

Abstract
Genetic polymorphisms in natural Leishmania populations have been reported in endemic areas. Microsatellite typing is a useful tool to elucidate the genetic variability of parasite strains, due to its capability for high-resolution mapping of genomic targets. The present study employed multilocus microsatellite typing (MLMT) to explore the genotypic composition of Leishmania infantum in naturally infected dogs by genotyping parasites infecting different tissues with or without in vitro expansion. Eighty-six samples were collected from 46 animals in an endemic region of visceral leishmaniasis (VL). MLMT was performed for 38 spleen samples and 48 L. infantum cultures isolated from different tissues. Of the 86 samples, 23 were effectively genotyped by MLMT, identifying nine multilocus genotypes (MLG; referred to as MLG A–I). MLGs A, B and C were detected in more than one type of tissue and in more than one sample. Conversely, MLG D-I were uniquely detected in one sample each. The results showed that multiple genotype infections occur within a single host and tissue. Paired sample analysis revealed the presence of different MLMT alleles in 14 dogs, while the same MLG allele was present in 15 animals. STRUCTURE analysis demonstrated the presence of two populations; 13 samples displayed a similar admixture of both ancestral populations, and these were not assigned to any population. Only samples for which Q ≥ 0.70 after CLUMPP alignment were considered to be part of Population 1 (POP1) or Population 2 (POP2). POP2 comprised the majority of samples (n = 54) compared to POP1 (n = 19). This study presents evidence of multiple genotype infections (caused by L. infantum) in dogs in an area with high VL transmission. Further investigations must be undertaken to determine the effects of multiple infection on the host immune response and disease dynamics and treatment.
Author summary
American visceral leishmaniasis (VL) is a parasitic disease caused by the protozoan Leishmania infantum. This parasite can infect humans and animals and is transmitted by sand flies. Domestic dogs are considered an important host, and like humans, they can manifest the disease or present asymptomatic infections. Studies have identified genetic variations among L. infantum parasites from different endemic regions in the American continent. For other parasitic diseases (e.g., malaria), studies have suggested that multigenetic infection predicts the development of symptoms and can lead to a high level of transmission. However, the effects of the genetic composition of Leishmania parasites on VL need to be ascertained. This study used highly variable microsatellite markers to investigate multigenotype L. infantum populations among naturally infected dogs living in an area in which VL is highly prevalent. Samples obtained from different tissues were examined to identify the occurrence of multiple genotypes in the same animal and even within the same tissue.
Introduction
The Leishmania donovani complex comprises two major species: Leishmania infantum (syn. Leishmania chagasi) and Leishmania donovani. These parasites cause visceral leishmaniasis (VL) around the world. L. infantum is associated with a zoonotic epidemiological cycle, while L. donovani is mostly associated with an anthroponotic cycle. In the zoonotic cycle observed in the Americas, the Middle East, Central Asia, China and the Mediterranean, dogs represent the most important domestic reservoir host and display clinical characteristics similar to those seen in humans with the disease [1].
In the Old World, both human and canine VL are associated with several L. infantum zymodemes [2]. Conversely, in the Americas, IOC/Z1 = MON1 is highly predominant. Using high-resolution genomic targets (such as microsatellites), which are useful in exploring variability among closely related strains, it is possible to identify different genotypes within the same zymodeme [3, 4]. Microsatellite-based analyses have revealed different multilocus genotypes (MLGs) circulating in the Americas [5] and have detected at least three different genetic populations (i.e., Population 1 [POP1], Population 2 [POP2] and Population 3 [POP3]) in Brazil [6]. POP1 is widely distributed in most Brazilian endemic regions. POP2 is well dispersed but predominant in the state of Mato Grosso. POP3 is less well dispersed and mainly comprises strains found in Mato Grosso do Sul. Several genotypes have been revealed to be infecting dogs in these regions [6].
Parasite infections are often characterized by a mix of distinct genotypes [7]. Such multiple genotype infections are likely more difficult for the immune defense of a host to control; thus, the probability that an infection will thrive is greater [8]. Distinct genotypes have been described that share the same milieu within a host, and such coexistence can either promote competition or cooperation among genotypes [8–10]. The genetic composition of the coinfecting pathogen population can significantly affect infection outcomes [11]. For instance, in malaria patients, studies have suggested that multiclonal infection predicts the risk of subsequent clinical malaria [12, 13] and that a high transmission intensity is maintained by a high level of mixed infection [14].
In canine visceral leishmaniasis (CVL), the infection outcome is a consequence of the interactions between the parasite and the host genetic background [15]; however, the direct effect of protozoan genotypes on disease dynamics remains unclear. Despite the relevance of multiple genotype infections, genetic characterization of Leishmania has rarely been attempted [16, 17], especially by using naturally infected animal and human tissues without in vivo or in vitro expansion.
Recently, whole genome sequencing (WGS) was employed as a proof-of-principle for the sequencing of L. donovani genomes directly from clinical samples of patients with VL [18]. Pairwise comparisons between the parasite genomes in clinical samples and the derived in vitro-cultured promastigotes showed genomic differences suggesting the presence of polyclonal infections in patients. However, the authors noted that direct analysis of the Leishmania genome in clinical samples is still hampered by the presence of high levels of human DNA and the large variation in the parasite load. Furthermore, this approach needs to be tested on samples from other hosts, including animal reservoirs and insect vectors, and in different host tissues. While many drawbacks still limit the use of WGS, microsatellite analysis represents an alternative for analyzing Leishmania parasites that infect different hosts without the potential bias caused by parasite culture, which may vary the average genomic content along in vitro maintenance [19]. The Leishmania genome is relatively rich in microsatellites [20], which represent important molecular markers to assess variability within species and between closely related strains. Thus, targeting microsatellites is a promising option for direct tissue studies [16]. Herein, we hypothesized that Leishmania-infected dogs from areas highly endemic for human VL are prone to polyclonal infection. We employed microsatellites to type DNA derived from spleen samples of dogs presenting different clinical scores and variable parasite loads. Furthermore, we typed DNA isolated from spleen and other tissues obtained from cultured parasites. Microsatellites are locus-specific and highly polymorphic. The results obtained herein portray polyclonal infections caused by Leishmania parasites in vertebrate hosts and depict differences between clinical samples and derived culture isolates.
Methods
Animals, sample collection, Leishmania parasite isolation, culture and species identification
This work was conducted with convenience samples. Samples were collected from 46 Leishmania-seropositive dogs destined to be euthanized (with the informed consent of the dogs’ owners) at the Center for Zoonosis Control in four endemic municipalities (i.e., Rondonópolis, Barra do Garças, Várzea Grande and Cuiabá) in the state of Mato Grosso, Brazil. These animals were euthanized, as they tested positive for Leishmania infection based on the rapid test (Dual Path Platform—DPP CVL, BioManguinhos, FIOCRUZ) and ELISA, according to the Brazilian Ministry of Health recommendations. The clearance of the Fundação Oswaldo Cruz Animal Ethics Committee was not required, as the samples were obtained postmortem, and the animals were not interfered for the purposes of this project. Fig 1 and S1 Table show all the samples included in this study in relation to the animal identification, the tissue collected and the type of sample (i.e., the tissue or parasite isolated in the culture) used for the molecular analysis. Information on clinical signs and parasite load in the spleen was gathered from our previous study [21].
Fig 1. Flowchart outlining the sample collection.

DNA was directly obtained from tissues, body fluid or Leishmania infantum parasites isolated and maintained in culture media. All samples were subjected to MLMT analysis. The number of tissues or Leishmania cultures analyzed is shown in parentheses.
Immediately after euthanasia, spleen and liver fragments, bone marrow aspirate and ascites were harvested for parasite isolation or DNA extraction. Leishmania parasites were isolated by maintaining spleen and liver fragments, bone marrow and ascites aspirates in culture conditions. The samples were seeded in a biphasic blood agar base and NNN-Schneider-Drosophila medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich), 1.8% (v/v) penicillin, streptomycin and amphotericin (Sigma-Aldrich) and maintained at 25°C. Positive and non-contaminated cultures were maintained in the previously mentioned biphasic medium until the number of parasites required to perform the first in vitro passage was reached. Parasites from passages 2–5 were grown in liquid medium in 25 cm3 flasks for 3 days and then harvested for DNA extraction.
Leishmania species identification was performed by multilocus enzyme electrophoresis for parasites successfully isolated and maintained in culture or by PCR-RFLP of the hsp70 gene, following well-established protocols [22, 23].
To obtain DNA directly from the tissues of positive animals, only spleen tissues were employed and were stored in a buffer solution (10 mM NaCl, 10 mM EDTA, and 10 mM Tris HCl) at -20°C.
DNA extraction from tissues and culture of parasites
Promastigotes from positive cultures were expanded at 25°C in Schneider’s insect medium, pH 7.2, supplemented with 10% (v/v) FBS and 2% (v/v) male human urine. The parasites were grown to a density of 1×109 cells/mL (late log phase) and washed twice with PBS (pH 7.2) before DNA extraction.
Total DNA was extracted from spleen and from L. infantum promastigotes kept in vitro. DNA extraction was carried out using the Wizard Genomic DNA Purification System (Promega, Madison, WI, USA) following the manufacturer's instructions, which included a prior digestion phase with 17.5 μL of proteinase K (20 mg/mL) for 12 h at 55°C. The DNA was dissolved in 100 μL of tris EDTA buffer (TE buffer).
PCR amplification and microsatellite markers
Fourteen primers conjugated with fluorophores HEX or 6FAM were used for microsatellite amplification (as previously described in [24]). PCR was performed in a volume of 25.0 μL containing 1X reaction buffer, 1.5 mM MgCl2, 2.5 mM deoxyribonucleotide triphosphate (dNTPs), 2U Taq polymerase (GoTaq, DNA Polymerase; Promega) and 100 ng DNA. Amplification conditions were achieved using Veriti equipment (Applied Biosystems, Foster City, CA/US) and consisted of an initial denaturation step at 95°C for 5 minutes, followed by 35 cycles at 95°C for 30 seconds, an annealing temperature for 30 seconds (S2 Table) and 72°C for 1 minute and a final extension at 72°C for 10 minutes.
Each amplicon (1.0 μL) was mixed with formamide (Hi-Di Formamide, Life Technologies) and a ladder marker (GeneScan 500 ROX Size Standard, Life Technologies). Fragment-length screening was performed with 500 ROX (Applied Biosystems) as the size standards using an ABI3130XL Genetic Analyzer (Applied Biosystems). The length analyses were performed on the Peak Scanner using Software v1.0 (available at http://www.appliedbiosystems.com). The results were standardized based on size profiles from the L. infantum reference strain IOC/L0579 (MHOM/BR/1974/PP75).
Data analysis
A multilocus genotype (MLG) was assigned only to samples presenting the complete 14 microsatellite marker profile. Differences in at least one marker were enough to assign samples to a specific MLG. Samples with one or more missing data were not assigned to any MLG.
Population genetics algorithms were used to describe the clustering characteristics of the MLGs based on a previous study (see [6]). STRUCTURE software (v2.3.4) was used to infer the population structure. Next, Bayesian statistics and Markov Chain Monte Carlo simulations were used to estimate the assigned proportion of each individual belonging to each population (membership coefficient Q). The data for each value of K (from 1 to 15) were submitted on the STRUCTURE Harvester software v0.6.1 (available at http://taylor0.biology.ucla.edu/structureHarvester/, accessed on July 2019) for the estimation of delta K and the preparation of the indfiles for the software CLUMPP (v1.1.2), which was used to perform alignments from the Q values of each K STRUCTURE population. The resulting bar plots were visualized using Microsoft Excel software, and individuals were assigned to the cluster for which they exhibited the highest Q value. Finally, the Microsatellite Analyzer software was used to determine the fixation index (FST) with significance (p) tested with 1,000 permutations.
Results and discussion
Overall, 86 samples collected from 46 animals were analyzed (Fig 1). MLMT was performed by employing DNA derived directly from 38 spleen samples from L. infantum-infected dogs and from 48 L. infantum cultures isolated from different tissues. For 29 dogs, more than one sample type (tissue or parasite culture) was analyzed. For five animals, MLMT was performed only on Leishmania cultured isolates from different tissues (S1 Table).
Of the 14 loci analyzed, nine were polymorphic: Li45–24, Li41–56, Lm2TG, Lm4TA, Li 71–70, Li 71–52, Klist 70–39, Li 71–33 and Li 22–35. Alleles for each sample and marker are presented in S1 Table.
All samples were typed as L. infantum before the MLMT analyses. DNA from tissues was subjected to Leishmania species identification by PCR-RFLP hsp70, and cultured parasites were identified by isoenzyme electrophoresis in accordance with the protocols adopted by the Leishmania Collection (http://clioc.fiocruz.br/) from Fiocruz [22, 23].
This study was conducted in a highly endemic region of VL where different genotypes and genetic populations of L. infantum have been detected previously [6, 25]. Therefore, exposure to multiple independent genotype infections during the lifetime of the host is expected. Twenty-three samples (23 of n = 86) were effectively genotyped by MLMT, and nine MLGs were detected, which were referred to as MLG A–I (S1 Table). MLGs were assigned only to samples for which all the MLMT markers were successfully amplified and determined. The same MLG was detected in different tissues (Fig 2). MLG A was the most frequently detected (n = 13) and was detected in spleen (tissue) and in the derived isolated promastigotes from spleen, bone marrow and liver. MLG B was detected in spleen (tissue) and in bone marrow-derived cultures. MLG C was detected in the derived cultures of spleen and ascites aspirate. Therefore, MLGs A, B and C were detected in different tissue types in 13, 2 and 2 samples, respectively. Other MLGs were exclusively detected in one tissue type but represented by only one sample (S1 Table). Thus, specific MLG-tissue tropism cannot be assumed.
Fig 2. Multilocus genotypes (MLGs) assigned by MLMT found in Leishmania infantum-infected dogs in an endemic area of visceral leishmaniasis in Brazil.

Microsatellite marker (MLMT) analysis was performed using DNA from tissue-derived L. infantum cultures or tissue DNA spleens. Due to missing data, 64 analyzed samples could not be genotyped. The remaining 22 samples were successfully assigned to an MLG (A to I). Three MLGs were found in more than one tissue. One genotype (A) was detected in all samples except those derived from ascites.
Four MLGs (A, B, E and G) were observed in bone marrow-derived Leishmania DNA, and two MLGs (A and F) were observed in the liver. Six MLGs were detected among the spleen samples, four of which (A, C, D and I) were found in spleen-derived cultures and three of which (A, B, and H) were identified directly from spleen tissue DNA. The results indicate that multiple genotype infections occur within a host and even within a single tissue sample (Fig 3).
Fig 3. Multiple genotype Leishmania infantum infections detected by MLMT in paired samples from dogs from an endemic area of visceral leishmaniasis in Brazil.
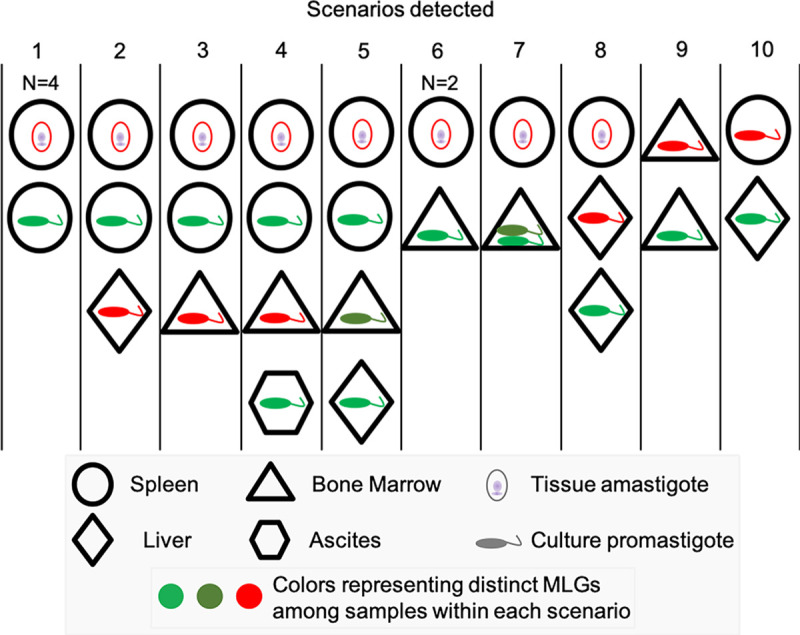
Microsatellite analyses were performed for 14 markers using DNA from tissue derived from cultured parasites or using tissue (spleen) DNA extracted directly from naturally infected animals. Each column represents one scenario observed among the analyzed paired samples. For example, Scenario 1 was observed in 4 dogs, in which the paired samples (spleen tissue and spleen-derived isolated promastigotes) exhibit distinct multilocus genotypes (MLGs). Scenario 2 reproduces Scenario 1 with the additional observation of the same MLG in spleen tissue and liver-derived cultures. Scenario 7 depicts distinct MLGs in spleen (tissue) and bone marrow-derived cultures, in which two distinct MLGs were codetected.
In the paired sample analysis, distinct MLMT alleles were detected in 14 dogs (Table 1 and S1 Table), reflecting 10 different scenarios (Fig 3) in which infection by distinct MLGs occurred. The observations within our sample revealed i) different MLGs in two cultures derived from the same tissue (Animals 113, 116 and 121—Table 1 and Scenarios 8 and 9—Fig 3); ii) different MLGs coinfecting the same tissue and the same MLGs infecting different tissues (Animals 244, 252 and 256—Table 1; Scenarios 2, 3, 4, 5 and 7—Fig 3). It is unlikely that the in vitro conditions favored a specific genotype, as we also observed paired sample genotype matching. Thus, the effect of culture conditions may reflect the random filtering of parasite cell populations.
Table 1. Animals displaying different microsatellite alleles representing Leishmania infantum parasites infecting distinct tissues.
For DNA isolation directly from tissues, only spleen samples were employed, but DNA was also isolated from cultured parasites isolated from diverse tissues from naturally infected dogs living in an endemic area of visceral leishmaniasis in Brazil.
| Animal ID | Sample type1 | Sample ID | Tissue2 | Marker | Scenario (depicted in Fig 3) | |
|---|---|---|---|---|---|---|
| Allele 1 |
Allele 2 | |||||
| 107 | Lm4TA | |||||
| Tissue | 107 | Spleen | 81 | 83 | ||
| Strain | 3130 | Bone Marrow | 79 | 83 | 6 | |
| 110 | Lm4TA | |||||
| Tissue | 110 | Spleen | 83 | 83 | 6 | |
| Strain | 3131 | Bone Marrow | 81 | 81 | ||
| 113* | Lm2TG | |||||
| Tissue | 113 | Spleen | 140 | 140 | 7 | |
| Strain | 3132 | Bone Marrow | 142 | 142 | ||
| Strain | 3158 | Bone Marrow | 142 | 142 | ||
| Lm4TA | ||||||
| Tissue | 113 | Spleen | - | - | ||
| Strain | 3132 | Bone Marrow | 81 | 81 | ||
| Strain | 3158 | Bone Marrow | 83 | 83 | ||
| 116 | Li41–56 | |||||
| Strain | 3133 – 1st | Bone Marrow | 88 | 90 | 9 | |
| Strain | 3133 – 2nd | Bone Marrow | 90 | 90 | ||
| 121 | Li22–35 | |||||
| Tissue | 121 | Spleen | 94 | 94 | 8 | |
| Strain | 3134 | Liver | 94 | 94 | ||
| Strain | 3135 | Liver | 96 | 96 | ||
| Lm4TA | ||||||
| Tissue | 121 | Spleen | 83 | 83 | ||
| Strain | 3134 | Liver | 83 | 83 | ||
| Strain | 3135 | Liver | 79 | 79 | ||
| Li 71–33 | ||||||
| Tissue | 121 | Spleen | 105 | 105 | ||
| Strain | 3134 | Liver | 105 | 105 | ||
| Strain | 3135 | Liver | 106 | 106 | ||
| Li 71–52 | ||||||
| Tissue | 121 | Spleen | - | - | ||
| Strain | 3134 | Liver | 110 | 110 | ||
| Strain | 3135 | Liver | 108 | 108 | ||
| Li71–70 | ||||||
| Tissue | 121 | Spleen | 100 | 100 | ||
| Strain | 3134 | Liver | 100 | 100 | ||
| Strain | 3135 | Liver | 98 | 98 | ||
| 124 | Lm2TG | |||||
| Tissue | 124 | Spleen | 138 | 142 | 1 | |
| Strain | 3137 | Spleen | 136 | 142 | ||
| 239 | Li22–35 | |||||
| Tissue | 239 | Spleen | 94 | 94 | 1 | |
| Strain | 3201 | Spleen | 94 | 96 | ||
| 244 | Li 71–33 | |||||
| Tissue | 244 | Spleen | 105 | 107 | ||
| Strain | 3206 | Spleen | 107 | 107 | 4 | |
| Strain | 3205 | Bone Marrow | 105 | 107 | ||
| Strain | 3207 | Ascites | 107 | 107 | ||
| Li22–35 | ||||||
| Tissue | 244 | Spleen | 94 | 94 | ||
| Strain | 3206 | Spleen | 94 | 96 | ||
| Strain | 3205 | Bone Marrow | 94 | 96 | ||
| Strain | 3207 | Ascites | 94 | 96 | ||
| 251 | Li22–35 | |||||
| Strain | 3210 | Spleen | 94 | 96 | 10 | |
| Strain | 3211 | Liver | 94 | 94 | ||
| 252 | Lm4TA | |||||
| Tissue | 252 | Spleen | 79 | 83 | ||
| Strain | 3213 | Spleen | 83 | 83 | 3 | |
| Strain | 3212 | Bone Marrow | 79 | 83 | ||
| 254 | Lm2TG | |||||
| Tissue | 254 | Spleen | 142 | 142 | 1 | |
| Strain | 3215 | Spleen | 128 | 142 | ||
| 256* | Lm2TG | |||||
| Tissue | 256 | Spleen | 142 | 142 | 2 | |
| Strain | 3217 | Spleen | 144 | 144 | ||
| Strain | 3218 | Liver | 142 | 142 | ||
| Lm4TA | ||||||
| Tissue | 256 | Spleen | 83 | 83 | ||
| Strain | 3217 | Spleen | 81 | 81 | ||
| Strain | 3218 | Liver | 83 | 83 | ||
| 257 | Lm2TG | |||||
| Tissue | 257 | Spleen | 142 | 142 | ||
| Strain | 3220 | Spleen | 142 | 144 | 5 | |
| Strain | 3219 | Bone Marrow | 142 | 146 | ||
| Strain | 3221 | Liver | 142 | 144 | ||
| 291* | Lm2TG | |||||
| Tissue | 291 | Spleen | 142 | 142 | 1 | |
| Strain | 3323 | Spleen | 142 | 144 | ||
| Lm4TA | ||||||
| Tissue | 291 | Spleen | 83 | 83 | ||
| Strain | 3323 | Spleen | 85 | 85 | ||
1Sample type—type of sample employed for DNA extraction; Tissue indicates that DNA was extracted directly from a spleen fragment and Strain indicates that DNA was extracted from cultured parasites.
2Tissue–the tissue from which DNA was extracted or the parasite (strain) from which DNA was isolated; *Different population assignment was made after STRUCTURE analysis
As examples: i) the same MLG occurred in the spleen tissue and bone marrow-derived promastigotes (Animal 252 and Scenarios 3 and 4 in Fig 3) or liver (see Animal 256 and Scenarios 2 in Fig 3); however, the spleen-derived isolate differed. ii) Two isolates from different liver fragments of the same animal were typed as distinct MLG, and one of them was identical to the MLG from the spleen tissue (see Animal 121).
The differences in paired samples were found either in one (see Animals 124, 239, 252, 254 and 257) or two loci (Animals 244, 256 and 291). The difference in only one marker may be due to the selection of a less frequent, genetically diverse clone from the initial population. Errors cannot be ruled out, although microsatellite marker stability has already been demonstrated [26]. All mismatches in the same dog genotype were triple-checked via independent PCR and fragment analyses.
MLMT analysis allocated the paired samples of three dogs in different populations (Table 1 and Fig 4): Samples from Animal 113 were typed as POP1 in spleen and POP2 in the bone marrow; Animal 256 was typed as POP1 in the spleen and POP2 in the liver-derived sample; Distinct spleen samples from Animal 291 were genotyped as distinct MLMT populations.
Fig 4. Population structure of Leishmania infantum infecting dogs in an endemic area of Visceral Leishmaniasis in Brazil.

Data were obtained on the STRUCTURE software and processed for visualization (see Material and Methods section for details). MLMT was performed in 86 samples for 14 microsatellite markers using DNA extracted from Leishmania isolated from tissues or using DNA extracted directly from the spleen of infected dogs. Two genetic clusters were identified corresponding to POP1 (dark blue horizontal bar) and POP2 (light blue horizontal bar). Each vertical line represents one sample and is divided into the estimated proportion (length of the colour) of membership in that population. 13 samples showed admixture and we considered as POP1 or POP2 when presenting Q≥0.70 (k = 2; Fst = 0.405753; p = 0.0001). Symbols +, # and * indicate dogs 256, 291 and 113, respectively.
Distinct promastigote cultures derived from the same tissue may present similar L. infantum MLG combinations, but only the most prevalent MLG is efficiently detected by MLMT. Therefore, although both MLGs originally coinfected the tissue and were successfully isolated, the culture conditions stochastically filtered the parasite cell populations, affecting the final detection of MLGs. More high-resolution approaches, such as single-cell sequencing or cloning, could be used to explore this phenomenon.
Samples analyzed herein came from dogs that mainly lived in the same municipality; thus, the low number of unique MLGs that was found is expected and suggests clonal propagation. Higher genotypic diversity was demonstrated in another study conducted in Brazil [6], but the analyzed strains were from different geographic regions and hosts. Our results are similar to those obtained by Motoie and coworkers [16] who also used MLMT to genotype L. infantum directly from dog tissue collected at different municipalities in the State of São Paulo. Of the 112 samples analyzed, Motoie et al. observed 33 MLGs (of which 20 represented unique MLGs) structured across two populations that corresponded to the Northwest and Southeast regions of the State.
The STRUCTURE analysis grouped the samples into two populations (k = 2; FST = 0.405753; p = 0.0001), which is supported by the FST value; 13 samples displayed a similar admixture for both ancestral populations. Only samples for which Q ≥ 0.70 after the CLUMPP alignments were considered to be part of POP1 or POP2 (4). In our previous study, at least two L. infantum populations were detected in Mato Grosso [6], which is the same region in which the present study was conducted. Due to missing data, comparisons with the samples analyzed in our previous study could not be performed. Even though this corroborated the results of our previous study, a higher proportion of POP2 samples (54 samples) was also observed. Conversely, POP1 comprised only 21 samples (see [6]). Based on the analysis of MLGs, there was no indication of tissue tropism in any of the populations.
In many parasitic diseases, acquired immunity due to primary infection does not always fully protect against secondary infection [27, 28]. Thus, the dogs could have been subject to multiple infections during their lifetime, as they had lived in an endemic region. Multiple genotype infections may also occur in sand flies ultimately transmitted to vertebrate hosts; however, Leishmania detection in sand flies is not an easy task, and methods such as MLMT are not commonly employed. It is known that sand flies can present natural mixed infections by distinct Leishmania species [29, 30], and experimental infections using L. major and L. turanica showed no sign of competition [31]. Recently, it was hypothesized that the particular composition of the sand fly fauna in one region in Italy may play a role in the selection of a particular L. infantum population that circulates in humans and vectors but not in dogs [32]. Although the methodology employed was different, and the results cannot be directly compared, studies conducted in Brazil indicated that the same L. infantum strain infected dogs and humans [33, 34].
In endemic areas in which different Leishmania genotypes are transmitted, the disease can be the result of a heterogeneous infective inoculum, which is likely the result of an accumulation of multiple independent infections [35]. Superinfections, particularly by strains with different genotypes, have been shown to be important in the pathogenesis of some parasitic diseases [27, 36]. In malaria, parasite genetic diversity and multiplicity of infection affect clinical outcomes, responses to drug treatment and naturally acquired or vaccine-induced immunity [37]. The spleen parasite loads and clinical scores of the dogs examined in the present study had been previously determined [21], but no difference in clinical signs or parasite load was observed between multiple MLG infection and single MLG infection groups. Of the 29 dogs from which paired samples were obtained, 15 presented the same MLG (the single MLG infection group). Of those, eight had high clinical scores, five had medium scores and two had low scores. Similarly, of the 14 dogs that had multiple MLG infections, six had high clinical scores, another six had medium scores and two had low scores. In both groups, most animals had high parasite loads (a parameter associated with modifications in the structure of splenic lymphoid microarchitecture and deficiencies in cytokine expression) [21].
Dogs in high endemic CVL areas are prone to multiple infections, i.e., may be coinfected by multiple L. infantum genotypes. However, such coinfections are not easily detected due to the low resolution in the technical approaches available. This could lead to the underestimation of the frequency and diversity of multiple genotype infections. Studies on malaria patients have suggested that the multiplicity of infection usually reflects the degree of transmission intensity, although the association is not linear [38, 39]. In endemic regions for malaria, mixed infections represent more than 70% of infections [14]. In a population infected by Plasmodium species, multiclonal infections were detected in 62.2% of the samples by long-fragment deep sequencing and 45.9% of the samples by short-fragment deep sequencing, but only 5.2% of the samples were successfully identified by microsatellite markers [37]. The detection of different MLMT profiles in parasites collected from human patients who have presented more than one episode of VL might reflect a clinical relapse or reinfection [40]; also, multiclonal infections cannot be ruled out. To date, very few studies have considered the occurrence of mixed infections in Leishmania. Recently, chromosome copy number variation was demonstrated among clones isolated from an HIV-VL patient. Variation was also observed in the k26-PCR amplicon, suggesting mixed infection. In addition, different karyotypes were observed in strains isolated from different organs (i.e., the spleen, skin and bone marrow) from the same patient [41]. Differences in the chromosome ploidy of strains isolated from different organs may be the result of clonal selection of parasites in the host due to specific selective pressures at the infection site. Genetically diverse infections may be common in many VL endemic areas; however, it is important to address the effects that coinfected strains have on each other in a natural host-parasite system.
The present descriptive study found evidence of multiple MLG natural infections by L. infantum in hosts living in a high-transmission area for VL. Further investigations on the effects of multiple infections are needed. Studies on other pathogens have shown that coinfections affect pathogen transmission and virulence, thus influencing disease dynamics [42]. Multiple genotype infections may have an impact on host immune responses [8, 43] and the effectiveness of disease control [44]. Notably, as observed in the antimony deposition in different organs of glucantime-treated rhesus monkeys, the pharmacokinetics of leishmanicidal drugs may vary depending on the organ [45]. This could lead to the development of resistance or selection if a more resistant genotype is found in an organ showing reduced levels of drug accumulation.
The results presented herein confirm the hypothesis raised in this study by showing the presence of multiple infections (multilocus genotypes, as inferred by microsatellite analysis) in dogs infected by L. infantum living in a highly endemic area for VL, which may contribute to the evolution of virulence in naturally infected dogs. Additionally, differences between clinical samples and derived culture isolates were also described. However, the understanding of the physiological and evolutionary consequences of these infections remains limited.
Supporting information
The genotypic composition of Leishmania infantum in naturally infected dogs was evaluated in parasites obtained from different tissues with or without in vitro expansion. The reference size in length of each allele of the microsatellite markers is presented. The observed length is presented for each animal.
(XLSX)
(DOCX)
Acknowledgments
We would like to thank Aline Moreira from the Fragment Analysis Platform of the Oswaldo Cruz Institute (Plataforma Genômica—Sequenciamento de DNA—PDTIS/FIOCRUZ RPT01A).
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by grants from CNPq - National Council for Scientific and Technological Development (Elisa Cupolillo: 421850/2017-5, 302622/2017-9; Renato Porrozzi: 307772/2015-2), FAPERJ - Carlos Chagas Filho Research Foundation of Rio de Janeiro State (Elisa Cupolillo: 202.983/2016), PAEF-IOC-FIOCRUZ, and Capes- Brazilian Federal Agency for Support and Evaluation of Graduate Education (Finance code 001)- Programa Institucional de Internacionalização (PrInt). Elisa Cupolillo is a CNPq fellow (302622/2017-9). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.OPAS. Manual de procedimientos para vigilancia y control de las leishmaniasis en las Américas. Washington: Organización Panamericana de la Salud; 2019. [Google Scholar]
- 2.Gramiccia M, Gradoni L. The current status of zoonotic leishmaniases and approaches to disease control. International journal for parasitology. 2005;35(11–12):1169–80. Epub 2005/09/16. 10.1016/j.ijpara.2005.07.001 . [DOI] [PubMed] [Google Scholar]
- 3.Ochsenreither S, Kuhls K, Schaar M, Presber W, Schonian G. Multilocus microsatellite typing as a new tool for discrimination of Leishmania infantum MON-1 strains. Journal of clinical microbiology. 2006;44(2):495–503. Epub 2006/02/04. 10.1128/JCM.44.2.495-503.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oddone R, Schweynoch C, Schonian G, de Sousa Cdos S, Cupolillo E, Espinosa D, et al. Development of a multilocus microsatellite typing approach for discriminating strains of Leishmania (Viannia) species. Journal of clinical microbiology. 2009;47(9):2818–25. Epub 2009/07/10. 10.1128/JCM.00645-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhls K, Alam MZ, Cupolillo E, Ferreira GE, Mauricio IL, Oddone R, et al. Comparative microsatellite typing of new world leishmania infantum reveals low heterogeneity among populations and its recent old world origin. PLoS Negl Trop Dis. 2011;5(6):e1155 Epub 2011/06/15. 10.1371/journal.pntd.0001155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira GE, dos Santos BN, Dorval ME, Ramos TP, Porrozzi R, Peixoto AA, et al. The genetic structure of Leishmania infantum populations in Brazil and its possible association with the transmission cycle of visceral leishmaniasis. PLoS One. 2012;7(5):e36242 Epub 2012/05/19. 10.1371/journal.pone.0036242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bose J, Kloesener MH, Schulte RD. Multiple-genotype infections and their complex effect on virulence. Zoology (Jena, Germany). 2016;119(4):339–49. Epub 2016/07/09. 10.1016/j.zool.2016.06.003 . [DOI] [PubMed] [Google Scholar]
- 8.Read AF, Taylor LH. The ecology of genetically diverse infections. Science (New York, NY). 2001;292(5519):1099–102. Epub 2001/05/16. 10.1126/science.1059410 . [DOI] [PubMed] [Google Scholar]
- 9.Balmer O, Tanner M. Prevalence and implications of multiple-strain infections. The Lancet Infectious diseases. 2011;11(11):868–78. Epub 2011/11/01. 10.1016/S1473-3099(11)70241-9 . [DOI] [PubMed] [Google Scholar]
- 10.Mideo N. Parasite adaptations to within-host competition. Trends in parasitology. 2009;25(6):261–8. Epub 2009/05/05. 10.1016/j.pt.2009.03.001 . [DOI] [PubMed] [Google Scholar]
- 11.Kinnula H, Mappes J, Sundberg L-R. Coinfection outcome in an opportunistic pathogen depends on the inter-strain interactions. BMC Evol Biol. 2017;17(1):77–. 10.1186/s12862-017-0922-2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bereczky S, Liljander A, Rooth I, Faraja L, Granath F, Montgomery SM, et al. Multiclonal asymptomatic Plasmodium falciparum infections predict a reduced risk of malaria disease in a Tanzanian population. Microbes and infection. 2007;9(1):103–10. Epub 2006/12/30. 10.1016/j.micinf.2006.10.014 . [DOI] [PubMed] [Google Scholar]
- 13.Mayor A, Saute F, Aponte JJ, Almeda J, Gomez-Olive FX, Dgedge M, et al. Plasmodium falciparum multiple infections in Mozambique, its relation to other malariological indices and to prospective risk of malaria morbidity. Tropical medicine & international health: TM & IH. 2003;8(1):3–11. Epub 2003/01/22. 10.1046/j.1365-3156.2003.00968.x . [DOI] [PubMed] [Google Scholar]
- 14.Soe TN, Wu Y, Tun MW, Xu X, Hu Y, Ruan Y, et al. Genetic diversity of Plasmodium falciparum populations in southeast and western Myanmar. Parasites & vectors. 2017;10(1):322 Epub 2017/07/06. 10.1186/s13071-017-2254-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maia C, Campino L. Biomarkers Associated With Leishmania infantum Exposure, Infection, and Disease in Dogs. Frontiers in cellular and infection microbiology. 2018;8:302 Epub 2018/09/22. 10.3389/fcimb.2018.00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motoie G, Ferreira GE, Cupolillo E, Canavez F, Pereira-Chioccola VL. Spatial distribution and population genetics of Leishmania infantum genotypes in Sao Paulo State, Brazil, employing multilocus microsatellite typing directly in dog infected tissues. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2013;18:48–59. Epub 2013/05/15. 10.1016/j.meegid.2013.04.031 . [DOI] [PubMed] [Google Scholar]
- 17.Alam MZ, Bhutto AM, Soomro FR, Baloch JH, Nakao R, Kato H, et al. Population genetics of Leishmania (Leishmania) major DNA isolated from cutaneous leishmaniasis patients in Pakistan based on multilocus microsatellite typing. Parasites & vectors. 2014;7:332 Epub 2014/07/18. 10.1186/1756-3305-7-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domagalska MA, Imamura H, Sanders M, Van den Broeck F, Bhattarai NR, Vanaerschot M, et al. Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PLoS Negl Trop Dis. 2019;13(12):e0007900 Epub 2019/12/13. 10.1371/journal.pntd.0007900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bussotti G, Gouzelou E, Côrtes Boité M, Kherachi I, Harrat Z, Eddaikra N, et al. Leishmania Genome Dynamics during Environmental Adaptation Reveal Strain-Specific Differences in Gene Copy Number Variation, Karyotype Instability, and Telomeric Amplification. mBio. 2018;9(6). Epub 2018/11/08. 10.1128/mBio.01399-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossi V, Wincker P, Ravel C, Blaineau C, Pages M, Bastien P. Structural organisation of microsatellite families in the Leishmania genome and polymorphisms at two (CA)n loci. Molecular and biochemical parasitology. 1994;65(2):271–82. Epub 1994/06/01. 10.1016/0166-6851(94)90078-7 . [DOI] [PubMed] [Google Scholar]
- 21.Cavalcanti AS, Ribeiro-Alves M, Pereira Lde O, Mestre GL, Ferreira AB, Morgado FN, et al. Parasite load induces progressive spleen architecture breakage and impairs cytokine mRNA expression in Leishmania infantum-naturally infected dogs. PLoS One. 2015;10(4):e0123009 Epub 2015/04/16. 10.1371/journal.pone.0123009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cupolillo E, Grimaldi G Jr., Momen H. A general classification of New World Leishmania using numerical zymotaxonomy. The American journal of tropical medicine and hygiene. 1994;50(3):296–311. Epub 1994/03/01. 10.4269/ajtmh.1994.50.296 . [DOI] [PubMed] [Google Scholar]
- 23.Graca GC, Volpini AC, Romero GA, Oliveira Neto MP, Hueb M, Porrozzi R, et al. Development and validation of PCR-based assays for diagnosis of American cutaneous leishmaniasis and identification of the parasite species. Memorias do Instituto Oswaldo Cruz. 2012;107(5):664–74. Epub 2012/08/02. 10.1590/s0074-02762012000500014 . [DOI] [PubMed] [Google Scholar]
- 24.Kuhls K, Keilonat L, Ochsenreither S, Schaar M, Schweynoch C, Presber W, et al. Multilocus microsatellite typing (MLMT) reveals genetically isolated populations between and within the main endemic regions of visceral leishmaniasis. Microbes and infection. 2007;9(3):334–43. Epub 2007/02/20. 10.1016/j.micinf.2006.12.009 . [DOI] [PubMed] [Google Scholar]
- 25.Mestre GL, Fontes CJ. [The spread of the visceral leishmaniasis epidemic in the State of Mato Grosso, 1998–2005]. Revista da Sociedade Brasileira de Medicina Tropical. 2007;40(1):42–8. Epub 2007/05/09. 10.1590/s0037-86822007000100008 . [DOI] [PubMed] [Google Scholar]
- 26.Kuhls K, Chicharro C, Canavate C, Cortes S, Campino L, Haralambous C, et al. Differentiation and gene flow among European populations of Leishmania infantum MON-1. PLoS Negl Trop Dis. 2008;2(7):e261 Epub 2008/07/10. 10.1371/journal.pntd.0000261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Araujo F, Slifer T, Kim S. Chronic infection with Toxoplasma gondii does not prevent acute disease or colonization of the brain with tissue cysts following reinfection with different strains of the parasite. The Journal of parasitology. 1997;83(3):521–2. Epub 1997/06/01. . [PubMed] [Google Scholar]
- 28.Porrozzi R, Teva A, Amaral VF, Santos da Costa MV, Grimaldi G Jr. Cross-immunity experiments between different species or strains of Leishmania in rhesus macaques (Macaca mulatta). The American journal of tropical medicine and hygiene. 2004;71(3):297–305. Epub 2004/09/24. . [PubMed] [Google Scholar]
- 29.Darvishi M, Yaghoobi-Ershadi MR, Shahbazi F, Akhavan AA, Jafari R, Soleimani H, et al. Epidemiological study on sand flies in an endemic focus of cutaneous leishmaniasis, bushehr city, southwestern iran. Frontiers in public health. 2015;3:14 Epub 2015/02/24. 10.3389/fpubh.2015.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rafizadeh S, Saraei M, Abaei MR, Oshaghi MA, Mohebali M, Peymani A, et al. Molecular Detection of Leishmania major and L. turanica in Phlebotomus papatasi and First Natural Infection of P. salehi to L. major in North-East of Iran. Journal of arthropod-borne diseases. 2016;10(2):141–7. Epub 2016/06/17. [PMC free article] [PubMed] [Google Scholar]
- 31.Chajbullinova A, Votypka J, Sadlova J, Kvapilova K, Seblova V, Kreisinger J, et al. The development of Leishmania turanica in sand flies and competition with L. major. Parasites & vectors. 2012;5:219 Epub 2012/10/04. 10.1186/1756-3305-5-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rugna G, Carra E, Bergamini F, Calzolari M, Salvatore D, Corpus F, et al. Multilocus microsatellite typing (MLMT) reveals host-related population structure in Leishmania infantum from northeastern Italy. PLoS Negl Trop Dis. 2018;12(7):e0006595 Epub 2018/07/06. 10.1371/journal.pntd.0006595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falqueto A, Ferreira AL, dos Santos CB, Porrozzi R, da Costa MV, Teva A, et al. Cross-sectional and longitudinal epidemiologic surveys of human and canine Leishmania infantum visceral infections in an endemic rural area of southeast Brazil (Pancas, Espirito Santo). The American journal of tropical medicine and hygiene. 2009;80(4):559–65. Epub 2009/04/07. . [PubMed] [Google Scholar]
- 34.Teixeira DG, Monteiro GRG, Martins DRA, Fernandes MZ, Macedo-Silva V, Ansaldi M, et al. Comparative analyses of whole genome sequences of Leishmania infantum isolates from humans and dogs in northeastern Brazil. International journal for parasitology. 2017;47(10–11):655–65. Epub 2017/06/14. 10.1016/j.ijpara.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gradoni L. Epizootiology of canine leishmaniasis in Southern Europe In: R K-K, editor. Canine leishmaniasis: an update Romainville: Hoechst Roussel Vet; 1999. p. 32–9. [Google Scholar]
- 36.Gleichsner AM, Reinhart K, Minchella DJ. The influence of related and unrelated co-infections on parasite dynamics and virulence. Oecologia. 2018;186(2):555–64. Epub 2017/12/14. 10.1007/s00442-017-4035-9 . [DOI] [PubMed] [Google Scholar]
- 37.Zhong D, Lo E, Wang X, Yewhalaw D, Zhou G, Atieli HE, et al. Multiplicity and molecular epidemiology of Plasmodium vivax and Plasmodium falciparum infections in East Africa. Malaria journal. 2018;17(1):185 Epub 2018/05/04. 10.1186/s12936-018-2337-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann EH, da Silveira LA, Tonhosolo R, Pereira FJ, Ribeiro WL, Tonon AP, et al. Geographical patterns of allelic diversity in the Plasmodium falciparum malaria-vaccine candidate, merozoite surface protein-2. Annals of tropical medicine and parasitology. 2001;95(2):117–32. Epub 2001/04/12. 10.1080/00034980120045833 . [DOI] [PubMed] [Google Scholar]
- 39.Paul RE, Brockman A, Price RN, Luxemburger C, White NJ, Looareesuwan S, et al. Genetic analysis of Plasmodium falciparum infections on the north-western border of Thailand. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93(6):587–93. Epub 2000/03/16. 10.1016/s0035-9203(99)90057-3 . [DOI] [PubMed] [Google Scholar]
- 40.Pomares C, Marty P, Banuls AL, Lemichez E, Pratlong F, Faucher B, et al. Genetic Diversity and Population Structure of Leishmania infantum from Southeastern France: Evaluation Using Multi-Locus Microsatellite Typing. PLoS Negl Trop Dis. 2016;10(1):e0004303 Epub 2016/01/26. 10.1371/journal.pntd.0004303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zackay A, Cotton JA, Sanders M, Hailu A, Nasereddin A, Warburg A, et al. Genome wide comparison of Ethiopian Leishmania donovani strains reveals differences potentially related to parasite survival. PLoS genetics. 2018;14(1):e1007133 Epub 2018/01/10. 10.1371/journal.pgen.1007133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alizon S, de Roode JC, Michalakis Y. Multiple infections and the evolution of virulence. Ecology letters. 2013;16(4):556–67. Epub 2013/01/26. 10.1111/ele.12076 . [DOI] [PubMed] [Google Scholar]
- 43.Cox FE. Concomitant infections, parasites and immune responses. Parasitology. 2001;122 Suppl:S23–38. Epub 2001/07/10. 10.1017/s003118200001698x . [DOI] [PubMed] [Google Scholar]
- 44.Li XX, Zhou XN. Co-infection of tuberculosis and parasitic diseases in humans: a systematic review. Parasites & vectors. 2013;6:79 Epub 2013/03/26. 10.1186/1756-3305-6-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedrich K, Vieira FA, Porrozzi R, Marchevsky RS, Miekeley N, Grimaldi G Jr., et al. Disposition of antimony in rhesus monkeys infected with Leishmania braziliensis and treated with meglumine antimoniate. Journal of toxicology and environmental health Part A. 2012;75(2):63–75. Epub 2011/12/02. 10.1080/15287394.2012.624826 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The genotypic composition of Leishmania infantum in naturally infected dogs was evaluated in parasites obtained from different tissues with or without in vitro expansion. The reference size in length of each allele of the microsatellite markers is presented. The observed length is presented for each animal.
(XLSX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.