

Abstract
Background
Rett syndrome (RTT) is a neurological disorder caused by mutation of the X-linked MECP2 gene, which results in the progressive disruption of excitatory and inhibitory neuronal circuits. To date, there is no effective treatment available for the disorder. Studies conducted in RTT patients and murine models have shown altered expression of N-methyl-D-aspartate receptors (NMDARs). Genetic deletion of the NMDAR subunit, GluN2A, in mice lacking Mecp2 is sufficient to prevent RTT phenotypes including regression of vision.
Methods
We performed a systematic, randomized pre-clinical trial of chronic administration of low dose (8 mg/kg, i.p.) ketamine, an NMDAR antagonist, starting either early in development or at the onset of RTT phenotype in Mecp2-null mice.
Results
Daily exposure to ketamine ameliorated RTT symptoms and extended lifespan of treated Mecp2-null mice without adverse side effects, Furthermore, significant improvement was observed in cortical processing and connectivity, which were fully restored to a wild-type (WT) level, particularly when treatment started at the onset of regression.
Conclusion
Our findings provide strong evidence that targeting NMDA receptors can be a safe and effective treatment for RTT.
Keywords: pre-clinical trial, behavior, cortical activity, parvalbumin connectivity, breathing, survival
One Sentence Summary:
Ketamine ameliorates Rett syndrome phenotypes.
Introduction
Rett syndrome (RTT) is a severe progressive neurological disorder that mainly affects girls and is characterized by early neurological regression followed by loss of acquired cognitive, social, and motor skills, together with development of autistic behavior (1), cardio-respiratory abnormalities and seizures (2–4). The disease typically manifests around 6–18 months and represents the second most common cause of severe intellective disability in the female gender (5) with a frequency of approximately 1 : 10,000 (6). Classic RTT is due to de novo mutations in an X-linked gene that encodes for methylCpG-binding protein 2 (MECP2) (5). MeCP2 is a multifunctional chromatin protein that regulates gene expression by either repressing or activating transcription, or by functioning at a post-transcriptional level (7).
Mecp2-null mice recapitulate key neurological phenotypes observed in RTT patients beginning at 5–6 weeks of age, whereas female heterozygous mice show delayed onset of overt signs (4–12 months) (8; 9). Similar to the neuroanatomical findings in humans, murine Rett models also display microcephaly without gross neuropathological changes or neurodegeneration (8; 9). A decrease in synaptic plasticity, or the ability of neurons to change their synaptic strength in response to activity, is also observed in many neuronal types (10; 11). The loss of Mecp2 mainly induces a progressive imbalance of excitatory-inhibitory synaptic activity across brain circuits (12–14), either in favor of excitation or inhibition depending on the brain region and the developmental time point. Notably, hyper-excitability is a widespread feature of brainstem nuclei (13; 15–17) and hippocampus (18; 19) while hypo-excitability characterizes forebrain regions (13; 14; 20–22). This overall network imbalance culminates with the onset of RTT regression.
Accumulating evidence suggests that NMDAR dysfunction significantly contributes to the regression process. Indeed, postmortem studies in RTT patients have shown irregular expression of NMDARs and an elevated glutamate/glutamine peak across brain regions (23; 24). Similar alterations in NMDAR expression and subunit composition are also present in Mecp2-null mice (10; 14; 25; 26). Renormalizing NMDAR composition in Mecp2-null mice by genetically deleting the NMDAR subunit, GluN2A, strikingly prevents regression of visual cortical function and preserves neuronal cortical activity (14). Moreover, acute administration of a sub-anesthetic dosage of ketamine, an ionotropic glutamatergic NMDAR antagonist, is sufficient to reverse forebrain hypofunction and rescue sensorimotor gating behavioral abnormalities in Mecp2-null mice (13). Ketamine induces cortical disinhibition in human and in behaving animal studies (27; 28) by preferentially modulating NMDAR activity onto parvalbumin (PV) fast spiking inhibitory neurons (29; 30). Notably, the loss of Mecp2 induces an early maturation of PV-positive cells, which leads to increased inhibitory innervations onto pyramidal cells and reduced neuronal activity in the visual cortex (14).
In the present study, we tested the hypothesis that a chronic pharmacological manipulation of the NMDAR can rescue or prevent onset of RTT phenotypes. Here, we evaluated the effects of low-dose, daily ketamine treatment in Mecp2-null mice. In particular, we assessed the safety and efficacy of two treatment paradigms starting at different key time points of development: 1) from eye opening at postnatal day 15 (P15), when the first alteration in the PV cortical circuits is observed but before the onset of RTT phenotypes and 2) from P30, at the onset of RTT symptoms and when the visual regression begins (14). Our results indicate that both prolonged ketamine treatment paradigms were well tolerated and significantly extended the lifespan of Mecp2-null mice. At the cortical level, PV-circuits were no longer hyper-connected and neuronal excitatory circuits exhibited WT level of neuronal activity.
Materials and Methods
Study Design.
The primary outcome measures of the study included (i) ketamine tolerance in healthy animals, (ii) behavioral analysis of ketamine treated Mecp2-null mice and (iii) analysis of visual cortical circuits.
The treatment and genotype were randomized and blinded from team members (figure S1). End points were predefined as the presence or absence of statistically significant differences of the above-listed parameters. No outliers were eliminated.
Animals.
All procedures were approved by IACUC at Boston Children’s Hospital and conducted in Mecp2-null mouse line (B6.129P2(C)-Mecp2tm1.1Bird/J) crossed with C57BL6 (9). In vivo recordings were also performed on GluN2A KO mice (from M. Mishina, University of Tokyo) (31).
Drug treatment.
Ketamine HCl (Ketaset, Fort Dodge) was dissolved in saline (0.9% NaCl) which also served as the vehicle control. Ketamine (8 mg/kg) was administered daily via intraperitoneal (i.p.) injection at the same time each day. Animals across multiple litters were randomly assigned to a treatment group. Each litter at least contributed a Mecp2 KO and a WT mouse to the study.
Mecp2 WT and KO mice were divided into two groups: 1) P15 to P55 paradigm (40 days); 2) P30 to P55 paradigm (25 days).
Pharmacokinetic (PK) analysis.
P15, P30 and adult C57BL/6J mice received a single intraperitoneal dose of ketamine (8 mg/kg; n = 3/dose/time point). Blood and brains were collected for analysis at specific time points (see Supplementary Materials).
Neurobehavioral characterization.
Weight and general condition of the animals were evaluated daily. All tests were performed at the same time of day and in the same dedicated observation room within the Neurodevelopmental Behavioral Core (NBC) at BCH (see Supplementary Materials).
Spontaneous locomotor activity.
The distance traveled (in centimeters) and the mean velocity (in centimeters/second) was recorded in 5 min periods with ActiTrack software (Panlab/Harvard Apparatus, Cornellà, Spain).
Phenotypic scoring.
Animals were scored using the RTT phenotypic severity scoring system described previously (11).
Rotarod.
Animals were placed on a rotating rod apparatus (Economex Enclosure, Columbus Instruments, Columbus, Ohio), at a constant speed of 4 rpm for 10 seconds for acclimatization. The test session ended when the animal fell off the rod.
Prepulse inhibition of the startle reflex (PPI).
PPI was defined as the percentage reduction in mean startle response magnitude for each mouse at each prepulse and control trials. % PPI = 100 × [(pulse alone) − (prepulse + pulse)] / pulse alone.
Optomotor Task.
Visual threshold acuity was evaluated using the optomotor task (32) (Cerebral Mechanics, Lethbridge, Alberta). Vehicle and ketamine-treated mice were tested at P30, P40 and P55.
Whole-body Plethysmography.
Breathing was recorded from unrestrained awake mice at P30 and between P48-P55 using a constant flow whole-body plethysmograph (200 ml chamber) (EMKA Technologies, Paris, France) (33). Mice were kept for 1 hour in the chamber. Only periods of quiet breathing during the last 20 min were analyzed to measure the number of apneas per minute. Apneas were defined when the breath holding was longer than 2 normal respiratory cycles.
In vivo single unit recordings.
In vivo recordings were performed at P55–60, under Nembutal (50 mg/kg, i.p.) / chlorprothixene (0.025 mg/kg, i.m.) anesthesia using standard techniques (14). Cortical activity in binocular zone of primary visual cortex was recorded using multichannel probes (A1×16−3mm50−177; Neuronexus technologies, Ann Arbor, Michigan, Supplementary Materials).
Immunohistochemistry.
Primary antibodies and dilutions are detailed in Supplementary Materials. Quantitative analyses of the binocular zone of visual cortex across all layers were performed blind to genotype and treatment. Mean pixel intensity (at 1003) of the PV signal in each field (1,024 × 1,024) was measured using MacBiophotonics ImageJ software. The number of perisomatic synapses (at 100X) was determined on triple-stained images (PV, GAD65, DAPI) using the “particle analysis” function (ImageJ). NeuN-positive cell density was quantified per area by using ImageJ software and per volume with Volocity (version 5.5; PerkinElmer, Cambridge, Massachusetts).
Western Blot.
Mecp2 WT and KO mice were acutely injected with ketamine 8 mg/kg either at P15 or P30. Visual cortices were dissected an hour later (Supplementary Materials).
Statistical analysis.
All data are presented as mean ± standard error. Behavioral differences between treatment groups were carried out using Kruskal-Wallis test, Kaplan-Meier, Chi-Square and two-way ANOVA as appropriate. In vivo recordings and immunohistochemistry quantification were compared using Kruskal-Wallis and Kolmogorov-Smirnov tests as appropriate. p < 0.05 was used to define statistical significance. All statistics were performed using GraphPad Prism (version 5.0) software.
Results
Low dose ketamine does not induce negative behavioral outcomes.
Despite the fact ketamine is widely used as an anesthetic and analgesic in pediatric clinical practice, it is well known that it may cause adverse effects when administered at high doses (34). Therefore we decided to perform a series of studies to evaluate brain penetrance and exclude any detrimental effect of the low-dosage of ketamine (8 mg/kg).
We first conducted a pharmacokinetic analysis to quantify ketamine penetrance in the brain following a single intraperitoneal injection of 8 mg/kg in adult WT mice (Figure 1A). As previously reported, ketamine showed preferential distribution with a brain to plasma ratio of approximately 2 to 1 (Figure 1A and Table 1) (35) and was quickly eliminated (plasma t1/2,β = 1.1 ± 0.8 h).
Figure. 1.
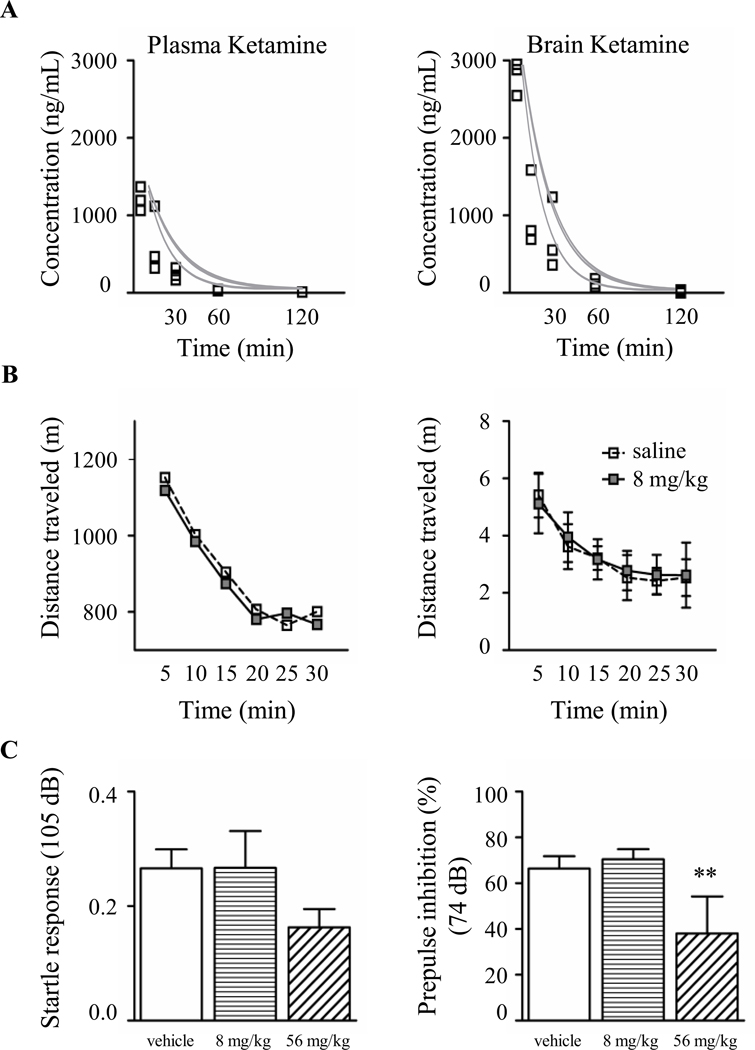
Ketamine 8 mg/kg is rapidly absorbed in the brain and does not induce side effects. (A) Pharmacokinetic analysis of total ketamine concentration in plasma (left) and brain (right) in WT adult mice after a single intraperitoneal injection (n = 3 mice per time point). (B) A single dose of ketamine 8 mg/kg did not affect spontaneous locomotor activity (Two-way ANOVA, p ≥ 0.05. Empty square: WT-saline, n = 6 mice; gray square: WT-ketamine, n = 15 mice) (C) High dose of ketamine (56 mg/kg) significantly reduced PPI response (Kruskal-Wallis, ** p ≤ 0.01, Dunn’s post-test. WT–saline, n = 10; WT-ketamine 8 mg/kg, n = 10; WT-ketamine 56 mg/kg, n = 8 mice).
Table 1.
Mouse pharmacokinetic parameters after 8 mg/kg i.p. administration of ketamine.
| Cmax (ng/mL) | V (L) | AUCinf (ng.h/L) | t1/2,α (h) | t1/2,β (h) | CLd (L/h) | CL (L/h) | B/P ratio | |
|---|---|---|---|---|---|---|---|---|
| P15 | 463 ± 87 | 115 ± 38 | 122 ± 26 | 0.05 ± 0.03 | 0.28 ± 0.17 | 157 ± 78 | 71 ± 12 | 14.7 ± 1 |
| P30 | 1760 ± 231 | 81 ± 12 | 296 ± 48 | 0.09 ± 0.03 | 0.25 ± 0.13 | 284 ± 86 | 30 ± 7.2 | 8.7 ± 3.7 |
| Adults | 1209 ± 152.5 | 69 ± 4 | 366 ± 59 | 0.07 ± 0.01 | 1.11 ± 0.8 | 326 ± 51 | 48 ± 3 | 2.7 ± 0.5 |
Cmax : maximum concentration, V: volume of distribution, AUCinf: area under the concentration time curve (AUC) with the last concentration extrapolated based on the elimination rate constant, t1/2,α: distribution of half-life, t1/2,β: elimination half-life, CLd: distribution clearance, CL: systemic clearance, B/P: brain to plasma ratio. Data are expressed as mean ± SEM.
Major side effects of ketamine administered at high, sub-anesthetic doses include agitation, confusion and psychosis (36). Similarly in rodents, acute administration of a high dose of ketamine (more than 20 mg/kg) produces behavioral abnormalities such as disruption of sensorimotor gating, impaired cognitive function, and hyperlocomotor activity (38–40). Importantly, lower dosages (2.5 – 10 mg/kg) elicit few if any of these adverse effects (39; 40).
To confirm that 8 mg/kg of ketamine does not trigger major behavioral side effects in WT mice, we measured spontaneous locomotor activity and prepulse inhibition (PPI) of the acoustic startle reflex (41). We used an open field to quantify the distance traveled (in centimeters) and the mean velocity (in cm/sec) for 30 minutes immediately after a single injection of ketamine and found no significant difference compared with control mice injected with vehicle (Figure 1B). We then analyzed acoustic startle and PPI responses after an acute administration of ketamine or vehicle. Responses in both tests were unaffected by ketamine when administered at 8 mg/kg (Figure 1C). On the contrary, at a higher dose (56 mg/kg), ketamine induced a slight decrease in the startle response and a significant reduction of PPI (Figure 1C).
Next, we evaluated chronic ketamine treatments in WT mice that started either at P15 or P30 until P55 (WT-k15, WT-k30, respectively). Both treatments failed to elicit any disruption of the basic acoustic startle and PPI responses (figure S2). In addition, prolonged ketamine administration in WT mice did not evoke weight loss (figure S3A) (42) or neuronal cell-death (figure S3B and C) (43).
Finally we tested whether acute administration of low-dosage ketamine would enhance neuronal activity of excitatory cortical circuits as previously reported (28; 29). We found that 8 mg/kg of ketamine was sufficient to increase spontaneous and evoked neurotransmission in pyramidal neurons as revealed by single unit recordings from primary visual cortex (figure S4A). Interestingly, we did not see such potentiation in GluN2A KO mice (figure S4B) supporting the notion that ketamine mainly acts through NMDAR containing GluN2A subunit.
Overall, our data demonstrate that ketamine dosed at 8 mg/kg rapidly accumulates in the brain, does not induce negative side effects after acute or chronic administration, and significantly increases cortical activity by targeting NMDAR.
Randomized pre-clinical study with chronic ketamine administration.
Mecp2-null (KO) mice were injected daily intraperitoneally (i.p.) with either vehicle (KO-v) or ketamine (KO-k) starting at P15 (KO-15) or P30 (KO-30). The treatment was continued until P55 (figure S1A). Control groups were Mecp2 WT littermates treated with vehicle (WT-v15 and WT-v30). A total of 67 KO and 37 WT mice were used in the study.
Littermates from multiple litters were randomly allocated to the different treatment groups. Drugs were administered at the same time each day. Every 15 days, starting at P30, a tailored battery of validated behavioral tests was carried out 23 hours after the last injection (figure S1, see Material and Methods). Each behavioral test was performed at the same time of day by the same person. Individuals caring for the animals, administering the drug, performing the experiments, and quantifying the outcomes were blinded to the genotype and treatments (figure S1B).
Effect of prolonged ketamine treatment on RTT symptoms.
To determine whether ketamine could prevent or delay onset of RTT phenotype, we injected Mecp2-null mice with either vehicle (KO-v15; KO-v30) or ketamine (KO-k15; KO-k30) and evaluated multiple outcomes up to P55. Both groups of KO-v mice showed markedly reduced survival compared to WT-v mice, as expected for this RTT mouse model (9) (figure S5A; median survival P53). On the contrary, both groups of KO-k mice showed significantly extended survival compared with KO-v mice (median survival P83 for both KO-k15 and KO-k30). Interestingly, at P80, 47% of the KO-k15 and 36 % of the KO-k30 mice were alive while all of the KO-v were already deceased (Figure 2A, insert). Ketamine-treated Mecp2 KO mice still exhibited reduced body weight compared to WT littermates (Figure 2B).
Figure. 2.
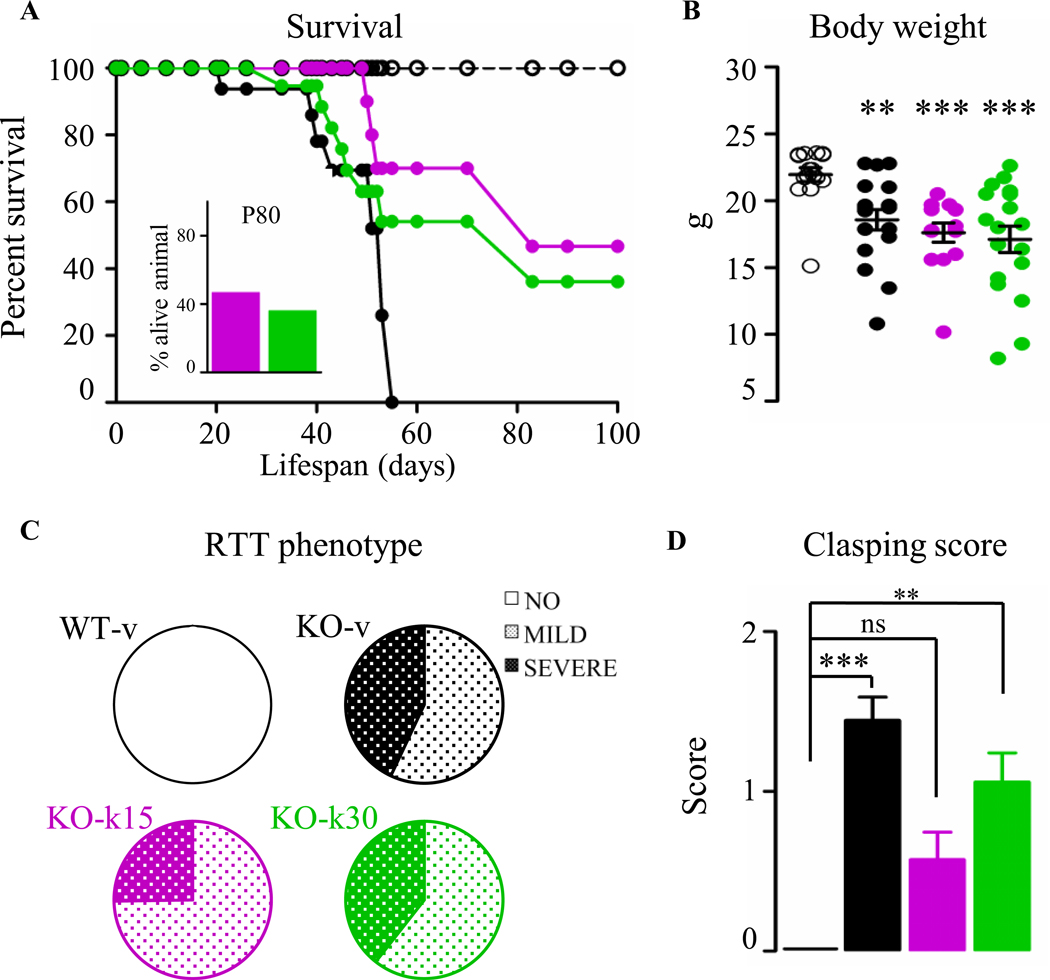
Prolonged ketamine treatment improves key RTT-like phenotypes. (A) Survival curves in WT (empty circle) and in Mecp2 KO treated with vehicle (black-filled circle) or ketamine from P15 (magenta) or from P30 (green) revealed prolonged ketamine treatment improved the lifespan of Mecp2 KO mice. (B) No improvement of the body weight of P55 treated Mecp2 KO mice was observed (Kruskal-Wallis, ** p ≤ 0.01; *** p ≤ 0.001, Dunn’s post-test). Data are expressed as mean ± SEM. (C) The phenotypic severity score spans three groups: absent (white), mild (light polka dots), severe RTT phenotype (dark polka dots). Notably, almost 80% of the adult KO-k15 mice had a mild score compared to their age matched KO-v. KO-k30 mice did not show an improvement in the RTT score severity. (D) The clasping phenotypic score was improved mainly in the KO-k15 mice (WT-v, n = 13; KO-v, n = 18; KO-k15, n = 14; KO-k30, n = 17 mice). Data are expressed as mean ± SEM.
The prolonged lifespan was instead accompanied by overall improved health particularly in KO-k15 mice, as measured using an established observational scoring system (Figure 2C and S5B) (11; 44). At P55, all WT-v had a score lower than 2 while 43% of the KO-v exhibited severe RTT-like symptoms (score > 6). Interestingly, ketamine treatment from P15 shifted the RTT score towards a mild RTT phenotype with only 25% of animals displaying the highest level of phenotypic severity (Figure 2C). Among the several RTT phenotypes scored, we found that the hindlimb clasping phenotype, an indication of neurological and motor dysfunction (45), was significantly reduced in KO-k15 (Figure 2D).
We then evaluated the effect of ketamine treatments on motor performance and apnea incidence, two prominent features of RTT (3; 4). Mice were tested on a non-accelerating rotarod in four consecutive trials (figure S6A and B). As expected, motor performance of WT-v mice improved with repetition and age, whereas KO-v did not improve and instead became significantly worse with age. Both ketamine treatments partially improved motor coordination. Indeed, KO-k15 mice stayed longer on the rod compared to KO-v mice at both ages. KO-k30 started at the same level as the KO-v at P30 but did not regress with age (figure S6A). Learning performance, evaluated by testing the mice over successive trials showed that KO-v did not show any improvement between the first and the last trial. KO-k15 mice significantly increased their motor performances at the last trial only at P45 whereas KO-k30 mice improved their performance by P55 but not at P45 (figure S6B).
Finally, we evaluated breathing in awake, unrestrained mice using whole body plethysmography at P30 and P55 (Figure 3A and B). The total respiratory cycle duration was measured for at least 230 cycles (mean 1303+/− 160 cycles) acquired during a period of quiet breathing. The count of apneas per minute (> 2 normal total respiratory cycle durations) was then quantified as previously described (33). The number of apneas per minute dramatically increased in KO-v by P50 (33; 46). Interestingly, ketamine treatment delayed the onset of apneas in the KO-k15 mice while preventing worsening of apneas episodes when treatment started from P30 (Figure 3B).
Figure. 3.
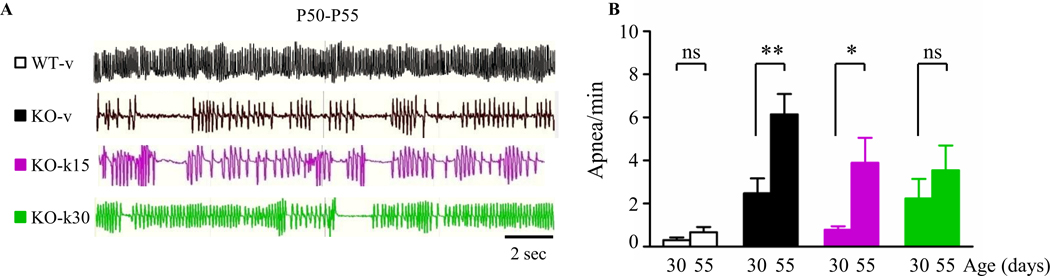
Ketamine delays the worsening of the respiratory function. (A) Representative plethysmography traces illustrating breathing pattern. (B) Quantification of the number of apneas per minute. Ketamine treatment from P30 prevented the developmental increase of apneic episodes (Wilcoxon signed-rank test, * p < 0.05, ** p ≤ 0.01. WT-v, n = 8; KO-v, n = 11; KO-k15, n = 7; KO-k30, n = 8 mice).
Overall, these data suggest that both chronic ketamine treatments significantly ameliorate RTT neurological phenotypes without detrimental effects
Effect of ketamine treatment on visual cortical processing.
We have previously demonstrated that the visual system is a reliable biomarker of regression in Mecp2-null mice (14). We therefore chose to investigate the effect of ketamine treatments on visual function using an optomotor (OPT) task to assess the response of unrestrained animals to high contrast moving gratings. As previously shown (14), all mice reached adult acuity by P30, but then KO-v mice exhibited a rapid regression between P30 and P55. Consequently, at P45 and P55, the OPT visual acuity in KO-v mice was significantly lower than age-matched WT-v (Figure 4B). Interestingly, both ketamine treatments slowed down regression of visual acuity. At P45, the OPT visual acuity was not different between WT and KO treated mice. At P55, although KO-k mice displayed a lower OPT acuity than WT-v, it was significantly higher than in KO-v (Figure 4B).
Figure. 4.
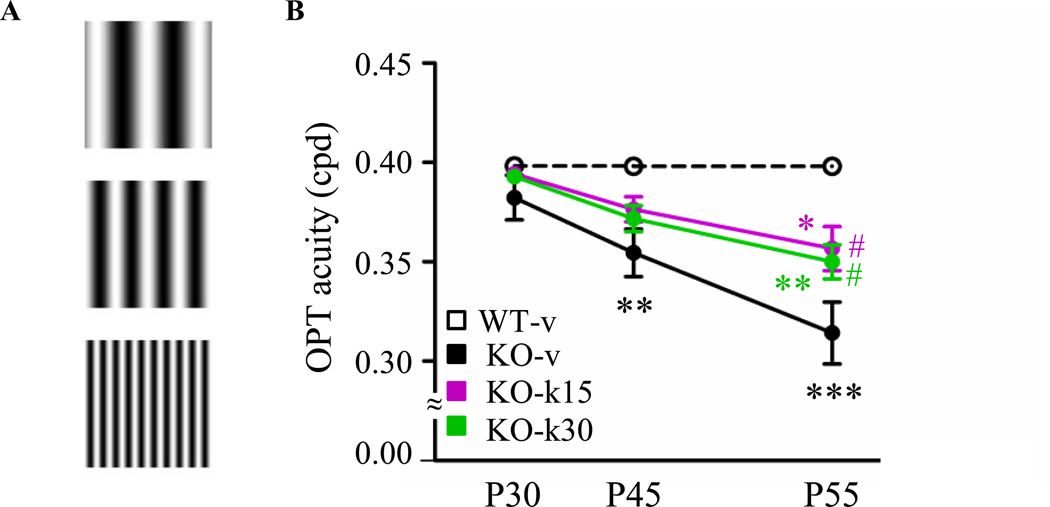
Ketamine delays regression of the visual acuity. (A) Example of stimulation used to measure visual acuity by optomotor task (OPT). (B) Average OPT visual in WT-v (n = 14), KO-v (n = 20), KO-k15 (n =11) and KO-k30 (n = 21) mice. Visual acuity was significantly reduced in KO-v (Two-way ANOVA, ** p ≤ 0.01,*** p ≤ 0.001, Bonferroni post-test), whereas ketamine treatment delayed the visual regression. At P55, KO-k15 and KO-k30 acuity was significantly higher than KO-v (Two-way ANOVA, # p ≤ 0.05, Bonferroni post-test), but still statistically lower than WT-v (Two-way ANOVA, * p ≤ 0.05,** p ≤ 0.01, Bonferroni post-test).
We then performed in vivo extracellular recordings of spontaneous and maximal evoked responses of pyramidal cells in response to drifting sinusoidal gratings. At P5560, both the maximal-evoked response and spontaneous activity were significantly reduced in KO-v mice, confirming that visual circuits are largely silent in Mecp2-null mice compared to WT littermates (Figure 5B and C) (14). In addition, neuronal responses exhibited higher variability to repeated presentation of the preferred stimulus (coefficient of variation, CV; Figure 5D and E), suggesting less reliable signal processing and cognitive impairments (47). Remarkably, both ketamine treatments increased the evoked and spontaneous activities to WT level. However, only the treatment starting at P30 was effective to renormalize the coefficient of variation to WT level (Figure 5E). Together, these results suggest that the treatment starting from P30 was more effective for renormalizing visual cortical processing in Mecp2-null mice.
Figure. 5.
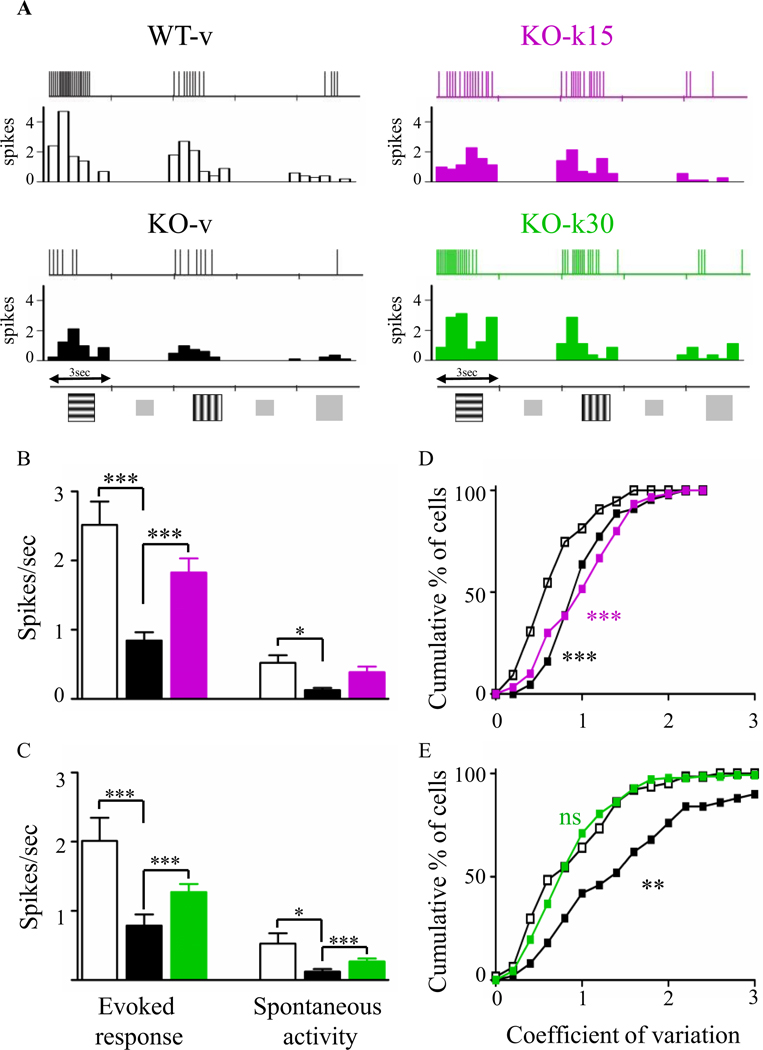
Ketamine increases neuronal activities and response reliability in visual cortex. (A) Representative spike trains and corresponding peristimulus time histogram (PSTH) in response to two oriented gratings or a uniform gray stimulus (8 presentations each). (B, C) Averages of maximal evoked and spontaneous activities. Ketamine treatment significantly increased both types of activity in both treatment paradigms (Kruskal-Wallis, * p ≤ 0.05, ** p ≤ 0.01,*** p ≤ 0.001, Dunn’s post-test). (D, E) Coefficient of variation of the response to the preferred gratings. Only KO-k30 had a restored coefficient of variation value (Kolmogorov-Smirnov test, ** p ≤ 0.01; *** p ≤ 0.001. WT-v15, n = 74; KO-v15, n = 66; KO-k15, n = 44; WT-v30, n = 57; KO-v30, n = 60; KO-k30, n = 138 cells).
Effect of prolonged ketamine on parvalbumin connectivity.
The spiking activity of cortical pyramidal cells are controlled by PV-positive inhibitory cells. Importantly, PV circuits are particularly sensitive to the inhibiting effects of ketamine and determine the overall disinhibition of cortical activity (29; 30; 48). In the absence of Mecp2, PV circuits exhibit a significant increase in neurite complexity and number of axonal perisomatic boutons onto pyramidal cells starting as early as eye opening (P15) which contributes to the silencing of cortical circuits (14; 20; 49). Consequently, we hypothesized that ketamine treatments may have restored the normal wiring of PV-circuits allowing WT level of cortical activity.
PV immunofluorescence intensity was significantly upregulated in KO-v compared with WT-v age-matched mice (figure S7; 14). Both ketamine treatments normalized PV intensity similar to WT-v levels in adult Mecp2-null mice (figure S7). This decrease was not linked to a change in PV-cell density as no change was observed between different genotypes and drug treatments despite a reduction in cortical thickness and soma volume (Table S1). We then quantified the density of PV-positive perisomatic boutons onto pyramidal and PV-cell somata (50). While KO-v exhibited a significant increase of PV-positive boutons onto pyramidal but not on PV cells, both KO-k groups were indistinguishable from WT-v controls (Figure 6). All together, these data demonstrate that both treatments were sufficient to rewire PV-circuitry in the Mecp2-null visual cortex.
Figure. 6.
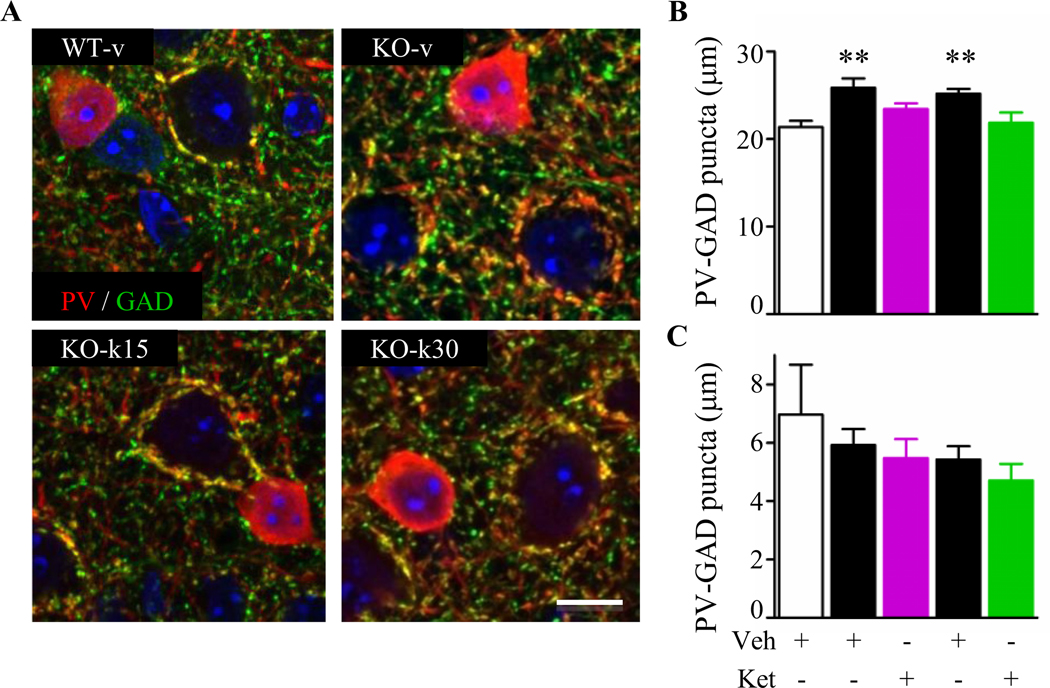
Prolonged ketamine treatment restores parvalbumin-circuit inputs onto pyramidal cells. (A) Representative confocal high magnification images showing parvalbumin (PV, green) and GAD65 (GAD, red) in WT-v, KO-v, KO-k15 and KO-k30. Scale bar 10 mm. (B) PV-cell innervations of pyramidal cell somata were statistically increased in KO-v compared to WT-v (Kruskal-Wallis, ** p ≤ 0.01, Dunn’s post-test). Both ketamine treatments reduced PV-innervations towards WT levels. (C) PV-PV connections were not affected by the loss of Mecp2 or by the ketamine treatments. (WT-v15, n = 3; KO-v15, n = 3; KO-k15, n = 4; WT-v30, n = 3; KO-v30, n = 4; KO-k30, n = 4 mice). Data are expressed as mean ± SEM.
Discussion
Our study provides the first in vivo evidence that prolonged treatment with a low dose of the NMDAR antagonist ketamine significantly extended life span and renormalized cortical circuits without detrimental side effects. Administration of ketamine slowed down the regression of optomotor visual acuity, prevented the silencing of neuronal cortical circuits and rewired PV circuit connectivity to WT levels. Importantly, even a treatment starting at the onset of the regression (P30) was sufficient to rescue RTT phenotype.
Ketamine’s mechanism of action is complex, affecting not only NMDAR but also neurotransmitter and neuromodulatory systems particularly when administered at anesthetic concentrations (51). Among them, ketamine and in particular one of its metabolites, dehydronorketamine (DHNK), may act on ion channels such as the nicotinic acetylcholine receptor (AChR) by inhibiting the receptor in a non-competitive and voltage-dependent manner (52; 53). Although we cannot exclude this possible mechanism of action in our experiments, we believe that this is not a major contributor due to the fact our pharmacokinetic analysis revealed that norKetamine (norKet) and not DHNK was the major circulating metabolite at both ages (figure S8; 53; 54) likely targeting the NMDAR (55).
As a use-dependent noncompetitive antagonist, ketamine binds within the open ion channel, blocking calcium influx, and working preferentially at sites of excess NMDAR activation (56). Because PV-cells are continuously depolarized and exhibit high levels of firing (57), their NMDARs are likely to escape Mg2+ blockade. Thus, a low dose of ketamine may be sufficient to dampen their firing activity (30; 58), decrease overall inhibition of cortical circuits and tip the balance of synaptic transmission toward excitation (figure S4A; 58).
In the absence of Mecp2, PV circuits form exuberant somatic connections onto pyramidal neurons that increase throughout development and significantly contribute to the progressive silencing of cortical circuits (13; 14). PV circuits develop in an activity-dependent manner and sensory deprivation can halt such anatomical and functional maturation (59). Similar to dark rearing from birth (14; 60), prolonged ketamine treatment may reduce the excitatory drive onto PV-cells. This results in a rapid loss of local inhibition, a long-lasting rewiring of PV-connectivity onto pyramidal neurons, and the eventual restoration of cortical activity (figure S4 and Figure 5) (29; 30; 48; 58).
The indirect modulation of excitatory neuron firing rate may contribute to the activity-dependent release of brain derived neurotrophic factor (BDNF) (61). However, we cannot exclude the possibility that ketamine also acts directly on pyramidal cells. These two mechanisms are not mutually exclusive and could work together to exert a long-lasting effect. Indeed, the anti-depressant action of ketamine occurs by blocking tonic activation of GluN2B-containing NMDARs in pyramidal neurons (62) and by modulating several downstream signaling components including mammalian target of rapamycin (mTOR) pathway, glycogen synthase kinase-3 (GSK3) and eukaryotic elongation factor 2 (eEF2) (63). Influencing each of these targets leads to an up-regulation of protein synthesis including BDNF (64; 65) which then contributes to strengthened synaptic plasticity (66). Indeed, we found that acute ketamine was sufficient to activate mTOR pathway by upregulating pAKT/AKT ratio in both Mecp2 WT and KO visual cortex at P30 but not at P15 (figure S9 A–D). Considering that in the absence of Mecp2, both mTOR signaling (figure S9 E–F; 67) and BDNF expression are severely down-regulated across brain regions as RTT progresses (68), ketamine treatment may alter this trajectory.
The hypothesis that NMDAR modulation may have beneficial effects on the RTT phenotype has been proposed by multiple independent studies published in recent years. Memantine, a weak NMDAR blocker, partially reverses short-term plasticity in hippocampal slices of Mecp2-null mice, but fails to halt the progression of RTT phenotypes (44). It is noteworthy that there are several differences in the way memantine and ketamine interact with NMDAR including their ability to change glutamate binding at rest (69), the trapping properties of the drugs (70), and their unique effects on synaptic and extrasynaptic receptors (71). Moreover, ketamine and memantine activate different downstream intracellular pathways, as memantine does not inhibit the phosphorylation of eukaryotic elongation factor 2 (eEF2), nor does it augment subsequent expression of BDNF (69).
Although re-activation of Mecp2 expression in fully symptomatic mice can reverse at least some of the deficits caused by the loss of gene function during development (11), the timing with which Mecp2-downstream signaling is altered could play a critical role in determining the effectiveness of potential therapeutic interventions. Overall our results indicate that both treatments were effective in ameliorating multiple RTT phenotypes. However, the degree of efficacy varied depending on the features analyzed and most likely the neuronal circuit affected. The treatment starting at P15 was more effective in improving subcortical phenotype while P30 treatment fully recovered normal cortical visual processing by increasing the reliability of visual responses in addition to the normal evoked and spontaneous activity (Figure 5C and E).
A factor that may have contributed to these outcomes is represented by the higher brain exposure to ketamine at P15 compared to P30 and adulthood (figure S8). This may have impacted the maturation of excitatory cortical circuits by interfering with their synaptic development resulting in an incomplete reliability of the evoked neuronal responses (72). Although brain circuit alterations in pre-symptomatic Mecp2-null mice have been identified (14; 20; 73), NMDAR subunit composition becomes disrupted in a cell-specific manner only at onset of regression (10; 24–26). By P30, pyramidal cells still exhibit immature GluN2B-containing NMDARs while PV-cells have already completed their switch to GluN2A and are hyper-connected onto pyramidal cells, actively silencing cortical circuits (26). A low dosage of ketamine at P30 may then be the ideal intervention to specifically target such differential GluN2 expression in cortical circuits with minimal or no off-target effects. The net result of these effects is a progressive renormalization of the wiring and function of neuronal circuits and RTT phenotype at large (Figure 7).
Figure. 7.
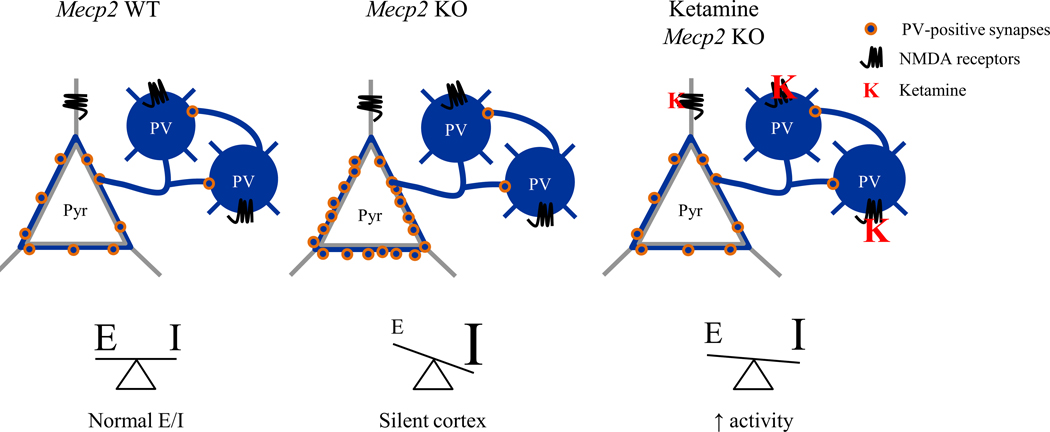
Ketamine preferentially modulates parvalbumin cortical circuits leading to a rebalancing of cortical activity in Mecp2 KO mice. Excitatory/inhibitory (E/I) imbalance underlies impairment in cortical processing. Increased parvalbumin (PV) puncta density onto pyramidal cells leads to a silent cortex. The NMDAR antagonist ketamine primarily acts on NMDARs localized on PV-cells. It reduces their spiking activity, results in the disinhibition of pyramidal cells, and thereby renormalizes the E/I balance.
In summary, our study demonstrated that NMDARs are new and innovative therapeutic targets for the treatment of RTT-related symptoms. Future pre-clinical studies are required to explore whether the therapeutic benefits of ketamine can extend to Mecp2 Het female mice and eventually allow the design of effective and safe clinical trial in RTT patients.
Supplementary Material
Acknowledgments:
We thank G. Solinap and J. Ciarrusta for early contributions to the study, E. Chang for helping with Western blot experiments, A.D. Hill and Boston Children’s Hospital IDDRC imaging core (NIH-P30-HD-18655) for imaging software, L.M. Pereira and Quantitative Clinical Pharmacology and Pharmacokinetics Laboratory at Boston Children’s Hospital for performing PK analysis, Neurodevelopmental Behavioral Core (CHB IDDRC, P30 HD18655) for behavioral analyses and Dr. Hensch and members of the Fagiolini and Hensch labs for helpful discussion. This work was supported by International Rett Syndrome Foundation and Boston Children’s Hospital Translational Research Program.
Footnotes
Competing interests: All authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes:
- 1.Hagberg B (2001): Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev, 8:61–6. [DOI] [PubMed] [Google Scholar]
- 2.Chahrour M, Zoghbi HY (2007): The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron, 56: 422–437. [DOI] [PubMed] [Google Scholar]
- 3.Katz DM, Dutschmann M, Ramirez JM, Hilaire G (2009): Breathing disorders in Rett syndrome: Progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol, 168: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. (2010): Rett syndrome: Revised diagnostic criteria and nomenclature. Ann Neurol, 68: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999): Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet, 23: 185–188. [DOI] [PubMed] [Google Scholar]
- 6.Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. (2006): Rett syndrome in Australia: a review of the epidemiology. J Pediatr, 148:347–352. [DOI] [PubMed] [Google Scholar]
- 7.Lyst MJ, Bird A (2015): Rett syndrome: a complex disorder with simple roots. Nat Rev Genet, 16: 261–274. [DOI] [PubMed] [Google Scholar]
- 8.Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001): Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet, 27: 327–331. [DOI] [PubMed] [Google Scholar]
- 9.Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001): A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet, 27: 322–326. [DOI] [PubMed] [Google Scholar]
- 10.Asaka Y, Jugloff DGM, Zhang L, Eubanks JH, Fitzsimonds RM (2006): Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis, 21: 217–227. [DOI] [PubMed] [Google Scholar]
- 11.Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007): Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315: 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shepherd GMG, Katz DM (2011): Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: Focus on Mecp2 and Met. Curr Opin Neurobiol, 21: 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, Katz DM (2012): Brain Activity Mapping in Mecp2 Mutant Mice Reveals Functional Deficits in Forebrain Circuits, Including Key Nodes in the Default Mode Network, that are Reversed with Ketamine Treatment. J Neurosci, 32: 13860–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, et al. (2012): NMDA Receptor Regulation Prevents Regression of Visual Cortical Function in the Absence of Mecp2. Neuron, 76: 1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kline DD, Ogier M, Kunze DL, Katz DM (2010): Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci, 30: 5303–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W (2007): Early defects of GABAergic synapses in the brainstem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol, 99: 112–121. [DOI] [PubMed] [Google Scholar]
- 17.Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB (2009): Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci, 29: 12187–12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, He J, Jugloff DGM, Eubanks JH (2008): The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus, 18: 294–309. [DOI] [PubMed] [Google Scholar]
- 19.Calfa G, Hablitz JJ, Pozzo-Miller L (2011): Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J Neurophysiol, 105: 1768–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB (2005): Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A, 102: 12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GMG (2009): Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci, 29: 12440–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood L, Shepherd GMG (2010): Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiol Dis, 38: 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blue ME, Naidu S, Johnston M V. (1999): Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol, 45: 541–545. [DOI] [PubMed] [Google Scholar]
- 24.Blue ME, Naidu S, Johnston M V (1999): Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol, 156: 345–352. [DOI] [PubMed] [Google Scholar]
- 25.Blue ME, Kaufmann WE, Bressler J, Eyring C, O’driscoll C, Naidu S, Johnston M V. (2011): Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat Rec, 294: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mierau S, Patrizi A, Hensch T, Fagiolini M (2015): Cell-specific regulation of NMDA receptor maturation by MeCP2 in cortical circuits. Biol Psychiatry, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D (1997): Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry, 154: 805–811. [DOI] [PubMed] [Google Scholar]
- 28.Jackson ME, Homayoun H, Moghaddam B (2004): NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A, 101: 8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM (2006): A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci, 26: 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL (2007): Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science, 318: 1645–1647. [DOI] [PubMed] [Google Scholar]
- 31.Fagiolini M, Katagiri H, Miyamoto H, Mori H, Grant SGN, Mishina M, Hensch TK (2003): Separable features of visual cortical plasticity revealed by N-methyl-Daspartate receptor 2A signaling. Proc Natl Acad Sci U S A, 100: 2854–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prusky GT, Alam NM, Beekman S, Douglas RM (2004): Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci, 45: 4611–4616. [DOI] [PubMed] [Google Scholar]
- 33.Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G (2008): Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol, 160: 116–121. [DOI] [PubMed] [Google Scholar]
- 34.Morgan CJA, Curran HV (2012): Ketamine use: A review. Addiction, 107: 27–38. [DOI] [PubMed] [Google Scholar]
- 35.Shaffer CL, Osgood SM, Smith DL, Liu J, Trapa PE (2014): Enhancing ketamine translational pharmacology via receptor occupancy normalization. Neuropharmacology, 86: 174–180. [DOI] [PubMed] [Google Scholar]
- 36.Rowland LM (2005): Subanesthetic ketamine: How it alters physiology and behavior in humans. Aviat Sp Environ Med, 76: C52–58. [PubMed] [Google Scholar]
- 37.Irifune M, Shimizu T, Nomoto M (1991): Ketamine-induced hyperlocomotion associated with alteration of presynaptic components of dopamine neurons in the nucleus accumbens of mice. Pharmacol Biochem Behav, 40: 399–407. [DOI] [PubMed] [Google Scholar]
- 38.Verma A, Moghaddam B (1996): NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci, 16: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cilia J, Hatcher P, Reavill C, Jones DNC (2007): (+/−) Ketamine-induced prepulse inhibition deficits of an acoustic startle response in rats are not reversed by antipsychotics. J Psychopharmacol, 21: 302–311. [DOI] [PubMed] [Google Scholar]
- 40.Imre G, Fokkema DS, Boer JA, Ter Horst GJ (2006): Dose-response characteristics of ketamine effect on locomotion, cognitive function and central neuronal activity. Brain Res Bull, 69: 338–345. [DOI] [PubMed] [Google Scholar]
- 41.Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR (2001): Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res, 49: 171–178. [DOI] [PubMed] [Google Scholar]
- 42.Venâncio C, Magalhães A, Antunes L, Summavielle T (2011): Impaired spatial memory after ketamine administration in chronic low doses. Curr Neuropharmacol, 9: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C, Zheng D, Xu J, Lam W, Yew DT (2013): Brain damages in ketamine addicts as revealed by magnetic resonance imaging. Front Neuroanat, 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weng SM, McLeod F, Bailey MES, Cobb SR (2011): Synaptic plasticity deficits in an experimental model of rett syndrome: Long-term potentiation saturation and its pharmacological reversal. Neuroscience, 180: 314–321. [DOI] [PubMed] [Google Scholar]
- 45.Auerbach W, Hurlbert MS, Hilditch-Maguire P, Wadghiri YZ, Wheeler VC, Cohen SI, et al. (2001): The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet, 10: 2515–2523. [DOI] [PubMed] [Google Scholar]
- 46.Viemari J-C, Roux J- C, Tryba AK, Saywell V, Burnet H, Peña F, et al. (2005): Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci, 25: 11521–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell VA, Oades RD, Tannock R, Killeen PR, Auerbach JG, Johansen EB, Sagvolden T (2006): Response variability in Attention-Deficit/Hyperactivity Disorder: a neuronal and glial energetics hypothesis. Behav Brain Funct, 2: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeevakumar V, Kroener S (2014): Ketamine Administration During the Second Postnatal Week Alters Synaptic Properties of Fast-Spiking Interneurons in the Medial Prefrontal Cortex of Adult Mice. Cereb Cortex, 1–13. [DOI] [PubMed] [Google Scholar]
- 49.Tomassy GS, Morello N, Calcagno E, Giustetto M (2014): Developmental abnormalities of cortical interneurons precede symptoms onset in a mouse model of Rett syndrome. J Neurochem, 131: 115–127. [DOI] [PubMed] [Google Scholar]
- 50.Hensch TK (2005): Critical period plasticity in local cortical circuits. Nat Rev Neurosci, 6: 877–888. [DOI] [PubMed] [Google Scholar]
- 51.Sleigh J, Harvey M, Voss L, Denny B (2014): Ketamine - more mechanisms of action than just NMDA blockade. Trends Anaesth Crit Care, 4: 76–81. [Google Scholar]
- 52.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. (2013): Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Eur J Pharmacol, 698: 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamakura T, Chavez-Noriega LE, Harris RA (2000): Subunit-dependent inhibition of human neuronal nicotinic acetylcholine receptors and other ligand-gated ion channels by dissociative anesthetics ketamine and dizocilpine. Anesthesiology, 92: 1144–1153. [DOI] [PubMed] [Google Scholar]
- 54.Craft JB, Coaldrake LA, Yonekura ML, Dao SD, Co EG, Roizen MF, et al. (1983): Ketamine, catecholamines, and uterine tone in pregnant ewes. Am J Obstet Gynecol, 146: 429–434. [DOI] [PubMed] [Google Scholar]
- 55.Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM (1997): Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol, 333: 99–104. [DOI] [PubMed] [Google Scholar]
- 56.Mealing GA, Lanthorn TH, Murray CL, Small DL, Morley P (1999): Differences in degree of trapping of low-affinity uncompetitive N-methyl-D-aspartic acid receptor antagonists with similar kinetics of block. J Pharmacol Exp Ther, 288: 204–210. [PubMed] [Google Scholar]
- 57.Tseng KY, O’Donnell P (2007): Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex, 17: 1235–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Homayoun H, Moghaddam B (2007): NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci, 27: 11496–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ (2004): Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci, 24: 9598–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK (2008): Experience-Dependent Transfer of Otx2 Homeoprotein into the Visual Cortex Activates Postnatal Plasticity. Cell, 134: 508–520. [DOI] [PubMed] [Google Scholar]
- 61.Kuczewski N, Porcher C, Gaiarsa J-L (2010): Activity-dependent dendritic secretion of brain-derived neurotrophic factor modulates synaptic plasticity. Eur J Neurosci, 32: 1239–44. [DOI] [PubMed] [Google Scholar]
- 62.Miller OH, Yang L, Wang C-C, Hargroder E a, Zhang Y, Delpire E, Hall BJ (2014): GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife, 3: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Naughton M, Clarke G, Oleary OF, Cryan JF, Dinan TG (2014): A review of ketamine in affective disorders: Current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action. J Affect Disord, 156: 24–35. [DOI] [PubMed] [Google Scholar]
- 64.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, et al. (2011): NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature, 475: 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duncan WC, Sarasso S, Ferrarelli F, Selter J, Riedner BA, Hejazi NS, et al. (2012): Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol, 16: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kavalali ET, Monteggia LM (2012): Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry, 169: 1150–1156. [DOI] [PubMed] [Google Scholar]
- 67.Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, et al. (2011): Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet, 20: 1182–1196. [DOI] [PubMed] [Google Scholar]
- 68.Li W, Pozzo-Miller L (2014): BDNF deregulation in Rett syndrome. Neuropharmacology, 76: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gideons ES, Kavalali ET, Monteggia LM (2014): Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A, 111: 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kotermanski SE, Wood JT, Johnson JW (2009): Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J Physiol, 587: 4589–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou X, Hollern D, Liao J, Andrechek E, Wang H (2013): NMDA receptormediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis, 4: e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ko H, Cossell L, Baragli C, Antolik J, Clopath C, Hofer SB, Mrsic-Flogel TD (2013): The emergence of functional microcircuits in visual cortex. Nature, 496: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gadalla KKE, Ross PD, Riddell JS, Bailey MES, Cobb SR (2014): Gait Analysis in a Mecp2 Knockout Mouse Model of Rett Syndrome Reveals Early-Onset and Progressive Motor Deficits. PLoS One, 9: e112889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.