

Abstract
Preserving mitochondrial activity is crucial in rescuing cardiac function following acute myocardial ischemia/reperfusion (I/R). The sex difference in myocardial functional recovery has been observed after I/R. Given the key role of mitochondrial connexin43 (Cx43) in cardiac protection initiated by ischemic preconditioning, we aimed to determine the implication of mitochondrial Cx43 in sex-related myocardial responses and to examine the effect of estrogen (17β-estradiol, E2) on Cx43, particularly mitochondrial Cx43-involved cardiac protection following I/R. Mouse primary cardiomyocytes and isolated mouse hearts (from males, females, ovariectomized females, and doxycycline-inducible Tnnt2-controlled Cx43 knockout without or with acute post-ischemic E2 treatment) were subjected to simulated I/R in culture or Langendorff I/R (25-min warm ischemia/40-min reperfusion), respectively. Mitochondrial membrane potential and mitochondrial superoxide production were measured in cardiomyocytes. Myocardial function and infarct size were determined. Cx43 and its isoform, Gja1–20k, were assessed in mitochondria. Immunoelectron microscopy and co-immunoprecipitation were also used to examine mitochondrial Cx43 and its interaction with estrogen receptor-α by E2 in mitochondria, respectively. There were sex disparities in stress-induced cardiomyocyte mitochondrial function. E2 partially restored mitochondrial activity in cardiomyocytes following acute injury. Post-ischemia infusion of E2 improved functional recovery and reduced infarct size with increased Cx43 content and phosphorylation in mitochondria. Ablation of cardiac Cx43 aggravated mitochondrial damage and abolished E2-mediated cardiac protection during I/R. Female mice were more resistant to myocardial I/R than age-matched males with greater protective role of mitochondrial Cx43 in female hearts. Post-ischemic E2 usage augmented mitochondrial Cx43 content and phosphorylation, increased mitochondrial Gja1–20k, and showed cardiac protection.
Keywords: Sex-based mitochondrial performance, Estrogen, Cardiac dysfunction, Mitochondrial connexin 43, Myocardial ischemia reperfusion
Introduction
Despite significant advances in current therapy, myocardial ischemia continues to be a leading cause of mortality in developed countries for both men and women [5]. During occlusion of a coronary artery, timely restoration of blood flow to the heart (reperfusion) remains the primary treatment of choice for myocardial ischemia, which in turn results in ischemia reperfusion (I/R) injury. Mitochondria, one of the most important subcellular organelles in cardiomyocytes, are a central player in determining the severity of myocardial damage during I/R injury [45]. Preserving mitochondrial integrity and activity is critical to lessening myocardial impairment after I/R. Mitochondrial modulation has thus become an area of growing therapeutic interest. Of note, clinical studies have shown that women are at an advantage on cardiac recovery after myocardial ischemia, as demonstrated by smaller infarct size, better myocardial salvage, enhanced myocardial perfusion, and improved survival in women compared to age-matched men [14, 23, 24, 41, 53–55]. However, few studies have focused on investigating sex-related cardiac mitochondrial function, particularly during acute I/R injury.
On the other hand, although sex differences are owing to multiple factors, female hormone–estrogen is critical in mediating cardiac protection in the cardiovascular system. Our previous work has shown that female rodent hearts exhibited better cardiac recovery than male hearts following acute I/R injury [81, 83, 85, 86] and this salutary effect was attributable to the rapid action of estrogen [73, 79, 86]. It is noteworthy that most studies related to estrogen therapy in the cardiovascular system employ an approach of preventive usage of estrogen in clinical research or preischemic treatment in animal experiments [12, 38, 39, 48, 76]. Given clinical therapeutic potential in the treatment of acute myocardial ischemia, a strategy of post-ischemic administration of estrogen will be more practical compared to pretreatment. In fact, post-injury treatment with estrogen has been tested in other acute injury models including trauma hemorrhage [1, 36, 46], burn [92], and sepsis [44, 47]. However, it is unknown whether post-ischemic estrogen usage is able to provide mitochondrial preservation, thus protecting myocardium against I/R.
Connexin-43 (Cx43 or Gja1), a gap junction protein, is well documented on maintaining the cell-to-cell (intercellular) electrical coupling for synchronized cardiac contraction [31, 33, 59, 74, 75]. Emerging evidence has indicated that Cx43 is present in cardiomyocyte subsarcolemmal mitochondria (SSM, one major population of mitochondria that locate directly beneath the sarcolemma and are in contrast to another population of mitochondria–interfibrillar mitochondria [IFM] that are aligned among the myofibrils) [11, 71, 72]. Mitochondrial Cx43 significantly contributes to protecting myocardial function during I/R [7, 11, 57, 62, 72, 93]. Cardiac protective approaches to increasing myocardial SSM Cx43 mediate mitochondria-derived protection [7, 11, 62, 65, 71, 72]. Interestingly, total and mitochondrial Cx43 content is reduced in aged mouse hearts, likely contributing to the age-associated loss of cardiac protection [8]. Cx43 expression is also affected by sex with the higher level in female rat hearts compared to male rats [43, 77]. Furthermore, chronic estrogen supplementation prior to injury preserves myocardial Cx43 levels [16], suggesting a link between sex/estrogen and Cx43. Therefore, in this study, we determined the implication of sex in safeguarding mitochondrial function upon acute cardiac stress and examined the effect of post-ischemic estrogen (17β-estradiol, E2) treatment on Cx43, particularly mitochondrial Cx43-involved cardiac protection following acute I/R.
Methods and materials
Animals
Male and female C57BL/6J mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). Female C57BL/6J mice were ovariectomized at 5–6 weeks old and purchased as surgically modified animals. Our previous study has shown that ovariectomy (OVX) significantly decreased serum levels of E2 in female animals [86]. All mice were acclimated for at least 5 days with a standard diet before experiments. Mice at 10–18 weeks old were used for the experiments. Doxycycline-inducible Tnnt2-controlled Cx43 knockout (Cx43-ic-KO) was generated by using doxycycline-inducible Tnnt2-cre mice [90, 96] bred with Cx43flox/flox mice (the Jackson Laboratories). Cx43flox/flox (Tnnt2-Cre negative) mice were used as control mice. The Cx43 deletion was carried out by feeding doxycycline-containing chow (doxycycline 200 mg/kg, Big-Serv) for 10 days. The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Indiana University. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85–23, revised 1996).
Adult mouse cardiomyocyte isolation and treatments
Single cardiomyocytes were isolated from adult male and female mouse hearts using a Langendorff perfusion isolation system. After mice were injected with heparin (100 IU, i.p.), they were euthanized with isoflurane overdose. The hearts were excised rapidly and placed in ice-cold calcium-free perfusion buffer containing (in mM): NaCl 113, NaH2PO4 0.6, NaHCO3 1.6, KCl 4.7, KH2PO4 0.6, MgSO4 1.2, HEPES 10, taurine 30, 2,3-butanedione monoxime (BDM) 10, and glucose 20 (pH 7.4). Hearts were retrogradely perfused and digested with collagenase II (1.5 mg/ml) as described previously [40]. After isolated cardiomyocytes were sequentially restored in perfusion buffer containing calcium (100, 250, 500, or 1000 μmol/L CaCl2), they were seeded into laminin (20 μg/ml)-precoated 96-well plate with cardiomyocyte plating medium (MEM with glutamine + 2.5% FBS, 10 mM BDM, and 1% Pen/Strep) and cultured for 2 h at 37 °C, 5% CO2 for adherence. After that, both male and female cardiomyocytes were treated with vehicle, E2 (100 nM), H2O2 (50 μM), or E2 + H2O2 for 1 h. Doses for E2 and H2O2 were selected based on the previous work from our group [80, 84] and Dr. Sen’s laboratory [64]. The final H2O2 dose was also determined by a dose–response study (Figure S1A and S1B). We further performed simulated I/R on cardiomyocytes isolated from male and female mice. We pelleted primary mouse cardiomyocytes in hypoxic solution (NaCl 119, KCl 5.4, MgSO4 1.3, NaH2PO4 1.2, HEPES 5, MgCl2 0.5, CaCl2 0.9, lactate 20, in mM, BSA 0.1%, pH = 6.5), covered with a layer of mineral oil [13, 34, 51], and incubated them at 37 °C for 20 min. The period for simulated ischemia was selected on a preliminary study (Figure S1C). After simulated ischemia, cells were cultured in perfusion buffer (+ CaCl2 1 mM, BSA 0.5%) without or with E2 (100 nM) for 60 min (re-oxygenation or simulated reperfusion). Mitochondrial membrane potential and mitochondrial superoxide production were determined in these cardiomyocytes afterward.
Assessment of mitochondrial membrane potential
Isolated mouse cardiomyocytes after treatments were incubated with cardiomyocyte plating medium or perfusion buffer (+ CaCl2 1 mM, BSA 0.5%) containing a fluorescent probe JC-1 (1 μM, G-Biosciences, St. Louis, MO, USA) at 37 °C. JC-1 enters into the cytosol as monomers showing green fluorescence, while it goes into mitochondria forming dimers/aggregates and turns into red fluorescence. After a 30-min incubation, live cell imaging on cardiomyocytes was done using an Axio Observer Z1 motorized microscope (Zeiss, Oberchoken, Germany) with a 10× objective. Red and green fluorescence intensity in individual cardiomyocyte was quantified using ImageJ (NIH). The red to green fluorescence intensity ratio indicates the mitochondrial membrane potential.
Measurement of mitochondrial superoxide production
After the treatments, isolated mouse cardiomyocytes were loaded with MitoSOX Red (5 μM, Thermo Fisher Scientific, USA) in cardiomyocyte plating medium or perfusion buffer (+ CaCl2 1 mM, BSA 0.5%) and incubated at 37 °C for 20 min. MitoSOX Red specifically targets mitochondria in live cells. The more superoxide production in mitochondria, the greater is the fluorescence intensity observed. After 20-min incubation, cells were washed two times with cardiomyocyte plating medium. The live cell imaging on cardiomyocytes was done using an Axio Observer Z1 motorized microscope (Zeiss, Oberchoken, Germany) with a 10× objective. Red fluorescence intensity in an individual cardiomyocyte was quantified using ImageJ (NIH), which indicated mitochondrial superoxide production.
Isolated mouse heart preparation (Langendorff model)
Mouse hearts were isolated and subjected to Langendorff I/R as we previously described [37, 80, 82, 83, 85, 87]. Briefly, mice were anesthetized with isoflurane and heparinized (100 IU i.p.), and hearts were rapidly excised. The isolated heart was placed in ice-cold KH perfusion buffer for aorta annulation under a dissection microscope. The heart was then perfused in the isovolumetric Langendorff mode (70 mmHg) and paced at 420 bpm/min during the whole experimental time except ischemia. Data were continuously recorded using a PowerLab 8 preamplifier/digitizer (AD Instruments Inc., Milford, MA). The maximal positive and negative values of the first derivative of pressure (+ dP/dt and −dP/dt) were calculated using PowerLab software.
Isolated mouse hearts were subjected to the I/R protocol as follows: at least 15-min equilibration followed by 25-min global ischemia (37 °C) and 40-min reperfusion. Post-ischemic treatment with vehicle or 2 nM of E2 [73] during the entire period of reperfusion was performed on male, normal female, and OVX female mouse hearts.
Infarct size measurement
The left ventricle (LV) from another set of male and OVX female mouse hearts without or with post-ischemic E2 treatment, as well as normal female mouse hearts after 25-min global ischemia (37 °C) and 40-min reperfusion, was frozen for 30 min at − 20 °C and transversely sectioned into ~ 1 mm-thick slices (along the long axis) using a mouse heart slicer. The heart slices were stained with 1% 2,3,5-triphenyltetrazolium chloride (TTC) for 10 min at 37 °C and then transferred into 10% neutral formalin overnight. Images were taken and each slice was weighted the day after. The infarct area and the LV area were analyzed in each slice using ImageJ (NIH). The infarct size (percentage) was calculated as total infarct weight per total LV weight from all slices [13, 95]. The researcher performing infarct size measurement was blinded to the experimental groups.
Mitochondria isolation from mouse hearts
Mitochondrial and cytosolic fractions were separated by differential centrifugation using mitochondria isolation kit for tissue (Thermo Fisher Scientific) according to the manufacturer’s protocols. SSM and IFM were isolated using the methods described previously [71, 72]. Following experiments, the heart was minced in mitochondrial isolation buffer (pH 7.2, in mM: mannitol 225, sucrose 75, MOPS 10, Tris–HCl 10, EGTA 1, and 0.1% bovine serum albumin [BSA] and homogenized with a D1000 handheld homogenizer (Benchmark Scientific, Atkinson, NH). After centrifugation at 500g, 4 °C for 5 min, the supernatant was collected and added with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific) for the SSM preparation, while the pellet was digested in isolation buffer containing trypsin (5 mg/g pellet) for 10 min on ice and homogenized using a Teflon pestle. Digestion was stopped by adding Halt protease and phosphatase inhibitor cocktail. Each fraction was spun at 500g for 5 min at 4 °C and the supernatant was centrifuged at 3000g at 4 °C for 10 min to pellet SSM and IFM, respectively. The crude mitochondrial pellets from either SSM or IFM were resuspended in isolation buffer with protease and phosphatase inhibitors, and protein content was determined using a Bradford assay.
Western blotting
The heart tissues were lysed in cold RIPA buffer containing Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The protein extracts (10–30 μg) from heart tissue or mitochondria were subjected to electrophoresis on a 4–15% Criterion TGX Precast midi protein gel (Bio-Rad, Hercules, CA, USA) and transferred to a nitrocellulose membrane. The membranes were incubated with the following primary antibodies, respectively: phospho-Cx43 (ser368, #3511), Cx43 (#3512), GAPDH (#5174), LAMP1 (#3243), ATP2A2/SERCA2 (#9580), Na, K-ATPase (#3010), Golgin-97 (#13192), Cox IV (#4850), VDAC (#4661) (Cell Signaling Technology, Beverly, MA, USA), Cx43 C-terminus (AB1728, MilliporeSigma, Burlington, MA, USA), his-tone H3 (PA5–16183, Thermo Fisher Scientific), followed by horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibody. Detection was conducted using SuperSignal West Pico stable peroxide solution (Thermo Fisher Scientific). Immunoblotting band density measurement was performed using the Image J software (NIH).
Co‑immunoprecipitation assay (Co‑IP)
Mitochondria extracts were obtained from OVX F hearts without or with E2 treatment using mitochondria isolation kit for tissue (Thermo Fisher Scientific) and added in ice-cold IP lysis/wash buffer. Co-IP was performed on mitochondrial lysates using the Co-IP kit (Thermo Fisher Scientific) based on the manufacturer’s instructions. Briefly, antibody immobilization was achieved by using AminoLink plus coupling resin incubated with 5 μg of anti-ERα antibody (MA5–13065, Thermo Fisher Scientific). Lysates were then incubated with anti-ERα resin at 4 °C overnight. After elusion, proteins were probed by Western blotting with antibodies against ERα and Cx43.
Transmission electron microscopy (TEM)
The heart tissue was fixed with 3% glutaraldehyde in 0.1 M sodium cacodylate (SC) buffer and rinsed with 0.1 M SC buffer, followed by post-fixation with 1% osmium tetroxide in 0.1 M SC buffer for 1 h. After the second rinse, the tissue specimens were dehydrated through a series of graded ethyl alcohols from 70 to 100% and then infiltrated with two changes of 100% acetone and a 50:50 mixture of acetone and embedding resin (Embed 812, Electron Microscopy Sciences, Hatfield, PA, USA) for over the weekend. Specimen vial lids were popped and acetone allowed to evaporate off for 3 h. Then specimens were embedded in a fresh change of 100% embedding media. Following polymerization overnight at 60 °C, the blocks were then ready to section. Thin sections were cut (80–90 nm), stained with UA replacement stain (Electron Microscopy Sciences), and viewed on a Tecnai Spirit (ThermoFisher, Hillsboro, OR, USA). Digital images were taken with an AMT (Advanced Microscope Techniques, Danvers, MA, USA) CCD camera in a blinded fashion. The mitochondrial content was determined by quantifying the number and the size of each mitochondrion per field using the Image J software (NIH).
Immunoelectron microscopy
The heart tissue was fixed with 4% paraformaldehyde in 0.1 M SC buffer. After being rinsed in 0.1 M SC buffer and dehydrated through a series of graded ethyl alcohols from 70 to 100% ETOH, the specimens were placed in half 100% ETOH and half embedding resin, Unicryl (Electron Microscopy Sciences, Hatfield, PA, USA), overnight at 4 °C. The specimens were then embedded in fresh resin and polymerized at 4 °C using a UV light (360 nm wavelength). Thin sections were cut (80–90 nm), placed on formvar/carbon-coated grids, dried, and readied for immunostaining. After blocking and permeabilization, the sections were incubated with anti-Cx43 antibody (1:5, 71–0700, Thermo Scientific) over the weekend, rinsed three times in incubation buffer, and subsequently, incubated with anti-rabbit colloidal gold 10 nm (1:50, Aurion, Electron Microscopy Sciences) overnight at 4 °C. After being rinsed, the sections were put in 2% glutaraldehyde for post-fixation (5 min), in 1% OsO4 (2 min) and then stained with UA replacement stain (Electron Microscopy Sciences) for 10 s. The sections were viewed on Tecnai Spirit (ThermoFisher). Digital images were randomly taken with an AMT (advanced microscope techniques) CCD camera in a blinded fashion.
Statistical analysis
All reported results were mean ± SEM. Data were evaluated using unpaired t test or analysis of variance (ANOVA) followed by multiple comparison test. Difference was considered statistically significant when p < 0.05. All statistical analyses were performed using the GraphPad Prism (Graph-Pad, La Jolla, CA, USA).
Results
Sex differences in cardiomyocyte mitochondrial function
We first determined the mitochondrial membrane potential and mitochondrial superoxide production between male and female mouse cardiomyocytes. We observed a higher level of mitochondrial membrane potential in female cardiomyocytes than in male counterparts as determined by higher red/ green fluorescence ratio in female cells (Fig. 1a), but no significant difference was noticed in sex-based mitochondrial superoxide production under normal condition (Fig. 1b). Considering substantial amount of reactive oxygen species (ROS) produced following I/R [18], we selected H2O2 to mimic I/R and to stress cardiomyocytes. Our results revealed that H2O2 markedly decreased mitochondrial membrane potential in both male and female cardiomyocytes (Fig. 1c). However, male cardiomyocyte mitochondria generated more superoxide than female ones in response to H2O2 (Fig. 1d). To better represent clinical I/R injury [13, 34, 51], we also performed simulated I/R experiments on isolated cardiomyocytes. We found that simulated I/R significantly damaged mitochondrial membrane potential in the male and female cardiomyocytes (Fig. 1e) and triggered more mitochondrial superoxide generation in male cardiomyocytes than female ones (Fig. 1f), suggesting less oxidative stress in female cardiac mitochondria than male ones in response to I/R.
Fig. 1.
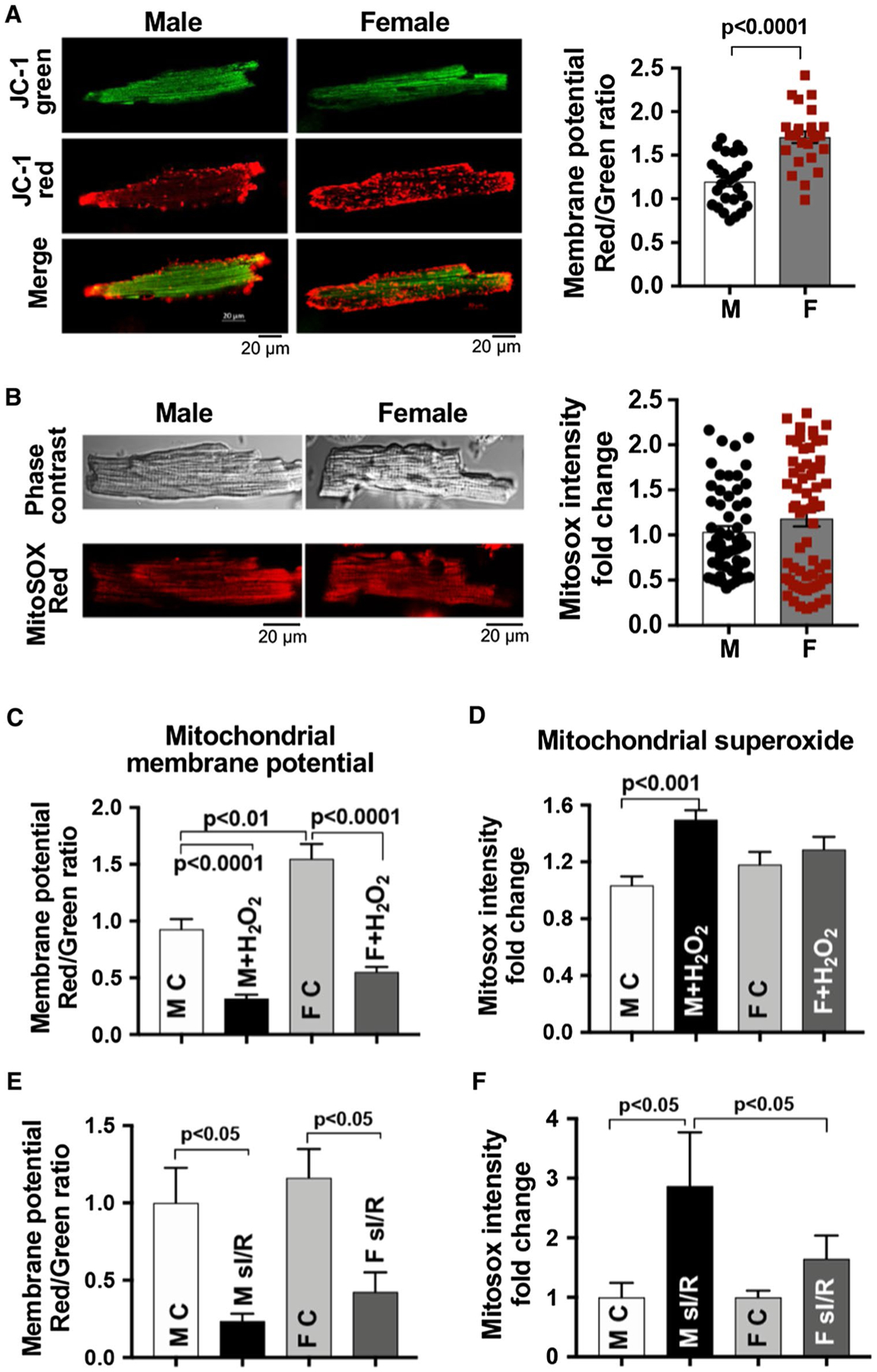
Sex-dependent cardiomyocyte mitochondrial responses. a Mitochondrial membrane potential using JC-1 in cardiomyocytes from adult male (M) and female (F) mouse hearts. b Mitochondrial superoxide production using MitoSOX Red in male and female mouse cardiomyocytes. c, d Changes of mitochondrial membrane potential and mitochondrial superoxide production in response to H2O2 (50 μM, 1 h) in male and female mouse cardiomyocytes. e, f Simulated ischemia/reperfusion (sI/R)-damaged mitochondrial membrane potential and -induced mitochondrial superoxide production in male and female mouse cardiomyocytes. Twenty-min ischemia followed by 1-h reperfusion (re-oxygenation) was used. Mean ± SEM, > 25 cardiomyocytes/group/trial, and at least three trials repeated; results analyzed using unpaired t test
E2 treatment preserves mitochondrial membrane potential and reduces mitochondrial superoxide production in cardiomyocytes
Considering the importance of estrogen in mediating sex-dependent responses in the cardiovascular system, we next assessed the effect of E2 on mitochondrial performance in both male and female mouse cardiomyocytes that were subjected to H2O2 or simulated I/R. E2 preserved H2O2-disrupted mitochondrial membrane potential and decreased H2O2-induced mitochondrial superoxide production in male mouse cardiomyocytes (Fig. 2a). Intriguingly, E2 treatment starting at the onset of reperfusion and present at the complete period of re-oxygenation significantly protected male cardiomyocyte mitochondria (Fig. 2b) against simulated I/R as demonstrated by preserved mitochondrial membrane potential and decreased mitochondrial superoxide generation in part. Furthermore, female cardiomyocyte mitochondrial membrane potential was also protected by E2 usage following H2O2 or simulated I/R (Fig. 2c).
Fig. 2.
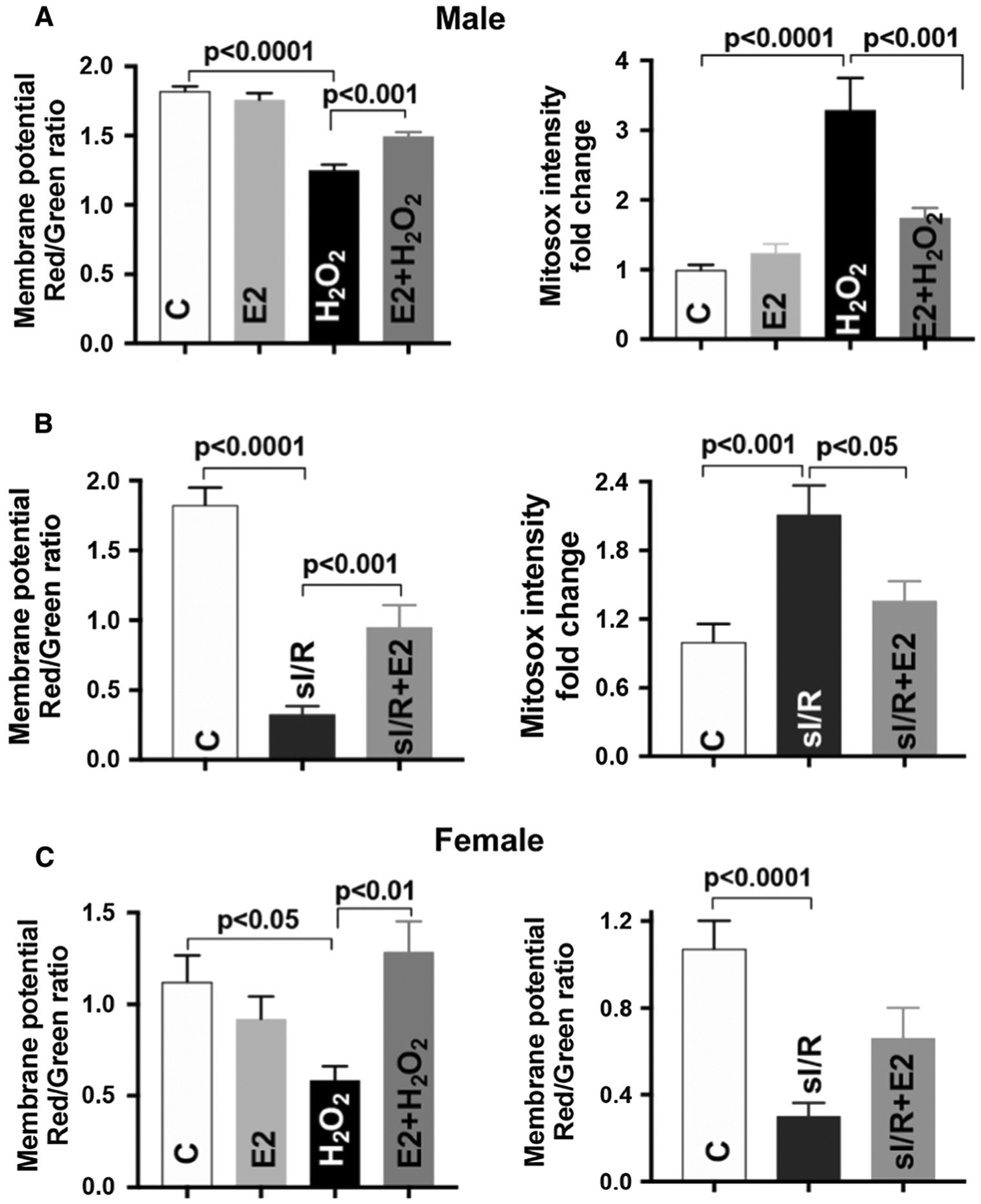
Estrogen improves cardiomyocyte mitochondrial function. a 17β-estradiol (E2, 100 nM) preserves H2O2-disrupted mitochondrial membrane potential and reduces mitochondrial superoxide production in male mouse cardiomyocytes. b Post-ischemic E2 treatment improves mitochondrial membrane potential and decreases mitochondrial superoxide production in male cardiomyocytes subjected to simulated ischemia/reperfusion (sI/R). c E2 treatment preserves mitochondrial membrane potential in female cardiomyocytes in response to H2O2 or sI/R. Mean ± SEM, > 25 cardiomyocytes/group/trial and at least four trials were repeated; results analyzed using one-way ANOVA with multiple comparisons test
Acute post‑ischemic infusion of E2 improved myocardial functional recovery and reduced infarct size following I/R
Post-ischemic E2 usage may be beneficial in a clinical setting. We observed that post-ischemic administration of E2 significantly improved myocardial functional recovery (left ventricular developed pressure [LVDP] and ± dP/dt) in male (Fig. 3a–c) and ovariectomized female (OVX F) mouse hearts (Fig. 3e–g) following I/R compared to vehicle groups. To further estimate a degree of I/R injury [13], we measured myocardial infarct size. We found E2 treatment during reperfusion markedly reduced infarct size in male (Fig. 3d) and OVX F hearts (Fig. 3h) after I/R. We also noticed a significant decrease in infarct size for female mouse hearts compared to male hearts subjected to I/R (Fig. 3i). No significant change of LVDP was noticed in male and OVX F stability controls (Fig. 3j). Not surprisingly, we did not observe any difference in female hearts without or with post-ischemic E2 treatment (Fig. S2).
Fig. 3.
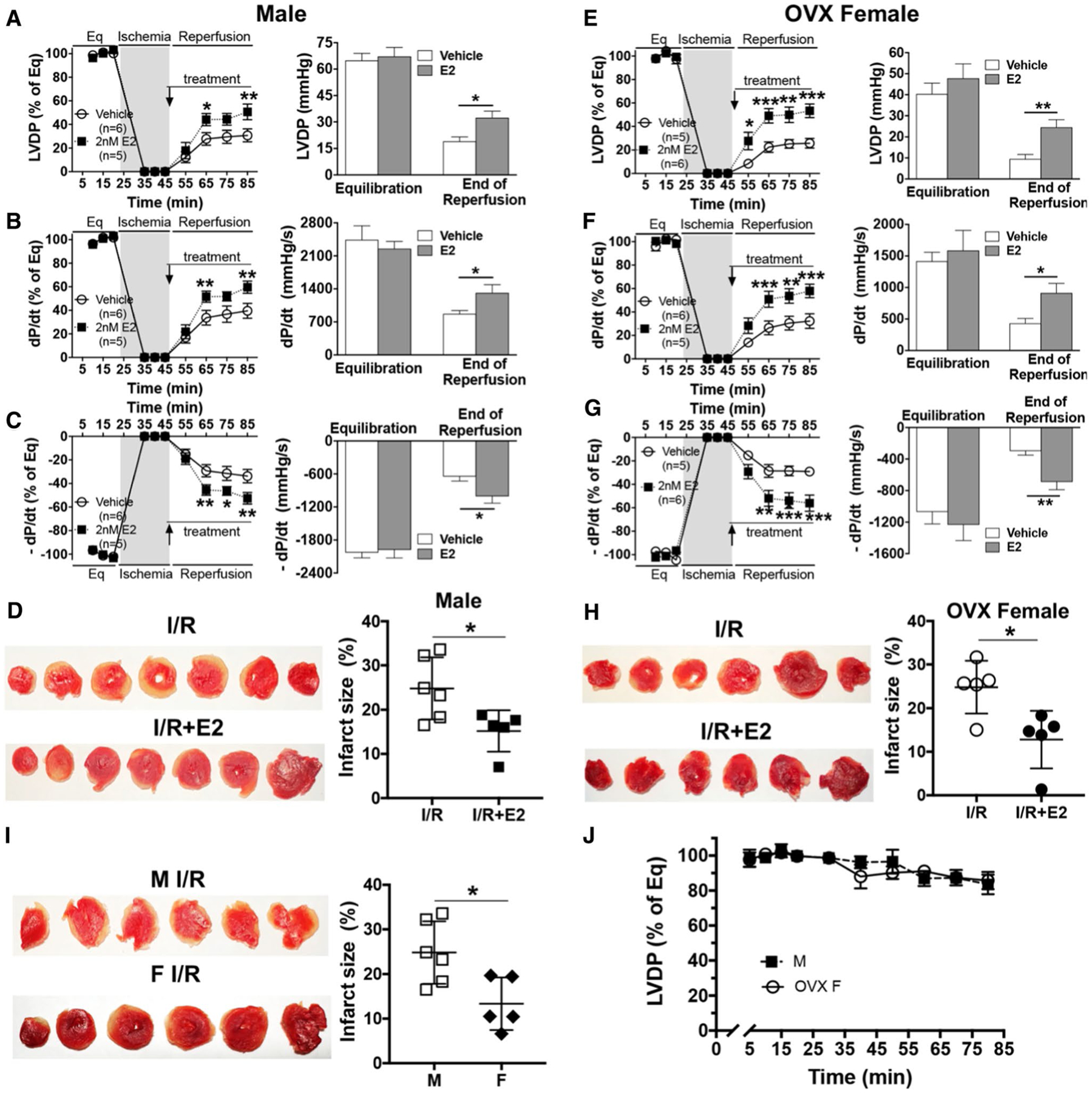
Post-ischemic 17β-estradiol (E2) treatment improves myocardial function and reduces infarct size following acute ischemia/ reperfusion (I/R). Post-ischemic infusion of E2 improves myocardial functional recovery in male (M) mouse hearts: a LVDP, b +dP/dt, c – dP/dt, and in ovariectomized female (OVX F) mouse hearts: e LVDP, f +dP/dt, g – dP/dt. Representative photographs of transverse slices with TTC staining and infarct size comparisons between post-ischemic E2 treatment and their untreated counterparts in male (d) and OVX female (h) mouse hearts following I/R. i Infarct size comparison between male and female mouse hearts after I/R. j Changes of LVDP in stability controls of male and OVX F mouse hearts perfused for 85 min (n = 4/group). LVDP: left ventricular developed pressure = LV systolic pressure − diastolic pressure; ± dP/dt: maximum/minimum value of first derivative of LV pressure; Eq equilibration. Mean ± SEM, results analyzed using two-way ANOVA with multiple comparisons of Sidak test in a–c and e–g, unpaired t test in bar graphs, d, h and i, *p < 0.05, **p < 0.01, ***p < 0.001
Sex‑dependent mitochondrial levels of Cx43 following acute I/R
Given better myocardial functional recovery in female hearts than in male hearts following I/R from our previous studies [81, 83, 85, 86] and the importance of mitochondrial Cx43 in cardiac protection [7, 11, 57, 62, 72, 93], we next determined myocardial levels of Cx43 between male and female. Without I/R there was no sex-specific difference in Cx43 expression in mouse hearts and cardiac mitochondria (Fig. 4a, b, S4A). However, higher levels of Cx43 were observed in female mouse hearts than in male hearts following I/R (Fig. 4c, S4B). Similarly, increased mitochondrial content of Cx43 was noticed in female myocardium compared to male myocardium after I/R (Fig. 4d, S4C), suggesting a potential role of mitochondrial Cx43 in sex-dependent mitochondrial responses. Isolated mitochondrial morphology was detected by TEM (Fig. 4e). The mitochondrial purity was also determined by Western blotting. The mitochondrial preparations were free of contamination of cytosol and other subcellular organelles (Fig. 4f).
Fig. 4.
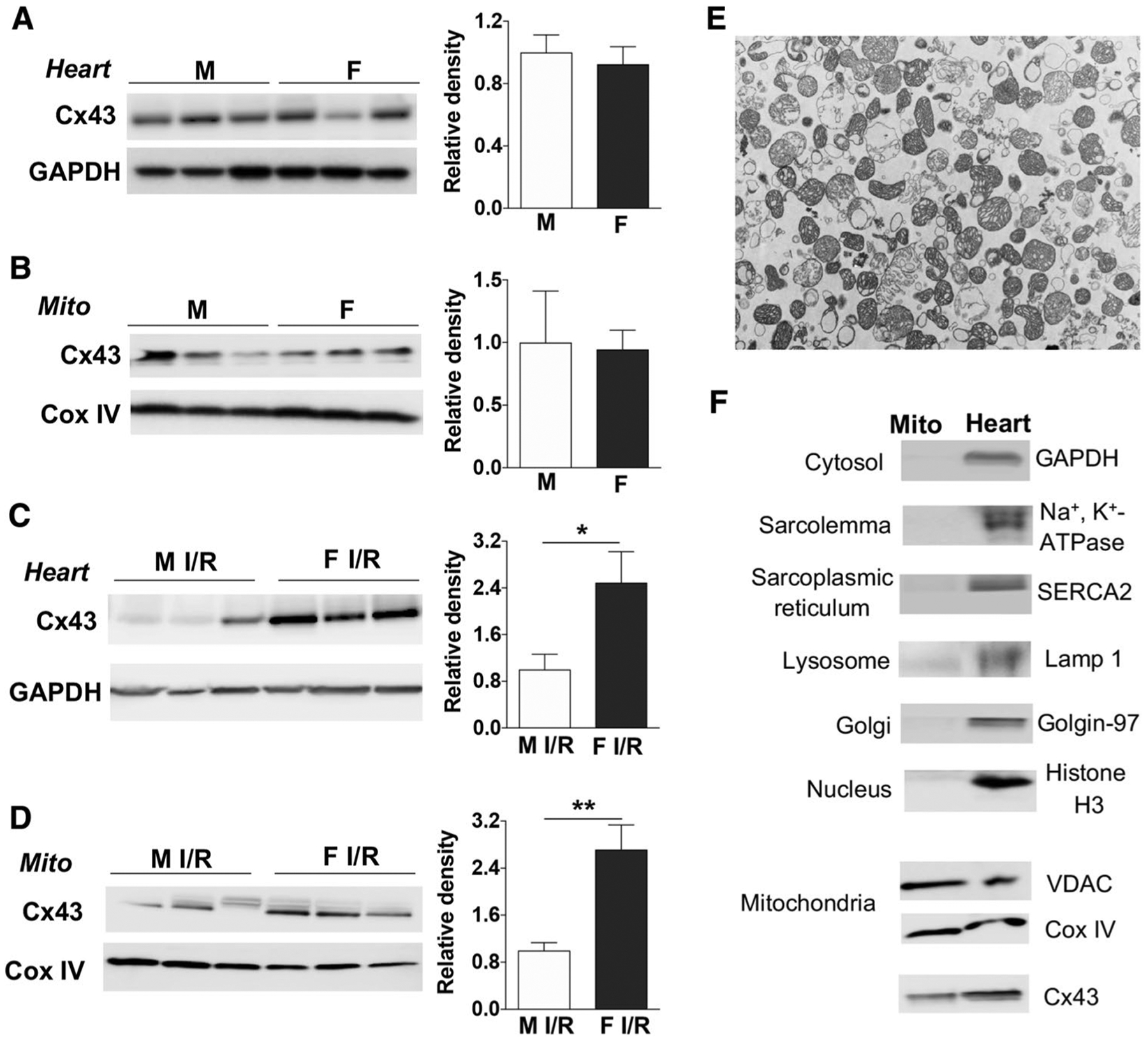
Sex differences in Cx43 expression after I/R. Cx43 expression in male (M) and female (F) mouse heart lysate (a) and cardiac mitochondria (Mito) (b) without I/R. Following acute Langendorff I/R, Cx43 levels in male and female mouse heart lysate (c) and cardiac mitochondria (Mito) (d). Isolated mitochondria from mouse hearts detected by transmission electron microscopy (e). Analysis of isolated mitochondria and total cardiac lysates (heart) (f). VDAC and Cox IV are used as mitochondrial markers. Cyto: cytosol. Western blots in a–d show individual samples from one trial (other original Western blots shown in Fig. S4A, S4B and S4C). Bar graphs (relative density) are combined analysis for immunoblotting band intensity normalized to GAPDH or Cox IV, respectively. Mean ± SEM, unpaired t test, n = 3–6/group, *p < 0.05, **p < 0.01
Estrogen modulated Cx43 levels and phosphorylation in the I/R heart
Next, we determined whether endogenous or exogenous estrogen plays a role in mediating myocardial Cx43 expression following acute I/R. We observed that depletion of endogenous estrogen by OVX significantly reduced Cx43 expression in female heart tissue (Fig. 5a, S4D) and female cardiac mitochondria (Fig. 5b, S4E) after I/R, whereas post-ischemic E2 treatment markedly increased Cx43 levels in heart tissue (Fig. 5c) and cardiac mitochondria (Fig. 5d, S4F) in OVX females following I/R. Additionally, post-ischemic E2 usage augmented the content of Cx43 and Gja1–20k, a smaller isoform of Cx43, to SSM (but not IFM) in OVX F mouse hearts after acute I/R (Fig. 5d, S4F, S4G). Post-ischemic administration of E2 increased Gja-20k in male heart mitochondria as well (Fig. 5e, S4J). We further found Cx43 mostly present in the mitochondria compared to cytosol (Fig. S4K).
Fig. 5.
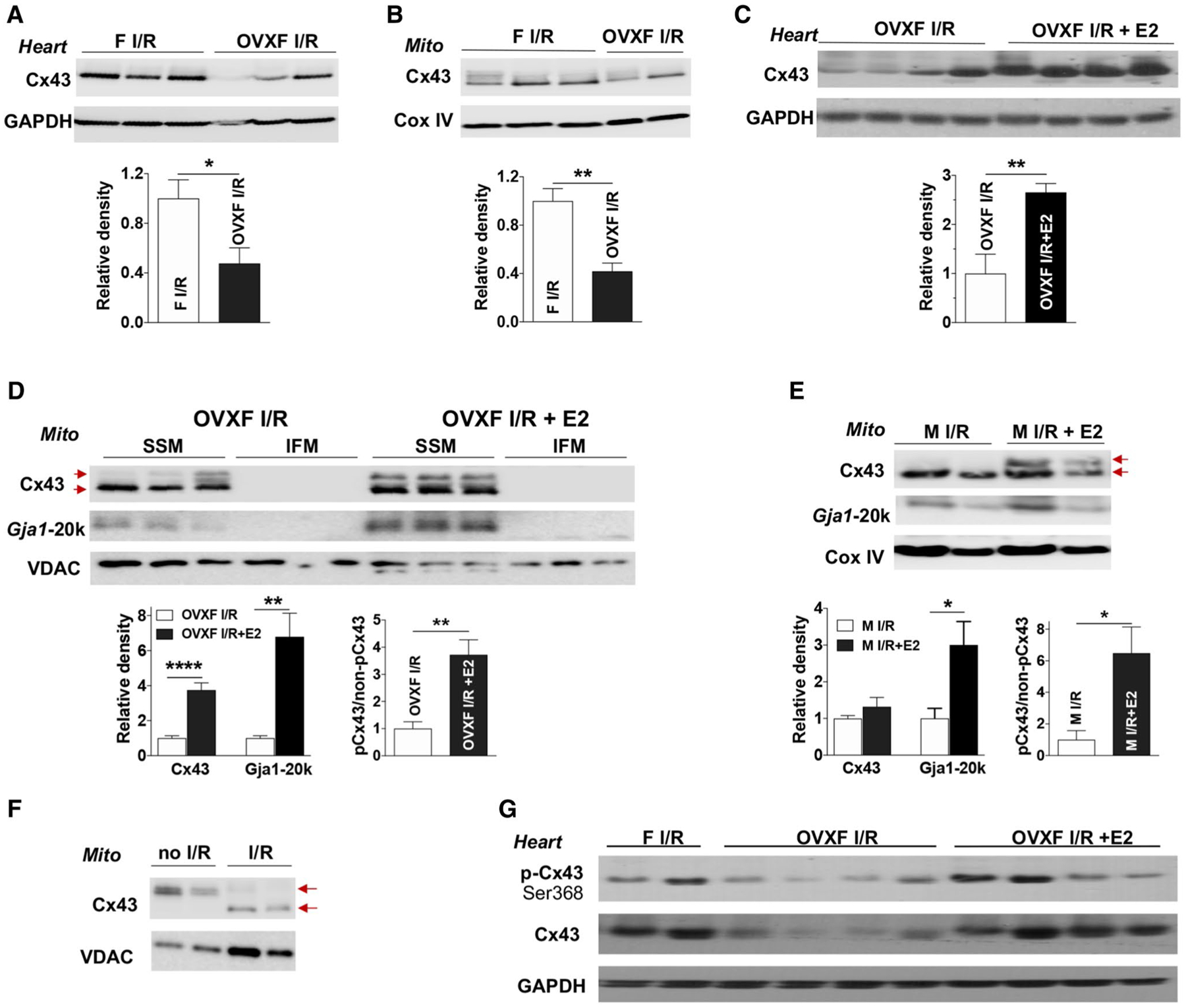
The role of endogenous and exogenous estrogen in myocardial Cx43 expression following acute I/R. Depletion of endogenous estrogen by ovariectomy (OVX) reduces Cx43 levels in female (F) heart tissue (a) and cardiac mitochondria (Mito) (b) after myocardial I/R. Post-ischemic E2 infusion increases Cx43 content in OVX F heart tissue (c) and in cardiac subsarcolemmal mitochondria (SSM) (d) following I/R, as well as augmented Gja1–20k in SSM. d Cx43 and Gja1–20k are preferably present in SSM compared to interfibrillar mitochondrial (IFM). Post-ischemic E2 usage augments Gja1–20k in mitochondria of male hearts after acute I/R (e). d, e Higher levels of phosphorylated Cx43 (pCx43, upper arrow) vs. non-phosphorylated Cx43 (non-pCx43, lower arrow) in post-ischemic treatment with E2 in OVX F and male mouse hearts following I/R. f De-phosphorylated Cx43 (lower arrow) by I/R in mitochondria compared to phosphorylated mitochondrial Cx43 (upper arrow) in mouse hearts without I/R. g Post-ischemic E2 infusion regulates myocardial Cx43 phosphorylation at Ser368 in OVX F hearts after I/R. Western blots in a–e show individual samples from one trial (other original Western blots shown in Fig. S4D–S4 J). Densitometry data (relative density) are analyzed by immunoblotting band intensity normalized to loading control– GAPDH, Cox IV, or VDAC, respectively, and pCx43 vs. non-pCx43 in (d) and (e). Mean ± SEM, n = 4–7/group, unpaired t test, *p < 0.05, **p < 0.01
Notably, Cx43 electrophoreses are multiple bands between 41 and 46 kDa, including a faster migrating form (non-phosphorylated Cx43 at around 41 kDa) and at least one slower migrating form (primarily phosphorylated Cx43 at about 45 kDa) [4, 69]. It is documented that the majority of Cx43 is phosphorylated in the heart under normal conditions [3, 4]. Ischemia results in significant dephosphorylation of Cx43, whereas reperfusion variably modulates Cx43 phosphorylation and/or de-phosphorylation in isolated hearts [4, 70]. Generally, approach to preventing dephosphorylation of Cx43 is cardioprotective [50]. We found an overall dephosphorylation of Cx43 in mouse myocardial mitochondria subjected to acute I/R compared to perfusion control (Fig. 5f), which was consistent with previous finding on acute I/R dephosphorylating Cx43 in rat heart mitochondria [30]. Most intriguingly, higher levels of phosphorylated Cx43 in mitochondria were observed in male and OVX female mouse hearts treated with E2 during reperfusion compared to their untreated counterparts (Fig. 5d, e, S4F, S4H). Also, post-ischemic E2 treatment increased Cx43 phosphorylation at Ser368 in OVX female heart tissue after I/R (Fig. 5g).
Furthermore, Immunoelectron microscopy indicated colloidal gold-marked Cx43 particles present in OVX F hearts (Fig. 6a), whereas no particles were detected in control preparations without the Cx43 antibody. E2 usage significantly increased the presence of Cx43 particles in OVX F heart mitochondria (Fig. 6b).
Fig. 6.
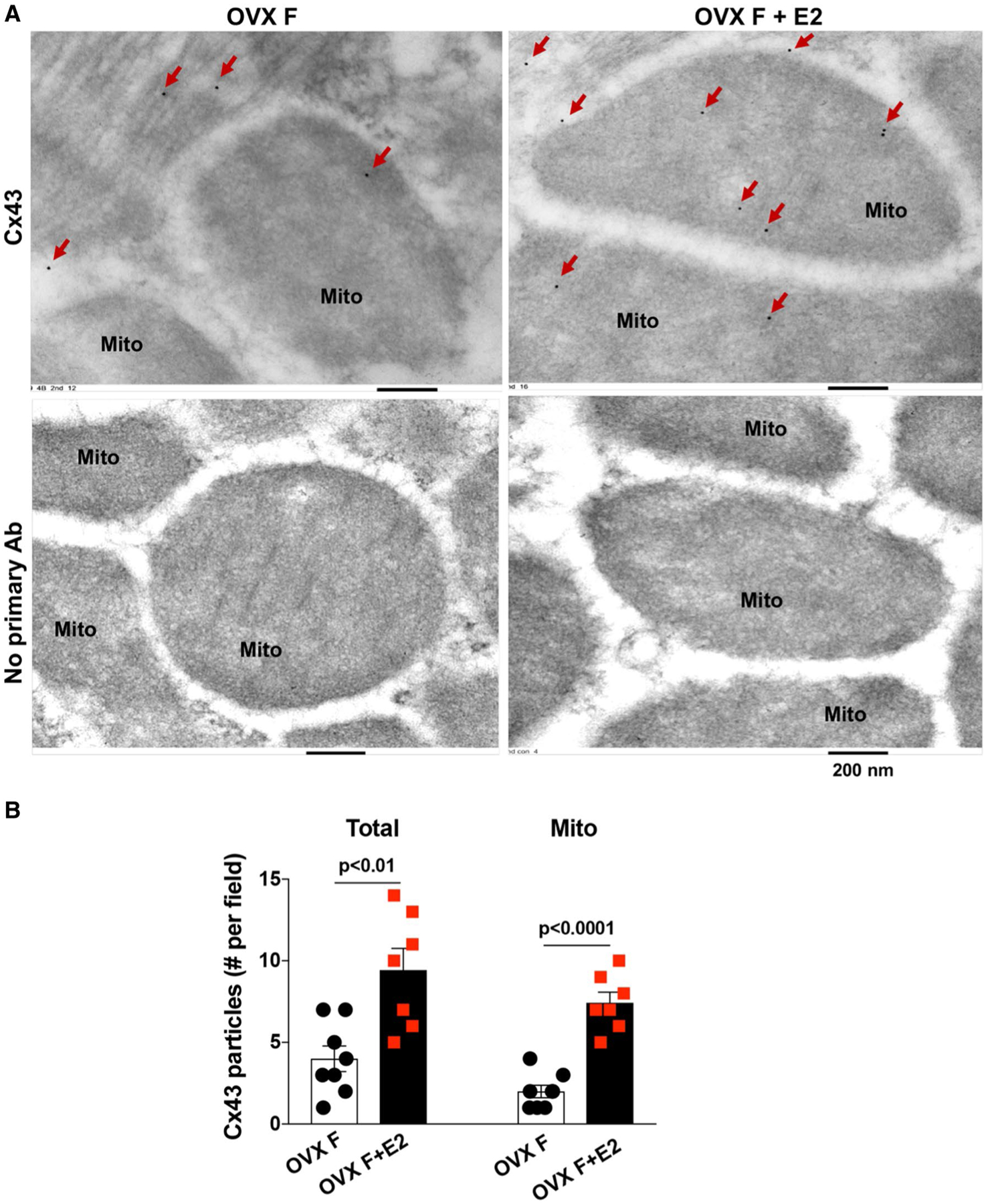
Mitochondrial Cx43 content determined by immunoelectron microscopy. a Representative images of immunoelectron microscopy show gold-marked Cx43 particles (red arrows) present in OVX F hearts ± E2. b Bar graph shows total number of gold–labeled Cx43 particles in the field and number in the mitochondria. Mean ± SEM, unpaired t test
The interaction of estrogen receptor (ER)α and Cx43 by E2 in myocardial mitochondria
ERα is present in the plasma membrane [19, 63] and such localization of ERα enables the rapid action of E2 via ERα-initiated non-genomic signaling. It has been reported that membrane-associated ERα is involved in regulating Cx43 function in rat cardiomyocytes during metabolic inhibition [19]. Therefore, we determined mitochondrial levels of ERα and the potential binding of ERα/Cx43 initiated by estrogen treatment. Significantly enhanced ERα levels were observed in mitochondria of OVX F hearts with post-ischemic E2 usage compared to their untreated counterparts following I/R (Fig. 7a, S4K). In addition, as shown in Fig. 7b, binding of Cx43 to ERα was noticed in mitochondria isolated from OVX F hearts following E2 treatment, indicating that Cx43 was an interacting partner of ERα and that using E2 increased the interaction of Cx43 with ERα in myocardial mitochondria.
Fig. 7.
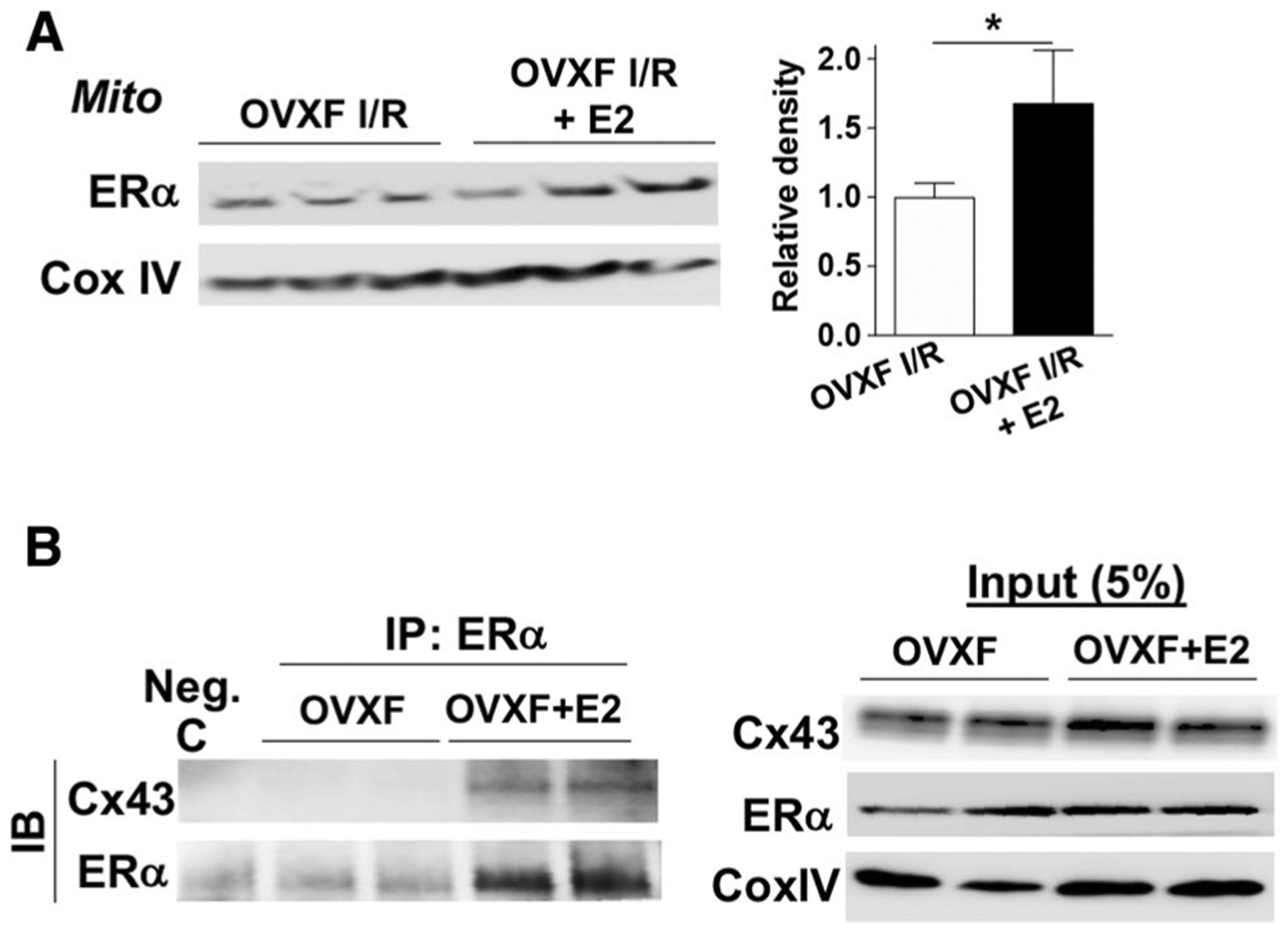
The binding of Cx43 to ERα in mouse heart mitochondria by estrogen. a Western blotting for ERα in mitochondrial extracts from OVX F mouse hearts± post-ischemic E2 treatment after I/R (another set of original Western blots shown in Fig. S4K). Bar graph (relative density) is combined analysis for band intensity normalized to loading control—Cox IV or VDAC, respectively. Mean ± SEM, n = 6/group, unpaired t test, *p < 0.05. b Binding of Cx43 to ERα is observed in mitochondria isolated from OVX F mouse hearts with E2 infusion by co-IP. Mitochondrial extracts were immunoprecipitated using anti-ERα antibody or control IgG (Neg. c) and blotted with antibodies of anti-Cx43 and anti-ERα. 5% input as positive control. Experiment was repeated two times (n = 4 hearts/group)
Aggravated mitochondrial damage and impaired myocardial functional recovery in Cx43‑ic‑KO mice following acute I/R
The Cx43-null mice die shortly after birth [59]. Homozygous cardiac MHC-controlled deletion of Cx43 mice show abnormal conduction properties starting at 2–3 weeks of age with sudden cardiac death, resulting in no longer than 2 months of life [33]. A tamoxifen-inducible recombination is not suitable for our study due to tamoxifen interfering with the effect of estrogen. Therefore, we generated doxycycline-inducible Tnnt2-controlled deletion of Cx43 mice (Cx43-ic-KO) in our study. After 10-day doxycycline-containing chow feeding, there was no difference in body weight (Fig. S5A) and cardiac function (LV ejection fraction and fractional shorting by echocardiography, Fig. S5B, S5C) between male adult (16–24-week old) WT and Cx43-ic-KO mice. However, a change in the QRS shape from ECG recording was noticed in Cx43-ic-KO mice (Fig. S5D), which was in line with previous observation from cardiomyocyte-restricted deletion of Cx43 mouse line (MyHC-Cre:Cx43flox/flox) [26]. Reduced cardiac Cx43 expression was detected in Cx43-ic-KO mice after 10-day doxycycline-containing chow feeding (Fig. S5E). With Cx43 expression present in cardiac fibroblasts, vascular smooth muscle cells and endothelial cells, we did not observe complete deletion of Cx43 in the myocardium. Similarly, decreased Cx43 content in mitochondria was observed in the Cx43-ic-KO hearts (Fig. S5F). Immunoelectron microscopy also indicated immunogold dots of Cx43 within male WT heart mitochondria, whereas gold-marked Cx43 particles were barely detected in Cx43-ic-KO cardiac mitochondria (Fig. 8a).
Fig. 8.
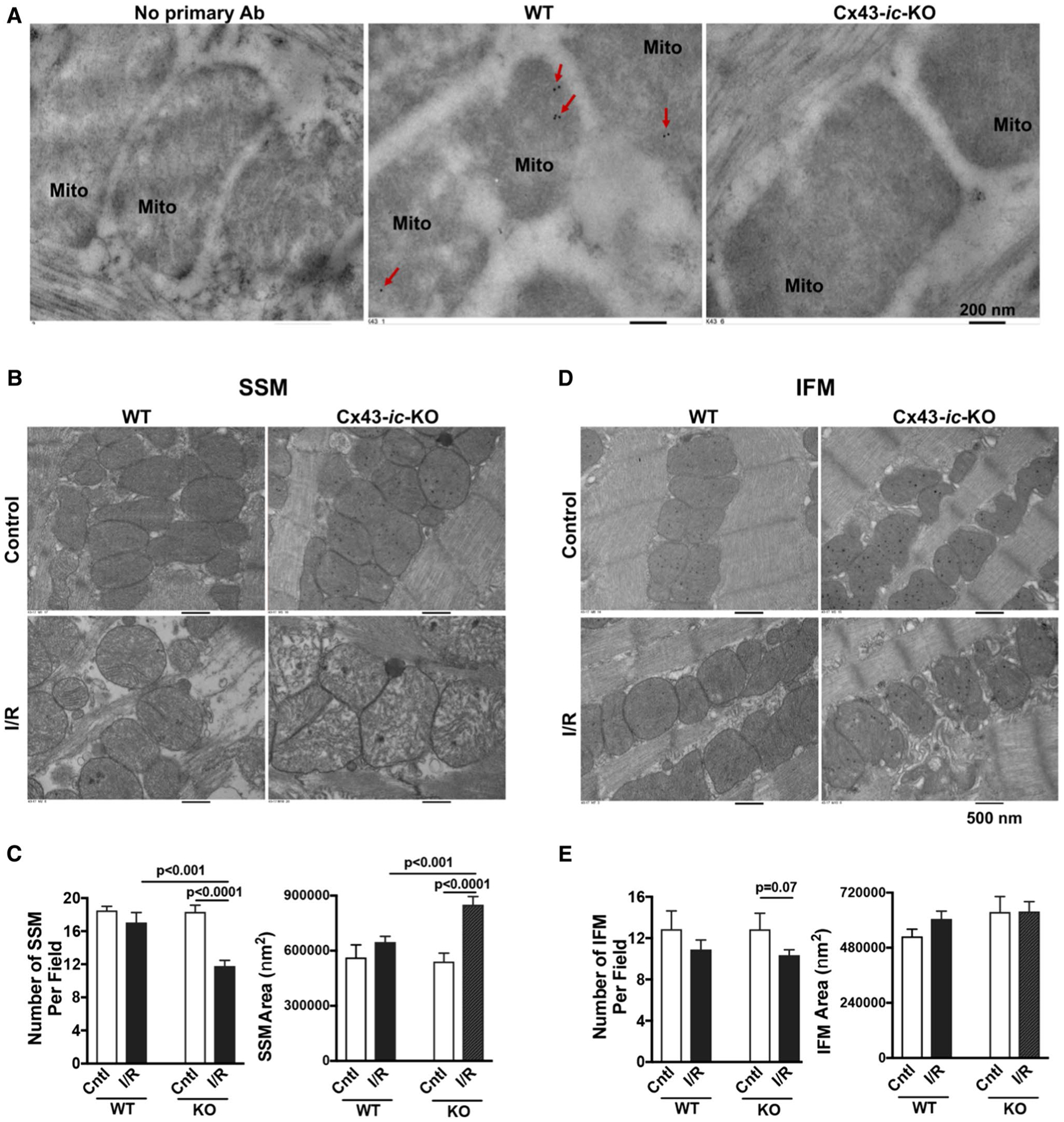
Mitochondrial damage in Cx43-ic-KO mouse hearts following acute I/R. a Representative images of immunoelectron microscopy show immunogold dots of Cx43 (red arrows) within male WT heart mitochondria, but neither male Cx43-ic-KO heart mitochondria nor heart sections in the absence of primary antibody incubation. b Transmission electron microscopy micrographs of subsarcolemmal mitochondria (SSM) in male WT and male Cx43-ic-KO mouse hearts without or with acute I/R. c Quantification of SSM number and size. d Transmission electron microscopy micrographs of interfibrillar mitochondria (IFM) in male WT and male Cx43-ic-KO mouse hearts without or with acute I/R. e Quantification of IFM number and size. A total of > 10 fields per condition were analyzed using the Image J software (NIH). Mean ± SEM, unpaired t test
Of note, there were no differences in terms of mitochondrial number and size between WT and Cx43-ic-KO mouse hearts without I/R (Fig. 8b–e). However, more mitochondrial damage (reduced number and enlarged mitochondria) for SSM was noticed in Cx43-ic-KO mouse hearts than in WT hearts following I/R (Fig. 8b, c), but not for IFM (Fig. 8d, e). Acute I/R caused more impairment in SSM compared to IFM in Cx43-ic-KO mouse heart (Fig. S6). Additionally, we barely detected recovery of LV function (LVDP) following I/R in Cx43-ic-KO hearts compared to their WT counterparts (Fig. 9a). Considering that not all Cx43-ic-KO hearts were able to catch up the paced heart rate, we also evaluated rate pressure product (RPP = LVDP*BPM [beats per minute]) and observed similar results (Fig. 9b) as LVDP did. Importantly, Cx43-ic-KO abolished the protective effect of post-ischemic E2 treatment on myocardial function following I/R (Fig. 9c, d), whereas improved LV functional recovery was observed in WT hearts treated with post-ischemic E2 (Fig. 9e), suggesting the critical role of Cx43 in mediating estrogen-initiated acute cardiac protection.
Fig. 9.

Ablation of cardiac Cx43 abolishes E2-induced protection for myocardial function following acute I/R. a, b LVDP and RPP in male WT and Cx43-ic-KO mouse hearts following I/R. c, d LVDP and RPP in male Cx43-ic-KO mouse hearts without or with post-ischemic E2 treatment. e LVDP in male WT mouse hearts ± E2 treatment starting at the onset of reperfusion. Mean ± SEM, n = 4–5/group, unpaired t test, *p < 0.05, **p < 0.01, ****p < 0.0001
Discussion
In this study, we employed a new strategy using post-ischemic administration of estrogen instead of preventive usage as a potential therapeutic option for treating ischemic heart disease. Here, our data clearly indicated that E2 protected mitochondrial performance in mouse primary cardiomyocytes against simulated I/R when given at the onset of reperfusion/re-oxygenation. More importantly, post-ischemic treatment of E2 significantly improved LV functional recovery and reduced infarct size in both male and endogenous estrogen-depleted female mouse hearts following acute myocardial I/R. This estrogen-derived acute cardiac protection was related to Cx43, particularly mitochondrial Cx43, at least in part.
It is well documented that there is the intrinsic resistance of isolated female hearts to post-ischemic contractile dysfunction [15, 21, 48, 81, 83, 85, 86]. Mitochondria, present in thousands of copies per cardiomyocyte, are critical to ensuring cardiomyocyte contractility and viability. Although information is limited, sex differences are observed in cardiomyocyte mitochondrial function. Female heart mitochondria are more resistant to calcium-induced mitochondrial permeability transition pore (mPTP) opening than males [56, 61]. In addition, there is less mitochondrial H2O2 generation and lower mitochondrial oxidative damage in female rat hearts compared to males [22, 42, 48]. In the present study, our finding that less mitochondrial ROS (superoxides) were generated in female cardiomyocytes compared to male ones following H2O2 exposure or simulated I/R is consistent with those previous studies [22, 42, 48]. However, we did not observe sex differences in mitochondrial membrane potential in response to H2O2 or simulated I/R, suggesting that this important index of mitochondrial hemostasis is comparable between male and female cardiomyocytes following cardiac stress.
Accumulating evidence has indicated that Cx43 protein is implicated in multiple aspects of mitochondrial function including mitochondrial complex I-mediated oxygen consumption and ATP production [9], potassium handling [57], mPTP opening [71], and ROS formation [68]. Mitochondrial Cx43 exists in a hemichannel (HC)-like configuration, which is mostly closed under physiological status and opened during ischemia-like conditions [50, 60]. Ischemic preconditioning (IPC), the sub-lethal injury, triggers small amounts of mitochondrial Cx43 HCs opening, thus initiating downstream protective pathways following I/R [50]. Reducing mitochondrial Cx43 content abolishes IPC-mediated cardiac protection [35, 62, 66]. However, in the absence of IPC, mitochondrial Cx43 HCs seem to play a deleterious role in the heart following ischemia [28]. The excessive opening of mitochondrial Cx43 HCs facilitated calcium entering mitochondria, leading to mitochondrial calcium overload and cell death, whereas pharmacological inhibition of mitochondrial Cx43 HCs prevented cell death in cardiomyocytes subjected to simulated I/R [28]. Both inhibition of Cx43 HCs and genetic ablation of Cx43 strongly reduced infarct size in the heart following I/R [28]. Notably, dephosphorylation of Cx43 by ischemia contributes to Cx43 HC opening [50, 60]. IPC is also noticed to prevent ischemia-induced Cx43 dephosphorylation [50]. In this study, higher levels of non-phosphorylated mitochondrial Cx43 was observed in the heart after I/R, whereas more phosphorylated Cx43 proteins were present in the post-ischemic E2-treated mouse heart mitochondria. This E2-prevented dephosphorylation of mitochondrial Cx43 might lead to Cx43 HCs’ remaining closure in mitochondria during reperfusion, thus conferring cardiac protection. Our data regarding increased interaction of ERα and Cx43 in mitochondria by estrogen also supported this hypothesis, because E2 pretreatment-activated ERα was found to initiate the PKC pathway, thereby preventing metabolic inhibition-induced dephosphorylation of Cx43 in rat cardiomyocytes [19, 20].
Cx43 is primarily present in SSM [11, 72], but not in IFM. Our findings that post-ischemic E2 treatment augmented SSM Cx43 levels in OVX F hearts were in line with these studies. SSM are more vulnerable to I/R injury compared with IFM likely due to their being less resistant to I/R-induced mitochondrial permeability transition and their higher exposure to oxygen gradient than IFM [17]. In this study, our results (Fig. S6) that more damaged mitochondria occurred in SSM compared to IFM in Cx43-ic-KO hearts subjected to myocardial I/R confirmed this. However, we noticed more severe SSM injury in Cx43-ic-KO hearts compared to their WT counterparts after I/R. This finding is in contrast to that mitochondrial Cx43-formed HCs play a detrimental role during ischemia-like conditions [28]. Notably, the relative role of Cx43 HCs in ischemia depends on the duration and severity of I/R, varying from benefit at early injury to a later cell death accelerator in severely damaged heart. The opening of mitochondrial Cx43 HCs during ischemia and at early reperfusion may release excessive ions or toxic by-products from mitochondria and maintain mitochondrial health [50, 60]. In our study, we used a 25-min ischemia followed by 40-min reperfusion, the course and degree of which was much less severe than those of the previously reported one (30-min ischemia and 120-min reperfusion) [28]. This less severe I/R injury might not lead to excessive opening of mitochondrial Cx43 HCs and the adaptive opening/closure of mitochondrial Cx43 HCs could provide protective effect following I/R. Therefore, it was possible that ablation of Cx43 resulted in more severe damage in SSM. In addition, our data showed that post-ischemic E2 treatment augmented mitochondrial Cx43 content, associated with improved myocardial functional recovery and reduced infarct size, implying that mitochondrial Cx43 might not always play a deleterious role in the inured heart. In fact, pharmacological inhibition of mitochondrial Cx43 HCs significantly reduces SSM calcium retention capacity [71] and increases calcium-induced mPTP opening [10], suggesting a protective role of Cx43 HCs in SSM [50]. Furthermore, Cx43 has many other non-channel functions attributable to Cx43 c-terminus interacting with signaling and scaffolding proteins. Mitochondrial Cx43 has been shown to interact with the apoptosis-inducing factor, which plays a role in cell death when released into cytoplasm [25]. Collectively, it deserves further investigation regarding the role of mitochondrial Cx43 in myocardial responses to acute injury.
Cx43-formed gap junctions between cells and HCs at plasma membrane are also involved in modulating myocardial responses to I/R. On the one hand, reduced Cx43 protein or increased dephosphorylation of Cx43 leads to electrical cell uncoupling, thus contributing to ischemia-induced arrhythmogenesis [4, 49]. The approach to preventing ischemia-induced Cx43 gap junction uncoupling is cardioprotective. On the other hand, blockade of gap junction uncoupling/closure has been reported to increase hypercontracture spreading and cell death [29]. Pharmacological inhibition of Cx43 HCs by Gap19 at plasma membrane, particularly sarcolemma, improves cell survival and decreases the infarct size following I/R [88]. In this study, post-ischemic E2 treatment increased cardiac Cx43 phosphorylation at Ser368 (Fig. 5g), suggesting that E2 might confer protection partially via preserving Cx43 gap junction communication or blocking Cx43 HCs at plasma membrane in the heart against I/R. However, the detailed mechanism underlying how E2 acts on Cx43 gap junctions and sarcolemmal Cx43 HCs, as well as mitochondrial Cx43 HCs, requires further investigation, particularly considering that the cardiac protective effect provided by ZP1609 (Gap134 or danegaptide) could be due to its modulation of Cx43 gap junctions or sarcolemmal Cx43, but independent of mitochondrial Cx43 [6].
Smaller isoforms of Cx43 C-terminus have been shown to play an important role in regulating Cx43 function [27, 67]. Overexpression of Gja1–20k is sufficient to rescue mitochondrial localization/organization during oxidative stress [27]. Exogenous Gja1–20k delivery improves mitochondrial biogenesis and metabolic quiescence, thus protecting myocardium from ischemic and I/R injury [2]. In this study, augmented mitochondrial Gja1–20k was noticed in post-ischemic E2-treated mouse hearts after I/R. Therefore, it is possible that the increased mitochondrial Gja1–20k could mediate estrogen-induced mitochondrial protection against myocardial I/R as well.
Interestingly, we found higher levels of overall and mitochondrial Cx43 content in E2-treated mouse hearts (40-min treatment) compared to their untreated counterparts following I/R. Although estrogen could increase Cx43 expression via directly binding to its promoter region [94], our data (Fig. S3) did not support estrogen-regulated Cx43 expression at a transcriptional level in the current study. Notably, there is a rapid turnover of Cx43 with its half-life as short as 1–2 h in the heart [3]. Estrogen/estrogen receptor has been implicated in reducing Cx43 ubiquitination in breast cancers [78]. In this regard, increased Cx43 levels might be due to estrogen-decreased degradation of myocardial Cx43 during I/R.
There is an inconsistent finding that female hearts are not protected against I/R injury [52]. In this paper, Lieder et al. showed similar myocardial functional recovery and infarct size in female Lewis rat hearts and male counterparts after acute I/R. It is noted that different rat strains (Sprague–Dawley [48, 81, 86] and Wister [21] vs. Lewis) were employed in those studies which demonstrated cardiac protection in female hearts compared to male hearts in response to I/R. Given that genetic background is one of major factors impacting on myocardial infarct size [32], inconsistent results from Lieder et al. might be attributable to a different rat strain/genetic background used in their study. Also, female animals in different stages of estrus cycle could be another potential reason for the inconsistent finding. In the present study, a difference in LVDP and dP/dt under baseline condition was observed between male and OVX female hearts with reduced myocardial function in OVX female. Decreased cardiac function has been reported in the OVX mice from other groups as well [89, 91]. One explanation for this is likely due to depletion of endogenous estrogen by OVX affecting calcium transients, excitation–contraction coupling gain, and sarcoplasmic reticulum content in cardiomyocytes [58].
In conclusion, we have shown that post-ischemic E2 treatment protects mitochondrial performance in cardiomyocytes against simulated I/R in culture, improves myocardial functional recovery and reduces the infarct size following acute I/R. The protective effect of E2 likely occurs at multiple aspects involving Cx43: inhibition of mitochondrial Cx43 HCs, modulation of mitochondrial Cx43 content, augmentation of mitochondrial Gja1–20k, preventing gap junction uncoupling or blockade of Cx43 HCs at the plasma membrane.
Supplementary Material
Acknowledgements
This study was supported by the National Institutes of Health (NIH) R56 HL139967 (M.W.) and Showalter Trust Fund (M.W.). The authors thank Dr. Weinian Shou at IU School of Medicine for providing us doxycycline-inducible Tnnt2-Cre mice and for expert assistance in breeding Cx43-ic-KO mouse line. The authors thank Dr. Teresa A. Zimmers at IU School of Medicine for allowing access to the equipment for taking fluorescent images. The authors thank Dr. Subhadip Ghatak for contributions in editing the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Conflict of interest None.
References
- 1.Ba ZF, Hsu JT, Chen J, Kan WH, Schwacha MG, Chaudry IH (2008) Systematic analysis of the salutary effect of estrogen on cardiac performance after trauma-hemorrhage. Shock 30:585–589. 10.1097/SHK.0b013e31816f1a45 [DOI] [PubMed] [Google Scholar]
- 2.Basheer WA, Fu Y, Shimura D, Xiao S, Agvanian S, Hernandez DM, Hitzeman TC, Hong T, Shaw RM (2018) Stress response protein GJA1–20 k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight. 10.1172/jci.insight.121900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beardslee MA, Laing JG, Beyer EC, Saffitz JE (1998) Rapid turnover of connexin43 in the adult rat heart. Circ Res 83:629–635. 10.1161/01.res.83.6.629 [DOI] [PubMed] [Google Scholar]
- 4.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE (2000) Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87:656–662. 10.1161/01.res.87.8.656 [DOI] [PubMed] [Google Scholar]
- 5.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C, Stroke Statistics S (2017) Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 10.1161/CIR.0000000000000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boengler K, Bulic M, Schreckenberg R, Schluter KD, Schulz R (2017) The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. Br J Pharmacol 174:2060–2073. 10.1111/bph.13804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R (2005) Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res 67:234–244. 10.1016/j.cardiores.2005.04.014 [DOI] [PubMed] [Google Scholar]
- 8.Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R (2007) Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol 292:H1764–H1769. 10.1152/ajpheart.01071.2006 [DOI] [PubMed] [Google Scholar]
- 9.Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, Cabestrero A, Fernandez-Sanz C, Semenzato M, Di Lisa F, Rohrbach S, Garcia-Dorado D, Heusch G, Schulz R (2012) Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 16:1649–1655. 10.1111/j.1582-4934.2011.01516.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boengler K, Schulz R (2017) Connexin 43 and mitochondria in cardiovascular health and disease. Adv Exp Med Biol 982:227–246. 10.1007/978-3-319-55330-6_12 [DOI] [PubMed] [Google Scholar]
- 11.Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. 10.1007/s00395-009-0007-5 [DOI] [PubMed] [Google Scholar]
- 12.Booth EA, Obeid NR, Lucchesi BR (2005) Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 289:H2039–H2047. 10.1152/ajpheart.00479.2005 [DOI] [PubMed] [Google Scholar]
- 13.Botker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femmino S, Garcia-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhauser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schluter KD, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G (2018) Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol 113:39 10.1007/s00395-018-0696-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canali E, Masci P, Bogaert J, Bucciarelli Ducci C, Francone M, McAlindon E, Carbone I, Lombardi M, Desmet W, Janssens S, Agati L (2012) Impact of gender differences on myocardial salvage and post-ischaemic left ventricular remodelling after primary coronary angioplasty: new insights from cardiovascular magnetic resonance. Eur Heart J Cardiovasc Imaging 13:948–953. 10.1093/ehjci/jes087 [DOI] [PubMed] [Google Scholar]
- 15.Casin KM, Fallica J, Mackowski N, Veenema RJ, Chan A, St Paul A, Zhu G, Bedja D, Biswal S, Kohr MJ (2018) S-Nitrosoglutathione reductase is essential for protecting the female heart from ischemia-reperfusion injury. Circ Res 123:1232–1243. 10.1161/CIRCRESAHA.118.313956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CC, Lin CC, Lee TM (2010) 17beta-Estradiol decreases vulnerability to ventricular arrhythmias by preserving connexin43 protein in infarcted rats. Eur J Pharmacol 629:73–81. 10.1016/j.ejphar.2009.11.050 [DOI] [PubMed] [Google Scholar]
- 17.Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ (2012) Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J Cardiovasc Pharmacol 59:101–108. 10.1097/FJC.0b013e31823827cc [DOI] [PubMed] [Google Scholar]
- 18.Chen YR, Zweier JL (2014) Cardiac mitochondria and reactive oxygen species generation. Circ Res 114:524–537. 10.1161/CIRCRESAHA.114.300559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung TH, Wang SM, Liang JY, Yang SH, Wu JC (2009) The interaction of estrogen receptor alpha and caveolin-3 regulates connexin43 phosphorylation in metabolic inhibition-treated rat cardiomyocytes. Int J Biochem Cell Biol 41:2323–2333. 10.1016/j.biocel.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 20.Chung TH, Wang SM, Wu JC (2004) 17beta-estradiol reduces the effect of metabolic inhibition on gap junction intercellular communication in rat cardiomyocytes via the estrogen receptor. J Mol Cell Cardiol 37:1013–1022. 10.1016/j.yjmcc.2004.08.003 [DOI] [PubMed] [Google Scholar]
- 21.Ciocci Pardo A, Scuri S, Gonzalez Arbelaez LF, Caldiz C, Fantinelli J, Mosca SM (2018) Survival kinase-dependent pathways contribute to gender difference in the response to myocardial ischemia-reperfusion and ischemic post-conditioning. Cardiovasc Pathol 33:19–26. 10.1016/j.carpath.2017.12.003 [DOI] [PubMed] [Google Scholar]
- 22.Colom B, Oliver J, Roca P, Garcia-Palmer FJ (2007) Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc Res 74:456–465. 10.1016/j.cardiores.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 23.De Luca G, Parodi G, Sciagra R, Bellandi B, Verdoia M, Vergara R, Migliorini A, Valenti R, Antoniucci D (2013) Relation of gender to infarct size in patients with ST-segment elevation myocardial infarction undergoing primary angioplasty. Am J Cardiol 111:936–940. 10.1016/j.amjcard.2012.12.011 [DOI] [PubMed] [Google Scholar]
- 24.Dehghan A, Leening MJ, Solouki AM, Boersma E, Deckers JW, van Herpen G, Heeringa J, Hofman A, Kors JA, Franco OH, Ikram MA, Witteman JC (2014) Comparison of prognosis in unrecognized versus recognized myocardial infarction in men versus women > 55 years of age (from the Rotterdam Study). Am J Cardiol 113:1–6. 10.1016/j.amjcard.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 25.Denuc A, Nunez E, Calvo E, Loureiro M, Miro-Casas E, Guaras A, Vazquez J, Garcia-Dorado D (2016) New protein-protein interactions of mitochondrial connexin 43 in mouse heart. J Cell Mol Med 20:794–803. 10.1111/jcmm.12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckardt D, Kirchhoff S, Kim JS, Degen J, Theis M, Ott T, Wiesmann F, Doevendans PA, Lamers WH, de Bakker JM, van Rijen HV, Schneider MD, Willecke K (2006) Cardiomyocyte-restricted deletion of connexin43 during mouse development. J Mol Cell Cardiol 41:963–971. 10.1016/j.yjmcc.2006.07.017 [DOI] [PubMed] [Google Scholar]
- 27.Fu Y, Zhang SS, Xiao S, Basheer WA, Baum R, Epifantseva I, Hong T, Shaw RM (2017) Cx43 Isoform GJA1–20 k promotes microtubule dependent mitochondrial transport. Front Physiol 8:905 10.3389/fphys.2017.00905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadicherla AK, Wang N, Bulic M, Agullo-Pascual E, Lissoni A, De Smet M, Delmar M, Bultynck G, Krysko DV, Camara A, Schluter KD, Schulz R, Kwok WM, Leybaert L (2017) Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res Cardiol 112:27 10.1007/s00395-017-0618-1 [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M (2004) Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res 61:386–401. 10.1016/j.cardiores.2003.11.039 [DOI] [PubMed] [Google Scholar]
- 30.Gorbe A, Varga ZV, Kupai K, Bencsik P, Kocsis GF, Csont T, Boengler K, Schulz R, Ferdinandy P (2011) Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: the role of connexin 43. Am J Physiol Heart Circ Physiol 300:H1907–H1913. 10.1152/ajpheart.01242.2010 [DOI] [PubMed] [Google Scholar]
- 31.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE (1997) Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Investig 99:1991–1998. 10.1172/JCI119367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y, Flaherty MP, Wu WJ, Tan W, Zhu X, Li Q, Bolli R (2012) Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Res Cardiol 107:288 10.1007/s00395-012-0288-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI (2001) Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res 88:333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G (2005) Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 97:583–586. 10.1161/01.RES.0000181171.65293.65 [DOI] [PubMed] [Google Scholar]
- 35.Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y, Ishikawa Y, Insel PA, Roth DM (2008) Caveolin-3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol 44:123–130. 10.1016/j.yjmcc.2007.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Bland KI, Chaudry IH (2009) Mechanism of salutary effects of estrogen on cardiac function following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. Crit Care Med 37:2338–2344. 10.1097/CCM.0b013e3181a030ce [DOI] [PubMed] [Google Scholar]
- 37.Huang C, Gu H, Yu Q, Manukyan MC, Poynter JA, Wang M (2011) Sca-1 + cardiac stem cells mediate acute cardioprotection via paracrine factor SDF-1 following myocardial ischemia/reperfusion. PLoS One 6:e29246 10.1371/journal.pone.0029246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutchens MP, Nakano T, Kosaka Y, Dunlap J, Zhang W, Herson PS, Murphy SJ, Anderson S, Hurn PD (2010) Estrogen is renoprotective via a nonreceptor-dependent mechanism after cardiac arrest in vivo. Anesthesiology 112:395–405. 10.1097/ALN.0b013e3181c98da9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeanes HL, Tabor C, Black D, Ederveen A, Gray GA (2008) Oestrogen-mediated cardioprotection following ischaemia and reperfusion is mimicked by an oestrogen receptor (ER)alpha agonist and unaffected by an ER beta antagonist. J Endocrinol 197:493–501. 10.1677/JOE-08-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabaeva Z, Zhao M, Michele DE (2008) Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol 294:H1667–H1674. 10.1152/ajpheart.01144.2007 [DOI] [PubMed] [Google Scholar]
- 41.Kay J, Dorbala S, Goyal A, Fazel R, Di Carli MF, Einstein AJ, Beanlands RS, Merhige ME, Williams BA, Veledar E, Chow BJ, Min JK, Berman DS, Shah S, Bellam N, Butler J, Shaw LJ (2013) Influence of sex on risk stratification with stress myocardial perfusion Rb-82 positron emission tomography: results from the PET (positron emission tomography) prognosis multicenter registry. J Am Coll Cardiol 62:1866–1876. 10.1016/j.jacc.2013.06.017 [DOI] [PubMed] [Google Scholar]
- 42.Khalifa AR, Abdel-Rahman EA, Mahmoud AM, Ali MH, Noureldin M, Saber SH, Mohsen M, Ali SS (2017) Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol Rep. 10.14814/phy2.13125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knezl V, Bacova B, Kolenova L, Mitasikova M, Weismann P, Drimal J, Tribulova N (2008) Distinct lethal arrhythmias susceptibility is associated with sex-related difference in myocardial connexin-43 expression. Neuro Endocrinol Lett 29:798–801 [PubMed] [Google Scholar]
- 44.Knoferl MW, Angele MK, Diodato MD, Schwacha MG, Ayala A, Cioffi WG, Bland KI, Chaudry IH (2002) Female sex hormones regulate macrophage function after trauma-hemorrhage and prevent increased death rate from subsequent sepsis. Ann Surg 235:105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohro S, Hogan QH, Nakae Y, Yamakage M, Bosnjak ZJ (2003) Repeated or prolonged isoflurane exposure reduces mitochondrial oxidizing effects. Anesthesiology 98:275–278 [DOI] [PubMed] [Google Scholar]
- 46.Kozlov AV, Duvigneau JC, Hyatt TC, Raju R, Behling T, Hartl RT, Staniek K, Miller I, Gregor W, Redl H, Chaudry IH (2010) Effect of estrogen on mitochondrial function and intracellular stress markers in rat liver and kidney following trauma-hemorrhagic shock and prolonged hypotension. Mol Med 16:254–261. 10.2119/molmed.2009.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuebler JF, Jarrar D, Toth B, Bland KI, Rue L 3rd, Wang P, Chaudry IH (2002) Estradiol administration improves splanchnic perfusion following trauma-hemorrhage and sepsis. Arch Surg 137:74–79 [DOI] [PubMed] [Google Scholar]
- 48.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E (2010) Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res 106:1681–1691. 10.1161/CIRCRESAHA.109.213645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE (2000) Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 101:547–552. 10.1161/01.cir.101.5.547 [DOI] [PubMed] [Google Scholar]
- 50.Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, Laird DW, Naus CC, Green CR, Schulz R (2017) Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol Rev 69:396–478. 10.1124/pr.115.012062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Heinzel FR, Boengler K, Schulz R, Heusch G (2004) Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol 36:161–163. 10.1016/j.yjmcc.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 52.Lieder HR, Irmert A, Kamler M, Heusch G, Kleinbongard P (2019) Sex is no determinant of cardioprotection by ischemic preconditioning in rats, but ischemic/reperfused tissue mass is for remote ischemic preconditioning. Physiol Rep 7:e14146 10.14814/phy2.14146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mehilli J, Kastrati A, Dirschinger J, Pache J, Seyfarth M, Blasini R, Hall D, Neumann FJ, Schomig A (2002) Sex-based analysis of outcome in patients with acute myocardial infarction treated predominantly with percutaneous coronary intervention. JAMA 287:210–215 [DOI] [PubMed] [Google Scholar]
- 54.Mehilli J, Ndrepepa G, Kastrati A, Nekolla SG, Markwardt C, Bollwein H, Pache J, Martinoff S, Dirschinger J, Schwaiger M, Schomig A (2005) Gender and myocardial salvage after reperfusion treatment in acute myocardial infarction. J Am Coll Cardiol 45:828–831. 10.1016/j.jacc.2004.11.054 [DOI] [PubMed] [Google Scholar]
- 55.Meller SM, Lansky AJ, Costa RA, Soffler M, Costantini CO, Brodie BR, Cox DA, Stuckey TD, Fahy M, Grines CL, Stone GW (2013) Implications of myocardial reperfusion on survival in women versus men with acute myocardial infarction undergoing primary coronary intervention. Am J Cardiol 112:1087–1092. 10.1016/j.amjcard.2013.05.052 [DOI] [PubMed] [Google Scholar]
- 56.Milerova M, Drahota Z, Chytilova A, Tauchmannova K, Houstek J, Ostadal B (2016) Sex difference in the sensitivity of cardiac mitochondrial permeability transition pore to calcium load. Mol Cell Biochem 412:147–154. 10.1007/s11010-015-2619-4 [DOI] [PubMed] [Google Scholar]
- 57.Miro-Casas E, Ruiz-Meana M, Agullo E, Stahlhofen S, Rodriguez-Sinovas A, Cabestrero A, Jorge I, Torre I, Vazquez J, Boengler K, Schulz R, Heusch G, Garcia-Dorado D (2009) Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res 83:747–756. 10.1093/cvr/cvp157 [DOI] [PubMed] [Google Scholar]
- 58.Parks RJ, Bogachev O, Mackasey M, Ray G, Rose RA, Howlett SE (2017) The impact of ovariectomy on cardiac excitation-contraction coupling is mediated through cAMP/PKA-dependent mechanisms. J Mol Cell Cardiol 111:51–60. 10.1016/j.yjmcc.2017.07.118 [DOI] [PubMed] [Google Scholar]
- 59.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J (1995) Cardiac malformation in neonatal mice lacking connexin43. Science 267:1831–1834 [DOI] [PubMed] [Google Scholar]
- 60.Retamal MA, Schalper KA, Shoji KF, Orellana JA, Bennett MV, Saez JC (2007) Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia-reperfusion. J Membr Biol 218:49–63. 10.1007/s00232-007-9043-y [DOI] [PubMed] [Google Scholar]
- 61.Ribeiro RF Jr, Ronconi KS, Morra EA, Do Val Lima PR, Porto ML, Vassallo DV, Figueiredo SG, Stefanon I (2016) Sex differences in the regulation of spatially distinct cardiac mitochondrial subpopulations. Mol Cell Biochem 419:41–51. 10.1007/s11010-016-2748-4 [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, Konietzka I, Miro E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D (2006) Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res 99:93–101. 10.1161/01.RES.0000230315.56904.de [DOI] [PubMed] [Google Scholar]
- 63.Ropero AB, Eghbali M, Minosyan TY, Tang G, Toro L, Stefani E (2006) Heart estrogen receptor alpha: distinct membrane and nuclear distribution patterns and regulation by estrogen. J Mol Cell Cardiol 41:496–510. 10.1016/j.yjmcc.2006.05.022 [DOI] [PubMed] [Google Scholar]
- 64.Roy S, Khanna S, Nallu K, Hunt TK, Sen CK (2006) Dermal wound healing is subject to redox control. Mol Ther 13:211–220. 10.1016/j.ymthe.2005.07.684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruiz-Meana M, Nunez E, Miro-Casas E, Martinez-Acedo P, Barba I, Rodriguez-Sinovas A, Inserte J, Fernandez-Sanz C, Hernando V, Vazquez J, Garcia-Dorado D (2014) Ischemic preconditioning protects cardiomyocyte mitochondria through mechanisms independent of cytosol. J Mol Cell Cardiol 68:79–88. 10.1016/j.yjmcc.2014.01.001 [DOI] [PubMed] [Google Scholar]
- 66.See Hoe LE, Schilling JM, Tarbit E, Kiessling CJ, Busija AR, Niesman IR, Du Toit E, Ashton KJ, Roth DM, Headrick JP, Patel HH, Peart JN (2014) Sarcolemmal cholesterol and caveolin-3 dependence of cardiac function, ischemic tolerance, and opioidergic cardioprotection. Am J Physiol Heart Circ Physiol 307:H895–H903. 10.1152/ajpheart.00081.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smyth JW, Shaw RM (2013) Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep 5:611–618. 10.1016/j.celrep.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soetkamp D, Nguyen TT, Menazza S, Hirschhauser C, Hendgen-Cotta UB, Rassaf T, Schluter KD, Boengler K, Murphy E, Schulz R (2014) S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res Cardiol 109:433 10.1007/s00395-014-0433-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solan JL, Lampe PD (2007) Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol 217:35–41. 10.1007/s00232-007-9035-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Srisakuldee W, Jeyaraman MM, Nickel BE, Tanguy S, Jiang ZS, Kardami E (2009) Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res 83:672–681. 10.1093/cvr/cvp142 [DOI] [PubMed] [Google Scholar]
- 71.Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KB, Kardami E (2014) The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res 103:72–80. 10.1093/cvr/cvu066 [DOI] [PubMed] [Google Scholar]
- 72.Sun J, Nguyen T, Aponte AM, Menazza S, Kohr MJ, Roth DM, Patel HH, Murphy E, Steenbergen C (2015) Ischaemic preconditioning preferentially increases protein S-nitrosylation in subsarcolemmal mitochondria. Cardiovasc Res 106:227–236. 10.1093/cvr/cvv044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Terrell AM, Crisostomo PR, Markel TA, Wang M, Abarbanell AM, Herrmann JL, Meldrum DR (2008) Postischemic infusion of 17-beta-estradiol protects myocardial function and viability. J Surg Res 146:218–224. 10.1016/j.jss.2007.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME, Saffitz JE (1998) Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction. Circulation 97:686–691 [DOI] [PubMed] [Google Scholar]
- 75.Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG (2003) Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res 92:1209–1216. 10.1161/01.RES.0000074916.41221.EA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trentzsch H, Stewart D, De Maio A (2003) Genetic background conditions the effect of sex steroids on the inflammatory response during endotoxic shock. Crit Care Med 31:232–236. 10.1097/01.CCM.0000045185.37551.19 [DOI] [PubMed] [Google Scholar]
- 77.Tribulova N, Dupont E, Soukup T, Okruhlicova L, Severs NJ (2005) Sex differences in connexin-43 expression in left ventricles of aging rats. Physiol Res 54:705–708 [PubMed] [Google Scholar]
- 78.Tsai CF, Cheng YK, Lu DY, Wang SL, Chang CN, Chang PC, Yeh WL (2018) Inhibition of estrogen receptor reduces connexin 43 expression in breast cancers. Toxicol Appl Pharmacol 338:182–190. 10.1016/j.taap.2017.11.020 [DOI] [PubMed] [Google Scholar]
- 79.Vornehm ND, Wang M, Abarbanell A, Herrmann J, Weil B, Tan J, Wang Y, Kelly M, Meldrum DR (2009) Acute postischemic treatment with estrogen receptor-alpha agonist or estrogen receptor-beta agonist improves myocardial recovery. Surgery 146:145–154. 10.1016/j.surg.2009.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang L, Gu H, Turrentine M, Wang M (2014) Estradiol treatment promotes cardiac stem cell (CSC)-derived growth factors, thus improving CSC-mediated cardioprotection after acute ischemia/ reperfusion. Surgery 156:243–252. 10.1016/j.surg.2014.04.002 [DOI] [PubMed] [Google Scholar]
- 81.Wang M, Baker L, Tsai BM, Meldrum KK, Meldrum DR (2005) Sex differences in the myocardial inflammatory response to ischemia-reperfusion injury. Am J Physiol Endocrinol Metab 288:E321–E326. 10.1152/ajpendo.00278.2004 [DOI] [PubMed] [Google Scholar]
- 82.Wang M, Crisostomo P, Wairiuko GM, Meldrum DR (2006) Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol 290:H2204–H2209. 10.1152/ajpheart.01219.2005 [DOI] [PubMed] [Google Scholar]
- 83.Wang M, Crisostomo PR, Markel TA, Wang Y, Meldrum DR (2008) Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 118:S38–S45. 10.1161/CIRCULATIONAHA.107.756890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang M, Tan J, Coffey A, Fehrenbacher J, Weil BR, Meldrum DR (2009) Signal transducer and activator of transcription 3-stimulated hypoxia inducible factor-1alpha mediates estrogen receptor-alpha-induced mesenchymal stem cell vascular endothelial growth factor production. J Thorac Cardiovasc Surg 138:163–171. 10.1016/j.jtcvs.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang M, Tsai BM, Crisostomo PR, Meldrum DR (2006) Tumor necrosis factor receptor 1 signaling resistance in the female myocardium during ischemia. Circulation 114:I282–I289. 10.1161/CIRCULATIONAHA.105.001164 [DOI] [PubMed] [Google Scholar]
- 86.Wang M, Tsai BM, Reiger KM, Brown JW, Meldrum DR (2006) 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J Mol Cell Cardiol 40:205–212. 10.1016/j.yjmcc.2005.06.019 [DOI] [PubMed] [Google Scholar]
- 87.Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR (2009) Estrogen receptor beta mediates increased activation of PI3 K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol 296:R972–R978. 10.1152/ajpregu.00045.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang N, De Vuyst E, Ponsaerts R, Boengler K, Palacios-Prado N, Wauman J, Lai CP, De Bock M, Decrock E, Bol M, Vinken M, Rogiers V, Tavernier J, Evans WH, Naus CC, Bukauskas FF, Sipido KR, Heusch G, Schulz R, Bultynck G, Leybaert L (2013) Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res Cardiol 108:309 10.1007/s00395-012-0309-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, Lu L, Tan Y, Jiang L, Zhao M, Gao E, Yu S, Liu J (2019) GPR 30 reduces myocardial infarct area and fibrosis in female ovariectomized mice by activating the PI3 K/AKT pathway. Life Sci 226:22–32. 10.1016/j.lfs.2019.03.049 [DOI] [PubMed] [Google Scholar]
- 90.Wu B, Zhou B, Wang Y, Cheng HL, Hang CT, Pu WT, Chang CP, Zhou B (2010) Inducible cardiomyocyte-specific gene disruption directed by the rat Tnnt2 promoter in the mouse. Genesis 48:63–72. 10.1002/dvg.20573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yan Y, Miao D, Yang Z, Zhang D (2019) Loss of p27(kip1) suppresses the myocardial senescence caused by estrogen deficiency. J Cell Biochem 120:13994–14003. 10.1002/jcb.28674 [DOI] [PubMed] [Google Scholar]
- 92.Yao X, Wigginton JG, Maass DL, Ma L, Carlson D, Wolf SE, Minei JP, Zang QS (2014) Estrogen-provided cardiac protection following burn trauma is mediated through a reduction in mitochondria-derived DAMPs. Am J Physiol Heart Circ Physiol 306:H882–H894. 10.1152/ajpheart.00475.2013 [DOI] [PubMed] [Google Scholar]
- 93.Chung YW, Chen Y, Sun J, Tong G, Hockman SC, Faiyaz Ahmad SGE, Bae DH, Polidovitch N, Jian Wu, Rhee DK, Lee BS, Gucek M, Daniels MP, Brantner CA, Backxg PH, Murphy E, Manganiello VC (2015) Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci USA 112:E2253–E2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu W, Dahl G, Werner R (1994) The connexin43 gene is responsive to oestrogen. Proc Biol Sci 255:125–132. 10.1098/rspb.1994.0018 [DOI] [PubMed] [Google Scholar]
- 95.Zhang H, Gong G, Wang P, Zhang Z, Kolwicz SC, Rabinovitch PS, Tian R, Wang W (2018) Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J Mol Cell Cardiol 123:38–45. 10.1016/j.yjmcc.2018.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang W, Qu X, Chen B, Snyder M, Wang M, Li B, Tang Y, Chen H, Zhu W, Zhan L, Yin N, Li D, Xie L, Liu Y, Zhang JJ, Fu XY, Rubart M, Song LS, Huang XY, Shou W (2016) Critical Roles of STAT3 in beta-adrenergic functions in the heart. Circulation 133:48–61. 10.1161/CIRCULATIONAHA.115.017472 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.