantage of different melanoma patient models, we provide evidence for the functional relevance of transcriptional HLA-I APM suppression. Our data show that patient-derived melanoma cells with silenced HLA-I APM are resistant toward cognate autologous CD8+ T cells. Seeking counteracting mechanisms, we demonstrated that tumor cell–intrinsic activation of the immunoreceptor RIG-I by its synthetic ligand 3pRNA induces de novo transcription of HLA-I APM genes, thereby overcoming T cell resistance. Consistently, a remarkable increase in HLA-I surface expression of 3pRNA-treated tumor cells going along with a strong enhancement in T cell activation was detected in different patient models. The HLA-Ilo to HLA-Ihi phenotypic switch could also be induced in vivo upon 3pRNA injection into human melanomas subcutaneously grown on NOD/SCID mice, using a clinical applied carrier system for dsRNA delivery.
Since RIG-I triggers IFN-I release, we initially expected the restoration of antigen presentation to be dependent on autocrine and paracrine IFN-I signaling in melanoma cells. Strikingly, RIG-I activation also allowed for de novo HLA-I APM transcription in IFN-I–resistant JAK1-deficient melanoma cells, indicating RLH-dependent stimulation of an IFN-I–independent salvage pathway, which resensitized tumor cells to T cells. In addition to demonstrating activation of the salvage pathway in JAK1 mutants, we also demonstrated activation of the salvage pathway in IFNAR2- and STAT1-deficient tumor cells. Previous studies, including our own, showed that a subset of melanomas acquires inactivating genetic alterations in different IFN-I and IFN-II signaling pathway components, contributing to primary and acquired ICB resistance (4, 27–29). Here, we demonstrate that IFN-I/IFN-II–resistant melanomas can still be forced to upregulate HLA-I APM expression in order to enhance responsiveness to immunotherapy.
To unravel the underlying mechanism we screened IFN-sensitive and IFN-resistant cell lines for RIG-I-controlled transcription factors and found IRF1, IRF3, and NLRC5 to be upregulated in all cases. NLRC5 is a known transcriptional activator of HLA-I, TAP, and LMP genes (34, 35) and its loss has been associated with cancer immune evasion (47). In the course of viral infection, RIG-I activation induces NLRC5 expression, mediating HLA-I upregulation (35). In our experimental setting however, siRNA knockdown of NLRC5 had no major impact on HLA-I APM upregulation by 3pRNA. In contrast, ablation of IRF1 or IRF3 by siRNA abrogated the effect. While activation of IRF3 downstream of RIG-I/MAVS signaling is well established, the connection to IRF1 is less studied (22, 33, 48). Both IRF1 and IRF3 have been implicated in the IFN-I–independent induction of virus defense genes (33, 49). Our study now extends this mechanistic link to genes coding for HLA-I APM components and T cell activation in tumor cells. Also, de novo expression of chemokines, attracting CD8+ T cells into transplanted autologous tumors in a chicken CAM model, efficiently occurred in a RIG-I–dependent but IFN-I–independent manner.
Consistent with our observations from distinct in vivo and patient models, we detected enhanced expression of chemokines, HLA-I APM genes, and markers of T cell activation in RIG-Ihi (DDX58hi) melanomas of the TCGA sample collection, further emphasizing the clinical relevance of our findings. Moreover, melanoma biopsies from anti–PD-1— and anti–CTLA-4–treated patient cohorts showed a strong correlation between expression of RIG-I (DDX58) pathway genes and HLA-I APM genes, nominating RIG-I as a druggable therapeutic target. Recent mouse model studies demonstrated that targeted RLH activation in the tumor microenvironment can transform a T cell–deficient melanoma metastasis into a T cell–inflamed lesion and enhance the efficacy of anti–CTLA-4 and anti–PD-1 therapies (23, 50–52). Notably, HLA-I APMlo patient tumors lack T cell infiltrates (11, 36) and absence of T cells in melanoma lesions has been described as a primary resistance mechanism in ICB (53, 54). Thus, injection of synthetic RIG-I ligands into accessible lesions of cancer patients could be a promising strategy to achieve both an increased infiltration of T cells into tumors and enhanced T cell recognition of tumor cells, which could improve clinical responses in ICB. In fact, we demonstrated that HLA-I APM upregulation by 3pRNA and checkpoint blocking antibodies synergistically enhanced the reactivity of CD8+ TILs toward autologous melanoma cells in an anti–PD-1 nonresponder model. Synergism was observed not only for combined 3pRNA/anti–PD-1 but also 3pRNA/anti-TIGIT treatment, in line with the positivity of TILs for PD-1 and TIGIT and with the expression of their ligands on melanoma cells. These findings provide a strong rational for already ongoing clinical trials combining intratumoral injection of synthetic RLH ligands (NCT03739138, NCT02828098, NCT02423863) with anti–PD-1 or anti–PD-L1 antibodies.
Recent studies demonstrated RLH activation by dsRNA derived from human endogenous retrovirus (hERV) transcripts, reexpressed in different cancers and associated with improved patient outcome (55–57). Our data from analyses on patient-derived tumor cells and melanoma biopsies indicate a clear benefit from direct targeting of RIG-I by optimized synthetic ligands in order to achieve efficient HLA-I APM induction. So far, RLH ligands are complexed with specific carrier systems and injected into accessible patient lesions, which induces not only local but also systemic immune responses (58). However, therapy efficacy would benefit from delivery of dsRNA to distant (micro)metastases, clearly demanding for the development targeted carrier systems such as antibody-coupled nanoparticles (59, 60).
Overall, our study links transcriptional HLA-I APM suppression in melanoma to primary resistance in anti–CTLA-4 and anti–PD-1 therapy. Tumor cell–intrinsic activation of the innate immune receptor RIG-I restores antigen processing and presentation and overcomes resistance to autologous CD8+ T cells. Though HLA-I APM upregulation is generally thought to be IFN-dependent, RIG-I efficiently induces de novo HLA-I APM expression even in IFN-resistant tumor cells. Our findings, which demonstrate synergistic effects on antitumor T cell responses of combined RIG-I activation and ICB, suggest that targeted tumor cell–intrinsic RLH activation presents a potent strategy to enhance the efficacy of immunotherapies in metastasized IFN-sensitive and IFN-resistant melanoma and other neoplasia.
Methods
Tumor cells.
Melanoma cell lines were established at the Department of Dermatology (University Hospital Essen, Essen, Germany) from surgically resected, mechanically dissected tumor tissues, as previously described (3, 29). Melanoma cells (Ma-Mel-47, Ma-Mel-54a, Ma-Mel-61b, Ma-Mel-61g, Ma-Mel-61h, Ma-Mel-62, Ma-Mel-66b, Ma-Mel-86c, UKE-Mel-105b, UKE-Mel-105c, UKE-Mel-154c) were authenticated by genetic profiling on genomic DNA at the Institute for Forensic Medicine (University Hospital Essen) using the AmpFLSTR-Profiler Plus kit (Applied Biosystems). Fibrosarcoma cell lines U3A and U5A (31, 32) and melanoma cells were cultured in RPMI1640 medium or DMEM (Gibco) with glutamine and confirmed to be mycoplasma-free in monthly intervals. Both media were supplemented with 10% (vol/vol) fetal calf serum (PAA Laboratories), 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained at 5% CO2, 37°C.
T cells.
Tumor-reactive bulk CD8+ T cells and tumor antigen–specific CD8+ T cell clones from peripheral blood of patients Ma-Mel-86 and Ma-Mel-61 were established as previously described (3, 29). TILs were isolated from tumor single-cell suspensions of lymph node metastasis UKE-Mel-154c using the gentleMACS Dissociator (Miltenyi Biotec) for tumor digestion. CD8+ TILs were positively selected from single-cell suspension using anti-CD8 beads (Miltenyi Biotec). Selected TILs (106 cells/well of 24-well plate) were cocultured with irradiated (100 Gy) autologous UKE-Mel-154c melanoma cells (105 cells/well) in 2 mL AIM-V medium (Gibco-BRL) supplemented with 10% (vol/vol) human AB serum (complete medium). Interleukin-2 (IL-2) was added on day 3 at 250 IU/mL (Chiron Corporation). TILs (106 cells/well) were restimulated in weekly intervals with 105 irradiated tumor cells in IL-2–supplemented complete medium. All cells were maintained at 5% CO2, 37°C.
Mice.
Ma-Mel-86c cells (2 × 106 cells) were injected subcutaneously into the left flank of 6- to 12-week-old NOD/SCID mice (Jackson Laboratory). On day 9 after inoculation, tumors with a volume of approximately 50 mm3 were injected with 50 μg control RNA or 3pRNA complexed with in vivo-jetPEI (Polyplus), according to the manufacturer’s protocol. After 24 hours, mice were sacrificed and tumors were collected, formalin-fixed, and paraffin-embedded for subsequent analysis by immunohistochemistry.
IFN treatment.
Tumor cells (1 × 105 to 2 × 105 cells/well) were seeded in 6-well culture plates 16 to 20 hours before treatment with IFNα-2b (1000 IU/mL Roferon-A, Roche) or IFN-γ (500 IU/mL Imukin, Boehringer Ingelheim). Control cells were left untreated. Following an incubation of 20 to 24 hours at 37°C, cells were subjected to further analyses.
Synthesis of 3pRNA.
For generation of DNA template–dependent in vitro–transcribed RNA (3pRNA), the T7-promoter region 5′-CAGTAATACGACTCACTATAG-3′ was hybridized with the promoter + template strand (5′-TTGTAATACGACTCACTATAGGGACGCTGACCCAGAAGATCTACTAGAAATAGTAGATCTTCTGGGTCAGCGTCCC) and directly used as a template for in vitro transcription reactions. 3pRNA was provided by Christoph Coch and Gunther Hartmann, (Institute of Clinical Chemistry and Clinical Pharmacology, University of Bonn, Bonn, Germany). Nonstimulatory control RNA (5′-CACACACACACACACACACA-3) was purchased from Biomers.
3pRNA and siRNA transfection.
Tumor cells (1 × 105 to 2 × 105 cells/well) were seeded in 6-well culture plates 16 to 20 hours before transfection with 100–200 ng/mL 3pRNA or control (ctrl) RNA, using Lipofectamine 2000 (Invitrogen). Where indicated, tumor cells were transfected with siRNA (10 nM) specific for IRF1, IRF3, NLRC5, or control siRNA (Dharmacon) using Viromer blue (Lipocalyx) 20 to 24 hours before 3pRNA or ctrl RNA (200 ng/mL) treatment. All transfections were performed according to the manufacturer′s instructions. Cells were subjected to further analyses following an incubation of 20 to 24 hours at 37°C.
Antibodies for immunoblot, immunocytochemistry, and immunohistochemistry.
The following anti-human antibodies were used for immunoblot: mouse anti-STAT1 (clone 9H2), rabbit anti-phospho STAT1 (Y701) (clone 58D6), rabbit anti-phospho IRF3 (S396) (clone 4D4G), rabbit anti-GAPDH (clone 14C10), rabbit anti–RIG-I (clone D14G6) were purchased from Cell Signaling; rabbit anti-IRF1 (clone H-205), rabbit anti-IRF3 (clone FL-425), rabbit anti-OAS3 (clone M-190) were obtained from Santa Cruz. TAP1 was detected with mouse mAb NOB-1 (61) and LMP2 with mouse mAb SY-1 (61). Both antibodies were provided by Soldano Ferrone (Department of Surgery, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA). NLRC5 was detected with rat mAb 3H8 (62), provided by Thomas A. Kufer (Institute of Nutritional Medicine, Department of Immunology, University of Hohenheim, Stuttgart, Germany). Mouse mAb HC10 (recognizing an epitope on specific B2M-free HLA-A and all B2M-free HLA-B and -C heavy chains) from Nordic-MUbio was used for immunoblots, immunocytochemistry, and staining of formalin-fixed paraffin-embedded (FFPE) tissue sections. Mouse anti-human Melan-A/MART-1 (clone M2-7C10) for FFPE tissue staining was purchased from von Zytomed. Additionally, cryostat tissue sections were stained with mouse antibodies specific for GP100 (clone HMB-45, Dako), HLA-I antigen complexes (clone W6/32, Dianova), B2M (clone BBM.1, Abcam), CD3 (clone HIT3a, BD Pharmingen).
Immunoblot.
Tumor cells were lysed in RIPA buffer for protein isolation. Proteins (10–50 μg) were separated by SDS-PAGE, electroblotted onto nitrocellulose membranes, and probed with the indicated primary anti–human antibodies. After washing, membranes were incubated with the appropriate secondary antibodies linked to horseradish peroxidase (HRP). Antibody binding was visualized using the ECL chemiluminescence system (Thermo Fisher Scientific). GAPDH levels served as loading control in all experiments.
Immunocytochemistry.
For immunocytochemistry, cells (1.5 × 104) were placed onto glass slides using the cytospin cytocentrifuge universal 30F (Hettich Zentrifugen). Cells were fixed with aceton for 15 minutes and slides were allowed to air-dry before storing at –20°C. Staining of fixed cells was carried out on the AutostainerLink48 (Dako) using the Real Detection System Alkaline Phosphatase/RED Rabbit/Mouse and EnVision FLEX Hematoxylin (Dako). Images of stained sections were acquired with the Axio Observer.Z1 microscope (Zeiss).
Immunohistochemistry.
Serial cryostat tissue sections were stained with indicated primary antibodies in combination with a polymer kit containing an AP-coupled secondary antibody (ZytoChem-Plus AP Polymer Kit, Zytomed). Serial FFPE sections were stained using the Real Detection System Alkaline Phosphatase/RED Rabbit/Mouse and EnVision FLEX Hematoxylin (Dako).
Quantitative real-time PCR (qPCR).
Total mRNA was isolated from tumor cells using the RNeasy plus Mini Kit (Qiagen), in combination with RNAse-free DNase Set (Qiagen) according to the manufacturer′s instructions. Reverse transcription, TaqMan-based real-time PCR and calculation of relative expression were performed as previously described (29). In all experiments, the amount of specific mRNA was normalized to endogenous GAPDH mRNA levels.
Flow cytometry.
Cells were stained for surface marker expression with either directly labeled or nonlabeled antibodies in combination with a secondary PE-conjugated goat anti-mouse F(ab’)2 (IgG (H+L) polyclonal) (Dianova). Background fluorescence was defined based on unstained cells or cells stained with or secondary antibody only. After fixation, cells were analyzed employing Gallios flow cytometer (Beckman Coulter) and Kaluza software. Surface expression was determined as mean fluorescence intensity (MFI), normalized to background MFI (unstained or secondary antibody only). Mouse antibodies used in flow cytometry were as follows: anti–CD54-PE (clone 84H10) from Beckman Coulter; anti–PD-L1-PE (clone 29E2A3), anti-CD155(PVR)-APC (clone SKIL4), anti–CD3-BV42 (clone UCHT1), anti–CD8-APC/Cy7 (clone SK1), anti–PD-1–APC (clone EH12.2H7) and anti–TIGIT-PE/Cy7 (clone A15153G) from BioLegend; anti-β2m–associated HLA class heavy chains (clone W6/32) from eBioscience.
T cell activation assays.
IFN-γ release by CD8+ T cells in the presence of 3pRNA- or control RNA–transfected melanoma cells was quantified by ELISpot assay as follows: 2 × 104 T cells were coincubated with 2 × 104 melanoma cells per well (96-well plate) at 37°C. Following an incubation of 24 hours, IFN-γ release was detected using anti–human IFN-γ capture mAb (clone 1-D1K), biotinylated detection mAb (clone 7-6B-1) (Mabtech), and ExtrAvidin alkaline phosphatase (MilliporeSigma). Where indicated, tumor cells were preincubated with anti–HLA-I blocking mAb W6/32 or isotype-matched control IgG (50 μg/mL).
Intracellular IFN-γ staining was performed to determine TIL reactivity toward autologous tumor cells under combined 3pRNA/ICB treatment in 2 different settings. Initially, 1 × 105 CD8+ TILs were incubated with anti–PD-1 (nivolumab [1 μg/mL], Bristol Myers Squibb [BMS]) or anti-TIGIT (BMS-986207 [1 μg/mL], BMS) antibodies in the presence of autologous control RNA– or 3pRNA-transfected tumor cells (1 × 105) in AIM-V complete medium containing 10 μg/mL Brefeldin A (MilliporeSigma). After 4 hours, cells were subjected to the Fixation/Permeabilization Concentrate and Diluent kit (eBioscience) followed by staining with an antibody cocktail containing anti–human CD3-BV421 (clone UCHT1), CD8-APC/Cy7 (clone SK1) and IFN-γ–PE (clone B27) antibodies (BioLegend). Gallios flow cytometer and Kaluza software were used for cell analyses and data processing, respectively (Beckman Coulter). In a second sequential approach, CD8+ TILs were preincubated with anti-TIGIT (BMS-986207 [1 μg/mL], BMS) or anti–PD-1 (nivolumab [1 μg/mL], BMS) antibodies in the presence of irradiated autologous tumor cells. After 7 days, 1 × 105 T cells were stimulated for 4 hours with the control RNA– or 3pRNA-transfected tumor cells (1 × 105) in AIM-V complete medium containing 10 μg/mL Brefeldin A (MilliporeSigma), followed by intracellular cytokine staining as described above.
ELISA.
Supernatants from 3pRNA- and control RNA–transfected melanoma cells were harvested and analyzed for the content of CCL5 and CXCL10 by ELISA (BioLegend) following the manufacturer’s instructions.
Migration assay.
CD8+ T cell migration was determined in a Boyden chamber assay, using 5.0-μm pore size polycarbonate membrane Transwells (Corning). The lower chamber of the Transwell plates was filled with 500 μL conditioned medium (CM) from 3pRNA- or control RNA–transfected Ma-Mel-61g cells. RPMI medium containing 10–100 ng/mL recombinant CCL5 or CXCL10 was used as positive control. A quantity of 2 × 105 CD8+ T cells was resuspended in RPMI and then loaded to the upper side of the chamber (200 μL/well). After 4 hours, migration was stopped with 0.5 M EDTA and migrated cells were counted in a Neubauer chamber.
Chicken chorioallantoic membrane (CAM) assay.
Fertilized chicken eggs were obtained from Sörries-Trockels. Upon arrival, eggs were incubated in a thermostat under rotation at 37.5°C and 70% humidity for 3 days. For ex ovo culture fertilized eggs were cleaned with 3% Incidin on day 4 of embryonic development and cracked into sterile plastic reservoirs (63). The reservoirs were incubated at 37.5°C and 70% humidity without movement for 6 days to enable development of the embryos. For generation of on-plants on embryonic day 10, Ma-Mel-86c cells (3 × 106) were resuspended in 30 μL culture medium and transferred onto a 3 mm3 gelatin sponge (SMI AG). After the cell solution had been entirely absorbed, 2 cell-soaked sponges were transferred on distant sites of each chicken CAM and subsequently incubated for 4 days until solid tumors had formed. The 2 tumors on the same embryo were treated with either 10 μg 3pRNA or control RNA complexed with in vivo-jetPEI (Polyplus), every 24 hours for 2 consecutive days. Tumors were enclosed with a silicon ring to form a reservoir to constrain the 3pRNA or control RNA solution at the tumor side. A quantity of 1 × 105 T cells (autologous to Ma-Mel-86c) was injected with an omnican insulin syringe (Braun) into the blood vessels of each embryo. Twenty hours after T cell injection the tumors were harvested together with the attached CAM and fixed in 10% formalin.
TCGA melanoma data set.
Harmonized RNA-Seq data for TCGA SKCM (skin cutaneous melanoma) samples (n = 462) were obtained using the TCGAbiolinks package (64) in R (queried on 12/28/2018; project = “TCGA-SKCM”, data.category = “Transcriptome Profiling”, data.type = “Gene Expression Quantification”, workflow.type = “HTSeq - FPKM”). FPKM counts were then converted to TPM (transcript per million) normalized counts. HLA-I APM expression was defined as the geometric mean of the TPM expression for HLA-A, HLA-B, HLA-C, TAP1, TAP2, TAPBP, B2M, LMP2 (PSMB9), and LMP7 (PSMB8). RIG-I (DDX58) and RIG-I (DDX58) pathway gene expression was defined as geometric mean of the TPM (transcript per million) expression for DDX58 and DDX58, IRF1, IRF3, respectively.
Anti–CTLA-4– and anti–PD-1–treated patient cohorts.
Tumor pretreatment RNA-Seq data from the anti–CTLA-4–treated (n = 42) (30) and anti–PD-1–treated (n= 121) (39) patient cohorts were analyzed for differential expression of HLA-I APM genes. As previously described, clinical response was defined using both Response Evaluation Criteria In Solid Tumors (RECIST) and OS criteria. Briefly, clinical responders were defined as patients with a complete or partial response as well as patients with stable disease and OS of at least 1 year. Clinical nonresponders were defined as patients with progressive disease as best response as well as patients with stable disease and OS less than 1 year. As previously described, the long-survival group (n = 5) of the anti–CTLA-4 cohort (30) and 2 samples with a mixed RECIST response in the anti–PD-1 clinical cohort (39) were excluded from differential expression analysis.
Statistics.
For analyses of TCGA RNA-Seq data, high and low HLA-I APM expression groups were defined relative to median expression in the TCGA SKCM cohort. Differences in expression and survival between high and low expression groups were tested using the Mann-Whitney U test and the log-rank test, respectively. For analyses of RNA-Seq data from anti–CTLA-4– and anti–PD-1–treated patient cohorts, statistical testing for differential expression was performed using the Mann-Whitney U test. For survival analyses, high and low HLA-I APM expression groups were defined relative to the median TPM expression across the entire cohort. The difference in survival between high and low expression groups was tested using the log-rank test. For experimental data analyses, data from independent experiments were plotted as means plus standard error of the mean. For comparison between experimental groups, the 2-tailed paired t test was carried out using the GraphPad Prism 5.03 software (GraphPad). Experimental groups were considered to be significantly different at levels of less than P less than 0.05.
Study approval.
Patient samples, including tumor tissue and peripheral blood cells, were collected after written informed consent. Studies on human material were approved by the institutional review board (Ethikkommission, Universitätsklinikum Essen; vote SCABIO_114715). Animal experiments were approved by the Veterinäramt Nordrhein Westfalen (Düsseldorf, Germany) and performed in accordance with the German law for animal protection.
Author contributions
LS conducted experiments, acquired and analyzed data, and wrote the manuscript. FZ, BT, VTKLT, NP, S Howe, HB, HK, CR, and RB performed experiments and acquired and analyzed data. S Horn and JK conducted bioinformatic data analyses. Derek Liu and David Liu performed bioinformatic and statistical data analyses and reviewed the manuscript. CC, TAK, GH, and SF provided critical reagents. AS, SU, and DS collected clinical samples, annotated clinical data, and reviewed the manuscript. US, UD, MS, KSL, JCB, and EMVA reviewed the manuscript. KG and MT edited the manuscript and contributed to experimental design. AP designed the study, reviewed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
This work was partly funded by the Deutsche Krebshilfe (DKH, German Cancer Aid; Translational Oncology 70113455) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; BE 1394/12-1, HO 6389/2-1, PA 2376/1-1, SCHA 422/17-1 [KFO 337]).
Version 1. 05/19/2020
In-Press Preview
Version 2. 07/13/2020
Electronic publication
Version 3. 08/03/2020
Print issue publication
Footnotes
Conflict of interest: AP reports research grant support and provision of reagents from Bristol-Myers Squibb (BMS). EMVA reports advisory relationships and consulting with Tango Therapeutics, Genome Medical, Invitae, Illumina, and Ervaxx; research support from Novartis and BMS; equity in Tango Therapeutics, Genome Medical, Syapse, Ervaxx, and Microsoft; and travel reimbursement from Roche and Genentech. JCB has received speaker honoraria, advisory board honoraria, and/or research funding from Alcedis Amgen, Boehringer Ingelheim, BMS, CureVac, eTheRNA, IQVIA, Merck Serono, Novartis, Pfizer, Sanofi, ReProTher, and 4SC. GH is a cofounder of Rigontec GmbH. KSL is a cofounder of Abalos Therapeutics. DL reports funding by a postdoctoral fellowship from the Society for Immunotherapy of Cancers, which is funded in part by an educational grant from BMS. DS declares grants, personal fees, and/or nonfinancial support from BMS, Roche, Novartis, Regeneron, Sanofi, Merck Sharp & Dohme (MSD), Amgen, 4SC, Merck-EMD, Array, Pierre-Fabre, Philogen, Incyte, and Pfizer. MS has received speaker honoraria, advisory board honoraria, and/or research funding from AstraZeneca, BMS, Boehringer Ingelheim, MSD, Novartis, Pierre Fabre, Roche, and Takeda. AS reports personal fees from Novartis Adboard. SU declares research support, speaker honoraria, advisory board honoraria, and/or travel support from BMS, Merck Serono, MSD, Novartis, and Roche.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(8):4266–4281. https://doi.org/10.1172/JCI131572.
Contributor Information
Lina Such, Email: Lina.Such@gmx.de.
Fang Zhao, Email: fang.zhao@uk-essen.de.
Derek Liu, Email: Derek_Liu@hms.harvard.edu.
Beatrice Thier, Email: beatrice.thier@uk-essen.de.
Vu Thuy Khanh Le-Trilling, Email: Khanh.Le@uk-essen.de.
Antje Sucker, Email: Antje.Sucker@uk-essen.de.
Christoph Coch, Email: ccoch@uni-bonn.de.
Natalia Pieper, Email: natalia-pieper@gmx.de.
Sebastian Howe, Email: seb_howe@yahoo.de.
Hilal Bhat, Email: bhathilal673@gmail.com.
Halime Kalkavan, Email: Halime.Kalkavan@STJUDE.ORG.
Cathrin Ritter, Email: cathrin.ritter@merckgroup.com.
Robin Brinkhaus, Email: Robin.Brinkhaus@hotmail.com.
Selma Ugurel, Email: selma.ugurel@uk-essen.de.
Johannes Köster, Email: johannes.koester@protonmail.com.
Ulrike Seifert, Email: ulrike.seifert@uni-greifswald.de.
Ulf Dittmer, Email: ulf.dittmer@uni-due.de.
Martin Schuler, Email: martin.schuler@uk-essen.de.
Gunther Hartmann, Email: gunther.hartmann@ukb.uni-bonn.de.
Susanne Horn, Email: Susanne.Horn@medizin.uni-leipzig.de.
Soldano Ferrone, Email: SFERRONE@mgh.harvard.edu.
David Liu, Email: David_Liu@DFCI.HARVARD.EDU.
Eliezer M. Van Allen, Email: eliezerm_vanallen@dfci.harvard.edu.
Dirk Schadendorf, Email: Dirk.Schadendorf@uk-essen.de.
Klaus Griewank, Email: klaus.griewank@uk-essen.de.
Mirko Trilling, Email: mirko.trilling@uk-essen.de.
Annette Paschen, Email: annette.paschen@uk-essen.de.
References
- 1.Sucker A, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res. 2014;20(24):6593–6604. doi: 10.1158/1078-0432.CCR-14-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao F, et al. Melanoma lesions independently acquire T-cell resistance during metastatic latency. Cancer Res. 2016;76(15):4347–4358. doi: 10.1158/0008-5472.CAN-16-0008. [DOI] [PubMed] [Google Scholar]
- 4.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran E, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gettinger S, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7(12):1420–1435. doi: 10.1158/2159-8290.CD-17-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sade-Feldman M, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGranahan N, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–1271.e11. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chowell D, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grasso CS, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8(6):730–749. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perea F, et al. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int J Cancer. 2017;140(4):888–899. doi: 10.1002/ijc.30489. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen MH, Hood BL, Beck HC, Conrads TP, Ditzel HJ, Leth-Larsen R. Downregulation of antigen presentation-associated pathway proteins is linked to poor outcome in triple-negative breast cancer patient tumors. Oncoimmunology. 2017;6(5):e1305531. doi: 10.1080/2162402X.2017.1305531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anichini A, et al. Association of antigen-processing machinery and HLA antigen phenotype of melanoma cells with survival in American Joint Committee on Cancer stage III and IV melanoma patients. Cancer Res. 2006;66(12):6405–6411. doi: 10.1158/0008-5472.CAN-06-0854. [DOI] [PubMed] [Google Scholar]
- 14.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105(16):1172–1187. doi: 10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- 16.González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12(2):125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Megger DA, Philipp J, Le-Trilling VTK, Sitek B, Trilling M. Deciphering of the human interferon-regulated proteome by mass spectrometry-based quantitative analysis reveals extent and dynamics of protein induction and repression. Front Immunol. 2017;8:1139. doi: 10.3389/fimmu.2017.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 19.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103(22):8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato H, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlee M, et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31(1):25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo YM, Gale M. Immune signaling by RIG-I-like receptors. Immunity. 2011;34(5):680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bald T, et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4(6):674–687. doi: 10.1158/2159-8290.CD-13-0458. [DOI] [PubMed] [Google Scholar]
- 24.Yu X, et al. Activation of the MDA-5-IPS-1 viral sensing pathway induces cancer cell death and type I IFN-dependent antitumor immunity. Cancer Res. 2016;76(8):2166–2176. doi: 10.1158/0008-5472.CAN-15-2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pansky A, et al. Defective Jak-STAT signal transduction pathway in melanoma cells resistant to growth inhibition by interferon-alpha. Int J Cancer. 2000;85(5):720–725. doi: 10.1002/(SICI)1097-0215(20000301)85:5<720::AID-IJC20>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 26.Respa A, et al. Association of IFN-gamma signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clin Cancer Res. 2011;17(9):2668–2678. doi: 10.1158/1078-0432.CCR-10-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin DS, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sucker A, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440. doi: 10.1038/ncomms15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKendry R, John J, Flavell D, Müller M, Kerr IM, Stark GR. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci USA. 1991;88(24):11455–11459. doi: 10.1073/pnas.88.24.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lutfalla G, et al. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J. 1995;14(20):5100–5108. doi: 10.1002/j.1460-2075.1995.tb00192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixit E, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141(4):668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meissner TB, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA. 2010;107(31):13794–13799. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, et al. Respiratory syncytial virus infection upregulates NLRC5 and major histocompatibility complex class I expression through RIG-I induction in airway epithelial cells. J Virol. 2015;89(15):7636–7645. doi: 10.1128/JVI.00349-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Batran SE, et al. Intratumoral T-cell infiltrates and MHC class I expression in patients with stage IV melanoma. Cancer Res. 2005;65(9):3937–3941. doi: 10.1158/0008-5472.CAN-04-4621. [DOI] [PubMed] [Google Scholar]
- 37.Duewell P, et al. Targeted activation of melanoma differentiation-associated protein 5 (MDA5) for immunotherapy of pancreatic carcinoma. Oncoimmunology. 2015;4(10):e1029698. doi: 10.1080/2162402X.2015.1029698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engel C, et al. RIG-I resists hypoxia-induced immunosuppression and dedifferentiation. Cancer Immunol Res. 2017;5(6):455–467. doi: 10.1158/2326-6066.CIR-16-0129-T. [DOI] [PubMed] [Google Scholar]
- 39.Liu D, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med. 2019;25(12):1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chauvin JM, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manguso RT, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel SJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. doi: 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodig SJ, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. 2018;10(450):eaar3342. doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 44.Donia M, et al. Acquired immune resistance follows complete tumor regression without loss of target antigens or IFNγ signaling. Cancer Res. 2017;77(17):4562–4566. doi: 10.1158/0008-5472.CAN-16-3172. [DOI] [PubMed] [Google Scholar]
- 45.Ugurel S, et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer Immunol Immunother. 2019;68(6):983–990. doi: 10.1007/s00262-019-02341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paulson KG, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. 2018;9(1):3868. doi: 10.1038/s41467-018-06300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshihama S, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci USA. 2016;113(21):5999–6004. doi: 10.1073/pnas.1602069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Odendall C, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15(8):717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasan M, et al. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14(1):61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagato T, Lee YR, Harabuchi Y, Celis E. Combinatorial immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8 T-cell responses against established tumors. Clin Cancer Res. 2014;20(5):1223–1234. doi: 10.1158/1078-0432.CCR-13-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elion DL, et al. Therapeutically active RIG-I agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res. 2018;78(21):6183–6195. doi: 10.1158/0008-5472.CAN-18-0730. [DOI] [PubMed] [Google Scholar]
- 52.Heidegger S, et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci Immunol. 2019;4(39):eaau8943. doi: 10.1126/sciimmunol.aau8943. [DOI] [PubMed] [Google Scholar]
- 53.Taube JM, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiappinelli KB, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith CC, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018;128(11):4804–4820. doi: 10.1172/JCI121476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cañadas I, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018;24(8):1143–1150. doi: 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyi C, et al. Therapeutic immune modulation against solid cancers with intratumoral poly-ICLC: a pilot trial. Clin Cancer Res. 2018;24(20):4937–4948. doi: 10.1158/1078-0432.CCR-17-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi: 10.1038/s41467-018-03705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127(3):761–771. doi: 10.1172/JCI84424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, et al. A method to generate antigen-specific mAb capable of staining formalin-fixed, paraffin-embedded tissue sections. J Immunol Methods. 2005;299(1–2):139–151. doi: 10.1016/j.jim.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Neerincx A, et al. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285(34):26223–26232. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deryugina EI, Quigley JP. Chapter 2. Chick embryo chorioallantoic membrane models to quantify angiogenesis induced by inflammatory and tumor cells or purified effector molecules. Meth Enzymol. 2008;444:21–41. doi: 10.1016/S0076-6879(08)02802-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colaprico A, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.