

Abstract
Posttranslational modifications are a common feature of proteins associated with neurodegenerative diseases including prion protein (PrPC), tau, and α-synuclein. Alternative self-propagating protein states or strains give rise to different disease phenotypes and display strain-specific subsets of posttranslational modifications. The relationships between strain-specific structure, posttranslational modifications, and disease phenotype are poorly understood. We previously reported that among hundreds of PrPC sialoglycoforms expressed by a cell, individual prion strains recruited PrPC molecules selectively, according to the sialylation status of their N-linked glycans. Here we report that transmission of a prion strain to a new host is accompanied by a dramatic shift in the selectivity of recruitment of PrPC sialoglycoforms, giving rise to a self-propagating scrapie isoform (PrPSc) with a unique sialoglycoform signature and disease phenotype. The newly emerged strain has the shortest incubation time to disease and is characterized by colocalization of PrPSc with microglia and a very profound proinflammatory response, features that are linked to a unique sialoglycoform composition of PrPSc. The current work provides experimental support for the hypothesis that strain-specific patterns of PrPSc sialoglycoforms formed as a result of selective recruitment dictate strain-specific disease phenotypes. This work suggests a causative relationship between a strain-specific structure, posttranslational modifications, and disease phenotype.
Keywords: Infectious disease, Neuroscience
Keywords: Innate immunity, Neurodegeneration, Prions

Introduction
Prion diseases are a group of fatal neurodegenerative diseases of humans and other mammals that can arise spontaneously or via transmission (1). The transmissible agent of prion disease consists of a prion protein in β-sheet–rich self-propagating states referred to as the scrapie isoform (PrPSc) that serves as a template for conversion of the same protein in its normal, cellular form (PrPC) into disease-related states (2–6). In disease-related states, prion protein elicits multiple disease phenotypes that are often characterized by different clinical symptoms, cell tropism, affected brain regions, PrPSc deposition patterns, and incubation times to disease (7–9). The diversity of disease phenotypes within the same host has been attributed to the ability of PrPC to acquire multiple, conformationally distinct, self-replicating PrPSc states referred to as prion strains (10–15). Although the fact that individual PrPSc strains are conformationally different has been well established (14, 16–20), how individual strain-specific structures elicit multiple disease phenotypes remains puzzling. Currently, the relationship between PrPSc structure and CNS response remains empirical, whereas a mechanism that would describe this relationship in a predictable manner is lacking (21, 22). Moreover, it is becoming evident that the same concept of different disease phenotypes being associated with individual pathogenic strains of the same protein is applicable to other neurodegenerative diseases (23–26), yet the relationship between structure and disease phenotype remains poorly understood.
PrPC is posttranslationally modified with the glycosylphosphatidylinositol (GPI) anchor and 1 or 2 sialylated N-linked glycans (27–30). Previously, we showed that among hundreds of PrPC sialoglycoforms expressed by a cell, individual prion strains selectively recruit those PrPC sialoglycoforms that can be accommodated within a strain-specific structure (31–33). The selectivity is dictated by a strain-specific structure on one hand, and steric constraints associated with the charge, size, and a number of the N-linked glycans in individual PrP molecules on the other hand (34). In addition, the host appears to play a role in setting up the limits of selective recruitment. For instance, all hamster strains tested displayed only minor preferences toward specific sialoglycoforms, whereas mouse-adapted strains exhibited a much wider range of selectivity by excluding hypersialylated diglycosylated PrPC molecules in a strain-specific manner (32).
To explain the diversity in disease phenotypes, we proposed that as a result of selective recruitment of sialoglycoforms, unique strain-specific patterns of carbohydrate epitopes are formed on the PrPSc surface, and that the response of the CNS and disease phenotype are dictated by strain-specific carbohydrate patterns (34). This hypothesis highlights the role of posttranslational modifications and, specifically, in the case of prion diseases, the role of N-linked glycans in establishing a causative relationship between strain-specific structure and disease phenotype. As long as a strain is transmitted within the same host, the strain-specific structure and its carbohydrate pattern propagate faithfully, ensuring inheritance of strain-specific disease phenotypes. However, transmission to a new host is expected to alter the criteria for selective recruitments of PrPC glycoforms, resulting in a new carbohydrate pattern and disease phenotype.
To test the above hypothesis, Syrian hamster strain SSLOW, which is characterized by a minimal selectivity in recruitment, was serially transmitted to C57 black mice, a host in which prion strains display a broad range of strain-specific selectivities (32). To overcome the species barrier, SSLOW was first adapted to mouse PrPC substrate using serial protein misfolding cyclic amplification with beads (sPMCAb) before transmission to the new host. Despite adaptation to mouse substrate, 5 serial passages were required to stabilize the incubation time to clinical disease in mice. Remarkably, shortening of the incubation time to the disease was mirrored by a dramatic change in selectivity of recruitment and PrPSc conformational stability. Over the course of adaptation, the composition of PrPSc sialoglycoforms changed gradually from predominantly hypersialylated diglycosylated to hyposialylated monoglycosylated and unglycosylated isoforms. Upon adaptation to a new host, a unique signature of PrPSc sialoglycoform along with a new disease phenotype emerged. In comparison with other mouse-adapted strains, the newly emerged strain had the shortest incubation time to disease, was characterized by widespread PrPSc deposition across all brain areas, had an abundance of PrPSc colocalized with microglia, and showed a very strong, widespread proinflammatory response. We attributed the colocalization of PrPSc with microglia and the strong neuroinflammation to a unique sialoglycoform composition of PrPSc, namely a high proportion of hyposialylated and unglycosylated isoforms. The current study suggests the existence of a causative relationship between PrPSc sialoglycoform composition and disease phenotype.
Results
Previous studies illustrated that adaptation of a prion strain to a substrate of a new host in PMCA can significantly reduce or completely abrogate the species barrier of transmission to this host (35–38). For adapting the Syrian hamster strain SSLOW to mouse substrate, sPMCAb reactions were seeded with hamster brain–derived SSLOW and conducted using mouse normal brain homogenate (BH). Steady amplification was observed, suggesting that the SSLOW-specific PrPSc structure was effective in recruiting and converting mouse PrPC (Figure 1A). As negative controls, (a) serial dilutions of SSLOW material without amplification and (b) nonseeded PMCAb reactions were conducted in parallel. Both negative controls showed lack of PrPSc amplification (Figure 1A).
Figure 1. Adaptation of SSLOW to mice.

(A) Adaptation of hamster SSLOW to mouse PrPC in sPMCAb seeded with 103-fold-diluted SSLOW BHs and conducted using mouse normal BHs. Corresponding nonseeded sPMCAb and serial dilutions of SSLOW BHs in the absence of amplification are shown as references. (B) Left: Western blot analysis of PrPSc in mice inoculated with the hamster-derived SSLOW, mouse sPMCAb-adapted SSLOW, or noninoculated aged-matched controls. Products of the tenth sPMCAb round were used for inoculation. Right: Comparison of the PrPSc amounts in passages 1 and 2 of sPMCAb-adapted SSLOW. Animal number 3 was used for serial transmission. In A and B, all samples, with the exception of lane 1 (–PK), were treated with PK. (C) Western blot analysis of PrPSc from passages 2–6. (D) Change in the percentage of di-, mono-, and unglycosylated PrPSc as a function of serial passage. Data presented as individual animals and mean ± SD (n = 3 for passage 2 and n = 6 for the other groups). (E) Box-and-whisker plot of incubation time to clinical disease in i.c.-inoculated animals as a function of serial passage. The midline of the box-and-whisker plot denotes the median, the x represents the mean, and the ends of the box plot denote the 25th and 75th percentiles. For passage 2, only females (F) were used (n = 8); both males (M) and females were used for the remaining passages: n = 8 F + 5 M in passage 3, n = 4 F + 5 M in passage 4, n = 5 F + 5 M in passage 5, and n = 8 F + 5 M in passage 6. Data presented as the mean ± SD. ****P ≤ 0.0001 by 1-way ANOVA with Dunnett’s T3 multiple-comparisons test. (F) Kaplan-Meier survival plot for mice from serial passages 2–6. The survival curve for SSLOW-inoculated hamsters is provided as reference (41, 81). Western blots (A) were stained with antibody SAF-84 and in B and C with antibody ab3531.
Next, C57BL/6 mice were inoculated intracerebrally (i.c.) with hamster brain–derived SSLOW or mouse sPMCAb–derived SSLOW. None of the animals, including those inoculated with sPMCAb-derived SSLOW, developed clinical signs of disease. Nevertheless, 2 out of 3 animals challenged with hamster-derived SSLOW and all 8 animals inoculated with mouse sPMCAb–derived SSLOW were positive on Western blot (Figure 1B). Although preadaptation of hamster PrPSc to mouse substrate in sPMCAb seemed to help overcome the barrier, the lack of clinical disease in passage 1 raised the possibility that adaptation to a mouse substrate in sPMCAb did not completely abrogate the transmission barrier. In part, lack of clinical diseases could be due to a possible decline in specific prion infectivity in the course of sPMCAb, as reported in previous studies (39). To examine this possibility, animals were inoculated with brain-derived mouse strain 22L and sPMCAb-derived 22L produced in sPMCAb consisting of 10 rounds. Animals that received brain- or sPMCAb-derived 22L succumbed to the diseases at 161 ± 1 or 173 ± 5 days after inoculation, respectively. Although the sPMCAb-derived 22L group showed a slightly longer incubation time, this experiment suggests that the sPMCAb procedure used cannot account in full for the lack of clinical disease in animals inoculated with mouse sPMCAb-adapted SSLOW.
Serial transmission of sPMCAb-derived SSLOW revealed clinical signs of the disease in passage 2 at 285 ± 39 days after inoculation; then there was a significant reduction in the incubation time to the first signs in passage 3, and further modest reductions in passages 4 and 5 (Figure 1E). The incubation time to clinical disease was only 95 days in both passages 5 and 6, which was the shortest among currently known mouse-adapted strains (Figure 1, E and F). Brains of animals from all serial passages were PrPSc positive (Figure 1C). Starting from passage 2, the key pathological hallmarks of transmissible spongiform encephalopathies including PrPSc deposition, spongiosis, reactive astrogliosis, and microgliosis were apparent (Supplemental Figure 1 and Figure 2; supplemental material available online with this article; https://doi.org/10.1172/JCI138677DS1). Reminiscent of the large PrPSc plaques in SSLOW-infected hamsters (40–42), large plaques and areas with a high density of smaller PrPSc deposits were seen in passage 2 (Figure 2). Coimmunostaining for PrPSc and Iba1, a microglia-specific marker, revealed that reactive microglia surrounded or penetrated large PrPSc plaques (Figure 3, B and C, and Supplemental Figure 2). Astrocytes were also observed surrounding large PrPSc plaques (Figure 3A). Colocalization of small PrPSc deposits with microglia, and to a lesser extent with astrocytes, suggested that both types of cells phagocytose PrPSc (Figure 3, A–C, and Supplemental Figure 2). By passage 4, large PrPSc plaques completely disappeared, yet areas with high densities of PrPSc deposits could still be seen (Figure 2). Fine granular PrPSc deposits, while visible starting from passage 2, emerged as a main histopathological feature by passage 4 (Figure 2). In passage 6, the fine granular PrPSc deposits were prominent in most brain areas including the thalamus, hypothalamus, cortex, hippocampus, stem, and cerebellum (Supplemental Figure 3). The strain that emerged as a result of adaptation of SSLOW to mice will be referred to hereafter as SSLOW-Mo. Although previously characterized mouse-adapted strains showed tropism to specific brain regions (8, 9, 43, 44), SSLOW-Mo PrPSc deposits were widespread across the brain and affected most brain regions (Supplemental Figure 3). In summary, 5 serial passages were required for stabilizing the disease phenotype in mice. Lengthy adaptation to a new host suggests that preadaptation of SSLOW to a mouse substrate in sPMCAb was not sufficient to fully eliminate the transmission barrier, and that factors other than differences in amino acid sequence between PrPSc and PrPC contribute to the transmission barrier.
Figure 2. Change in PrPSc deposition upon serial passaging.
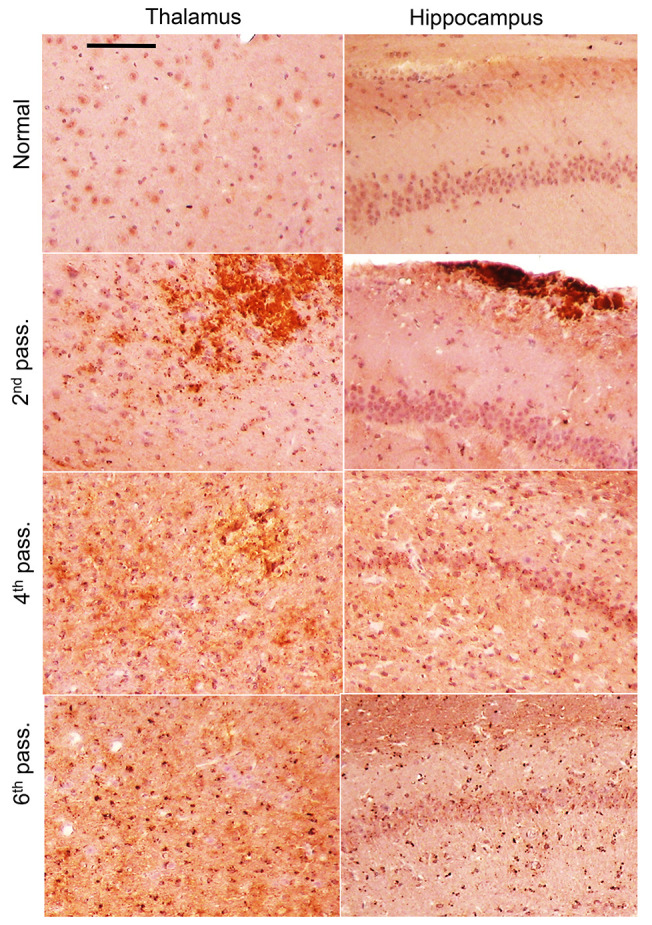
Representative images of PrPSc deposition in the thalamus and hippocampus of animals from passages 2, 4, and 6 of SSLOW-Mo and normal controls. Antibody SAF-84 was used for staining. Scale bar: 100 μm. Brains of normal-age mice (337–405 days old) were used for reference.
Figure 3. Histopathological analysis of reactive microglia, astrocytes, and PrPSc in passage 2.
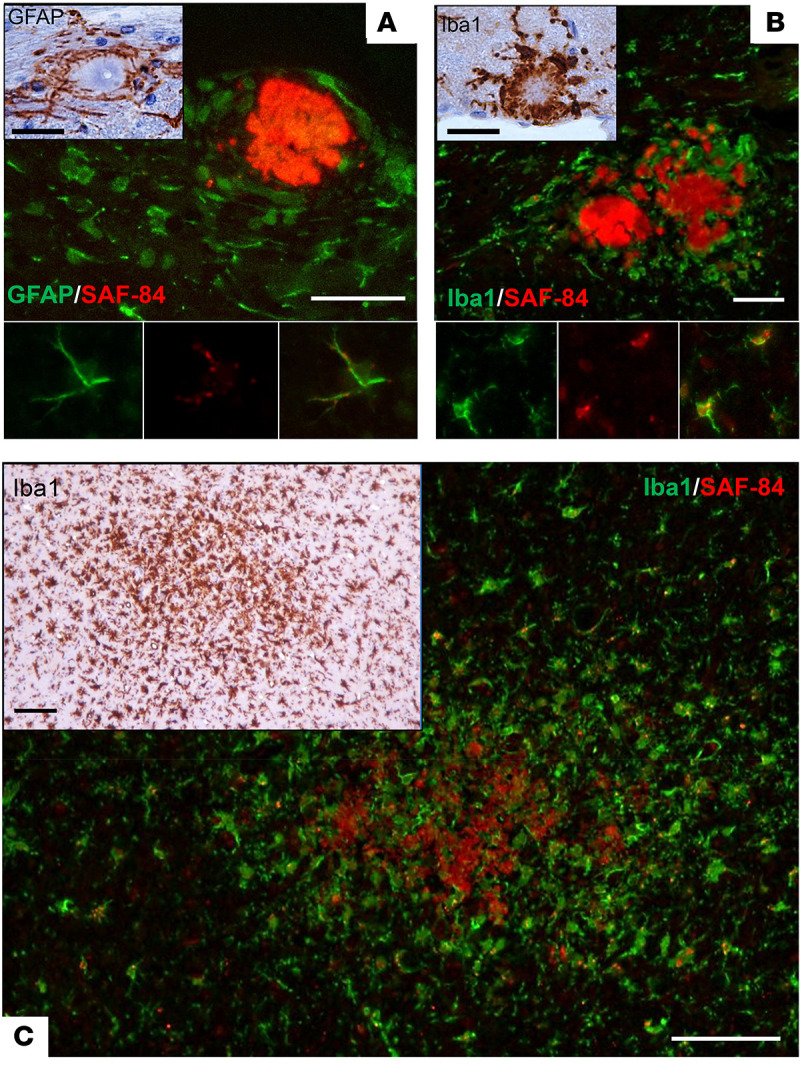
(A) Upper panel: PrPSc deposits (antibody SAF-84, red) and astrocytes (GFAP, green) in hippocampus. Insert: GFAP staining of astrocytes surrounding a plaque. Lower panel images: Colocalization of small PrPSc deposits with an astrocyte. (B) Upper panel: PrPSc plaques (antibody SAF-84, red) and microglia (Iba1, green) in hippocampus. Insert: Iba1 staining of microglia surrounding a plaque. Lower panel images: Colocalization of small PrPSc deposits with microglia. (C) Coimmunostaining of diffuse PrPSc deposits (antibody SAF-84, red) and microglia (Iba1, green) in thalamus. Insert: Iba1 staining of the same area. Scale bars: 25 μm in immunofluorescence images in A and B and 30 μm in DAB-stained inserts; 50 μm in immunofluorescence image in C and 100 μm in DAB-stained insert.
Analysis of glycoform ratios revealed that adaptation of SSLOW to a new host was accompanied by an increase in percentage of mono- and unglycosylated isoforms at the expense of diglycosylated isoforms (Figure 1D). The glycoform ratio stabilized by passage 6, mirroring the dynamics of the incubation time to disease (Figure 1, D and E). A drift in glycoform ratios suggests that selectivity of recruitment changes with strain adaptation, yet, in the absence of the knowledge about PrPSc sialylation status, it provided limited insight. The original SSLOW strain recruits PrPC sialoglycoforms proportionally to their expression levels, which results in a vast majority of PrP molecules being diglycosylated and heavily sialylated (Figure 4A, Supplemental Figure 4, and ref. 32). In contrast to SSLOW, known mouse-adapted strains display a wide range of strain-specific preferences for monoglycosylated and unglycosylated isoforms (32). To examine whether the selectivity of recruitment changes in a new host, the composition of SSLOW-Mo PrPSc was examined using 2-dimensional electrophoresis and Western blots (2D) (Supplemental Figure 4). On 2D, sialoglycoforms were divided arbitrarily into 5 groups corresponding to unglycosylated (group 1), hypo- and hypersialylated monoglycosylated (groups 2 and 3, respectively), and hypo- and hypersialylated diglycosylated (groups 4 and 5, respectively) isoforms (Figure 4B). Multiple-charge isoforms in the unglycosylated group were previously attributed to the heterogeneity of the GPI anchor (45). In the course of serial transmission, the profile of sialoglycoforms was transformed dramatically (Figure 4, A, C, and D). The relative proportion of the unglycosylated group increased substantially (Figure 4, A, C, and D). Among monoglycosylated isoforms, the contribution of hyposialylated isoforms grew gradually, whereas representation of the hypersialylated group dropped after a transient increase at passage 3 (Figure 4, A, C, and D). Contribution of both diglycosylated groups decreased, although more so for the hypersialylated relative to the hyposialylated group (Figure 4, A, C, and D). The transformation of the selective recruitment was particularly dramatic upon comparison with the original SSLOW strain (Figure 4E). Overall, the recruitment of hypersialylated isoforms decreased markedly, whereas the relative presentation of unglycosylated and hyposialylated monoglycosylated isoforms increased with strain adaptation (Figure 4C). In fact, in comparison with other mouse-adapted strains tested (22L, ME7, or RML), passage 6 of SSLOW-Mo had the highest proportion of unglycosylated isoforms, yet the lowest amounts of hypersialylated monoglycosylated isoforms (Figure 4F). In comparison with ME7 or 22L, SSLOW-Mo also had a lower proportion of hypersialylated diglycosylated isoforms. In summary, a unique signature of PrPc sialoglycoforms emerged as a result of adaptation to a new host. Notably, changes in selective recruitment stabilized after passage 5, mirroring the dynamics of the incubation time to disease (Figure 4, C and D).
Figure 4. Change in selective recruitment of PrPSc sialoglycoforms over the course of SSLOW-Mo adaptation.

(A) Representative intensity profiles of di- (blue lines), mono- (red lines), and unglycosylated (green lines) sialoglycoforms of brain-derived PrPSc of hamster SSLOW, SSLOW-Mo from serial passages 1–6, and mouse-adapted 22L. Profiles were built as described in Methods using 2D Western blots. (B) An example of 2D Western blot of SSLOW-Mo PrPSc showing classification of sialoglycoforms into 5 groups: 1, unglycosylated; 2 and 3, hypo- and hypersialylated monoglycosylated, respectively; 4 and 5, hypo- and hypersialylated diglycosylated, respectively. Western blot was stained with antibody ab3531. (C and D) Change in relative populations of 5 sialoglycoform groups of SSLOW-Mo PrPSc over the course of 6 serial passages. Two individual animals for each group are shown. (E) Comparison of relative populations of 5 sialoglycoform groups in passages 1 and 6 of SSLOW-Mo (blue and red circles, respectively) with those of hamster SSLOW (black triangles). (F) Comparison of relative populations of 5 sialoglycoform groups in passage 6 of SSLOW-Mo with those of mouse-adapted ME7, 22L, or RML. In panels C–F, serial passages of SSLOW-Mo: 1st, dark blue; 2nd, light blue; 3rd, dark green; 4th, brown; 5th, orange; 6th, red circles; hamster SSLOW, black triangles; mouse-adapted ME7, gray squares; 22L, bright green diamonds; RML, olive triangles.
Transformation of PrPc sialoglycoform composition over the course of adaptation suggests that the PrPc structure continued to evolve for 5 serial passages. To test whether this is the case, conformational stability, which was previously found to be informative for tracking structural changes, was analyzed (40, 46). As judged by guanidine hydrochloride–induced (GdnHCl-induced) denaturation experiments, conformational stability was the same for PrPSc from SSLOW-Mo passage 2 and the original, hamster-derived SSLOW (Figure 5, A and B). Nevertheless, a major structural change took place during the third serial passage, which also showed the largest drop in the incubation time to disease. After passage 3, relatively minor, gradual changes in PrPSc conformational stability could be observed, mirroring modest drops in the incubation time to the disease. The same stability was observed for PrPSc from passages 5 and 6, which was slightly higher than that of the mouse-adapted strain 22L. In summary, changes in conformational stability also mirrored drops in incubation time to disease along with changes in selective recruitment of sialoglycoforms.
Figure 5. Analysis of the conformational stability of PrPSc over the course of SSLOW-Mo adaptation.

(A) Western blot analysis of hamster SSLOW brain material, SSLOW-Mo brain materials from serial passages 2–6, or mouse 22L brain material. Brain materials were incubated with increasing concentrations of GdnHCl from 0.4 to 4 M for 1 hour, and then diluted out of GdnHCl, digested with PK, and analyzed by Western blot using antibody ab3531 for mouse and 3F4 for hamster brain materials. Undigested brain material (–PK) is provided as a reference. (B) Conformational stability profiles of brain-derived PrPSc built using densitometry analysis of the data presented in Western blots. Data presented as the mean ± SD (n = 3 animals).
The unique signature of sialoglycoform seen in SSLOW-Mo PrPSc, in combination with its short incubation time to disease, raised the question of whether a causative link between sialoglycoform composition and the disease phenotype exists. Short incubation time suggests that SSLOW-Mo PrPSc is highly toxic to neurons, assuming colocalization of PrPSc with neurons. To test whether this is the case, coimmunostaining for PrPSc and markers of neurons (MAP2), astrocytes (GFAP), microglia (Iba1), and oligodendrocytes (MBP) was performed. Surprisingly, pronounced colocalization of small PrPSc puncta with microglia, and to a lesser extent with astrocytes, was observed, while very minor if any association with neurons or oligodendrocytes was found (Figures 6 and 7). Occasionally, PrPSc could be observed in close proximity to neurons (Figure 6). However, it was not clear whether these PrPSc particles were indeed on the cell surface of neurons or were associated with microglia abundantly present across the brain (Figure 7). Unlike neurons or astrocytes, microglia do not replicate PrPSc. Nevertheless, extensive colocalization with microglia suggests that microglia efficiently phagocytose PrPSc, but may not be able to digest it. Very limited colocalization of PrPSc with neurons points to non–cell-autonomous toxicity as a primary mechanism responsible for the disease.
Figure 6. Coimmunostaining of PrPSc and neurons, astrocytes, or oligodendrocytes.

Representative images of coimmunostaining for PrPSc (antibody SAF-84, red) and neurons (MAP2, green), astrocytes (GFAP, green), or oligodendrocytes (MBP, green) in animals from passage 6 of SSLOW-Mo (A) or normal controls (B). Arrows in A point to small PrPSc deposits in close vicinity to neurons or astrocytes. Scale bars: 20 μm.
Figure 7. Colocalization of PrPSc and microglia.

Representative images of coimmunostaining for PrPSc (antibody SAF-84, red) and microglia (Iba1, green) in animals from passage 6 of SSLOW-Mo (A and B) or normal control (C). Scale bars: 20 μm (A and C) and 10 μm (B).
Neuroinflammation, as an important contributor to non–cell-autonomous toxicity, has been previously implicated in neurodegenerative diseases including prion diseases (47–49). To examine neuroinflammation status, expression of genes reporting on reactive phenotypes of microglia and astrocytes was examined in 4 brain regions (thalamus, hypothalamus, cortex, and hippocampus) in the animals of passage 6. Mice inoculated intraperitoneally (i.p.) were analyzed in parallel with the i.c.-inoculated group to test the extent to which the neuroinflammation status might be affected by priming microglia with injury associated with i.c. inoculation. Animals inoculated via the i.p. route developed the first signs of the disease at 124 ± 7.7 days after inoculation and were terminally sick at 148 ± 6.6 days after inoculation, again showing the shortest incubation time to disease among mouse-adapted strains. Both i.p.- and i.c.-inoculated groups showed equally strong upregulation of proinflammatory genes (Tnfa, Il1A, Cxcl10, and Ccl2), genes involved in innate immune response (Cd68, C3, and Tlr2), and genes associated with A1-, A2-, or PAN-reactive astrocytes (Figure 8, A and B). Remarkably, SSLOW-Mo animals showed a much stronger response relative to the 22L- or ME7-infected groups (Figure 8, A–C). Moreover, all 4 brain regions tested were affected more severely in SSLOW-Mo relative to the corresponding regions in 22L or ME7 animals, documenting widespread neuroinflammation in SSLOW-Mo (Figure 8, A–C). In fact, at least 3 regions in SSLOW-Mo (hippocampus, thalamus, and cortex) were affected more severely relative to the thalamus in the 22L and ME7 groups, which was the most affected region in these 2 strains (43). Astrocytes showed the same pattern of activation as proinflammatory genes or genes of innate immune response (Figure 8B). Consistent with previous studies (43, 50), genes associated with all 3 A1-, A2-, and PAN-reactive phenotypes were upregulated in astrocytes of SSLOW-Mo animals (Figure 8B).
Figure 8. Analysis of region-specific neuroinflammation in passage 6 of SSLOW-Mo.

Grouped-samples heatmap analysis of the expression of proinflammatory genes, genes reporting on neuroinflammation, and astrocyte-specific genes (A), or A1-, A2-, and PAN-specific markers (B) in 4 brain regions (Ctx, cortex; HTh, hypothalamus; Th, thalamus; Hp, hippocampus) of ME7 or 22L animals infected via the i.p. route (n = 6), passage 6 of SSLOW-Mo animals infected via the i.p. (n = 6) or i.c. route (n = 3), and the following normal control groups: group i (n = 3) is for SSLOW-Mo and 22L animals, and group ii (n = 6) is for ME7. (C) Normalized expression of Cd68, Tlr2, Cxcl10, Tnfa, C3, Il1a, and Serpina3n genes in 4 brain regions (Ctx, cortex; Hp, hippocampus; Th, thalamus; HTh, hypothalamus) in ME7 (orange lines), 22L (yellow lines), passage 6 of SSLOW-Mo (red lines) animals infected via the i.p. route, or normal control animals (green lines, groups i and ii combined data). Data presented as the mean ± SD. * and # indicate significant differences between SSLOW-Mo and 22L, or SSLOW-Mo and ME7, respectively (**** or ####P < 0.0001; *** or ###P < 0.001; ** or ##P < 0.01; * or #P < 0.05; n = 6 for SSLOW-Mo, 22L and ME7 groups, n = 6 for SSLOW-Mo, 22L and ME7 groups, n = 9 for the control group). P values were calculated using ordinary 2-way ANOVA with Tukey’s multiple-comparisons test.
Discussion
Although posttranslational modifications have been recognized as a common feature of proteins associated with neurodegenerative diseases, a substantial gap exists in our understanding of the role of posttranslational modifications in defining strain-specific structure and disease phenotype. Our previous studies established that among hundreds of PrPC sialoglycoforms expressed by a cell, prion strains recruit sialoglycoforms selectively (32, 33). Strain-specific structure of PrPSc, on one hand, and electrostatic repulsion between sialic acid residues of the N-linked glycans along with their size, on the other hand, impose electrostatic and steric constraints that dictate the selectivity of recruitment (34). Hamster strains, including SSLOW, display minimal constraints, as they easily accommodate diglycosylated and highly sialylated PrPC molecules (32, 51). In contrast, mouse-adapted strains exhibit much stronger constraints and preferentially exclude hypersialylated and diglycosylated PrPC isoforms in a strain-specific manner (32, 51). The fact that mouse-adapted strains prefer mono- and unglycosylated PrPC as a substrate has been well established, as was evident from both animal and in vitro studies (51, 52). Remarkably, selective preferences for mono- and unglycosylated substrates were completely lost upon desialylation of PrPC N-linked glycans, arguing that sialic acid residues impose major constraints preventing recruitment of diglycosylated PrPC (32). The current study demonstrated that the adaptation of a hamster strain to a new host was accompanied by dramatic changes in selective recruitment of PrPC sialoglycoforms, giving rise to a new strain with unique sialoglycoform composition and disease phenotype.
Among hamster strains, SSLOW displays one of the longest incubation times to disease (40, 41, 53). Upon adaptation to mice, a new strain with the shortest incubation time to disease among the mouse-adapted strains emerged. Besides incubation time, widespread deposition of PrPSc across brain regions, colocalization of PrPSc with microglia, and very intense, widespread neuroinflammation were among other distinctive features of SSLOW-Mo. A unique sialoglycoform signature along with a highly distinctive disease phenotype suggested that a causative link between PrPSc sialoglycoform composition and disease phenotype exists. Previously we showed that lectins specific for sialic acid show robust staining of PrPSc plaques, documenting localization of sialic acid residues on PrPSc surfaces (34). In the current study, shortening of the incubation time over the course of serial transmission was mirrored by changes in the selective recruitment of sialoglycoforms and PrPSc conformational stability.
The process of strain adaptation to a new host appeared to be complex and could consist of several steps or processes. Over the course of passages 1 and 2, significant changes in the sialylation pattern of SSLOW-Mo relative to that of the hamster SSLOW, yet very minor changes in PrPSc conformational stability, were observed. It is difficult to conclude without a doubt whether PrPSc structure changed at this step, as structurally different PrPSc might have the same stability. A major drop in the incubation time between passages 2 and 3 coincided with a major change in PrPSc conformation, yet a modest shift in the sialylation profile. More significant changes in sialylation patterns along with gradual and modest changes in conformational stability and incubation time were observed over the course of passages 4 and 5. The 2D approach employed in the current study tracks only major trends in dynamics of sialoglycoforms, leaving nuances behind. Moreover, we chose a very conservative way for analyzing 2D results. In calculating the relative contribution of the 5 groups, each spot within a group is given the same weight, regardless of its position on 2D blots and a number of sialic acid residues per PrP molecule. However, the number of sialic residues per PrP molecule increases progressively with a shift of the pI to acidic pH. As a result, changes in the relative contribution of hyper- versus hyposialylated groups that might be modest on the plots in Figure 4, C–F, in fact, could reflect significant changes in the sialylation levels and glycoform composition. Nevertheless, current work supports the hypothesis that changes in selective recruitment of sialoglycoforms are driven by a change of the host during the first serial passage and then by conformational changes in PrPSc during subsequent passages.
While structural change within PrPSc acts as one of the driving forces behind shifts in selective recruitment, a reverse feedback of N-glycan composition on the conformational stability of PrPSc and its aggregation state also exists. Changes in selective recruitment shift the pI of PrPSc toward basic pH, which is expected to alter its solubility, conformational stability, and aggregation states at physiological pH. Indeed, in parallel with the changes in the sialoglycoform composition over the course of adaptation, PrPSc aggregation status underwent a dramatic transformation as well. Reminiscent of the large PrPSc plaques in SSLOW-inoculated hamsters (40–42), PrPSc plaques were still present at passage 2 in mice along w=ith small granular deposits. In subsequent passages, large plaques were completely replaced by diffuse PrPSc deposits and, eventually, by small granular aggregates. How does PrPSc sialylation status explain changes in its aggregation states? In hamster SSLOW PrPSc, a high proportion of heavily sialylated and negatively charged N-glycans compensates the net positive charge of PrP polypeptide chains, making SSLOW PrPSc prone to aggregation into large plaques. Over the course of strain adaptation, a steady increase in the relative proportion of the unglycosylated and hyposialylated molecules shifts the pI of PrPSc particles from neutral to basic pH. Basic pI is expected to prevent PrPSc particles from forming large aggregates. Notably, glia have a different strategy of dealing with large plaques versus small particles. Microglia and astrocytes attempt to seclude large plaque-forming structures that could be compared with glial scars (as seen in Figure 3, A and C), whereas small PrPSc particles can be efficiently phagocytosed.
What makes microglia overly reactive in SSLOW-Mo in comparison with other strains? Hyposialylated PrPSc exposes galactose instead of sialic acid residues at the terminal position of the N-linked glycans. An increase in hyposialylated PrPSc over the course of adaptation of SSLOW-Mo is expected to intensify the “eat me” phagocytic response in microglia, which is known to be activated by exposed galactose (54–57). Previously we showed that partial desialylation of PrPSc enhanced the proinflammatory response of primary microglia in vitro (58). Moreover, the most intense proinflammatory response was observed in brain regions with the lowest sialylation status of PrPSc, also suggesting that a link between sialoglycoform composition and microglia response does exist (59). In SSLOW-Mo, the widespread proinflammatory phenotype of microglia was characterized by a robust upregulation of Il1A, Tnfa, and Cxcl10, the proinflammatory signaling molecules that can contribute to neurotoxicity and apoptosis. Moreover, significantly stronger expression levels of Cd68, a gene that reports on phagocytic activity, and C3, a component of the complement system, were observed across brain regions in SSLOW-Mo relative to ME7 or 22L groups, illustrating that a robust activation of “eat me” signaling occurred in SSLOW-Mo animals. Recent studies showed an elevation of C3 in Creutzfeldt-Jakob disease in humans (60). C3 plays a critical role in synapse pruning during normal brain development and chronic neurodegeneration (61–65). The synaptic pruning involves the tagging of synapses by C1q, then their opsonization by C3, followed by their engulfment and phagocytosis via an interaction with the C3 receptor expressed by microglia. One might suggest that a similar, complement-dependent mechanism is involved in the phagocytosis of PrPSc by microglia and/or by infiltrating macrophages. Consistent with this hypothesis, C1q was found to form a complex with prion protein oligomers in vitro (66). Moreover, the ability of C1q to interact with PrPSc was documented by a study in which C1q was found to mediate uptake and trafficking of PrPSc by cells of the peripheral immune system (67). Several components of a complement cascade including C1qa, C1qb, C1qc, and C3ar1 (receptor for C3a) that are normally involved in synaptic pruning were found to be strongly upregulated in brains of prion-infected mice (43). In the current study, the vast majority of small PrPSc aggregates were found to be colocalized with microglia, supporting the idea that microglia can efficiently phagocytose SSLOW-Mo PrPSc. Whether microglia can digest SSLOW-Mo PrPSc as efficiently is not clear. We propose that chronic exposure to PrPSc overactivates phagocytic and C3-dependent pruning pathways in microglia and astrocytes, which not only target PrPSc, but also synapses. Recent studies revealed that, in addition to microglia, reactive astrocytes might also play a significant role in neuroinflammation and neuronal toxicity (43, 68). Notably, in the current study, the same ranking order of astrocyte activation was observed between 4 brain regions in 3 strains as those for microglia. These results suggest that activation of microglia and astrocytes is tightly coupled. To summarize, phenotypic changes in both cell types, microglia and astrocytes, might contribute to non–cell-autonomous neuronal death. Among the 3 strains tested here, SSLOW-Mo displayed the strongest proinflammatory response and the shortest incubation time to disease, suggesting that neuroinflammation drives disease pathogenesis. It would be very interesting to use a panel of strains in a future study to examine whether a reverse correlation between the degree of neuroinflammation and incubation time exists.
Cross-species transmission of prions is controlled by a species barrier that manifests itself in a low attack rate, prolonged incubation time to clinical disease, or lack of disease. Traditionally, the magnitude of a barrier is believed to be determined by the extent to which the strain-specific conformation of donor PrPSc can accommodate the primary structure of the new host PrP (6, 69–71). When cross-species transmission is followed by serial passaging in a new host, strains can gradually adapt to a new species, a phenomenon known as prion strain adaptation (71). Notably, previous work documented a substantial barrier upon transmission of the new variant Creutzfeldt-Jakob disease from human patients to transgenic mice expressing human PrPC of identical amino acid sequences (72, 73). Other studies demonstrated the remarkable susceptibility of the bank vole for efficient transmission of prions from a variety of species in the absence of a significant species barrier (74, 75). These studies suggested that some hosts can replicate a range of prion strains very efficiently, despite sequence differences between donor PrPSc and host PrPC. On the other hand, a species barrier could be observed even in the absence of such differences, suggesting that other factors contribute to the barrier. It is expected that amplification of prions in vitro using PrPC from a new host would significantly reduce or completely abrogate the species barrier. Indeed, previous studies, which employed several animal species, including hamsters and WT or transgenic mice, and a diverse range of prion strains from a number of species, demonstrated that the species barrier could be significantly or fully abrogated by interspecies PMCAs (35–38).
In the current study, SSLOW did not show a significant barrier in interspecies sPMCAb, arguing that this strain can effectively use mouse PrPC as a substrate. However, a transmission barrier was observed upon serial passaging of mouse sPMCAb–derived SSLOW to mice, pointing out that a barrier still exists despite identity in the amino acid sequences of donor PrPSc and host PrPC. There are several reasons why PMCA might not faithfully recapitulate the adaptation process. Prion replication in PMCA and in vivo are dictated by different sets of rules. In PMCA, those PrPSc variants receive selective advantages that are sufficiently fragile to fragment under given sonication conditions, yet conformationally stable enough to avoid denaturation. Moreover, in PMCA, PrPSc is exposed equally well to PrPC substrates and cofactors expressed in different brain regions. Upon transmission to animals, PrPSc variants that were selected in PMCA may not be fit to propagate in vivo. First, fragmentation of PrPSc aggregates has to occur in the absence of sonication. Second, for propagation to persist, PrPSc should be resistant to clearance by glia. Third, PrPSc species have to elicit biological responses. Replication of transmissible, yet clinically silent PrPSc states would not result in a clinical disease (76, 77). Fourth, a spread of prions between brain regions is likely to be affected by region-specific differences in PrPC, such as differences in their sialylation status, and expression of strain-specific cofactors (59, 78).
In the current study, lengthy adaptation to mice was accompanied by an equally lengthy process of transformation in selectivity of recruitment of sialoglycoforms. This work argues that, in addition to congruency between the amino acid sequences of host PrPC and donor PrPSc, constraints generated by N-linked glycans dictate a parallel set of rules that govern strain adaptation. Electrostatic and steric constraints associated with N-linked glycans are believed to limit the range of folding patterns and quaternary structures accessible to PrP polypeptide chains within PrPSc (33). Knowledge on strain-specific selective recruitment of sialoglycoforms could be used for designing experimental conditions for selectively amplifying individual prion strains from a mixture in vitro (79). Generation of prion diseases de novo by PrP amyloid fibrils offered another example of how posttranslational modifications impose structural constraints and limit the set of PrPSc structures (40, 80, 81). In the absence of posttranslational modifications, multiple self-propagating states with different cross–β-sheet folding patterns could be formed in vitro using recombinant PrP (15, 82–85). However, serial transmission of recombinant PrP amyloid fibrils in WT animals displayed a considerable barrier followed by strain adaptation, despite identity in amino acid sequences of the inoculated PrP material and host PrPC (40, 41, 71, 80, 81). In fact, multiple serial passages were required to stabilize disease phenotypes (41). This barrier was attributed to the transformation of self-replicating structures from PrP fibril–specific, that N-linked glycans prohibit, to a PrPSc-specific structure that emerged in vivo under constraints imposed by posttranslational modifications (77, 86, 87).
Prion-like propagation of misfolded protein states is not limited to the prion protein (24, 88). In a manner similar to prions, a number of amyloidogenic proteins and peptides associated with neurodegenerative diseases can spread from cell to cell, be transmitted from animal to animal, or from human to animal, manifesting a strain-like phenomenon (23–26). Most of the proteins or peptides that spread in a prion-like fashion are not glycosylated, yet several types of posttranslational modifications, including phosphorylation of tau and α-synuclein, have been described. Unique strains of tau that display different subsets of posttranslational modifications are found in different tauopathies (89, 90). The role of posttranslational modifications in shaping strain structures and defining disease phenotypes is poorly understood. The results of the current study provide support for the hypothesis that strain-specific structure dictates selective recruitment of PrPC sialoglycoforms, resulting in a strain-specific pattern of carbohydrate epitopes on the PrPSc surface and a strain-specific disease phenotype. The mechanism of selective, strain-specific recruitment of differentially modified protein isoforms by self-propagating states and the role of posttranslational modification in dictating disease phenotypes might be worth considering in other neurodegenerative diseases.
Methods
Preparation of BH.
BH (10% [w/v]) was prepared in PBS, pH 7.4, using glass/Teflon homogenizers attached to a cordless 12-V compact drill, as previously described (41).
PMCAb.
As a source of SSLOW for sPMCAb, brain-derived materials from serial passage 4 of SSLOW in hamsters were used (41). Normal 10% BH from healthy hamsters was prepared as described previously (80) and used as a substrate for sPMCAb (91). The standard sonication program consisted of 5-second sonication pulses at approximately 200 W applied every 10 minutes during a 24-hour period, and the reactions were carried out in the presence of 2 3/32–inch Teflon beads (McMaster-Carr) (91). For each subsequent round, 20 μL of the reaction from the previous round was added to 80 μL of fresh substrate. To analyze production of PrPSc in sPMCAb, 10 μL of each sample was supplemented with 5 μL of SDS and 5 μL of Proteinase K (PK) (New England BioLabs) to the final concentrations 0.25% and 50 μg/mL, respectively, followed by incubation at 37°C for 1 hour. The digestion was terminated by addition of SDS-containing sample buffer and heating the samples for 10 minutes in a boiling water bath.
Bioassay.
SSLOW and SSLOW-Mo brain–derived materials were inoculated as 10% BH, prepared as described above in PBS. Immediately before inoculation, each inoculum was further dispersed by 30 seconds of indirect sonication at approximately 200 W in a microplate horn of a sonicator (Qsonica). sPMCAb-derived material was diluted 10-fold in PBS supplemented with 1% (w/v) bovine serum albumin before inoculation, to reduce the amount of detergent in the inoculum. Each C57BL/6 mouse received 20 μL of inoculum i.c. or 200 μL i.p. under 2% isoflurane anesthesia. After inoculation, animals were observed daily for signs of neurological disorders. Clinical signs included clasping hind legs, difficulty walking, abnormal gate, nesting problems, and weight loss. The animals were euthanized when they were unable to rear and/or lost 20% of their weight. For the Nanostring experiments, each C57BL/6 mouse received 200 μL of 1% 22L or ME7 BHs prepared in PBS and inoculated i.p. under 2% isoflurane anesthesia. After inoculation, animals were observed daily for signs of neurological disorders as previously described (43), and euthanized when they were unable to rear and/or lost 20% of their weight. 22L i.p.-inoculated animals were euthanized at 168–225 days postinoculation (dpi) (n = 6) and ME7 at 258–363 dpi (n = 6). Control groups were i.p. inoculated with PBS. Control group i was euthanized at 197–223 dpi (n = 3) and control group ii at 203–363 dpi (n = 6). Nanostring analysis of SSLOW-Mo was performed using i.p.- and i.c.-inoculated mice (n = 6 and 3, respectively).
Antibodies.
Primary antibodies used for immunoblotting, immunohistochemistry and immunofluorescence were as follows: anti-prion protein clone 3F4 (Covance SIG-39620, BioLegend), polyclonal anti-prion (ab3531, Abcam), anti-prion protein monoclonal clone SAF-84 (189775, Cayman Chemical), polyclonal anti-Iba1 (019-19741, FUJIFILM Wako Chemicals), anti-GFAP clone D1F4Q (12389, Cell Signaling Technology) for DAB staining, polyclonal anti-GFAP (AB5541, Millipore MilliporeSigma) for immunofluorescence, polyclonal anti-MAP2 (ab5392, Abcam), and anti-MBP clone EPR21188 (ab218011, Abcam). The secondary antibodies used for immunoblotting and immunohistochemistry were goat anti–rabbit IgG–HRP (474-1506, KPL) and goat anti–mouse IgG–HRP (474-1806, KPL). The secondary antibodies for immunofluorescence were goat anti–mouse IgG (Alexa Fluor 546, A-11003, Thermo Fisher Scientific), goat anti–rabbit IgG (Alexa Fluor 488, A-11008, Thermo Fisher Scientific), and goat anti–chicken IgG (Alexa Fluor 488, A-11039, Thermo Fisher Scientific).
PK digestion of BHs for Western blotting.
BHs (10%) were mixed with an equal volume of 4% sarcosyl in PBS, supplemented with 50 mM Tris, pH 7.5, digested with 20 μg/mL PK (New England BioLabs) for 30 minutes at 37°C with 1000 rpm shaking using a DELFIA plate shaker (PerkinElmer), and placed in a 37°C incubator. PK digestion was stopped by adding SDS sample buffer and heating the samples for 10 minutes in a boiling water bath. Samples were loaded onto NuPAGE 12% Bis-Tris gels, transferred to PVDF membranes, and probed with anti-prion antibody ab3531 or SAF-84, as indicated.
Analysis of conformational stability.
BHs (10%) were diluted 5-fold into PMCAb conversion buffer, and then supplemented with an equal volume of GdnHCl solution in PBS to a final concentration of GdnHCl ranging from 0.4 to 4 M, and incubated at room temperature for 1 hour. Next, 9 volumes of 2% sarkosyl in PBS were added to all samples followed by a 1-hour incubation at room temperature, and then the samples were treated with 20 μg/mL PK for 1 hour at 37°C with shaking. The digestion was stopped by adding 2 mM PMSF, and the proteins were precipitated in 4 volumes of ice-cold acetone, incubated overnight at –20°C, and centrifuged at 16,000 g for 30 minutes. Pellets were dried for 30 minutes, resuspended in 1× SDS sample buffer, loaded into NuPAGE 12% Bis-Tris gels, and then transferred to PVDF membranes and stained with antibody 3F4 or ab3531 for hamster or mouse samples, respectively. Conformational stability was performed for 3 animals from each passage.
2D electrophoresis.
Samples (25 μL) digested with PK, supplemented with gel loading buffer, and heated as described above, were solubilized for 1 hour at room temperature in 200 μL solubilization buffer (8 M urea, 2% [w/v] CHAPS, 5 mM TBP, 20 mM Tris-HCl, pH 8.0), and then alkylated by adding 7 μL of 0.5 M iodoacetamide and incubating for 1 hour at room temperature in the dark. Then, 1150 μL of ice-cold methanol was added and samples were incubated for 2 hours at –20°C. After centrifugation at 16,000 g at 4°C, the supernatant was discarded, and the pellet was resolubilized in 160 μL rehydration buffer (7 M urea, 2 M thiourea, 1% [w/v] DTT, 1% [w/v] CHAPS, 1% [w/v] Triton X-100, 1% [v/v] ampholyte, and a trace amount of bromophenol blue). Fixed, immobilized, precast IPG strips (ZM0018, Life Technologies) with a linear pH gradient from 3 to 10 were rehydrated in 155 μL of the resulting mixture overnight at room temperature inside IPG Runner cassettes (ZM0008, Life Technologies). Isoelectrofocusing (first-dimension separation) was performed at room temperature with rising voltage (175 V for 15 minutes, then 175–2000 V linear gradient for 45 minutes, then 2000 V for 30 minutes) on a Life Technologies Zoom Dual Power Supply using an XCell SureLock Mini-Cell Electrophoresis System (EI0001, Life Technologies). The IPG strips were then equilibrated for 15 minutes consecutively in (i) 6 M urea, 20% (v/v) glycerol, 2% SDS, 375 mM Tris-HCl pH 8.8, 130 mM DTT and (ii) 6 M urea, 20% (v/v) glycerol, 2% SDS, 375 mM Tris-HCl pH 8.8, 135 mM iodoacetamide, and loaded on 4%–12% Bis-Tris ZOOM SDS-PAGE precast gels (NP0330BOX, Life Technologies). For the second dimension, SDS-PAGE was performed for 1 hour at 170 V. Immunoblotting was performed with antibody 3F4 or ab3531, as indicated.
Western blot densitometry analysis.
1D or 2D Western blot signals were visualized using a FlourChem M imaging system (ProteinSimple). Densitometry was performed using AlphaView software (ProteinSimple) and analyzed as previously described (32). For generation of individual sialylation profiles, 2D gels were rotated about 90˚, to allow di-, mono-, and unglycosylated sets of spots to be defined as 3 vertical lanes using the “Lane Profile” function of AlphaView. Intensity profiles of di-, mono-, and unglycosylated “lanes” were imported to Excel for building graphs shown in Figure 4A.
Individual sialoglycoforms were divided into 5 groups according to their positions on 2D Western blots: unglycosylated, monoglycosylated hyposialylated, monoglycosylated hipersialylated, diglycosylated hyposialylated, and diglycosylated hypersialylated. The position of the demarcation line for separating isoforms into hyper- and hyposialylated groups was determined based on previous work that employed a panel of sialidases for desialylating PrPSc and establishing a boundary between the 2 groups (92). Quantification of intensities of the 5 groups in 2D blots was done with the “Multiplex band analysis” option. Within each 2D gel, 5 rectangular boxes of the same area were drawn around each group of spots (Figure 4B). The position of the rectangles was consistent between all 2D gels. The sixth rectangle of the same area was drawn on an empty part of the gel and used for background correction. The intensities of each set of spots were normalized by the sum of intensities in all 5 regions and plotted in SigmaPlot. 2D gels were run for 2 animals from each passage.
Histopathology and immunofluorescence.
Formalin-fixed brains (sagittal or coronal 3-mm slices) were treated for 1 hour in 96% formic acid before being embedded in paraffin. Sections (4 μm) produced using a Leica RM2235 microtome (Leica Biosystems) were mounted on slides and processed for hematoxylin and eosin or immunohistochemistry. Detection was performed by using HRP-labeled secondary antibodies and DAB Quanto chromogen and substrate (VWR), or Alexa Fluor 488–and Alexa Fluor 546–labeled secondary antibodies in the case of immunofluorescent detection.
To expose epitopes, slides were subjected to 20 minutes of hydrated autoclaving at 121°C in trisodium citrate buffer, pH 6.0, with 0.05% Tween 20. For detection of disease-associated PrP, an additional treatment in 88% formic acid was applied. PrP was stained with anti-prion antibody SAF-84. To detect astrocytes, rabbit anti-GFAP or chicken anti-GFAP was used. For microglia, neurons, and oligodendrocytes, rabbit anti-Iba1, chicken anti-MAP2, and rabbit anti-MBP were used, respectively. An autofluorescence eliminator (MilliporeSigma) was used according to the original protocol to reduce background fluorescence. Fluorescence images were collected using an inverted Nikon Eclipse TE2000-U microscope (Nikon Instruments Inc.) equipped with an X-cite 120 illumination system (EXFO Photonics Solutions Inc.) and a cooled 12-bit CoolSmap HQ CCD camera (Photometrics). Images were processed using WCIF ImageJ software (NIH).
Analysis of gene expression.
After euthanasia by CO2 asphyxiation, brains were immediately extracted and kept ice-cold during dissection. Brains were sliced using a rodent brain slicer matrix (Zivic Instruments). Central coronal sections (2 mm) of each brain were used to analyze the hypothalamus, thalamus, hippocampus, and cortex. The Allen Brain Atlas digital portal (http://mouse.brain-map.org/static/atlas) was used as a reference. RNA isolation was performed as described previously (43). RNA samples were processed by the Institute for Genome Science at the University of Maryland School of Medicine using the nCounter custom-designed Nanostring gene panel (Nanostring Technologies). Only samples with an RNA integrity number (RIN) greater than 7.2 were used for Nanostring analysis. All data passed quality control, with no imaging, binding, positive control, or CodeSet content normalization flags. The analysis of data was performed using nSolver Analysis Software 4.0. Ten housekeeping genes (Xpnpep1, Lars, Tbp, Mto1, Csnk2a2, CCdc127, Fam104a, Aars, Tada2b, and Cnot10) were used for normalization of gene expression.
Statistics.
Statistical analyses were performed using GraphPad Prism software, version 8.4.2 for Windows. For data analysis in Figure 1E, Brown-Forsythe and Welch’s 1-way ANOVA were applied followed by Dunnett’s T3 multiple-comparisons test. The differences in gene expression between 3 strains across 4 brain regions (Figure 8C) were analyzed by ordinary 2-way ANOVA with Tukey’s multiple-comparisons test.
The box-and-whisker plot of incubation time to clinical disease was built using Microsoft Excel. The midline denotes the median, the x represents the mean, and the ends of the box-and-whisker plot denote the 25th and 75th percentiles. The whiskers extend from the ends of the box to the minimum and maximum values. The interquartile range (IQR) was defined as the distance between the first quartile and the third quartile. A data point was considered an outlier if it exceeded a distance of 1.5 times the IQR below the first quartile or 1.5 times the IQR above the third quartile. P values of less than 0.05 were considered significant.
Study approval.
The study was carried out in strict accordance with the recommendations in the NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (assurance number: A32000-01; permit number: 0215002).
Author contributions
IVB and NM designed the study. KM and JCYC performed animal procedures and scored the disease signs. NM performed PMCAb reactions, 2D electrophoresis, analysis of conformational stability and brain dissection. JCYC performed isolation of RNAs. NM and JCYC prepared brain slices and performed immunohistochemistry. NM and IVB analyzed the data. IVB wrote the manuscript. All authors read edited and approved the final manuscript.
Supplementary Material
Acknowledgments
Financial support for this study was provided by NIH grants R01 NS045585 and R01 AI128925 to IVB.
Version 1. 06/02/2020
In-Press Preview
Version 2. 07/13/2020
Electronic publication
Version 3. 08/03/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(8):4382–4395.https://doi.org/10.1172/JCI138677.
Contributor Information
Natallia Makarava, Email: nmakarava@som.umaryland.edu.
Jennifer Chen-Yu Chang, Email: Jennifer_11294@hotmail.com.
Kara Molesworth, Email: kmolesworth@som.umaryland.edu.
Ilia V. Baskakov, Email: baskakov@som.umaryland.edu.
References
- 1.Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278(5336):245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Legname G, et al. Synthetic mammalian prions. Science. 2004;305(5684):673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 4.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104(23):9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baskakov, Breydo L. Converting the prion protein: what makes the protein infectious. Biochim Biophys Acta. 2007;1772(6):692–703. doi: 10.1016/j.bbadis.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 8.Carroll JA, et al. Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: comparison of three prion strains. PLoS Pathog. 2016;12(4):e1005551. doi: 10.1371/journal.ppat.1005551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karapetyan YE, et al. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4(5):e5730. doi: 10.1371/journal.pone.0005730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73(pt 2):329–334. doi: 10.1099/0022-1317-73-2-329. [DOI] [PubMed] [Google Scholar]
- 11.Peretz D, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10(4):854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safar J, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 13.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 2011;7(3):e1001317. doi: 10.1371/journal.ppat.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Montalban N, Makarava N, Savtchenko R, Baskakov Relationship between conformational stability and amplification efficiency of prions. Biochemistry. 2011;50(37):7933–7940. doi: 10.1021/bi200950v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klimova N, Makarava N, Baskakov The diversity and relationship of prion protein self-replicating states. Virus Res. 2015;207:113–119. doi: 10.1016/j.virusres.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998;273(48):32230–32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 17.Thomzig A, Spassov S, Friedrich M, Naumann D, Beekes M. Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. J Biol Chem. 2004;279(32):33847–33854. doi: 10.1074/jbc.M403730200. [DOI] [PubMed] [Google Scholar]
- 18.Spassov S, Beekes M, Naumann D. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim Biophys Acta. 2006;1760(7):1138–1149. doi: 10.1016/j.bbagen.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Tixador P, et al. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 2010;6(4):e1000859. doi: 10.1371/journal.ppat.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morales R, et al. Strain-dependent profile of misfolded prion protein aggregates. Sci Rep. 2016;6:20526. doi: 10.1038/srep20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baskakov, Caughey B, Requena JR, Sevillano AM, Surewicz WK, Wille H. The prion 2018 round tables (I): the structure of PrPSc. Prion. 2019;13(1):46–52. doi: 10.1080/19336896.2019.1569450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevillano AM, et al. Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J Clin Invest. 2020;130(3):1350–1362. doi: 10.1172/JCI131564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soto C, Estrada L, Castilla J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci. 2006;31(3):150–155. doi: 10.1016/j.tibs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015;38:87–103. doi: 10.1146/annurev-neuro-071714-033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau A, et al. α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci. 2020;23(1):21–31. doi: 10.1038/s41593-019-0541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woerman AL, et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019;137(3):437–454. doi: 10.1007/s00401-019-01959-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51(2):229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 28.Bolton DC, Meyer RK, Prusiner SB. Scrapie PrP 27-30 is a sialoglycoprotein. J Virol. 1985;53(2):596–606. doi: 10.1128/JVI.53.2.596-606.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry. 1989;28(21):8380–8388. doi: 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- 30.Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem. 1988;176(1):21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- 31.Katorcha E, Makarava N, Savtchenko R, D’Azzo A, Baskakov Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 2014;10(9):e1004366. doi: 10.1371/journal.ppat.1004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katorcha E, Makarava N, Savtchenko R, Baskakov Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep. 2015;5:16912. doi: 10.1038/srep16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baskakov, Katorcha E. Multifaceted role of sialylation in prion diseases. Front Neurosci. 2016;10:358. doi: 10.3389/fnins.2016.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baskakov, Katorcha E, Makarava N. Prion strain-specific structure and pathology: a view from the perspective of glycobiology. Viruses. 2018;10(12):E723. doi: 10.3390/v10120723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castilla J, Gonzalez-Romero D, Saá P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134(5):757–768. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green KM, et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008;4(8):e1000139. doi: 10.1371/journal.ppat.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chianini F, et al. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A. 2012;109(13):5080–5085. doi: 10.1073/pnas.1120076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Priem J, Langeveld JP, van Keulen LJ, van Zijderveld FG, Andreoletti O, Bossers A. Enhanced virulence of sheep-passaged bovine spongiform encephalopathy agent is revealed by decreased polymorphism barriers in prion protein conversion studies. J Virol. 2014;88(5):2903–2912. doi: 10.1128/JVI.02446-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klingeborn M, Race B, Meade-White KD, Chesebro B. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc Natl Acad Sci U S A. 2011;108(48):E1244–E1253. doi: 10.1073/pnas.1111255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makarava N, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119(2):177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makarava N, et al. Stabilization of a prion strain of synthetic origin requires multiple serial passages. J Biol Chem. 2012;287(36):30205–30214. doi: 10.1074/jbc.M112.392985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeffrey M, et al. Pathology of SSLOW, a transmissible and fatal synthetic prion protein disorder, and comparison with naturally occurring classical transmissible spongiform encephalopathies. Neuropathol Appl Neurobiol. 2014;40(3):296–310. doi: 10.1111/nan.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Makarava N, Chang JC, Molesworth K, Baskakov Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol Dis. 2020;137:104783. doi: 10.1016/j.nbd.2020.104783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makarava N, Chang JC, Kushwaha R, Baskakov Region-specific response of astrocytes to prion infection. Front Neurosci. 2019;13:1048. doi: 10.3389/fnins.2019.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katorcha E, Srivastava S, Klimova N, Baskakov Sialylation of glycosylphosphatidylinositol (GPI) anchors of mammalian prions is regulated in a host-, tissue-, and cell-specific manner. J Biol Chem. 2016;291(33):17009–17019. doi: 10.1074/jbc.M116.732040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Legname G, Nguyen HO, Baskakov, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A. 2005;102(6):2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gómez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci. 2013;33(6):2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998;8(3):449–457. doi: 10.1111/j.1750-3639.1998.tb00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Everbroeck B, et al. Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer’s disease. Acta Neuropathol. 2004;108(3):194–200. doi: 10.1007/s00401-004-0879-2. [DOI] [PubMed] [Google Scholar]
- 50.Hartmann K, et al. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol Commun. 2019;7(1):83. doi: 10.1186/s40478-019-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishina KA, et al. The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry. 2006;45(47):14129–14139. doi: 10.1021/bi061526k. [DOI] [PubMed] [Google Scholar]
- 52.Somerville RA. Host and transmissible spongiform encephalopathy agent strain control glycosylation of PrP. J Gen Virol. 1999;80(pt 7):1865–1872. doi: 10.1099/0022-1317-80-7-1865. [DOI] [PubMed] [Google Scholar]
- 53.Makarava N, Savtchenko R, Lasch P, Beekes M, Baskakov Preserving prion strain identity upon replication of prions in vitro using recombinant prion protein. Acta Neuropathol Commun. 2018;6(1):92. doi: 10.1186/s40478-018-0597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. 2012;32(3):946–952. doi: 10.1523/JNEUROSCI.3830-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linnartz-Gerlach B, Mathews M, Neumann H. Sensing the neuronal glycocalyx by glial sialic acid binding immunoglobulin-like lectins. Neuroscience. 2014;275:113–124. doi: 10.1016/j.neuroscience.2014.05.061. [DOI] [PubMed] [Google Scholar]
- 57.Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, Neumann H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia. 2016;64(1):35–47. doi: 10.1002/glia.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srivastava S, Katorcha E, Makarava N, Barrett JP, Loane DJ, Baskakov Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci Rep. 2018;8(1):11326. doi: 10.1038/s41598-018-29720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makarava N, Chang JC, Baskakov Region-specific sialylation pattern of prion strains provides novel insight into prion neurotropism. Int J Mol Sci. 2020;21(3):E828. doi: 10.3390/ijms21030828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. doi: 10.1093/braincomms/fcaa029. Ugalde CL, et al. Markers of A1 astrocytes stratify to molecular sub-types in sporadic Creutzfeldt-Jakob disease brain [published online March 17, 2020]. Brain Communications. [DOI] [PMC free article] [PubMed]
- 61.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 62.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15(4):209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 64.Lui H, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erlich P, et al. Complement protein C1q forms a complex with cytotoxic prion protein oligomers. J Biol Chem. 2010;285(25):19267–19276. doi: 10.1074/jbc.M109.071860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flores-Langarica A, Sebti Y, Mitchell DA, Sim RB, MacPherson GG. Scrapie pathogenesis: the role of complement C1q in scrapie agent uptake by conventional dendritic cells. J Immunol. 2009;182(3):1305–1313. doi: 10.4049/jimmunol.182.3.1305. [DOI] [PubMed] [Google Scholar]
- 68.Smith HL, et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron. 2020;105(5):855–866.e5. doi: 10.1016/j.neuron.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prusiner SB, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63(4):673–686. doi: 10.1016/0092-8674(90)90134-Z. [DOI] [PubMed] [Google Scholar]
- 70.Bartz JC, McKenzie DI, Bessen RA, Marsh RF, Aiken JM. Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J Gen Virol. 1994;75(pt 11):2947–2953. doi: 10.1099/0022-1317-75-11-2947. [DOI] [PubMed] [Google Scholar]
- 71.Baskakov The many shades of prion strain adaptation. Prion. 2014;8(2):27836. doi: 10.4161/pri.27836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bishop MT, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5(5):393–398. doi: 10.1016/S1474-4422(06)70413-6. [DOI] [PubMed] [Google Scholar]
- 73.Asante EA, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21(23):6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nonno R, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2(2):e12. doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Bari MA, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013;9(3):e1003219. doi: 10.1371/journal.ppat.1003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kovacs GG, Makarava N, Savtchenko R, Baskakov Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am J Pathol. 2013;183(5):1539–1547. doi: 10.1016/j.ajpath.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol. 2016;186(4):1006–1014. doi: 10.1016/j.ajpath.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burke CM, et al. Cofactor and glycosylation preferences for in vitro prion conversion are predominantly determined by strain conformation. PLoS Pathog. 2020;16(4):e1008495. doi: 10.1371/journal.ppat.1008495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Makarava N, Savtchenko R, Baskakov Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J Biol Chem. 2013;288(1):33–41. doi: 10.1074/jbc.M112.419531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Makarava N, et al. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011;7(12):e1002419. doi: 10.1371/journal.ppat.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Makarava N, et al. A new mechanism for transmissible prion diseases. J Neurosci. 2012;32(21):7345–7355. doi: 10.1523/JNEUROSCI.6351-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anderson M, Bocharova OV, Makarava N, Breydo L, Salnikov VV, Baskakov Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J Mol Biol. 2006;358(2):580–596. doi: 10.1016/j.jmb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 83.Makarava N, Baskakov The same primary structure of the prion protein yields two distinct self-propagating states. J Biol Chem. 2008;283(23):15988–15996. doi: 10.1074/jbc.M800562200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ostapchenko VG, et al. Two amyloid states of the prion protein display significantly different folding patterns. J Mol Biol. 2010;400(4):908–921. doi: 10.1016/j.jmb.2010.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ostapchenko VG, Makarava N, Savtchenko R, Baskakov The polybasic N-terminal region of the prion protein controls the physical properties of both the cellular and fibrillar forms of PrP. J Mol Biol. 2008;383(5):1210–1224. doi: 10.1016/j.jmb.2008.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Makarava N, Baskakov The evolution of transmissible prions: the role of deformed templating. PLoS Pathog. 2013;9(12):e1003759. doi: 10.1371/journal.ppat.1003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Makarava N, Savtchenko R, Baskakov Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathol Commun. 2015;3:69. doi: 10.1186/s40478-015-0248-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang W, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580(7802):283–287. doi: 10.1038/s41586-020-2043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arakhamia T, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell. 2020;180(4):633–644.e12. doi: 10.1016/j.cell.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonzalez-Montalban N, et al. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 2011;7(2):e1001277. doi: 10.1371/journal.ppat.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katorcha E, Baskakov Analyses of N-linked glycans of PrPSc revealed predominantly 2,6-linked sialic acid residues. FEBS J. 2017;284(21):3727–3738. doi: 10.1111/febs.14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.