

Abstract
Aging is associated with a high prevalence of hypertension due to elevated susceptibility of BP to dietary salt, but its mechanism is unknown. Serum levels of Klotho, an anti-aging factor, decline with age. We found that high salt (HS) increased BP in aged mice and young heterozygous Klotho-knockout mice and was associated with increased vascular expression of Wnt5a and p-MYPT1, which indicate RhoA activity. Not only the Wnt inhibitor LGK974 and the Wnt5a antagonist Box5 but Klotho supplementation inhibits HS-induced BP elevation, similarly to the Rho kinase inhibitor fasudil, associated with reduced p-MYPT1 expression in both groups of mice. In cultured vascular smooth muscle cells, Wnt5a and angiotensin II (Ang II) increased p-MYPT1 expression but knockdown of Wnt5a with siRNA abolished Ang II–induced upregulation of p-MYPT1, indicating that Wnt5a is indispensable for Ang II–induced Rho/ROCK activation. Notably, Klotho inhibited Wnt5a- and Ang II–induced upregulation of p-MYPT1. Consistently, Klotho supplementation ameliorated HS-induced augmentation of reduced renal blood flow (RBF) response to intra-arterial infusion of Ang II and the thromboxane A2 analog U46619, which activated RhoA in both groups of mice and were associated with the inhibition of BP elevation, suggesting that abnormal response of RBF to Ang II contributes to HS-induced BP elevation. Thus, Klotho deficiency underlies aging-associated salt-sensitive hypertension through vascular non-canonical Wnt5a/RhoA activation.
Keywords: Nephrology, Vascular Biology
Keywords: Hypertension
Introduction
Aging is a progressive accumulation of changes in the body that increases susceptibility to disorders such as Alzheimer’s disease, cerebrovascular disease, diabetes, and cardiovascular disease, including hypertension. Hypertension is the most important risk factor for death and disability worldwide (1). The incidence of hypertension is greatest among older adults; accordingly, aging is a potent and universal risk factor for cardiovascular disease, including heart attack, stroke, and heart failure (2). BP increases with age in most modern countries, possibly owing to elevated sensitivity of BP to dietary salt with aging (3, 4). Supporting this hypothesis, aging-associated BP elevation is not detected in the Yanomami people, a culture that does not consume salt (5, 6), suggesting that dietary salt is indispensable for aging-induced BP elevation and hypertension. Several investigators have reported plausible mechanisms for salt-sensitive hypertension in rodent models of obesity and hereditary hypertension (7–10). However, the mechanism underlying elevated susceptibility to dietary salt with aging remains unknown.
The anti-aging factor Klotho (11), which is mainly produced in the kidney, is secreted into the blood, and circulating Klotho acts as a hormone (12). The serum level of Klotho decreases with age (13), and Klotho deficiency induces vascular aging and arterial stiffness (14). Moreover, a recent study showed the inverse relationship between serum Klotho concentration and BP salt sensitivity in hypertensive patients (15), suggesting that salt-sensitive hypertension in the elderly is attributable to Klotho deficiency. In keeping with the mechanism of salt-sensitive hypertension induced by Klotho deficiency, homozygous Klotho-knockout mice showed the activation of Wnt signaling (16), indicating that endogenous Klotho serves as an antagonist of Wnt. Wnt signaling regulates diverse cellular responses during embryonic development, as well as various physiological and pathological processes in the adult, including mammalian aging. The pathway is generally dissected into 2 major pathways: the canonical (β-catenin–dependent) and non-canonical (β-catenin–independent) pathways, which have various branches (17). The canonical pathway requires Wnt ligand binding to frizzled (FZD) receptors and LDL receptor–related protein 5/6 (LRP5/6) coreceptors to initiate intracellular signaling via β-catenin nuclear translocation. On the other hand, the non-canonical pathway needs a ligand-receptor complex of Wnt, FZD, and receptor tyrosine kinase–like orphan receptor 1/2 (Ror1/2) to promote cell differentiation, migration, and polarization (18, 19) through small GTPases such as Rho, Rac, and Cdc42 (20, 21). Among the 19 Wnt family members, Wnt3a and Wnt5a activate the canonical and non-canonical Wnt pathways, respectively (22).
Notably, the aging phenotype in hematopoietic stem cells is induced by a shift from canonical Wnt signaling to non-canonical Wnt5a signaling (23). RhoA, which mediates key features of non-canonical Wnt signaling (21), is activated by Wnt5a (24), and aging enhances thromboxane A2–induced aortic contraction through activating RhoA (25). High salt augments the calcium-sensitization pathway through activation of RhoA/Rho kinase (ROCK) in rat aorta (26). Moreover, vasoconstriction mediated by the leukemia-associated Rho guanine nucleotide exchange factor G12/13-LARG via Rho/ROCK activation is required for the development of salt-sensitive hypertension (27). Together, these findings led us to hypothesize that high salt activates the Wnt5a/RhoA pathway in aging vasculature under Klotho deficiency and causes aging-associated hypertension through increased BP salt sensitivity.
Our initial purpose was to clarify the mechanism of increased BP salt sensitivity with age and the development of hypertension in the elderly. We show here that (a) dietary salt induces vascular non-canonical Wnt5a/RhoA activation under serum Klotho deficiency in aged mice, associated with salt-sensitive hypertension, but supplementation of Klotho suppresses salt-induced activation of the vascular Wnt5a/RhoA pathway, resulting in the inhibition of salt-sensitive hypertension; (b) activation of Rho/ROCK induced by angiotensin II (Ang II), an important vasoactive substance for BP regulation, depends on Wnt5a in in vitro study using vascular smooth muscle cells (VSMCs), and Ang II–induced activation of RhoA is inhibited by Klotho; (c) dietary high salt induces remarkable renal blood flow (RBF) reduction in response to intra-arterial infusion of Ang II in aged mice, but Klotho supplementation prevents it; and (d) not only the Rho inhibitor and Wnt inhibitor but Klotho supplementation can effectively suppress salt-induced hypertension under Klotho deficiency. In this series of studies, similar results were observed in young heterozygous Klotho-knockout (KL-KO) mice, which also have reduced serum levels of Klotho, and these results support the hypothesis that Klotho deficiency underlies the development of aging-associated salt-sensitive hypertension.
Results
Klotho deficiency induces salt-sensitive hypertension in aged WT and young KL-KO mice.
We examined serum levels of Klotho among aged WT (66–78 weeks old), young WT (18 weeks old), and young KL-KO (18 weeks old) mice fed a normal-salt (NS) diet. Aged WT mice had lower serum levels of soluble Klotho than young WT mice (Figure 1A), as previously reported (28). The levels of soluble Klotho in aged WT mice were similar to those in young KL-KO mice (Figure 1A). In agreement with previous reports indicating that the source of circulating Klotho is the kidney (29, 30), we also found that Klotho content was abundant in the kidney, but that it was scanty in the aorta (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI134431DS1), and that renal Klotho content, as well as serum Klotho, was lower in aged WT mice and young KO mice than in young WT mice, but that neither renal nor serum Klotho concentrations were changed by a high-salt (HS) diet (Supplemental Figure 1, B and C).
Figure 1. Dietary salt develops hypertension in Klotho-deficient mice.

(A) Left panel: Relative serum Klotho concentrations in young WT (n = 10), aged WT (n = 7), and young heterozygous Klotho-KO (KL-KO) mice (n = 8) fed a normal-salt (NS) diet. *P < 0.05 vs. young WT. Middle and right panels: Effects of Klotho supplementation (KL) on serum Klotho in aged WT (middle panel, n = 8 each) and KL-KO mice fed a high-salt (HS) diet (right panel, n = 9, 10, and 9 each from left to right). *P < 0.05 vs. aged WT HS diet–fed or KL-KO HS diet–fed mice. (B) Mean arterial BP (MBP) measured by telemetry in young WT (top), aged WT (middle), and KL-KO mice (bottom) that received NS diet, HS diet, or HS diet + KL. Arrows show the onset of the HS diet. Right panels show average MBP over a 24-hour period. n = 4 each. *P < 0.05 vs. HS diet–fed mice. Data are summarized as means ± SEM. Unpaired t tests were used for comparisons between 2 groups. For multiple comparisons, statistical analysis was performed using the Tukey-Kramer post hoc test. N.S., not significant.
In further studies, we tried to compare and examine levels of serum Klotho in aged WT and young KL-KO mice versus in young WT mice. In the early period of an HS diet, the BP of aged WT mice began to increase (Figure 1B, middle), but that of young WT mice did not (Figure 1B, top). On the second day of the HS diet, BP in HS diet–fed aged WT mice reached a plateau, and at the end of 2 weeks of the HS diet, the BP level was significantly higher than that in NS-fed aged WT mice (Figure 1B, middle), indicating the development of salt-sensitive hypertension in aged WT mice. Accordingly, an HS diet increased BP in young KL-KO mice as well as aged WT mice (Figure 1B, bottom), suggesting that Klotho deficiency underlies the increased salt sensitivity of BP. To further evaluate the role of Klotho deficiency in salt-sensitive hypertension, we supplemented Klotho to the HS-fed mice by in vivo transfection with a pcDNA3.1+/C-(K) DYK plasmid vector encoding full-length mouse α-Klotho using hemagglutination virus of Japan (HVJ) envelope vector, and found that Klotho supplementation, followed by successfully increased serum Klotho concentration (Figure 1A), prevented salt-induced BP elevation in aged WT mice and young KL-KO mice (Figure 1B). These results revealed that the development of salt-sensitive hypertension in aged WT mice is solely due to Klotho deficiency.
Vascular Rho/ROCK activation plays an important role in developing salt-sensitive hypertension.
To clarify the vascular mechanism of salt-sensitive hypertension induced by Klotho deficiency, we studied the vasoconstrictor response using the iliac artery. There were no significant differences in vasoconstrictive response to U46619, a thromboxane A2 analog that induces vasoconstriction by Rho/ROCK activation (31), among the iliac arteries of the 3 NS diet–fed mouse groups: young WT, aged WT, and young KL-KO mice (Figure 2A). However, an HS diet significantly increased vasoconstrictive responses to U46619 in aged WT and KL-KO mice, although it did not significantly change, but rather decreased, the response to U46619 in young WT mice (Figure 2A). In contrast with U46619, vasoconstrictive response to phenylephrine, which induces vasoconstriction mainly through the increased intracellular Ca2+ concentration (27), was not affected by HS diet among the 3 groups of HS diet–fed mice (Figure 2B). These results imply that HS activates RhoA specifically in vasculatures of the 2 Klotho-deficient groups of mice.
Figure 2. Dietary salt induces vasoconstriction with Rho activation under Klotho deficiency.

(A) Vasoconstrictive responses to U46619, a thromboxane A2 analog, in the iliac arteries of young WT, aged WT, and heterozygous KL-KO mice receiving NS diet, HS diet, or HS diet + KL. n = 6 each. *P < 0.05 vs. HS diet–fed mice. (B) Vasoconstrictive response to phenylephrine was not affected by HS in the iliac arteries of young WT, aged WT, and young KL-KO mice fed NS diet or HS diet. n = 6 each. No significant differences were observed between HS diet– and NS diet–fed mice in all groups. (C) p-MYPT1 expression in response to U46619 in the iliac arteries of aged WT mice and KL-KO mice fed NS or HS diets, relative to the level in young WT mice. n = 9 each. *P < 0.05 vs. young WT, aged WT, or KL-KO fed NS diet. (D) p-MYPT1 expression in response to U46619 in the iliac arteries of aged WT and KL-KO mice receiving NS diet, HS diet, or HS diet + KL. n = 6 each. *P < 0.05 vs. HS. Data are summarized as means ± SEM. Unpaired t tests were used for comparisons between 2 groups. For multiple comparisons, statistical analysis was performed using the Tukey-Kramer post hoc test. N.S., not significant.
Consistently with changes in vasoconstrictive response to U46619 with HS, the protein expression of phospho–myosin phosphatase target subunit (p-MYPT1), a marker of Rho/ROCK kinase activity, stimulated by U46619 was significantly increased by an HS diet in aged WT and KL-KO mice, but not in young WT mice (Figure 2C). Similar results were observed in salt-induced changes in active RhoA measured by pull-down assay (Supplemental Figure 2). These findings led us to the hypothesis that salt-induced BP elevation is mediated by Rho/ROCK activation in aged WT and KL-KO mice. Supporting this hypothesis, treatment with fasudil, a Rho/ROCK inhibitor, attenuated salt-induced BP elevation in aged WT and young KL-KO mice (Figure 3A), associated with reduced p-MYPT1 expression (Figure 3B), but fasudil did not affect BP in salt-loaded young mice (Supplemental Figure 3). Moreover, Klotho supplementation normalized the increased vasoconstrictive response to U46619 with HS in aged WT mice and KL-KO mice (Figure 2A), associated with reduced p-MYPT1 expression (Figure 2D), and resulted in the inhibition of salt-induced BP elevation (Figure 1B).
Figure 3. Treatment with fasudil, a Rho/ROCK inhibitor, suppresses salt-induced hypertension and elevation of p-MYPT1 expression in aged WT and heterozygous KL-KO mice.

(A) Circadian and average MBP of HS diet and HS diet + fasudil in aged WT (left) and KL-KO mice (right). n = 4 each. *P < 0.05 vs. HS diet–fed mice. (B) p-MYPT1 expression in response to U46619 in the iliac arteries of aged WT and KL-KO mice fed HS diet or HS diet + fasudil. n = 6 each. *P < 0.05 vs. HS diet–fed mice. Data are summarized as means ± SEM. Unpaired t tests were used for comparisons between 2 groups.
Non-canonical Wnt/RhoA pathway plays a key role in the development of salt-sensitive hypertension under Klotho deficiency.
Klotho serves as a potent suppressor of Wnt signaling (16). According to the mechanism of salt-induced RhoA activation by Klotho deficiency, it is possible that Wnt signaling regulates RhoA activity through the non-canonical Wnt pathway (32, 33). We next evaluated the effect of HS on the canonical and non-canonical Wnt pathways in aged WT and KL-KO mice. On the NS diet, active β-catenin expression in the aorta was significantly higher in aged WT mice than in young WT mice, in contrast with normal expression in KL-KO mice (Figure 4A). On the HS diet, active β-catenin expression in the aorta further increased in aged WT mice and also increased in KL-KO mice, but did not increase in young WT mice. Changes in dishevelled (Dvl) protein expression are in parallel with those in active β-catenin (Supplemental Figure 4A), possibly upon the formation of a Dvl–β-catenin complex induced by Wnt signaling. In contrast with those in Dvl, changes in dishevelled-associated activator of morphogenesis 1 (Daam1) and Ror2 expression (Supplemental Figure 4B) were consistent with those in p-MYPT1 expression (Figure 2C), but not in active β-catenin (Figure 4A). Together with the evidence that Dvl activation, as a branch point and an essential component of both arms of Wnt signaling, is involved in canonical and non-canonical signaling, but Ror2 activation and Daam1 activation are involved only in non-canonical Wnt signaling and induce RhoA activation through the formation of a Dvl-Daam1-RhoA complex by Ror2 (34, 35), these results strongly support our hypothesis that HS activates vascular RhoA through the non-canonical Wnt pathway.
Figure 4. Aortic active β-catenin expression increases in aged WT mice and is enhanced by HS diet.

Not β-catenin inhibitor but Wnt inhibitor suppressed salt-induced hypertension and vascular p-MYPT1 expression in HS diet–fed aged WT and heterozygous KL-KO mice. (A) Active β-catenin expression in the aortae of aged WT (left) and KL-KO mice (right) fed NS diet or HS diet, relative to the level in young WT mice. n = 9. *P < 0.05 vs. young WT, aged WT, or KL-KO fed NS diet. (B) Effects of LGK974 (blue) and ICG-001 (orange) on circadian changes in MBP in aged WT (left) and KL-KO mice fed HS diet (right). Right panels show average MBP. n = 4 each. *P < 0.05 vs. HS diet + LGK. (C and D) Effects of ICG-001 (C) and LGK974 (D) on p-MYPT1 expression stimulated by U46619 in the iliac arteries of aged WT and KL-KO mice fed HS diet. (C) Aged WT fed HS diet, n = 7; aged WT fed HS diet + ICG, n = 8; KL-KO fed HS diet and KL-KO fed HS diet + ICG, n = 6 each. (D) Aged WT fed HS diet and aged WT fed HS + LGK, n = 7 each. KL-KO fed HS diet and KL-KO fed HS diet + LGK, n = 8 each. *P < 0.05 vs. HS. Data are means ± SEM. Unpaired t tests were used for comparisons between 2 groups. For multiple comparisons, statistical analysis was performed using the Tukey-Kramer post hoc test. N.S., not significant.
There is an intricate relationship between Klotho and fibroblast growth factor 23 (FGF23), since the FGF23-KO mouse showed a phenotype similar to that of the KL-KO mouse (36). Given the crosstalk between FGF23 and Wnt/β-catenin signaling (37), moreover, we measured serum FGF23 concentration in mice fed NS and HS diets. Neither significant differences in serum FGF23 among young WT, aged WT, and young KL-KO mice fed the NS diet, nor changes in serum FGF23 with the HS diet (Supplemental Figure 5), suggest that salt-induced activation of the Wnt/RhoA pathway under Klotho deficiency is independent of FGF23.
In order to evaluate whether salt-induced BP elevation is mediated by the canonical Wnt/β-catenin pathway or the non-canonical Wnt/RhoA pathway, we investigated the effects of the β-catenin inhibitor ICG-001 (38) and the Wnt canonical/non-canonical inhibitor LGK974 (39) on vascular p-MYPT1 expression and BP in HS-fed aged WT and KL-KO mice. ICG-001 did not affect p-MYPT1 expression (Figure 4C) or BP (Figure 4B) in both HS-fed aged WT and KL-KO mice. In contrast with ICG-001, LGK974 could abolish salt-induced BP elevation in both mice (Figure 4B), and was associated with significant reduction of p-MYPT1 expression (Figure 4D). These findings indicate that HS-induced BP elevation can be attributed to activation of the non-canonical Wnt/RhoA pathway in the vasculature.
Wnt5a induces RhoA activation in human VSMCs.
Next, we evaluated the expression of Wnt family members in the aorta. Protein levels of Wnt3a, Wnt5a, and Wnt11 were significantly elevated by an HS diet in aged WT and KL-KO mice, whereas the levels of Wnt1 and Wnt4 were not (Figure 5, A and B, and Supplemental Figure 6). Notably, the expression patterns of Wnt3a (Figure 5A) and Wnt5a (Figure 5B) were similar to those of active β-catenin (Figure 3A) and p-MYPT1 (Figure 2C), respectively. In keeping with the previous study indicating that Wnt3a activates the Wnt canonical pathway (40), we found increased Wnt3a expression as well as active β-catenin expression in NS diet–fed aged WT mice, despite no increase in NS diet–fed KL-KO mice (Figure 5A). An HS diet further increased Wnt3a expression in aged WT mice, and also increased it in KL-KO mice. In contrast with Wnt3a, Wnt5a expression did not increase until the HS diet was fed to both groups of mice (Figure 5B), similarly to p-MYPT1 expression (Figure 2C). The present findings of the parallel changes either in Wnt3a and active β-catenin (Figure 4A) expression or in Wnt5a and p-MYPT1 expression (Figure 2C) led us to further investigate the individual roles of Wnt5a and Wnt3a in canonical and non-canonical Wnt pathways in an in vitro experiment using cultured VSMCs.
Figure 5. The expression patterns of vascular Wnt3a and Wnt5a are similar to those of active β-catenin and p-MYPT1 in aged WT and heterozygous KL-KO mice.

(A and B) Wnt3a (A) and Wnt5a (B) expression of aortae in aged WT (left) and KL-KO mice (right) fed NS or HS diet. n = 9 each. *P < 0.05 vs. young WT, aged WT, or KL-KO fed NS diet. Data are summarized as means ± SEM. For multiple comparisons, statistical analysis was performed using the Tukey-Kramer post hoc test. N.S., not significant.
In keeping with Wnt3a as an activator of the Wnt canonical pathway, recombinant (r) Wnt3a increased active β-catenin expression in cultured hVSMCs (Figure 6A). However, rWnt3a decreased p-MYPT1 expression (Figure 6B), consistently with the results of previous studies indicating that Wnt3a inhibited RhoA activation in platelets (41). In contrast to Wnt3a, rWnt5a did not change active β-catenin (Figure 6A), but increased p-MYPT1 expression (Figure 6B), as previously reported (32, 33). Thus, the respective effect of Wnt3a and Wnt5a on Wnt canonical β-catenin and non-canonical RhoA is mutually exclusive. Notably, the transfection of siRNA Wnt5a (Supplemental Figure 7) clearly decreased p-MYPT1 expression (Figure 6C), suggesting that not only exogenous but also endogenous Wnt5a increases RhoA activity. Moreover, the knockdown of Wnt5a with siRNA augmented the rWnt5a-induced increase in p-MYPT1 expression, the degree of which was significantly higher than that of the increase in cells with negative control siRNA (Figure 6C vs. Figure 6B). However, the treatment of Klotho inhibited the rWnt5a-induced increase in p-MYPT1 expression (Figure 6D).
Figure 6. Wnt5a- and Ang II–induced upregulation of p-MYPT1 in cultured human VSMCs.

(A) Recombinant (r) Wnt3a increased active β-catenin expression in cultured hVSMCs, but rWnt5a did not. n = 6 each. *P < 0.05 vs. control (Con). (B) rWnt3a decreased p-MYPT1 expression, whereas rWnt5a increased it. n = 6 each. *P < 0.05 vs. Con or rWnt5a. (C) Knockdown of Wnt5a with siRNA decreased p-MYPT1 expression relative to the negative control siRNA (siNC) (left), but the addition of rWnt5a markedly reversed it (right). n = 6 each. *P < 0.05. (D) rKlotho (KL) inhibited rWnt5a-induced upregulation of p-MYPT1 in cells with (right) or without siWnt5a (left). n = 6 each. *P < 0.05 vs. Con or rWnt5a + KL. (E) Effects of siWnt5a on Ang II–induced increase in p-MYPT1 expression. siWnt5a significantly decreased basal (third from left) and Ang II–induced (fourth) expression of p-MYPT1, but the decrease was reversed by rWnt5a (fifth); the additional KL again inhibited p-MYPT1 expression (sixth). n = 6 each. *P < 0.05 vs. siNC-Con or siWnt5a + Ang II + Wnt5a. Data are means ± SEM. Unpaired t tests were performed on comparisons between 2 groups. For multiple comparisons, statistical analysis was performed by Tukey-Kramer post hoc test. N.S., not significant.
Wnt5a dominantly contributes to salt-sensitive hypertension induced by Klotho deficiency through the non-canonical Wnt pathway.
In order to evaluate the involvement of Wnt5a in HS-induced RhoA activation and salt-sensitive hypertension, we investigated the effect of the Wnt5a antagonist Box5 (42) on vascular p-MYPT1 expression and BP in HS diet–fed aged WT and KL-KO mice. Treatment with Box5 attenuated the HS diet–induced increase in vascular p-MYPT1 expression in aged WT and KL-KO mice (Figure 7B), and was associated with the inhibition of salt-induced BP elevation (Figure 7A), despite little effect of Box5 on BP in the salt-loaded young WT mice (Supplemental Figure 3). The suppressive effect of Box5 was apparently weaker than that of LGK974 (Figure 4B); the average percentage decreases in mean BP were 6.1% and 6.7% by Box5 versus 10.8% and 9.3% by LGK974 in HS diet–fed aged WT and KL-KO mice, respectively. Given that the Wnt inhibitor LGK inhibits all Wnts, including Wnt5a and Wnt11, through the inhibition of porcupine (39), Wnt11 may also be involved in the development of salt-sensitive hypertension under Klotho deficiency. Indeed, expression of Wnt11 as well as Wnt5a significantly increased in HS diet–fed aged mice and KL-KO mice (Supplemental Figure 6). However, the present finding of definite inhibition by Box5 of vascular Rho/ROCK activation and salt-sensitive hypertension indicates that Wnt5a is mainly involved in developing salt-induced BP elevation in the 2 Klotho-deficient mouse models and provides evidence that the novel Klotho/Wnt5a/RhoA pathway plays a crucial role in aging-associated salt-sensitive hypertension.
Figure 7. Treatment with Wnt5a antagonist Box5 suppresses salt-induced hypertension and elevation of p-MYPT1 expression in aged WT and heterozygous KL-KO mice.

(A) Effects of Box5 treatment on circadian and average MBP in aged WT (left) and KL-KO mice with HS diet (right). n = 4 each. *P < 0.05 vs. HS diet–fed mice. (B) Effects of Box5 on p-MYPT1 expression stimulated by U46619 in the iliac arteries of aged WT (left) and KL-KO mice with HS diet (right). Aged WT fed HS diet, n = 7; aged WT fed HS diet + Box5, n = 8; KL-KO fed HS diet and KL-KO fed HS diet + Box5, n = 7 each. *P < 0.05 vs. HS diet–fed mice. Data are means ± SEM. Unpaired t tests were performed on comparisons between 2 groups.
Ang II–induced activation of the Wnt5a/RhoA pathway is suppressed by Klotho in cultured hVSMCs.
Ang II increases not only intracellular Ca2+ entry through G protein–coupled receptor Gq/11, but also Ca2+ sensitivity due to Rho/ROCK activation through G12/13 (27, 43), and leads to vasoconstriction; therefore, we investigated the involvement of Wnt5a in Ang II–induced RhoA activation in cultured hVSMCs. In the present study, Ang II increased p-MYPT1 expression in cultured hVSMCs (Figure 6E). Notably, knockdown of Wnt5a with siRNA abolished any Ang II–induced increase in p-MYPT1 expression, but the additional treatment of rWnt5a reversed this effect, suggesting that Wnt5a is indispensable for Ang II–induced activation of vascular Rho/ROCK. However, Klotho treatment suppressed Ang II–induced increase in p-MYPT1 expression in the presence of Wnt5a (Figure 6E).
Klotho reverses Ang II– and U46619-induced RBF reduction and BP elevation in HS diet–fed aged mice and KL-KO mice.
Ang II is a vasoactive substance that plays a key role in controlling vascular tone in renal vasculatures and RBF (44). Given the important role of RBF reduction in salt-sensitive hypertension (45, 46), we studied the response of RBF to Ang II and U46619 in the 2 Klotho-deficient mouse models fed an HS diet. We administered Ang II as bolus injections from the origin of the right renal artery in anesthetized aged WT mice and KL-KO mice. The injection of Ang II decreased RBF in both NS diet–fed aged WT and KL-KO mice, but responses were moderately augmented by HS in both groups (Figure 8, B and C), concomitant with a greater increase in BP (Figure 9, B and C). In contrast, changes in RBF and BP with intra-arterial injection of Ang II did not differ between NS diet–fed and HS diet–fed young WT mice (Figure 8A and Figure 9A). Notably, the increased responses to U46619, similarly to responses to Ang II, were observed in HS diet–fed aged WT mice and KL-KO mice (Figure 8, A–C, and Figure 9, A–C). Finally, Klotho supplementation reversed the augmented responses to Ang II and U46619 of RBF reduction and BP elevation (Figure 8, B and C, and Figure 9, B and C) in HS diet–fed aged WT and KL-KO mice. These results suggest that salt-induced augmentation of RBF response to Ang II is mediated by Rho/ROCK activation in aged WT and KL-KO mice, and strongly support our hypothesis that the Wnt5a/RhoA pathway plays a key role in the development of salt-sensitive hypertension in Klotho-deficient aged mice, possibly through reduced RBF in combination with elevated systemic vascular resistance (Figure 10).
Figure 8. Klotho supplement normalizes augmented response of RBF to Ang II and U46119 in salt-loaded aged WT mice and heterozygous KL-KO mice.

(A) Percent changes in RBF with the intra-arterial injection of Ang II (left) or U46619 (right) in young WT mice with NS or HS diet. Bar graphs show the area under the curve of changes in RBF compared with the baseline. For Ang II, n = 6 each; for U46619, n = 5 each. N.S., not significant. (B) Percent changes in RBF upon intra-arterial injection of Ang II (left) or U46619 (right) in aged WT mice receiving NS, HS, or HS diet + Klotho supplementation. Bar graphs show the area under the curve of changes in RBF relative to the baseline. n = 4 each. *P < 0.05 vs. HS diet–fed mice. (C) Percent changes in RBF upon intra-arterial injection of Ang II (left) or U46619 (right) in KL-KO mice with NS diet, HS diet, or HS diet + KL. Bar graphs show area under the curve of changes in RBF relative to the baseline. Ang II, n = 6 each; U46619, n = 6 (NS diet), n = 5 (HS diet), and n = 5 (HS diet + KL). *P < 0.05 vs. HS diet–fed mice. Data are means ± SEM. Unpaired t tests were performed on comparisons between 2 groups. For multiple comparisons, statistical analysis was performed by Tukey-Kramer post hoc test.
Figure 9. Klotho supplement normalizes Ang II– and U46619-induced BP elevation in salt-loaded aged mice and heterozygous KL-KO mice.

Changes in MBP from the baseline during injection of Ang II or U46619 in young WT (A) (Ang II, n = 6 each; U46619, n = 5 each), aged WT (B) (n = 4 each), and KL-KO (C) mice (Ang II, n = 6 each; U46619, n = 6 [NS diet], n = 5 [HS diet], and n = 5 [HS diet + KL]) with NS diet, HS diet, or HS diet + KL. *P < 0.05 vs. HS diet–fed mice. Data are means ± SEM. Unpaired t tests were performed on comparisons between 2 groups. For multiple comparisons, statistical analysis was performed by Tukey-Kramer post hoc test. N.S., not significant.
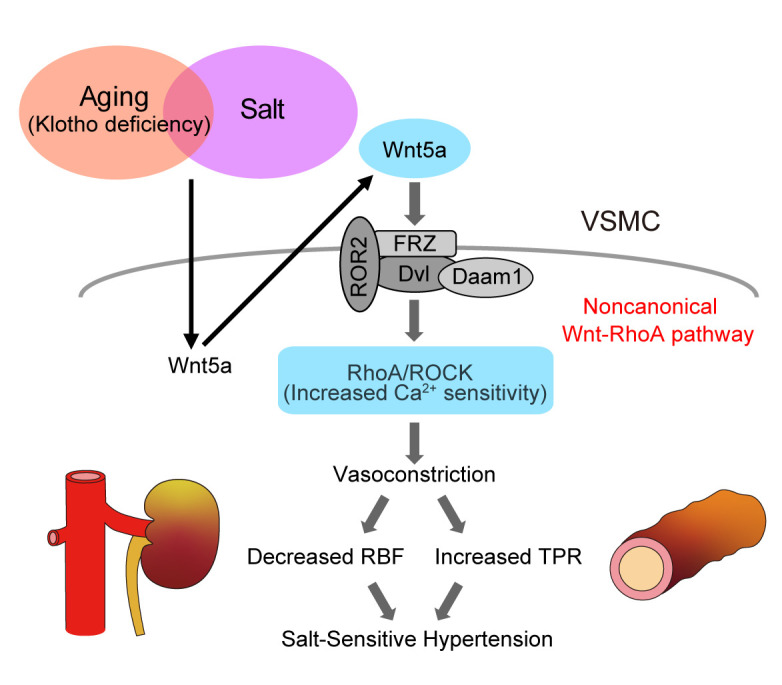
Figure 10. Hypothetical cartoon.

Activation of vascular Rho kinase via the non-canonical Wnt pathway contributes to salt-sensitive hypertension by Klotho deficiency. VSMCs, vascular smooth muscle cells; FRZ, frizzled receptor; Dvl, dishevelled; Daam1, dishevelled-associated activator of morphogenesis 1; ROR2, receptor tyrosine kinase–like orphan receptor 2; ROCK, Rho-associated coiled-coil–containing protein kinase; RBF, renal blood flow; TPR, total peripheral resistance.
Discussion
We present evidence demonstrating that Klotho deficiency underlies the development of salt-sensitive hypertension in aged mice through activation of the non-canonical Wnt5a/RhoA pathway (Figure 10). Specifically, we show that an HS diet increases vasoconstrictive response to U46619 associated with upregulation of p-MYPT1 expression in aged WT and young KL-KO mice, and in turn, vascular Rho/ROCK activation results in salt-induced BP elevation, possibly through vasoconstriction. Supporting this hypothesis, treatment with the ROCK inhibitor fasudil abolished salt-induced BP elevation. Moreover, we found a salt-induced increase in Wnt5a expression in parallel with that of p-MYPT in aged WT and KL-KO mice, and treatment with either the Wnt inhibitor LGK974 or the Wnt5a antagonist Box5 attenuated salt-induced BP elevation with reduced p-MYPT1 expression, implying that the non-canonical Wnt5a/RhoA pathway is involved in salt-induced BP elevation under Klotho deficiency. Notably, knockdown of Wnt5a with siRNA abolished Ang II–induced increase in p-MYPT1 expression in cultured hVSMCs, but this effect was reversed by pretreatment of rWnt5a. This result reveals that Wnt5a is essential for Ang II–induced Rho/ROCK activation. Moreover, consistently with the inhibition of Ang II–induced Rho activation by Klotho in hVSMCs, Klotho supplementation inhibited the augmented response to Ang II and U46619 of RBF reduction in HS diet–fed aged WT mice and young KL-KO mice, resulting in the absence of salt-induced BP elevation. These findings led us to the plausible hypothesis that salt-induced activation of the non-canonical Wnt5a/RhoA pathway, under the condition of circulating Klotho deficiency, contributes to the development of aging-associated salt-sensitive hypertension, possibly through decreased RBF in addition to increased total peripheral resistance (TPR).
According to the mechanism of BP salt sensitivity, previous studies have been focused on renal function (Guyton’s theory), and suggest that impaired renal capacity to excrete sodium in urine (47, 48) plays a key role in the development of salt-sensitive hypertension. Indeed, dietary HS increased cardiac output and BP in salt-sensitive hypertensive patients through sodium retention (49). However, BP elevation during salt repletion in salt-sensitive subjects was associated with a failure to lower TPR adequately when cardiac output rose (50, 51), and this finding led us to the plausible hypothesis that increased vascular resistance is more important than sodium retention for salt-induced BP elevation (52). Indeed, salt-induced BP elevation is associated with increased TPR in elderly hypertensive patients (53). It should be noted, however, that renal vascular resistance also increased in salt-loaded patients with salt-sensitive hypertension. Salt-sensitive hypertensive patients manifest a deranged renal hemodynamic adaptation to a dietary HS intake. On a low-salt diet, salt-sensitive and salt-resistant patients have similar RBF, but with HS intake RBF increases in salt-resistant but decreases in salt-sensitive patients (54, 55). Given the important role of Ang II in renal vascular contraction and RBF regulation, Ang II, but not phenylephrine, causes significant renal vasoconstriction in conscious rats (44), and an HS diet increases the response of RBF reduction to intrarenal injection of Ang II but not to norepinephrine in dogs (46). In the present study, an HS diet caused augmented responses of RBF reduction and BP elevation to intra-arterial injection of Ang II in both Klotho-deficient aged mice and KL-KO mice (Figure 8, B and C, and Figure 9, B and C), but these abnormal responses were reversed by Klotho supplementation. Interestingly, Ang II causes hypertension through the actions of Ang II type 1a (AT1a) receptors in the kidney, including renal vasculatures that reduce urinary sodium excretion, and the absence of AT1a receptors in the kidney alone is sufficient to protect from Ang II–dependent hypertension (56). Thus, it is possible that the abnormal response of RBF to Ang II, in addition to elevated TPR, contributes to salt-induced BP elevation under Klotho deficiency, although we did not measure central hemodynamics or urinary sodium excretion.
In keeping with the vascular mechanism for salt-sensitive hypertension induced by Klotho deficiency, HS increased the response to the thromboxane A2 analog, which induced vasoconstriction through Ca2+ sensitization that was mediated by Rho/ROCK activation in both Klotho-deficient aged WT and KL-KO mice. In contrast, the vasoconstrictive response to phenylephrine was not affected by salt loading in these mice, indicating salt-induced activation of RhoA specifically. The contraction of VSMCs is well known to depend on intracellular Ca2+ concentration and Ca2+ sensitization regulated by signaling pathways mediated by G protein receptors Gq/11 and G12/13, respectively. Wirth et al. reported that both Gq/11-mediated and G12/13-mediated signaling pathways in smooth muscle cells are required for salt-induced increase in vascular resistance and development of salt-sensitive hypertension (27). Interestingly, Gq/11-mediated signaling is necessary to maintain basal BP, but G12/13-mediated signaling is involved in the development of salt-induced hypertension without alteration of normal BP regulation. Wirth et al. also showed that phenylephrine α1-adrenergic effects on smooth muscle cells are exclusively mediated by Gq/11, whereas the vasoconstrictive effect of thromboxane A2 analog is due to the G12/13/RhoA pathway, which is intimately involved in salt-sensitive hypertension. Thus, our data imply that salt-induced activation of RhoA is mediated by the G12/13 pathway in Klotho-deficient aged WT and KL-KO mice.
Wnt proteins, which play a key role in mammalian aging, are a large family of cysteine-rich secreted molecules that are also essential in embryonic induction, cell polarity generation, and cell fate specification in various species (18). The canonical Wnt/β-catenin pathway is best understood to induce transcription of many target genes. Indeed, vascular expression of active β-catenin and Wnt3a was, in parallel, upregulated in aged mice on the NS diet and further increased by salt loading, but the β-catenin inhibitor ICG-001 did not affect salt-induced BP elevation in salt-loaded aged WT mice. In contrast, the Wnt inhibitor LGK974 normalized BP, and was associated with significant reduction of increased p-MYPT1 expression. Thus, the non-canonical Wnt pathway plays a crucial role in salt-dependent hypertension by Klotho deficiency in this study. Salt-induced upregulation of Wnt5a and p-MYPT1 also appeared, in parallel, in both aged WT mice and KL-KO mice. Wnt5a is one of the Wnt proteins that activate a β-catenin–independent pathway, which activates small G proteins, including Rac1 and RhoA, JNK, and Rho-associated kinases (22), whereas Wnt3a activates the β-catenin pathway (17, 57). In contrast to the inhibition of platelet aggregation by Wnt3a (41), Wnt5a potentiates U46619-induced platelet aggregation (24), indicating that the effects of Wnt5a and Wnt3a on RhoA activity oppose each other. Consistently, rWnt3a increases active β-catenin but decreases p-MYPT1 expression in hVSMCs, whereas rWnt5a increases p-MYPT1 expression without changes in active β-catenin expression. Moreover, treatment with the Wnt5a antagonist Box5, as well as the Wnt inhibitor LGK974, attenuated salt-induced BP elevation through reduced p-MYPT expression. Thus, Wnt5a plays an important role in salt-induced Rho/ROCK activation by Klotho deficiency. Despite the small contribution of the Wnt3a/β-catenin pathway to salt-induced BP elevation, however, upregulation of active β-catenin was observed in the NS diet–fed aged mice, which may be due to the other factors than Klotho (58) and might contribute to aging-related cardiac and vascular damage, heart failure, and arterial stiffness (59).
Notably, Ang II–induced Rho/ROCK activation disappeared when Wnt5a was knocked down with siWnt5a in hVSMCs, suggesting that Wnt5a is indispensable for Ang II–induced activation of Rho/ROCK. Thus, we discovered the Wnt non-canonical pathway to be involved in the action of Ang II, despite the presence of an interplay between the renin-angiotensin system and the canonical Wnt/β-catenin pathway (60). Moreover, the addition of Klotho inhibited Wnt5a- and Ang II–induced Rho/ROCK activation. These results imply that the inhibitory effect of Klotho on Ang II–induced Rho/ROCK activation is mediated by the interaction between Klotho and Wnt5a. Although several investigators demonstrated that Klotho supplementation inhibited Ang II–induced renal damage in rats (61), precisely how Klotho protects against Ang II remains unknown.
Klotho protein exists in several forms, including the full-length membrane form and a soluble circulating form. The membrane form complexes with FGF receptors to form coreceptors for FGF23, which allows it to participate in FGF23-mediated signal transduction and regulation of phosphate and calcium homeostasis (62). α-Klotho is predominantly expressed in the kidney, the parathyroid gland, and the choroid plexus of the brain (30), as confirmed by the present finding of abundant α-Klotho in the kidney but scanty expression in the vasculatures. However, homozygous KL-KO mice exhibit functional defects in organs that do not express α-Klotho, suggesting that circulating soluble Klotho can function as a hormone to act at a distance (12). Soluble Klotho, which is secreted from the kidney, is present in the blood with many functions including regulation of ion channels/transporters and growth factor signaling. How soluble Klotho exerts these pleiotropic functions is poorly understood. Given the activation of Wnt signaling in Klotho-deficient animals (16), Klotho appears to be a secreted Wnt antagonist. In a cell culture model, Wnt-Klotho interaction resulted in suppression of Wnt biological activity. In one plausible mechanism for the inhibitory effect of Klotho on Wnt signaling, Klotho binds to various Wnt family members and facilitates the closed- to open-state conformation of the Wnt-specific receptor FZD’s cysteine-rich domain binding cavity, resulting in the loss of FZD binding (63). Alternatively, Wnt5a is bound to the cell surface by heparan sulfate proteoglycans, leading to the activation of Wnt signaling, but Klotho, which has sialidase activity, cleaves Wnt5a from the proteoglycans, such that it cannot be internalized and signal (64, 65). In the present study, the Wnt5a inhibitor Box5 attenuated salt-induced BP elevation, although its inhibitory effect was apparently smaller than that of the Wnt inhibitor LGK974, possibly because of the involvement of the other Wnts besides Wnt5a, such as Wnt11, in salt-induced activation of the non-canonical Wnt/RhoA pathway. On the other hand, supplementation with Klotho serving as the antagonist to various Wnt family members including Wnt5a and Wnt11 could thoroughly inhibit salt-induced BP elevation in aged mice with Klotho deficiency.
It should be noted, however, that neither Wnt5a nor p-MYPT expression increased until administration of an HS diet in aged WT and KL-KO mice, suggesting that salt is needed for activation of the Wnt5a/RhoA pathway. Accumulating evidence indicates that inflammatory cytokines including IL-1β and IL-17 are involved in salt-sensitive hypertension (10), and that IL-1β activates RhoA/ROCK through upregulation of Wnt5a expression (66). Interestingly, HS increases salt and water storage in the interstitial compartment associated with increased TPR in salt-sensitive hypertensive patients, suggesting increased interstitial salt/water storage may relate to vascular dysfunction in salt-sensitive hypertension (67); notably, salt accumulation in muscle and skin increases with age (68). Taken together, the results show that HS diet–induced activation of the non-canonical Wnt5a/RhoA pathway might be attributable to overproduction of these cytokines with salt loading, and the sequence of events might be mediated by tissue salt accumulation. Further investigation is needed to clarify the precise mechanism of aging-associated salt-sensitive hypertension. Nevertheless, our results reveal that the non-canonical Wnt5a/RhoA pathway plays a crucial role in the development of salt-dependent hypertension by Klotho deficiency.
In conclusion, Klotho supplementation is not only a preventive for aging-induced hypertension, but additionally, the Klotho/Wnt/RhoA pathway may be a new target for the treatment of salt-sensitive hypertension in aged people.
Methods
Animal study protocols.
Animal care and treatment complied with the standards described in the Guidelines for the Care and Use of Laboratory Animals of the University of Tokyo. Mice were housed on a 12-hour light/12-hour dark cycle at 22°C. Male C57BL/6JJcl and heterozygous α-Klotho–knockout (KL+/–) mice (69), backcrossed onto the C57BL/6JJcl strain, were purchased from CLEA Japan. WT C57BL/6JJcl mice were used as controls. Sixteen-week-old WT mice (young WT), KL+/– mice of the same age (KL-KO), and 66- to 78-week-old (15- to 18-month-old) aged mice (aged WT) were used. Mice were randomly assigned to high-salt (HS; 8% wt/wt NaCl) or normal-salt (NS; 0.3% wt/wt NaCl) diet groups for 2 weeks, and some mice were supplied with Klotho by transfection of the Klotho gene. In addition, some KL-KO and aged WT mice received drugs while receiving HS, as described below, and were compared with nontreated animals in the HS group. Fasudil hydrochloride (100 mg/kg/d; Tokyo Chemical Industry) was dissolved with water and given to mice orally (70). ICG-001 (5 mg/kg/d; Cayman Chemical) or the corresponding amount of DMSO in PBS (as negative control) was injected i.p. once a day for 2 weeks (60). LGK974 (39) (3 mg/kg/d; Cayman Chemical) dissolved in 0.5% methylcellulose and 0.1% Tween 80 or a solvent control was given orally by gastric tube once a day for 2 weeks. The Wnt5a antagonist Box5 (13 mg/kg/d; Merck Millipore) dissolved in DMSO or a solvent control was administered subcutaneously by osmotic pump (Alzet) in each group. Two weeks later, blood was collected under isoflurane anesthesia, and the mice were humanely killed by cervical dislocation. After that, aortae and iliac arteries were harvested, stripped of connective tissues, and incubated in a water bath of Krebs solution at 37°C for 30 minutes. For measurement of the phospho-MYPT1 (p-MYPT1) level by Western blot, iliac arteries were additionally stimulated with 1 × 10–8 M U46619 for 3 minutes at the end of the incubation in the Krebs solution, frozen with liquid nitrogen, and preserved at –80°C. U46619, a thromboxane A2 receptor agonist, was used to stimulate RhoA and induce contraction in the strip of iliac arteries.
Measurement of BP by telemetry.
BP was measured by radiotelemetry as previously described (8) with some modifications. Model PA-C10 transmitters (Data Sciences International) were inserted into mice from the left carotid artery under isoflurane anesthesia, and mean arterial blood pressure (MBP) of conscious mice was measured directly. MBP was monitored on a Dataquest ART Silver 4.2 acquisition system (Data Sciences International). Continuous measurements of MBP were made for 10 seconds every 15 minutes, and data are presented as the average over 3 hours. Baseline measurements were recorded for 2 consecutive days before the mice began to receive HS.
Western blot analysis.
Western blot analysis was performed with a modified previous method (71). Harvested cells and arteries were lysed and homogenized with cold lysis buffer on ice and centrifuged at 17,700 g for 5 minutes. Lysis buffer consisted of 25 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 25 mM NaF, 1 mM sodium orthovanadate, 1% Igepal CA-630, 10% glycerol, protease inhibitor cocktail cOmplete (Roche), and distilled water. The supernatant fraction was collected, and protein concentrations were determined using RC DC protein assay kit (5000120JA, Bio-Rad) with NanoDrop (ND-2000c microvolume spectrophotometer, Thermo Fisher Scientific). Protein (4 μg/sample) was incubated with 5× Laemmli sample buffer at room temperature for more than 30 minutes and separated with 4%–15% precast gels by SDS-PAGE (Mini-Protean TGX Gels or Criterion TGX Gels, Bio-Rad). After transfer to PVDF membranes (TransBlot Turbo Transfer Pack, Bio-Rad), they the membranes were incubated with the following primary antibodies: anti–phospho-MYPT1 (Thr850) (36-003, Merck Millipore), anti–active β-catenin (05-665, Merck Millipore), anti-GAPDH (ab88245, Abcam), anti-Wnt1 (ab15251, Abcam), anti-Wnt3a (sc-136163, Santa Cruz Biotechnology), anti-Wnt4 (ab91226, Abcam), anti-Wnt5a (AF645, R&D Systems), anti-Wnt11 (ab31962, Abcam), anti-Dvl (sc-166303, Santa Cruz Biotechnology), anti-ROR2 (88639, Cell Signaling Technology), anti-Daam1 (sc-100942, Santa Cruz Biotechnology), or anti-RhoA (ARH05, Cytoskeleton Inc.). After membranes were incubated with the primary antibodies at 4°C overnight, they were washed with TBST for 5 minutes 4 times, and incubated with the following secondary antibodies for 2 hours at room temperature: HRP-conjugated donkey anti–rabbit IgG ECL antibody (NA9340-1ML, GE Healthcare UK Ltd.), HRP-conjugated sheep anti–mouse IgG ECL antibody (NA9310-1ML, GE Healthcare UK Ltd.), or donkey anti–goat IgG–HRP antibody (sc-2020, Santa Cruz Biotechnology). After membranes were washed with TBST for 5 minutes 4 times, signals were detected using enhanced chemiluminescence by ImageQuant LAS 4000 mini (GE Healthcare), and images were analyzed on an ImageQuant TL (GE Healthcare).
Klotho supplementation in mice.
Klotho protein was supplied to the mice by in vivo transfection with a plasmid vector encoding full-length mouse α-Klotho, using a reported method (72). pcDNA3.1+/C-(K) DYK plasmid vector containing a full-length open reading frame of mouse α-Klotho cDNA (NM_013823) was purchased from GenScript. For transformation, E. coli HST08 Premium Competent Cells (Takara) were mixed with a plasmid vector according to the manufacturer’s instructions, and the prepared mixture was seeded in the plate medium with ampicillin added to selectively harvest the transformed E. coli. After cultivation, transformed vector plasmid DNA was extracted and purified using NucleoBond Xtra Midi plus (Macherey-Nagel) according to the manufacturer’s instructions. To transfect the mice, 2.5 AU hemagglutination virus of Japan (HVJ) envelope vector (GenomeONE, Ishihara Sangyo Kaisha) (73) and 50 μg plasmid vector were mixed to generate HVJ carrying the Klotho plasmid. The mixtures were injected i.p. into the mice starting 4 days before salt loading and every 4 days thereafter. The efficiency of Klotho supplementation was assessed by monitoring of the level of Klotho protein in serum by Western blot. Full-length α-Klotho was posttranslationally cleaved, yielding 3 types of fragments: 1 long fragment consisting of the signal sequence and the KL1 and KL2 domains (130 kDa), and 2 short fragments, KL1 with the signal sequence and KL2 (~65 kDa each) (74). We detected soluble Klotho in serum using anti-KL1 antibody (ab154163, Abcam) and confirmed that the short soluble form of Klotho, consisting predominantly of KL1, was expressed in serum.
Measurement of serum Klotho levels.
Klotho protein in serum was immunoprecipitated and detected by Western blotting as previously reported, with some modifications (75). Mouse serum (50 μL) was mixed with 5 μL of 10% SDS, boiled at 100°C for 4 minutes, and cooled down at room temperature. The sample was then mixed with 0.4 mL of 1% Triton X-100 in PBS (pH 7.4) and centrifuged at 17,700 g for 15 minutes. The supernatant was collected and incubated at 4°C overnight with 1 μg anti-Klotho antibody (AF1819, R&D Systems). The next day, the sample was incubated at 4°C for 2 hours with 30 μL protein G-agarose (Thermo Fisher Scientific) to allow immunoprecipitation. The agarose beads were gently washed 4 times with 1% Triton X-100 in PBS (pH 7.4) and 1 time with PBS. The agarose beads were mixed with 2× Laemmli sample buffer and boiled at 95°C for 5 minutes. The resultant sample was analyzed by Western blot. Klotho was detected using anti-Klotho antibody (ab154163, Abcam).
Vascular function.
Vascular function was evaluated using a previous method with some modifications (76). Iliac arteries were immediately isolated, cleaned of adventitial fat, and cut transversally in ring segments with lengths of 4 mm. The endothelium was not removed. These preparations were performed in Krebs solution (120 mM NaCl, 25.0 mM NaHCO3, 5.9 mM KCl, 2.56 mM MgCl2, 1.26 mM NaH2PO4, 2.5 mM CaCl2, and 5.56 mM glucose), aerated with 5% CO2 in oxygen. For force measurements, the vascular rings were mounted between 2 tungsten wires in 37°C Krebs solution. One wire was stationary and the other was connected to a force transducer (TB-651T, Nihon Kohden Corp.). The rings were equilibrated with a resting force of 1.5 g for 30 minutes, and contraction was induced by 60 mM KCl twice with a 10-minute interval. After washing out of Krebs solution, the rings were incubated for 30 minutes to be equilibrated. First, cumulative application of phenylephrine (FUJIFILM Wako Pure Chemical) (1 × 10−10 to 1 × 10−5 mol/L) was performed. After the application was complete, the medium was exchanged 3 times, and the segments were incubated for 30 minutes to reestablish equilibrium. Next, cumulative application of U46619 (1 × 10−12 to 1 × 10−6 mol/L) was performed. Contraction induced by phenylephrine or U46619 was presented as a percentage of maximal contraction by KCl.
Cell culture and siRNA transfection.
Primary human aortic smooth muscle cells (AoSMCs) (CC-2571, Lonza) were cultured using the SmBM 2 kit (CC-3181, Lonza) with supplements and growth factors (CC-4149, Lonza) in ordinary flasks at 37°C and 5% CO2. When cells were passaged, they were seeded at a density of 5000 cells/cm2. These cells were previously reported to respond well to Ang II (77). Two days before experiments, cells of passages 2–4 were seeded at a density of 3500 cells/cm2. The medium was changed the day before, and cells were used at 80%–90% confluence. To examine expression of p-MYPT1, cells were incubated for 15 minutes with vehicle, 5.3 nM recombinant human Wnt3a (rWnt3a; 5036-WN, R&D Systems), and 5.3 nM recombinant human/mouse Wnt5a (rWnt5a; 645-WN, R&D Systems), and then harvested. To examine expression of active β-catenin, cells were incubated for 6 hours after the addition of rWnt3a or rWnt5a. For supplementation with Klotho, 20 nM recombinant human Klotho (5334-KL-25, R&D Systems) was added to the dishes 1 hour before the addition of rWnt3a or rWnt5a. For stimulation with Ang II, 1 × 10–7 M Ang II (A9525, MilliporeSigma) was added to the dishes and incubated for 10 minutes. In the siRNA experiments, human Wnt5a siRNA (catalog HSS111356, Thermo Fisher Scientific) and negative control (catalog 12935-300, Thermo Fisher Scientific) were added to dishes to a final concentration of 10 nM. For siRNA transfection, cells were incubated with a prepared mixture of siWnt5a or a negative control with Lipofectamine RNAiMAX regent (Thermo Fisher Scientific) in serum-reduced Opti-MEM (Thermo Fisher Scientific). A medium change was performed 18–21 hours later with normal medium, and cells were incubated for 48 hours before harvest. The efficiency of Wnt5a knockdown was evaluated by real-time reverse transcription PCR.
Quantitative real-time reverse transcription PCR.
After being washed with cool PBS, AoSMCs were mixed with 350 μL of RLT lysis buffer supplied in an RNeasy Mini Kit (QIAGEN), collected, and shredded by QIA shredder (QIAGEN). Total RNA of AoSMCs was purified using the RNeasy Mini Kit according to the manufacturer’s instructions. Final RNA was eluted with 40 μL water, and RNA concentrations were determined with NanoDrop (ND-2000c microvolume spectrophotometer, Thermo Fisher Scientific). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (catalog 4368813, Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative real-time reverse transcription PCR reactions were performed in a volume of 15 μL containing diluted cDNA and Power SYBR Green Master Mix (catalog 4367659, Thermo Fisher Scientific). Gene expression was measured using the Step One Plus Real-Time PCR System (Applied Biosystems), using the calibration curve method. Further analysis was performed in Microsoft Excel. Primers were as follows: human Wnt5a, TCTCAGCCCAAGCAACAAGG and GCCAGCATCACATCACAACAC; human β-actin, GCCAACCGCGAGAAGATGA and CATCACGATGCCAGTGGTA.
Measurement of RBF and direct BP.
Measurement of RBF was performed by modification of a technique from a previous report (78). All procedures were performed under 0.7%–1% isoflurane anesthesia on a heating pad. A dorsal incision was made in each mouse to clear the connective tissue around the right renal artery. After the dorsal incision was closed, the position was changed to supine. An intra-arterial pressure catheter (FTH-1211B-0018, Transonic Inc.) was placed into the right carotid artery, and direct measurement of BP was commenced using a Scisense SP200 pressure measurement system (FP095B, Transonic). Next, an abdominal midline incision was made to reach the abdominal aorta, from which the fat and connective tissue were gently removed. Intra-abdominal organs were covered with saline-soaked cotton to prevent drying. To administer drugs, a 34-gauge syringe needle for microinjection was introduced into the abdominal aorta, and the tip of the needle was placed just below the origin of the right renal artery (78). To measure RBF, a small probe for an ultrasonic blood flowmeter (MA0.5PSB, Transonic) was placed around the right renal artery and covered with viscous jelly to fill the gap between the artery and probes. The blood flow of the right renal artery was measured by ultrasonic transit-time technology using a TS420 perivascular flow meter (Transonic). The direct BP and RBF signals were sent to a PowerLab Data Acquisition converter (ADInstruments) and were monitored using LabChart Data Acquisition Software (ADInstruments). After recording of the baseline of stabilized RBF and BP, 2 ng Ang II was administered through the syringe needle, which was immediately flushed with 50 μL heparinized isotonic saline. For the stabilization of RBF and BP, a recovery period of at least 20 minutes was allowed. Next, after the stabilized RBF and BP were confirmed and recorded, 100 ng U46619 was administered. RBF is reported as 1-second averages, and baseline RBF was calculated as the average over 1 minute. Changes in RBF (%) were calculated as follows: (RBF at x seconds – RBF of the baseline)/RBF of the baseline × 100. Changes in RBF area were quantified (% • seconds) as the area under the curve of serial assessments from 21 seconds to 61 seconds as follows: Σ[changes in RBF (%) at k seconds + changes in RBF (%) at (k + 1) seconds] × 1/2, where k = 21, 22, … 60. ΔBP from the baseline was calculated as the average over 20 seconds at the peak of the change.
Measurement of active RhoA GTPases in aortae.
Activation of Rho GTPases in aortae of mice was investigated and quantified in aortic extracts using pull-down assays with a RhoA Activation Biochem Kit according to the standard protocol (BK036, Cytoskeleton Inc.). After collection of blood under isoflurane anesthesia, the mice were humanely killed by cervical dislocation, and thoracic aortae was harvested. Aortae were washed with cold Krebs solution, immediately frozen with liquid nitrogen, and preserved at –80°C. To prepare the aortic extract, 2 or 3 aortae were used per sample. Aortae were homogenized in cold Cell Lysis Buffer attached to the kit on ice and centrifuged at 4°C, 14,000 g, for 5 minutes, and supernatants were collected. Protein concentrations of samples were measured quickly, and samples containing the same amounts of protein were mixed with Rhotekin-RBD Protein beads and incubated on a rotator at 4°C for 1 hour. Pellets of the Rhotekin-RBD beads were made by centrifugation at 5000 g at 4°C for 1 minute, and 90% of the supernatants were removed. After the beads were washed with wash buffer and centrifuged again, the supernatants were carefully removed. Finally, beads were mixed with 20 μL 2× Laemmli buffer and boiled for 2 minutes. These samples were analyzed by SDS-PAGE and Western blot analysis. For detection of RhoA, anti-RhoA antibody (ARH05, Cytoskeleton Inc.) was used.
Measurement of serum FGF23 levels.
Serum FGF23 levels in mice were measured using an FGF23 ELISA kit according to standard protocol (CY-4000, Kainos).
Data and materials availability.
All data and materials supporting the findings of this study are available from WK upon request.
Statistics.
Data are summarized as means ± SEM. Two-tailed, unpaired t tests were used for comparisons between 2 groups. For multiple comparisons, statistical analysis was performed using the Tukey-Kramer post hoc test. P values less than 0.05 were considered significant.
Study approval.
This study was approved by the Animal Care and Use Committee of The University of Tokyo (approval numbers: RAC140803 and RAC190009). All the experimental procedures were performed in the laboratory animal facility accredited by the Animal Care and Use Committee of The University of Tokyo.
Author contributions
TF and WK conceived and designed the project. WK performed most of the experiments and analyzed and interpreted the data. RM performed the experiments on vascular function ex vivo and analyzed the data. MN, NA, DH, TM, KU, FKM, and SO discussed the results and interpreted the data. TF and WK wrote the manuscript, with key editing by RM, MN, NA, DH, KU, FKM, SO, and TM, and further input from all authors. WK and RM share the first authorship position. WK appears first in the list of authors because WK did more experiments and was primarily involved in the preparation and revision of the manuscript until publication.
Supplementary Material
Acknowledgments
We thank K. Kan and N. Ishibashi for their excellent technical assistance. This work was supported by Japan Society for the Promotion of Science KAKENHI (grants 18K08028, 15H05788, 15H02538, and 18K19533) and Japan Agency for Medical Research and Development–Advanced Research and Development Programs for Medical Innovation (AMED-CREST) (grant JP16gm0510009). The Division of Clinical Epigenetics, to which all the authors belong, is an endowment department and is supported with an unrestricted grant from EA Pharma Co. Ltd., MSD KK, Asahi Group Holdings Ltd., Astellas Pharma Inc., OMRON Healthcare Co. Ltd., Shionogi & Co. Ltd., Mochida Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., Mitsubishi Tanabe Pharma Corp., Toray Industries Inc., and Nippon Boehringer Ingelheim Co. Ltd.
Version 1. 06/29/2020
Electronic publication
Version 2. 08/03/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(8):4152–4166.https://doi.org/10.1172/JCI134431.
Contributor Information
Wakako Kawarazaki, Email: wkawarazaki-tky@umin.ac.jp.
Risuke Mizuno, Email: r-mizuno@vet.ous.ac.jp.
Mitsuhiro Nishimoto, Email: nishimoto-tky@umin.ac.jp.
Nobuhiro Ayuzawa, Email: ayuzawa-tky@umin.ac.jp.
Daigoro Hirohama, Email: hirohama-tky@umin.ac.jp.
Kohei Ueda, Email: koueda-tky@umin.net.
Fumiko Kawakami-Mori, Email: mori.fumiko322@gmail.com.
Shigeyoshi Oba, Email: shirokuma4627@gmail.com.
Takeshi Marumo, Email: tmarumo.npr@gmail.com.
Toshiro Fujita, Email: Toshiro.FUJITA@rcast.u-tokyo.ac.jp.
References
- 1.Lim SS, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckett NS, et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358(18):1887–1898. doi: 10.1056/NEJMoa0801369. [DOI] [PubMed] [Google Scholar]
- 3.Luft FC, Grim CE, Fineberg N, Weinberger MC. Effects of volume expansion and contraction in normotensive whites, blacks, and subjects of different ages. Circulation. 1979;59(4):643–650. doi: 10.1161/01.CIR.59.4.643. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger MH, Fineberg NS. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension. 1991;18(1):67–71. doi: 10.1161/01.HYP.18.1.67. [DOI] [PubMed] [Google Scholar]
- 5.Oliver WJ, Cohen EL, Neel JV. Blood pressure, sodium intake, and sodium related hormones in the Yanomamo Indians, a “no-salt” culture. Circulation. 1975;52(1):146–151. doi: 10.1161/01.CIR.52.1.146. [DOI] [PubMed] [Google Scholar]
- 6.Hajjar IM, Grim CE, George V, Kotchen TA. Impact of diet on blood pressure and age-related changes in blood pressure in the US population: analysis of NHANES III. Arch Intern Med. 2001;161(4):589–593. doi: 10.1001/archinte.161.4.589. [DOI] [PubMed] [Google Scholar]
- 7.Shibata S, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest. 2011;121(8):3233–3243. doi: 10.1172/JCI43124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mu S, et al. Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17(5):573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 9.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 10.Wilck N, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551(7682):585–589. doi: 10.1038/nature24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 12.Kurosu H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309(5742):1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koyama D, et al. Soluble αKlotho as a candidate for the biomarker of aging. Biochem Biophys Res Commun. 2015;467(4):1019–1025. doi: 10.1016/j.bbrc.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Chen K, Zhou X, Sun Z. Haplodeficiency of Klotho gene causes arterial stiffening via upregulation of scleraxis expression and induction of autophagy. Hypertension. 2015;66(5):1006–1013. doi: 10.1161/HYPERTENSIONAHA.115.06033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Citterio L, et al. Klotho gene in human salt-sensitive hypertension. Clin J Am Soc Nephrol. 2020;15(3):375–383. doi: 10.2215/CJN.08620719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317(5839):803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 17.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 18.Nusse R. Wnt signaling and stem cell control. Cell Res. 2008;18(5):523–527. doi: 10.1038/cr.2008.47. [DOI] [PubMed] [Google Scholar]
- 19.Mikels A, Minami Y, Nusse R. Ror2 receptor requires tyrosine kinase activity to mediate Wnt5A signaling. J Biol Chem. 2009;284(44):30167–30176. doi: 10.1074/jbc.M109.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai SL, Chien AJ, Moon RT. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 2009;19(5):532–545. doi: 10.1038/cr.2009.41. [DOI] [PubMed] [Google Scholar]
- 21.Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23(3):265–277. doi: 10.1101/gad.1760809. [DOI] [PubMed] [Google Scholar]
- 22.Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiol (Oxf) 2012;204(1):17–33. doi: 10.1111/j.1748-1716.2011.02294.x. [DOI] [PubMed] [Google Scholar]
- 23.Florian MC, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392–396. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SY, Kim S, Yun-Choi HS, Jho EH. Wnt5a potentiates U46619-induced platelet aggregation via the PI3K/Akt pathway. Mol Cells. 2011;32(4):333–336. doi: 10.1007/s10059-011-0134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novella S, et al. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age (Dordr) 2013;35(1):117–128. doi: 10.1007/s11357-011-9337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crestani S, Webb RC, da Silva-Santos JE. High-salt intake augments the activity of the RhoA/ROCK pathway and reduces intracellular calcium in arteries from rats. Am J Hypertens. 2017;30(4):389–399. doi: 10.1093/ajh/hpw201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirth A, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14(1):64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 28.Chen K, Sun Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell. 2018;17(4):e12762. doi: 10.1111/acel.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu MC, et al. Renal production, uptake, and handling of circulating αKlotho. J Am Soc Nephrol. 2016;27(1):79–90. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olauson H, Mencke R, Hillebrands JL, Larsson TE. Tissue expression and source of circulating αKlotho. Bone. 2017;100:19–35. doi: 10.1016/j.bone.2017.03.043. [DOI] [PubMed] [Google Scholar]
- 31.Pang H, Guo Z, Su W, Xie Z, Eto M, Gong MC. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2005;289(2):C352–C360. doi: 10.1152/ajpcell.00111.2005. [DOI] [PubMed] [Google Scholar]
- 32.Zhu S, Liu L, Korzh V, Gong Z, Low BC. RhoA acts downstream of Wnt5 and Wnt11 to regulate convergence and extension movements by involving effectors Rho kinase and Diaphanous: use of zebrafish as an in vivo model for GTPase signaling. Cell Signal. 2006;18(3):359–372. doi: 10.1016/j.cellsig.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Santos A, Bakker AD, de Blieck-Hogervorst JM, Klein-Nulend J. WNT5A induces osteogenic differentiation of human adipose stem cells via rho-associated kinase ROCK. Cytotherapy. 2010;12(7):924–932. doi: 10.3109/14653241003774011. [DOI] [PubMed] [Google Scholar]
- 34.Sharma M, Castro-Piedras I, Simmons GE, Pruitt K. Dishevelled: a masterful conductor of complex Wnt signals. Cell Signal. 2018;47:52–64. doi: 10.1016/j.cellsig.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oishi I, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8(7):645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- 36.Shimada T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meo Burt P, Xiao L, Hurley MM. FGF23 regulates Wnt/β-catenin signaling-mediated osteoarthritis in mice overexpressing high-molecular-weight FGF2. Endocrinology. 2018;159(6):2386–2396. doi: 10.1210/en.2018-00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henderson WR, et al. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107(32):14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ackers I, Malgor R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diab Vasc Dis Res. 2018;15(1):3–13. doi: 10.1177/1479164117738442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele BM, et al. Canonical Wnt signaling negatively regulates platelet function. Proc Natl Acad Sci U S A. 2009;106(47):19836–19841. doi: 10.1073/pnas.0906268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baarsma HA, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. 2017;214(1):143–163. doi: 10.1084/jem.20160675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guilluy C, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16(2):183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 44.Polichnowski AJ, Griffin KA, Long J, Williamson GA, Bidani AK. Blood pressure-renal blood flow relationships in conscious angiotensin II- and phenylephrine-infused rats. Am J Physiol Renal Physiol. 2013;305(7):F1074–F1084. doi: 10.1152/ajprenal.00111.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redgrave J, Rabinowe S, Hollenberg NK, Williams GH. Correction of abnormal renal blood flow response to angiotensin II by converting enzyme inhibition in essential hypertensives. J Clin Invest. 1985;75(4):1285–1290. doi: 10.1172/JCI111828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver JA, Cannon PJ. The effect of altered sodium balance upon renal vascular reactivity to angiotensin II and norepinephrine in the dog. Mechanism of variation in angiotensin responses. J Clin Invest. 1978;61(3):610–623. doi: 10.1172/JCI108972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campese VM, Karubian F. Salt sensitivity in hypertension: implications for the kidney. J Am Soc Nephrol. 1991;2(2 suppl 1):S53–S61. doi: 10.1681/ASN.V22s53. [DOI] [PubMed] [Google Scholar]
- 48.Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation. 2016;133(9):894–906. doi: 10.1161/CIRCULATIONAHA.115.018526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujita T, Henry WL, Bartter FC, Lake CR, Delea CS. Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med. 1980;69(3):334–344. doi: 10.1016/0002-9343(80)90002-9. [DOI] [PubMed] [Google Scholar]
- 50.Fujita T, Noda H, Ando K. Sodium susceptibility and potassium effects in young patients with borderline hypertension. Circulation. 1984;69(3):468–476. doi: 10.1161/01.CIR.69.3.468. [DOI] [PubMed] [Google Scholar]
- 51.Sullivan JM, Prewitt RL, Ratts TE, Josephs JA, Connor MJ. Hemodynamic characteristics of sodium-sensitive human subjects. Hypertension. 1987;9(4):398–406. doi: 10.1161/01.HYP.9.4.398. [DOI] [PubMed] [Google Scholar]
- 52.Morris RC, Schmidlin O, Sebastian A, Tanaka M, Kurtz TW. Vasodysfunction that involves renal vasodysfunction, not abnormally increased renal retention of sodium, accounts for the initiation of salt-induced hypertension. Circulation. 2016;133(9):881–893. doi: 10.1161/CIRCULATIONAHA.115.017923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimamoto H, Shimamoto Y. Time course of hemodynamic responses to sodium in elderly hypertensive patients. Hypertension. 1990;16(4):387–397. doi: 10.1161/01.HYP.16.4.387. [DOI] [PubMed] [Google Scholar]
- 54.Campese VM, Parise M, Karubian F, Bigazzi R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension. 1991;18(6):805–812. doi: 10.1161/01.HYP.18.6.805. [DOI] [PubMed] [Google Scholar]
- 55.Fujita T, Ando K, Ogata E. Systemic and regional hemodynamics in patients with salt-sensitive hypertension. Hypertension. 1990;16(3):235–244. doi: 10.1161/01.HYP.16.3.235. [DOI] [PubMed] [Google Scholar]
- 56.Crowley SD, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103(47):17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naito AT, Shiojima I, Komuro I. Wnt signaling and aging-related heart disorders. Circ Res. 2010;107(11):1295–1303. doi: 10.1161/CIRCRESAHA.110.223776. [DOI] [PubMed] [Google Scholar]
- 58.Naito AT, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012;149(6):1298–1313. doi: 10.1016/j.cell.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM. WNT signaling in cardiac and vascular disease. Pharmacol Rev. 2018;70(1):68–141. doi: 10.1124/pr.117.013896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou L, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol. 2015;26(1):107–120. doi: 10.1681/ASN.2014010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitani H, et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension. 2002;39(4):838–843. doi: 10.1161/01.HYP.0000013734.33441.EA. [DOI] [PubMed] [Google Scholar]
- 62.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 63.Fakhar M, Najumuddin, Gul M, Rashid S. Antagonistic role of Klotho-derived peptides dynamics in the pancreatic cancer treatment through obstructing WNT-1 and Frizzled binding. Biophys Chem. 2018;240:107–117. doi: 10.1016/j.bpc.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 64.Camilli TC, et al. Loss of Klotho during melanoma progression leads to increased filamin cleavage, increased Wnt5A expression, and enhanced melanoma cell motility. Pigment Cell Melanoma Res. 2011;24(1):175–186. doi: 10.1111/j.1755-148X.2010.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Behera R, et al. Inhibition of age-related therapy resistance in melanoma by rosiglitazone-mediated induction of Klotho. Clin Cancer Res. 2017;23(12):3181–3190. doi: 10.1158/1078-0432.CCR-17-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park SY, Kang MJ, Han JS. Interleukin-1β promotes neuronal differentiation through the Wnt5a/RhoA/JNK pathway in cortical neural precursor cells. Mol Brain. 2018;11(1):39. doi: 10.1186/s13041-018-0383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laffer CL, Scott RC, Titze JM, Luft FC, Elijovich F. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension. 2016;68(1):195–203. doi: 10.1161/HYPERTENSIONAHA.116.07289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kopp C, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61(3):635–640. doi: 10.1161/HYPERTENSIONAHA.111.00566. [DOI] [PubMed] [Google Scholar]
- 69.Takeshita K, et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109(14):1776–1782. doi: 10.1161/01.CIR.0000124224.48962.32. [DOI] [PubMed] [Google Scholar]
- 70.Shibata S, Nagase M, Fujita T. Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J Am Soc Nephrol. 2006;17(3):754–764. doi: 10.1681/ASN.2005050571. [DOI] [PubMed] [Google Scholar]
- 71.Kawarazaki W, et al. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J Am Soc Nephrol. 2012;23(6):997–1007. doi: 10.1681/ASN.2011070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J Am Soc Nephrol. 2013;24(5):771–785. doi: 10.1681/ASN.2012080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe T, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120(5):1645–1662. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Y, Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr Rev. 2015;36(2):174–193. doi: 10.1210/er.2013-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. 2015;26(5):1150–1160. doi: 10.1681/ASN.2014040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nishimoto M, Mizuno R, Fujita T, Isshiki M. Stromal interaction molecule 1 modulates blood pressure via NO production in vascular endothelial cells. Hypertens Res. 2018;41(7):506–514. doi: 10.1038/s41440-018-0045-1. [DOI] [PubMed] [Google Scholar]
- 77.Ozasa Y, et al. Notch activation mediates angiotensin II-induced vascular remodeling by promoting the proliferation and migration of vascular smooth muscle cells. Hypertens Res. 2013;36(10):859–865. doi: 10.1038/hr.2013.52. [DOI] [PubMed] [Google Scholar]
- 78.Ruan X, Oliverio MI, Coffman TM, Arendshorst WJ. Renal vascular reactivity in mice: AngII-induced vasoconstriction in AT1A receptor null mice. J Am Soc Nephrol. 1999;10(12):2620–2630. doi: 10.1681/ASN.V10122620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.