

Abstract
Compared to aryl–aryl π-stacking interactions, the analogous stacking of heteroarenes on amide π systems is less well understood and vastly underutilized in structure-based drug design. Recent theoretical studies have delineated the important geometric coordinates of the interaction, some of which have been confirmed with synthetic model systems based on Rebek imides. Unfortunately, a broadly useful and tractable protein–ligand model system of this interaction has remained elusive. Here we employed a known inhibitor scaffold to study π-stacking of diverse heteroarene substituents on the amide face of Gly238 in the cephalosporinases CTX-M-14 and CTX-M-27. Biochemical inhibition constants (Ki) and biophysical binding constants (Kd) were determined for nineteen new analogues against both enzymes, while multiple high-resolution co-crystal structures revealed remarkably consistent placement of the probe heteroarene on Gly238. The data presented support the predicted importance of opposing dipoles in amide-heteroarene interactions and should be useful for evaluating other theoretical predictions concerning these interactions.
Graphical Abstract

The binding of diverse heterocyclic probe ligands to a bacterial hydrolase reveals important features of amide–heteroarene π-stacking interactions.
Introduction
Mining of large crystallographic data sets has revealed the importance of non-canonical intermolecular interactions in protein structure and also in protein-ligand binding.1–4 One of these is the ability of protein backbone amides to participate in stacking interactions with their π surfaces. First noted in the stacking of aromatic side chains on backbone amides,1 the importance of the interaction in protein-ligand binding is becoming increasingly apparent. An important example is found in the S1 pocket of the serine protease factor Xa (fXa), which is lined by an amide backbone π surface that can engage heterocycles in the P1 side chain of fXa inhibitors.5, 6 In a recent computational study, Sherrill and co-workers concluded that the well-known affinity of chloroarene P1 moieties for the S1 pocket is better understood in terms of π stacking with the backbone amides than by a Cl–π interaction with Tyr228.
Recent computational studies by Imai,7 Diederich,8 and Wheeler9 have sought to define the optimal geometries and distances for amide-heteroarene interaction, using formamide7 or N-methylacetamide8, 9 (NMAC) as a model amide (Figure 1). In general these studies have suggested a preference for offset stacking in which the dipole moments or local electric field of the amide and heteroarene are roughly opposed (i.e. α~180o, Figure 1). Wheeler showed, however, that intermolecular N–H3C interactions could override the preference for opposed dipoles in some cases. Wheeler’s model9 therefore introduces additional molecular descriptors to better capture these local effects and has most recently10 been extended to stacking on the π surface of Arg-Asp salt bridges.
Figure 1.

Coordinate system employed in computational studies of amide-heteroarene interactions. Dipole moment vectors for the pyridyl ring (red arrow) and amide (blue arrow) are shown, using the physics convention with arrowhead electropositive.
Rebek introduced the use of cleft-like imides derived from Kemp’s triacid11 to model a variety molecular phenomena, from the stacking and H-bonding of adenine bases to abiotic self-replicating systems.12, 13 Recently, Diederich14 described an elegant application of this platform to interrogate amide-heteroarene π-stacking interactions. This system comprises a Rebek imide host and cognate 2,6-di(isobutyramido)pyridine guest that associate in non-polar solvents with their respective para substituents held in close proximity for interaction (Figure 2). Using double-mutant analyses to isolate incremental Gibbs free energies (ΔΔG) for the interacting distal substituents, this study confirmed the favourable effects of N-methyl carboxamide stacking on several different heteroarenes and confirmed N-Me amides as preferred stacking partners compared to phenyl, ethyl, or thiomethyl groups.
Figure 2.

A Rebek imide host bound through complementary hydrogen bonding to its cognate guest, placing pendant substituents (blue) in close proximity.
Factor Xa and its known ligands (Figure 3) would appear to be excellent models to study amide-heteroarene interactions in a more pharmacologically relevant context. A strong cation-π interaction in the S4 pocket along with a Cl–π interaction in S1 places an oxazole ring ~3.8 Å from the amide surface of Gln192, well placed for an amide-heteroarene interaction. However, a liability of this scaffold is the fact that the heteroarene ring being probed also serves as a linker to the P1 side chain. As noted by Diederich,6 replacement of 2,4-oxazole with related heteroarenes such as isoxazole or 2,5-oxazole significantly alters the angular relationship between the tricyclic core and the terminal chlorothiophene, an effect that is likely to overwhelm and confound any attempt to isolate and study the amide-heteroarene interaction within this system.
Figure 3.

Factor Xa inhibitor (left) and its interaction with Gln192 in the complex crystal structure (PDB: 2Y5G)
A more recent study by the same group15 employed reversible-covalent inhibitors of the cysteine protease cathepsin L in which the terminus of the P3 side chain was altered with diverse heteroarenes expected to interact with an amide π surface lining the S3 pocket. With a terminal heteroarene, this system avoids the problem of variable angles of departing bonds in different heteroarenes. However, study of the cathepsin inhibitors revealed a different complication – the targeted amides of Gly67 and Gly68 in the S3 pockets are arranged with opposite dipole orientations. Thus, while the expected preference of heteroarenes over simple arenes was confirmed, flexibility in the ligand’s P3 side chain allowed interaction with the amide π surface of either Gly67 or Gly68, rendering the effects of relative dipole-dipole angles impossible to discern.
Here we propose the serine hydrolase CTX-M, an extended-spectrum β-lactamase, as an improved model system for study of amide-heteroarene π-stacking under physiological conditions. The utility of this system hinges on a non-covalent, reversible inhibitor scaffold previously described by our laboratories,16, 17 and exemplified by 1 (Figure 4). Extensive crystallographic characterization of 1 and its congeners has revealed a highly conserved binding mode enforced by multiple polar and hydrophobic interactions in the active site of CTX-M enzymes. These interactions include H-bonding and stacking of the tetrazole ring with the β−3 strand, H-bonding with Asn132 and Asn104, and a hydrophobic interaction between the trifluoromethyl group and Pro167 (Supplementary Figure 1). Most relevant to the current study is Gly238 of the β−3 strand, which presents a π surface ideally positioned for stacking with probe heterocycles (i.e., R) in analogues such as 2-20 (Figure 4).
Figure 4.

Known CTX-M inhibitor 1 and new analogues 2–20 described herein.
Herein we describe the synthesis of 2–20 and the determination of Ki and Kd values for all analogues against CTX-M-14 and CTX-M-27. High-resolution complex crystal structures of select analogues confirmed the expected binding mode, which places the heteroarene substituent ~3.7–3.9Å from Gly238. The activity and binding data are interpreted in terms of suggested8, 9 ‘rules-of-thumb’ governing the interaction. We find these rules to be useful for predicting affinities in congeneric series, but caution that more rigorous computational analysis will be required to understand the subtleties of this interaction.
Results and discussion
The heteroarenes employed in analogues 2–20 were selected to encompass the majority of ring systems examined in earlier computational studies, and included regioisomeric pyridine, pyrimidine, furan, and thiophene analogues, as well nitrogen heterocycles of increasing N-atom count (Figure 5). The phenyl analogue 2 was prepared as a control and comparator. The synthesis of 2–20 involved the late-stage coupling of the relevant benzoic acid intermediates with commercial 3-(1H-tetrazol-5-yl)aniline using HATU. Benzoic acids for 2-13 and 15 were synthesized via Suzuki coupling of heterocyclic boronic acids or bromides with tert-butyl 3-bromo-5-(trifluoromethyl)benzoate or 3-bromo-5-(trifluoromethyl)benzoic acid, followed by cleavage of the t-butyl ester (when required). Benzoic acid intermediates for the preparation of analogues 14 and 16 were prepared via Ullman/Goldberg coupling, while compound 17 was obtained by reaction of tert-butyl 3-bromo-5-(trifluoromethyl)benzoate with imidazole in the presence of CuI and Pd(OAc)2.18 Benzoic acid intermediates used for the preparation of compounds 18-20 were obtained via cycloaddition or cyclization reactions of the corresponding azide, amine, or nitrile, respectively. All final analogues were purified by HPLC before evaluation. Full synthetic details and characterization of all analogues are provided in the Supporting Information.
Figure 5.
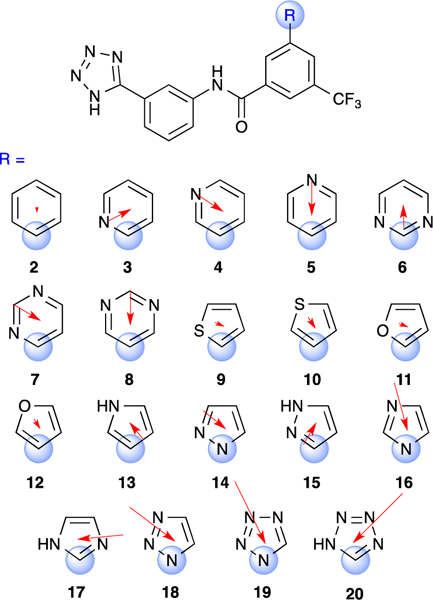
Structure of CTX-M inhibitors 2–20 bearing diverse heteroaryl substituents R. Blue spheres denote atom of attachment. Red arrows depict direction and magnitude of calculated dipole moments of corresponding methyl-substituted heteroarenes using B3LYP/6–31G** with PBF solvation (10.64 ε).
To determine whether the pendant heteroarenes in 2-20 are properly positioned to stack on Gly238 in the β−3 strand, the structures of representative analogues were solved in complex with both CTX-M-14 and CTX-M-27. These CTX-M isoforms differ only by the presence of Asp (CTX-M-14) or Gly (CTX-M-27) at position 240 of the β−3 strand, directly adjacent to the site of amide-heteroarene interaction with Gly238 (there is no residue 239 in the CTX-M sequence due to numbering conventions in Class A β-lactamases). Ligands 3, 14, and 20 were solved in complex with CTX-M-14 to resolutions of 1.4Å, 1.4Å, and 1.25Å, respectively. These structures revealed a highly conserved binding orientation that is exactly analogous to that of 1, with the pendant heteroarene involved in an apparent stacking interaction with Gly238, as posited (Figure 6 and Supplementary Figure 2). In all three structures, the side chain of Asp240 swings away from the terminal heteroarene ring towards solvent to accommodate the stacking interaction.
Figure 6.

Complex structures of analogues 3 (PDB: 6OOK; top), 14 (PDB: 6OOJ; middle) and 20 (PDB: 6OOF; bottom) bound to CTX-M-14 at 1.4Å, 1.4Å, and 1.25Å resolution. Unbiased Fo − Fc densities are shown at 3σ. Stacking distances are indicated, as measured from centroid of the heteroarene to the amide nitrogen atom.
It is notable that compound 20 binds in the canonical fashion when one considers that an unfavourable charge-charge interaction might have been expected between the tetrazol-5-yl ring and Asp240. Consistent with this, analogue 20 was both the weakest inhibitor of CTX-M-14, and the most potent inhibitor of CTX-M-27, Gly240 replacing Asp240 in the latter enzyme, thereby removing the putative charge-charge interaction (Table 1). Thus, a likely unfavourable interaction with Gly240 in CTX-M-14 is insufficient to produce a distinct binding mode for 20, thus suggesting that the remaining analogues in the series bind similarly.
Table 1.
Ki and Kd values for heterocycles 2–20 with CTX-M-14 and CTX-M-27.
| CTX-M-14 | CTX-M-27 | ||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R = | Kia (μM) | Kdb (μM) | Kia (μM) | Kdc (μM) | ΔΔGKid (kcal/mol) |
ΔΔGKde (kcal/mol) |
| 2 | phenyl | 79.6 ± 9.2 | N.D.f | 10.4 ± 0.4 | 40.3 ± 2.7g | -- | -- |
| 3 | pyrid-2-yl | 27.7 ± 0.9 | 8.14 ± 0.08 | 3.92 ± 0.48 | 1.79 ± 0.05 | 0.578 | 1.844 |
| 4 | pyrid-3-yl | 29.3 ± 1.9 | 43.5 ± 0.9 | 2.65 ± 0.31 | 3.85 ± 0.05 | 0.810 | 1.390 |
| 5 | pyrid-4-yl | 18.5 ± 1.3 | 30.3 ± 0.6 | 3.06 ± 0.24 | 5.90 ± 0.13 | 0.724 | 1.138 |
| 6 | pyrimidin-2-yl | 15.3 ± 2.1 | 4.07 ± 0.04 | 2.26 ± 0.28 | 0.834 ± 0.024 | 0.904 | 2.296 |
| 7 | pyrimidin-4-yl | 18.9 ± 2.1 | 8.34 ± 0.08 | 3.72 ± 0.23 | 2.91 ± 0.05 | 0.609 | 1.556 |
| 8 | pyrimidin-5-yl | 53.8 ± 4.8 | 42.1 ± 0.9 | 4.00 ± 0.36 | 4.41 ± 0.04 | 0.566 | 1.310 |
| 9 | thiophen-2-yl | 27.3 ± 1.8 | 50.2 ± 0.6 | 5.50 ± 0.27 | 13.8 ± 1.2g | 0.377 | 0.635 |
| 10 | thiophen-3-yl | 31.1 ± 2.2 | 51.0 ± 1.2 | 5.21 ± 0.28 | 11.4 ± 0.7g | 0.409 | 0.748 |
| 11 | furan-2-yl | 17.2 ± 0.3 | 15.7 ± 0.3 | 3.86 ± 0.37 | 2.50 ± 0.07 | 0.587 | 1.646 |
| 12 | furan-3-yl | 17.4 ± 0.4 | 26.1 ± 1.2 | 3.02 ± 0.45 | 3.28 ± 0.07 | 0.732 | 1.485 |
| 13 | 1H-pyrrol-3-yl | 10.6 ± 0.8 | 17.6 ± 0.7 | 2.07 ± 0.24 | 2.28 ± 0.06 | 0.956 | 1.701 |
| 14 | 1H-pyrazol-1-yl | 22.5 ± 2.7 | 19.4 ± 0.3 | 2.81 ± 0.33 | 2.19 ± 0.06 | 0.775 | 1.725 |
| 15 | 1H-pyrazol-3-yl | 12.6 ± 0.5 | 16.8 ± 0.5 | 1.69 ± 0.14 | 1.95 ± 0.07 | 1.076 | 1.793 |
| 16 | 1H-imidazol-1-yl | 30.5 ± 1.6 | 29.5 ± 0.6 | 3.63 ± 0.30 | 6.21 ± 0.07 | 0.623 | 1.107 |
| 17 | 1H-imidazol-2-yl | 16.9 ± 0.7 | 8.63 ± 0.17 | 4.04 ± 0.34 | 1.33 ± 0.03 | 0.560 | 2.020 |
| 18 | 1,2,3-triazol-1-yl | 32.7 ± 0.7 | 32.3 ± 0.9 | 3.65 ± 0.47 | 4.16 ± 0.10 | 0.620 | 1.345 |
| 19 | tetrazol-1-yl | 29.7 ± 0.7 | 29.8 ± 1.2 | 5.30 ± 0.23 | 7.24 ± 0.06 | 0.399 | 1.017 |
| 20 | tetrazol-5-yl | 82.0 ± 11.6 | 11.9 ± 0.1 | 1.49 ± 0.25 | 0.292 ± 0.004 | 1.151 | 2.918 |
Mean ± SEM of three replicates. Calculated from IC50 using Ki = IC50/(1 + [S]/Km). Km values were measured for each replicate.
Mean ± SEM of four replicates. SPR generated Kd values were measured at binding equilibrium.
Mean ± SEM of two replicates. SPR generated Kd values were measured at binding equilibrium.
Calculated from ΔΔG = -RTln(Ki,N/Ki,2) at 298 K, where N represents the compound to which reference compound 2 is being compared. Positive values indicate improved affinity.
Same as d, except Kd values were used relative to compound 2. Positive values indicate improved affinity.
Not determined due to solubility limits.
Mean ± SEM of four replicates.
High resolution (1.5Å and 1.25Å) structures were also solved of compounds 14 and 20 in complex with CTX-M-27 and revealed the expected binding mode and engagement of Gly238 (Figure 7). Amide-heteroarene stacking distances in the five new complex structures were measured from the centre of electron density of the heteroarene ring to the midpoint of the amide nitrogen atom and varied between 3.7–3.9Å, values that are consistent with those expected for an amide-heteroarene π-stacking interaction.
Figure 7.

Complex structures of analogues 14 (PDB: 6OOH; top) and 20 (PDB: 6OOE; bottom) bound to CTX-M-27 at 1.5Å and 1.25Å. Unbiased Fo − Fc densities are shown at 3σ. Stacking distances are indicated, as measured from centroid of the heteroarene to the amide nitrogen atom.
Compounds 2-20 were tested in 11-point dose response, in technical triplicate, for inhibition of both CTX-M-14 and CTX-M-27 using a nitrocefin substrate assay (Table 1). The inhibition curves for all replicates are provided as supporting information and speak to the precision of the measured Ki values. Consistent with the movement of Asp240 noted in the X-ray structures, analogues 2-20 returned Ki values that were ca. 5–10–fold weaker for CTX-M-14 than for CTX-M-27, the latter requiring no analogous movement of residue 240 to accommodate ligand binding (Figures 6 and 7). Similar isoform potency shifts across the analogue series provides additional evidence for a conserved binding mode.
Compared to phenyl congener 2, the heteroarene-bearing compounds 3-20 exhibited Ki values that were improved by ~3–6–fold (analogue 20 being an exception, for the reasons noted above). This was the expected result, and is consistent with the predictions of the computational studies that enhanced dipole-dipole and local electrostatic interaction favour heteroarenes as stacking partners over simple arenes. The magnitude of the potency shift was modest but statistically significant given the high precision of the Ki determinations. The compressed range of Ki values would be consistent with the expected weak nature of the amide-heteroarene interaction, and might further imply relatively small differences in desolvation energies across the congeneric series. If correct, this would be notable, given that previous model systems have exploited more hydrophobic binding pockets, in part to mitigate confounding desolvation effects.
To probe the binding affinities of 2-20 for CTX-M, we determined Kd values against both proteins by surface plasmon resonance (SPR) spectroscopy, employing the same avi-tagged proteins used for the Ki determinations. Standard error in the mean values (SEM) revealed the high precision of the Kd determinations, similar to the Ki data. We found the Kd values in absolute terms to be remarkably close to the biochemical Ki values for the majority of analogues, whereas some compounds like 3, 6, 7, 9 and 10 showed modest ~2–3–fold differences between Kd and Ki. In fact, only compound 20 exhibited Kd values that were more than 3-fold different from Ki against both proteins. Limited solubility of phenyl comparator 2 did not allow a Kd determination vs. CTX-M-14, while the Kd value of 2 vs. CTX-M-27 was ~4-fold weaker than the respective Ki. Overall, the biochemical and biophysical evaluation of 2-20 vs. CTX-M-14 and CTX-M-27 provided a robust data set for analyses of amide-heteroarene interactions. We used the CTX-M-27 Ki and Kd values to calculate ΔΔGKi and ΔΔGKd values for analogues 3-20 in reference to comparator 2 (Table 1).
Taken together, the earlier computational studies of Diederich and Wheeler predict that stronger amide-heteroarene π-stacking interactions should be expected when one or more of the following conditions are met.
-
1)
An antiparallel orientation of amide and heteroarene dipole moment vectors
-
2)
A greater magnitude of the dipole moment(s)
-
3)
The heteroarene is more electron-deficient
-
4)
Heteroarene ring nitrogens can participate in N---H–C interactions with the proximal amide function
We considered whether these simple ‘rules-of-thumb’, as might be applied by a practiced medicinal chemist, could be used to predict rank-order differences in the measured Ki and Kd values for 2-20. In performing this analysis, we assume that differences in ΔΔG values arise from the relative strength of the corresponding amide-heteroarene interaction, and ignore differences in desolvation penalties between analogues (though admittedly these may be important in certain cases). To eliminate the confounding effects of Asp240 and its interaction with the various heteroarenes, we limit the analysis to the data generated for CTX-M-27.
In computational studies of amide-heteroarene stacking the dipole interaction angle α (Figure 1) is varied systematically so as to identify the optimal value. In the present case, the binding mode of 2-20 in CTX-M places strict geometric constraints on the orientation of Gly238 and the interacting heteroarene, and producing what is predicted to be a sub-optimal α value in many cases. Unsurprisingly then, calculated dipole magnitude alone was a poor predictor of experimental ΔΔGKi and ΔΔGKd values (Figure 8). Discerning the effects of α on ΔΔG instead requires consideration of computed dipole magnitude and direction in the context of the established crystallographic binding mode.
Figure 8.

Computed dipole moments (μ) vs. A) ΔΔGKi and B) ΔΔGKd for 2-20. Dipole moments calculated from the corresponding methyl derivatives using B3LYP/6–31G** with PBF solvation (10.64 ε).
First, we considered regioisomeric series of pyridine (3–5) and pyrimidine (5–8) congeners, which are presented below in their predicted orientation relative to amide Gly238 when bound in CTX-M (Figures 9 and 10). Among these six analogues, the 2-pyridyl (3) and 2-pyrimidyl (6) heteroarenes are arranged with opposed dipole moments relative to Gly238 amide, while 4-5 and 7-8 have less favourable dipole-dipole orientations. In fact, the expected α values for 3 and 6 when bound to CTX-M are quite close to the optimal values of 105o (for 3) and 176o (for 6) reported by Wheeler for these heteroarenes in isolation.9 It is therefore significant that compounds 3 and 6 exhibited the best Kd values within each regioisomeric analogue set. The Ki values for 3-8 were more compressed and while the same rank-order trend holds for pyrimidines 6-8, the Ki values of 3-5 are very similar or within experimental error. Nevertheless, it was striking that rank-order binding affinities (Kd) of analogues 3-8 could be correctly predicted solely on the basis of the amide-heteroarene interaction.
Figure 9.
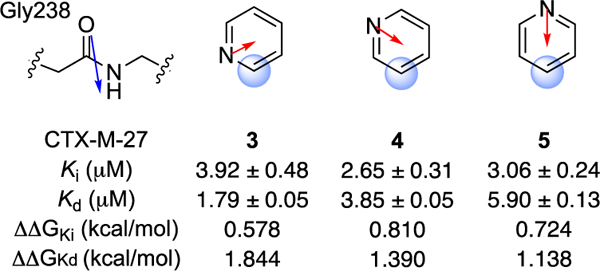
Relative orientation of Gly238 and heteroarene substituent R in analogues 3-5. Calculated dipole moments are shown in red; amide dipole in blue as reported by Diederich.15
Figure 10.
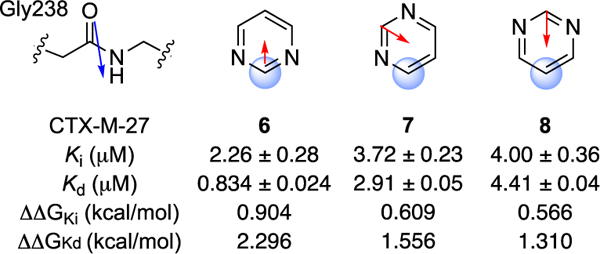
Relative orientation of Gly238 and heteroarene substituent R for analogues 6-8.
The predicted8 beneficial effect of additional ring nitrogen atoms was also reflected in the superior Kd values of pyrimidines 6-8 as compared to their corresponding pyridine regioisomers 3-5. The 3–5-fold differences in Kd across the two series are admittedly modest and one might be wary of over-interpreting these differences. On the other hand, a medicinal chemist applying a qualitative dipole-dipole analysis prospectively would have judged correctly which analogues to prioritize for synthesis and evaluation, and so such rules-of-thumb appear useful as applied to a rigid ligand scaffold and well behaved ligand-protein binding interaction such as that explored here.
In contrast to 6-8, the regioisomeric forms of the thiophene (9-10) and furan (11-12) analogues exhibited practically identical Ki and Kd values (Table 1). This finding is consistent with the similar magnitude and direction of dipole moments for these regioisomers (Figure 5). Also the Ki and Kd values of 11-12 were superior to 9-10 across all four data sets, consistent with a stronger amide-stacking interaction for the more electron-deficient furans as compared to thiophenes. The data for the remaining heterocyclic analogues 13-20 were not interpretable in terms of the rules of thumb applied. The presence of N-H donors in many of these analogues (13, 15, and 17) likely make polar interactions and desolvation penalties more significant, and these effects may overwhelm the more subtle contributions of dipole-dipole and local electrostatic interactions. A more rigorous analysis involving computed descriptions of local electrostatics and surface polarizability will likely be required to understand and make accurate predictions across a broader range of heterocycle-amide interactions present in 13-20. The exceptional binding affinity of tetrazole analogue 20 is however consistent with the predictions of Wheeler9 regarding tetrazole-amide interaction.
Finally, it is worth noting that analogues bearing axially unsymmetrical heteroarenes will have two distinct rotameric states capable of stacking on Gly238. The present crystal structures of 3 and 14 are of insufficient resolution to identify a preferred rotamer, but such analysis may be possible in the future, given that sub-Å resolution structures of 1 have been solved in which unambiguous heteroatom assignments are possible (Supplementary Figure 1).19 The identification of a preferred rotameric state in this way would enable a more refined understanding of how α values and other factors impact binding affinity in this model system.
Conclusions
Herein we present a new model system to study amide-heteroarene π-stacking in a pharmacologically relevant context. The bacterial hydrolase CTX-M-27 and inhibitor scaffold represented by 2-20 offer several advantages over previously employed model systems. These include: 1) a reversible and non-covalent ligand scaffold into which diverse heteroarenes can be incorporated in a terminal position, 2) a highly predictable and conserved binding mode that places the probe heterocycle unambiguously in contact with Gly238, and 3) a protein system that is highly amenable to X-ray crystallographic studies at high and ultra-high resolutions. The activity and binding data described herein appear to significantly report on the relative strength of amide-heteroarene stacking interactions between analogues, providing the first example where easily applied rules-of-thumb were used successfully to explain experimental binding affinity data, at least for congeneric series of analogues. In closing, we note that computational studies of the heteroarene-amide interaction present in this new model system should be facilitated by the geometric constraints imposed by the binding mode of 2-20 in CTX-M. It is our hope that a combination of theoretical and empirical study of this model system will produce new insights into the factors governing this intermolecular interaction.
Experimental
Synthesis and characterization
The syntheses and characterization of new compounds 2−20 are described in the Supporting Information. All compounds tested were judged to be of > 95% purity as assessed by a Waters Micromass ZQ 4000 equipped with Waters 2795 Separation Module, Waters 2996 Photodiode Array Detector (254 nm), and Waters 2424 ELS detector. Separations were carried out with an XBridge BEH C18, 3.5μm, 4.6 × 20 mm column, at ambient temperature (unregulated) using a mobile phase of water−methanol containing a constant 0.05% formic acid.
Protein expression and purification
All enzymes from this study were expressed using the BL21(DE3) cell line. Cells were grown on LB agar plates supplemented with 50 μg/mL kanamycin from cell stocks stored at −80°C. Single colonies were used to inoculate 50 mL of LB broth with 50 mg/mL kanamycin and grown at 37°C overnight. From the overnight culture, 10 mL of cells were used to inoculate 1 L of 2×YT broth for untagged and avitagged CTX-M-14 and CTX-M-27. The cells were grown at 37°C until an OD600 of 0.5 to 0.8 was reached at 600 nm. Overexpression of protein was induced with the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 20°C for 24 hours, and the cells were harvested with centrifugation at 5000 RPM for 15 minutes at 4°C.
For the untagged CTX-M-14 and CTX-M-27 cell pellets were resuspended in 50 mM MES pH 8.0 with 2 mM EDTA, while AviTag CTX-M-14 and CTX-M-27 were resuspended in 20 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol with 10 mM imidazole. Cells were lysed with sonication and cellular components separated via ultracentrifugation at 45,000 RPM for 1 hour. Untagged enzymes were loaded onto a CM sepharose column and eluted with an increasing NaCl gradient. The AviTag CTX-M was loaded onto a HisTrap affinity column and eluted with an increasing imidazole gradient. All enzymes were additionally purified using a size exclusion HiLoad 16/60 Superdex 75 column. Final protein purity was evaluated with SDS-Page to be at or greater than 95%.
β-lactamase inhibition assays
The hydrolytic activity of CTX-M-14 and CTX-M-27 was determined using the β-lactamase substrate nitrocefin in a reaction buffer containing 100 mM Tris pH 7.0, 20 mM NaCl, 0.02% Triton X-100, and 5% DMSO. Nitrocefin hydrolysis was monitored via absorbance (486 nm) using a FlexStation 3 microplate reader at 37 °C. The nitrocefin concentration was 50 μM for all inhibition assays. Compounds were tested for IC50 in 11-point dose response up to 2.5 mM and 500 μM for CTX-M-14 and CTX-M-27, respectively (as solubility allowed). The protein was added last to initiate the reaction; the final protein concentration was 0.1 nM for both enzymes. All compounds were tested as technical triplicates with three independent replicates. IC50 values were converted to Ki using Ki = IC50/(1 + [S]/Km). The Km of nitrocefin was measured for each replicate: 50 – 64 μM for CTX-M-14, and 14 – 22 μM for CTX-M-27 (data in Supporting Information). Nitrocefin was purchased from Sigma-Aldrich.
SPR binding assays
Compound Kd values were measured on a Biacore 4000 at 25 °C. Avi-CTX-M-14 and Avi-CTX-M-27 were immobilized on a Series S CM5 chip with EDC/NHS coupled Neutravidin using 10 mM HEPES pH 7.5, 150 mM NaCl, 0.05% Tween 20, and 250 uM TCEP-HCl. Protein immobilization levels varied between 3340–6166 RU and 4104–6634 RU for CTX-M-14 and CTX-M-27, respectively. Running buffer consisted of 10 mM HEPES pH 7.5, 150 mM NaCl, 0.05% Tween 20, 250 uM TCEP-HCl, and 5% DMSO. Compounds were flowed for 90s on and 120s off, with a 50% DMSO needle wash between injections. Compounds were tested in 10-point dose response with two internal blanks up to 500 μM and 50 μM for CTX-M-14 and CTX-M-27, respectively (as solubility allowed). Sensogram data was reference subtracted, solvent corrected, and blank subtracted; Kd values were measured at equilibrium binding between 65–85s. All compounds were tested in quadruplicate for CTX-M-14 and in duplicate for CTX-M-27. Representative sensograms and Kd fits can be viewed in the Supporting Information.
Crystallization and structure determination
All enzyme crystals were grown using the hanging drop approach, where both CTX-M-14 and CTX-M-27 protein stocks used were at 20 mg/ml. Equal parts well solution of 1 M potassium phosphate pH 7.9 were mixed with protein and incubated at 20°C. Complex structures were generated by soaking 5–10 mM ligand concentrations with protein crystals for 6–12 hours in 1 M potassium phosphate pH 7.9 or 1.44 M sodium citrate prior to cryoprotecting with 30% (wt/vol) sucrose supplemented crystal mother liquor. Crystal diffraction data sets were collected at the beamlines 22-ID, 22-BM, and 19-BM at Argonne National Lab Advanced Photon Source (APS). The data sets were indexed, integrated and scaled using the program HKL2000. Initial models were obtained via molecular replacement with the program Phaser in the Phenix suite. Refinement was carried out using phenix.refine, and ligand restraint files were generated with the program elBOW. The mFo-DFc and 2mFo-DFc maps were generated with the program phenix.mtz2map program for all structures. PDB codes for deposited structures are provided below.
CTX-M-14 with Compound 3: 6OOK
CTX-M-14 with Compound 14: 6OOJ
CTX-M-27 with Compound 14: 6OOH
CTX-M-14 with Compound 20: 6OOF
CTX-M-27 with Compound 20: 6OOE
Dipole calculations
Dipole moments were calculated with Jaguar in Maestro using the corresponding methyl derivatives, as done by Diederich in previous studies. Structures were first optimized using B3LYP/6–31G**, then dipoles were calculated using B3LYP/6–31G** with PBF solvation (10.64 ε). This dielectric was chosen as a surrogate for the local binding environment in CTX-M, which is moderately solvent exposed. Dipoles were also calculated for water (80.37 ε) and CHCl3 (4.806 ε), which had little effect on the magnitude and no effect on the relative rank order of dipole strength (Supplementary Figure 2). Model type also held no effect; M06–2X/6–31G** and B3LYP-D3/6–31G** with PBF solvation (10.64 ε) was the same as B3LYP/6–31G**, and gas phase B3LYP/6–31G** was the same as LMP2/6–31G** (Supplementary Figure 3). These results suggest that the reported dipole magnitude and rank order are accurate.
Supplementary Material
Acknowledgements
This work was funded in part by the National Institutes of Health (NIH) grant AI103158. We thank Dr. Wilian Cortopassi Coelho (Jacobson Laboratory, UCSF) for guidance in performing the dipole calculations.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Duan G, Smith VH and Weaver DF, The Journal of Physical Chemistry A, 2000, 104, 4521–4532. [Google Scholar]
- 2.Meyer EA, Castellano RK and Diederich F, Angew. Chem. Int. Ed. Engl, 2003, 42, 1210–1250. [DOI] [PubMed] [Google Scholar]
- 3.Bissantz C, Kuhn B and Stahl M, J. Med. Chem, 2010, 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salonen LM, Ellermann M and Diederich F, Angew. Chem. Int. Ed, 2011, 50, 4808–4842. [DOI] [PubMed] [Google Scholar]
- 5.Salonen LM, Bucher C, Banner DW, Haap W, Mary J-L, Benz J, Kuster O, Seiler P, Schweizer WB and Diederich F, Angew. Chem. Int. Ed, 2009, 48, 811–814. [DOI] [PubMed] [Google Scholar]
- 6.Salonen LM, Holland MC, Kaib PS, Haap W, Benz J, Mary JL, Kuster O, Schweizer WB, Banner DW and Diederich F, Chemistry, 2012, 18, 213–222. [DOI] [PubMed] [Google Scholar]
- 7.Imai YN, Inoue Y, Nakanishi I and Kitaura K, J. Comput. Chem, 2009, 30, 2267–2276. [DOI] [PubMed] [Google Scholar]
- 8.Harder M, Kuhn B and Diederich F, ChemMedChem, 2013, 8, 397–404. [DOI] [PubMed] [Google Scholar]
- 9.Bootsma AN and Wheeler SE, ChemMedChem, 2018, 13, 835–841. [DOI] [PubMed] [Google Scholar]
- 10.Bootsma AN and Wheeler SE, J Chem Inf Model, 2019, 59, 149–158. [DOI] [PubMed] [Google Scholar]
- 11.Kemp DS and Petrakis KS, The Journal of Organic Chemistry, 1981, 46, 5140–5143. [Google Scholar]
- 12.Rebek J, Acc. Chem. Res, 1990, 23, 399–404. [Google Scholar]
- 13.Wintner EA, Conn MM and Rebek J, JACS, 1994, 116, 8877–8884. [Google Scholar]
- 14.Harder M, Carnero MA Corrales N Trapp,. Kuhn B and Diederich F, Chemistry – A European Journal, 2015, 21, 8455–8463. [DOI] [PubMed] [Google Scholar]
- 15.Giroud M, Ivkovic J, Martignoni M, Fleuti M, Trapp N, Haap W, Kuglstatter A, Benz J, Kuhn B, Schirmeister T and Diederich F, ChemMedChem, 2017, 12, 257–270. [DOI] [PubMed] [Google Scholar]
- 16.Nichols DA, Jaishankar P, Larson W, Smith E, Liu G, Beyrouthy R, Bonnet R, Renslo AR and Chen Y, J. Med. Chem, 2012, 55, 2163–2172. [DOI] [PubMed] [Google Scholar]
- 17.Pemberton OA, Zhang X, Nichols DA, DeFrees K, Jaishankar P, Bonnet R, Adams J, Shaw LN, Renslo AR and Chen Y, Antimicrob. Agents Chemother, 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellina F, Cauteruccio S and Rossi R, The Journal of Organic Chemistry, 2007, 72, 8543–8546. [DOI] [PubMed] [Google Scholar]
- 19.Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL and Chen Y, J. Am. Chem. Soc, 2015, 137, 8086–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.