

Abstract
Bax and Bak, two functionally similar, pro-apoptotic proteins of the Bcl-2 family, are known as the gateway to apoptosis because of their requisite roles as effectors of mitochondrial outer membrane permeabilization (MOMP), a major step during mitochondria-dependent apoptosis. The mechanism of how cells turn Bax/Bak from inert molecules into fully active and lethal effectors had long been the focal point of a major debate centered around two competing, but not mutually exclusive, models: direct activation and indirect activation. After intensive research efforts for over two decades, it is now widely accepted that to initiate apoptosis, some of the BH3-only proteins, a subclass of the Bcl-2 family, directly engage Bax/Bak to trigger their conformational transformation and activation. However, a series of recent discoveries, using previously unavailable CRISPR-engineered cell systems, challenge the basic premise that undergirds the consensus and provide evidence for a novel and surprisingly simple model of Bax/Bak activation: the membrane (lipids)-mediated spontaneous model. This review will discuss the evidence, rationale, significance, and implications of this new model.
Keywords: Apoptosis, Bcl-2 family, Bax, Bak, BH3-only proteins, Bid, Bim, Bad, Bcl-xL, Mcl-1, Activator, Sensitizer, Direct activation, Indirect activation, Membrane-mediated Spontaneous activation, Mitochondrial outer membrane, auto-activation, de novo activation, retro-translocation, BH3 mimetics
Introduction
Apoptosis is a highly regulated form of cell death playing important roles in embryonic development, tissue homeostasis, and disease progression 1. In response to various death stimuli, cells often initiate a mitochondria-dependent apoptotic pathway that involves mitochondrial outer membrane permeabilization (MOMP), formation of the apoptosome, and the activation of effector caspases 2. Generally considered the “point of no return”, MOMP irreversibly commits cells to apoptosis through the actions of Bax and Bak, two functionally similar effector proteins of the Bcl-2 family 3.
Since the identification of Bcl-2 in human follicular lymphoma 4, 5 and its discovery as the first anti-apoptotic oncogene 6 over three decades ago, 17–18 proteins that share at least one of the four Bcl-2 homology domains (BH1–4) have been identified in the mammalian system as Bcl-2 family proteins 7, with the last member, Bmf, identified in 2001 8. The Bcl-2 family proteins are classified into anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1), pro-apoptotic effector (Bax, Bak, and Bok), and pro-apoptotic BH3-only proteins (Bad, Bid, Bik, Bim, Bmf, Hrk, Noxa, and Puma) 9 ( Figure 1). Three types of interactions—1) BH3-only–anti-apoptotic, 2) anti-apoptotic–Bax/Bak, and 3) BH3-only–Bax/Bak—appear to govern the functional relationships among the Bcl-2 family members during apoptosis 10, 11.
Figure 1. Schematic of Bcl-2 family proteins.
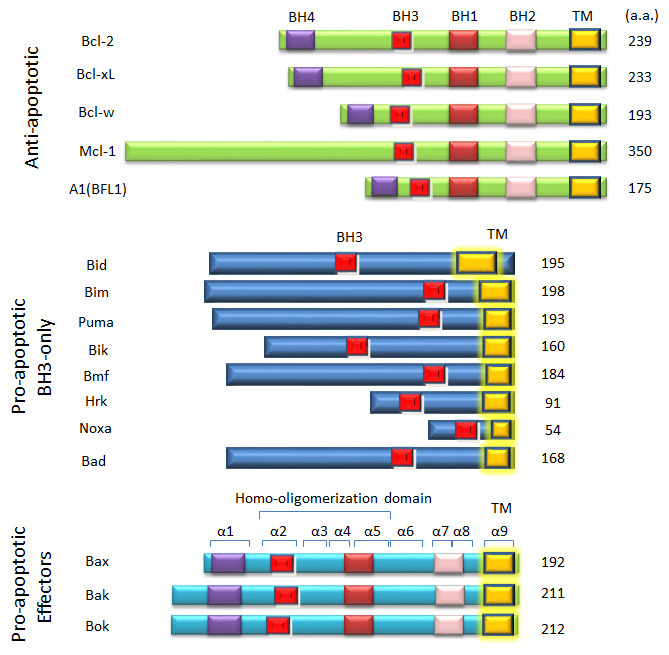
Bcl-2 family proteins (human) are classified into three groups: the pro-apoptotic BH3-only proteins, the pro-apoptotic effector proteins Bax, Bak, and Bok, and the anti-apoptotic Bcl-2 proteins. Each family member contains at least one of the four Bcl-2 homology (BH) domains, BH1–4. Bax has nine α-helices, with helix 9 serving as a mitochondrial-targeting sequence 13. Helices 2–5 constitute the homo-oligomerization domain of Bax 18, 42, 52. a.a., amino acids; TM, transmembrane.
As the requisite effectors of MOMP, both Bax and Bak are kept in check under normal conditions. Bax is primarily in the cytosol as a globular-shaped monomeric protein consisting of nine α-helices, with helix 9 fitting into a hydrophobic groove 12– 14. Bak has a similar structure 15 but is localized to the mitochondrial outer membrane (MOM) through its helix 9. Both Bax and Bak are activated during apoptosis 16. The active forms of Bax/Bak are known to homo-oligomerize in the MOM, forming proteinaceous/lipidic pores that are responsible for the release of cytochrome c and other apoptogenic factors 17, 18. The BH3-only and the anti-apoptotic Bcl-2 proteins regulate MOMP by promoting or suppressing the activation of Bax/Bak, respectively 9, 11, 19.
Bok has recently been shown to complement Bax/Bak in developmental cell death 20 and to function as a non-canonical effector of MOMP in response to proteasome inhibition 21. However, since the loss of both Bax and Bak is sufficient to block apoptosis induced by most apoptotic stimuli 22, the role of Bok in apoptosis remains unclear.
As both Bax and Bak undergo a dramatic transformation from benign proteins to lethal effectors during apoptosis, the mechanism of their activation has been the subject of intense debate and is considered to be the “holy grail” of apoptosis research 16, 23. Two competing but not mutually exclusive models, direct and indirect activation, had previously been proposed.
Direct activation
The first report of the direct interaction between a BH3-only protein and Bax/Bak was the identification of Bid as a BH3-containing death ligand that binds Bax. This finding led to the hypothesis that BH3-only proteins bind and activate Bax/Bak to initiate apoptosis 24. Subsequently, it was found that some BH3 domains, i.e. BidBH3 or BimBH3, have the ability to directly activate Bax/Bak and cause the release of cytochrome c from purified mitochondria. On the other hand, BH3 peptide from Bad (BadBH3) did not directly activate Bax/Bak but was able to bind to Bcl-2, displace BidBH3, and sensitize mitochondria to BidBH3-induced cytochrome c release 25. These findings formed the basis for the “direct activation” model, in which the BH3-only proteins are classified into two groups: the activators, which can directly activate Bax/Bak but can also be sequestered by the anti-apoptotic Bcl-2 proteins, and the sensitizers, which cannot directly activate Bax/Bak but are able to bind to the anti-apoptotic Bcl-2 proteins and sensitize cells to apoptosis. To induce apoptosis, the sensitizers bind the anti-apoptotic Bcl-2 proteins and displace the activators, which in turn directly bind to Bax/Bak, allosterically triggering their activation 25– 28 ( Figure 2A).
Figure 2. Comparison of the three models of BH3-only mediated Bax/Bak activation.
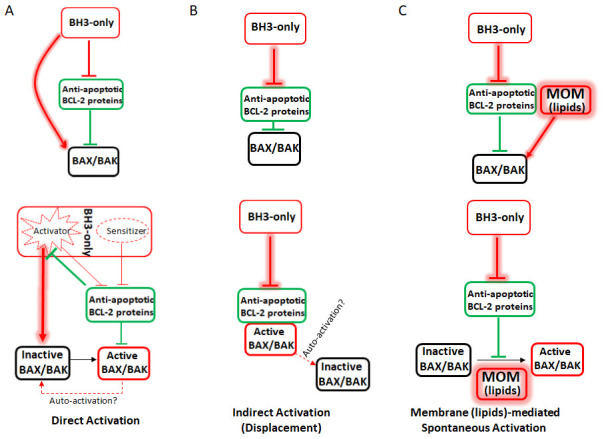
A) The direct activation model classifies BH3-only proteins as either activators (i.e. Bid, Bim, and Puma) or sensitizers (Bad). When not sequestered by anti-apoptotic Bcl-2 proteins, the activators directly bind to and activate Bax/Bak. Sensitizers cannot directly activate Bax/Bak but can bind to anti-apoptotic Bcl-2 proteins. To initiate apoptosis, the sensitizers bind to the anti-apoptotic Bcl-2 proteins, which release the activators from sequestration and allow them to directly activate Bax/Bak. B) The indirect activation model hypothesizes that Bax/Bak are constitutively active but are sequestered by the anti-apoptotic Bcl-2 proteins. During apoptosis, the BH3-only proteins bind to the anti-apoptotic Bcl-2 proteins and displace active Bax/Bak, which then damage the mitochondria. It is also hypothesized that active Bax can recruit inactive cytosolic Bax to the mitochondria through auto-activation. C) The membrane (lipids)-mediated spontaneous activation model hypothesizes that during apoptosis, the BH3-only proteins bind to and neutralize the anti-apoptotic Bcl-2 proteins. This allows the mitochondrial outer membrane (MOM) lipids to serve as the direct activator of Bax/Bak, as Bax freely migrates to the MOM through free diffusion. Helix 9 mediates membrane association for both Bax/Bak, and such association results in spontaneous activation of Bax/Bak in the lipid bilayer of the MOM. Similar to the indirect activation model, the BH3-only proteins’ only function is to inactivate the anti-apoptotic Bcl-2 proteins. However, in the membrane (lipids)-mediated spontaneous (MMS) model, both Bax and Bak are directly activated by the MOM. Figure 2c is adapted from Figure 5b of our previous paper in Cell Research 59.
Although the initial classification listed Bid and Bim as direct activators because of the ability of the BH3 domain or the full-length protein to release dyes of large molecular weight from liposomes in the presence of inactive Bax 25, 28– 32, the list of direct activators has grown over the years to include Puma, Noxa, Bik, Bmf, and Hrk, although a range of potency has been reported 26, 33– 39. On the other hand, Bad has been a classic sensitizer, as the BadBH3 peptide has been used as a negative control for direct activators in liposome assays 25, 28.
The interaction between a direct activator and Bax/Bak has been difficult to detect. This led to the hypothesis that activators only transiently engage Bax/Bak and activate them through a “hit and run” mechanism. The binding interface between an activator and Bax/Bak, while of great interest, has been controversial. NMR analysis provided evidence that the BH3 peptide from Bim transiently binds to a “trigger site” between helix 1 and helix 6 40, 41. However, other studies using Bax/Bak mutants that lack helix 9 found that a BidBH3 peptide binds the hydrophobic groove 42– 45. The discrepancy was largely resolved by new studies suggesting that the initial binding is at the trigger site and that the ensuing conformational changes allow a more stable interaction between the BH3 peptide and the hydrophobic groove 46, 47. However, the biological significance of binding at the trigger site has recently been challenged 48, 49.
Over the years, the direct activation model has evolved into several variants. The embedded together model emphasizes the importance of the membrane as a platform for all of the interactions among the Bcl-2 family proteins during Bax/Bak activation 50, 51, the unified model emphasizes that the anti-apoptotic Bcl-2 proteins not only sequester the activator BH3-only proteins but also suppress or sequester the activated Bax/Bak 30, and the interconnected model is highly analogous to the unified model but also contains a feed-forward mechanism of auto-activation 36. One unifying feature of these models is the direct attack and engagement of Bax/Bak by the activator BH3-only proteins as the initial triggering event leading to Bax/Bak activation.
Since the beginning of the last decade, direct activation has been widely accepted as the mechanism of Bax/Bak activation during apoptosis 10, 53, 54. However, as much of the supporting evidence has come from in vitro studies 25, 28, 31, 32, 41– 43, the physiological significance of this model has not been fully addressed.
Indirect activation
Indirect activation, or the “displacement” model, was first proposed in two important studies. First, Bak was shown to be normally sequestered by Bcl-xL and Mcl-1 but displaced by BH3-only proteins 55. Second, in direct opposition to the direct activation model, Willis et al. demonstrated that apoptosis can be induced by “sensitizer” BH3-only proteins or mutants of “activators” that are unable to bind Bax/Bak, even in cells that are deficient for the three activator BH3-only proteins Bid, Bim, and Puma 56. These results gave rise to the indirect activation model, which proposed that the effectors Bax/Bak are constitutively active but are normally sequestered by the anti-apoptotic Bcl-2 proteins. During apoptosis, BH3-only proteins bind to anti-apoptotic Bcl-2 proteins and indirectly activate Bax/Bak by displacing and releasing sequestered active Bax/Bak 57 ( Figure 2B, top).
Although this model is consistent with the activation of Bak, it has difficulty explaining the activation of Bax. It was therefore hypothesized that a small fraction of Bax is constitutively active but is sequestered by the anti-apoptotic Bcl-2 proteins on the mitochondria. The BH3-only proteins release the active Bax, which in turn recruits cytosolic Bax to the mitochondria and activates it through an auto-activation mechanism 58 ( Figure 2B, bottom). However, this hypothesis does not address the origin of the fraction of active Bax and the physiological relevance of auto-activation. Furthermore, as the indirect activation model was based on the key observation of Bax/Bak activation in Bid/Bim/Puma-deficient cells, the existence of additional activator BH3-only proteins made it more difficult to fully dissociate from the direct activation model 26, 33– 36. It is worth mentioning that in an elegant genetic knockin study in mice, Merino et al. compared the pro-apoptotic activities of wild-type Bim and its BH3-replacement mutants and presented data to suggest that neutralization of the anti-apoptotic Bcl-2 proteins is required but not sufficient for maximal apoptosis, suggesting the presence of both indirect and direct activation in vivo 60.
Functional redundancy and functional/physical connections: the Achilles’ heel of Bcl-2 research
A salient feature of the Bcl-2 family proteins is functional redundancy due to the presence of eight BH3-only, five anti-apoptotic, and two effector proteins. With multiple proteins sharing similar targets and functions in the same cell, it is difficult to pinpoint the real target, function, and mechanism of action of a particular protein 23, 61, 62.
As Bax and Bak are functionally redundant in many situations, our understanding of their functions was severely limited until a seminal study in which mouse embryonic fibroblasts (MEFs) doubly deficient for Bax and Bak were shown to be highly resistant to multiple apoptotic stimuli 22. This breakthrough ended years of frustration and established Bax and Bak as functionally equivalent and critical effectors of MOMP. However, the investigation into the functions and mechanisms of BH3-only proteins had been more challenging owing to the presence of eight BH3-only proteins, with seven of them potentially functioning as direct activators 33– 36. In efforts to differentiate direct and indirect activation, loss-of-function studies have been carried out to eliminate the direct activators by generating Bid/Bim double knockout (DKO), Bid/Bim/Puma triple knockout (TKO), Bid/Bim/Puma/p53 quadruple knockout (QKO), and Bid/Bim/Puma/Noxa QKO cells 36, 56, 63– 65. However, these cell models are insufficient to address this critical issue because of the presence of additional direct activators (i.e. Bik, Hrk, and Bmf) in those cells 33– 35.
The second challenge is the complex functional and physical connections among the Bcl-2 family proteins. The anti-apoptotic Bcl-2 proteins interact with at least some of the BH3-only proteins constitutively. Therefore, in cells deficient for certain BH3-only proteins—notably, the Bid/Bim/Puma TKO and Bid/Bim/Puma/Noxa QKO cells 36, 63—the loss of the BH3-only proteins should effectively increase the concentration of “unsequestered” or “active” Bcl-xL, Bcl-2, and Mcl-1, etc., whose total protein levels may not change. This increase would mean that these cells should have a greater capacity to sequester or suppress the active Bax/Bak and should at least in part explain the resistance of these TKOs and QKOs to apoptosis stimulation or the expression of the sensitizer protein Bad. Because of such complexity, vastly different conclusions have been drawn to either support or disprove the role of the activator proteins and the distinction between activators and sensitizers 26, 36, 59, 66.
“OctaKO” and “Bcl-2 allKO” cells: essential tools to reveal that direct activation is not required for BH3-only-mediated Bax/Bak activation and to establish the membrane (lipid)-mediated spontaneous model
In the past decade, the list of direct activator BH3-only proteins has grown to include Bid, Bim, Puma, Hrk, Noxa, Bik, and Bmf, which bind both Bax/Bak and the anti-apoptotic Bcl-2 proteins. Such a functional redundancy makes it near impossible to separate the functions of the direct activators and the sensitizers unless all of the direct activators are eliminated 61. The advent of CRISPR technology made it possible to remove this roadblock.
Through a sequential use of the CRISPR/Cas9 technology, HCT116 cells that lack all eight BH3-only proteins were generated and named OctaKO cells 66. This allowed for the interrogation of the function of individual BH3-only proteins without the participation of other BH3-only proteins. In this cell model, elimination of the two anti-apoptotic Bcl-2 proteins Bcl-xL and Mcl-1 by siRNA, pharmacological inhibition, or genetic elimination induced Bax/Bak activation and apoptosis efficiently. These results, for the first time, demonstrated that none of the direct activator BH3-only proteins is necessary for Bax/Bak activation once Bcl-xL/Mcl-1 are neutralized.
Similar strategies were used to generate the Bcl-2 allKO cells, which lost all eight pro-apoptotic BH3-only proteins, Bnip3 and Nix (non-apoptotic BH3-only proteins), the anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-xL, Mcl-1, A1, and Bcl-w), and Bax/Bak 66. These cells for the first time provided a clean slate to examine the functional relationship, through reconstitution, among Bcl-2 family proteins. Surprisingly, even minute amounts of Bax or Bak, re-introduced through a Dox-inducible expression system, were sufficient to kill these cells, and such killing was easily blocked by Bcl-xL. The highly efficient activation of Bax/Bak in Bcl-2 allKO cells is in stark contrast with the prediction from the direct activation model, which suggests that Bax should stay in the cytosol in the absence of an activator BH3-only protein. Importantly, mutants of Bax and Bak that lack their respective helix 9 (transmembrane [TM] domain) are incapable of homo-oligomerization and killing, and yet such activity was completely restored by the attachment of a heterologous TM domain, strongly suggesting that MOM association is sufficient for Bax/Bak activation 66.
These results formed the basis for a novel model of Bax/Bak activation, the membrane (lipid)-mediated spontaneous model (MMS), in which the BH3-only proteins activate Bax/Bak by inactivation or neutralization of the anti-apoptotic Bcl-2 proteins, instead of direct activation. This allows the Bax/Bak molecules to freely migrate to and diffuse in the MOM, homo-oligomerize, and form pores ( Figure 2C, Figure 3). In this model, the driving force of Bax/Bak activation is essentially free diffusion and the well-documented MOM association through helix 9 66. Consistent with this model, cytosolic Bax was reported to be constitutively targeted to the mitochondria in non-apoptotic cells 67, 68.
Figure 3. Membrane (lipids)-mediated spontaneous activation of Bax/Bak by the BH3-only proteins.
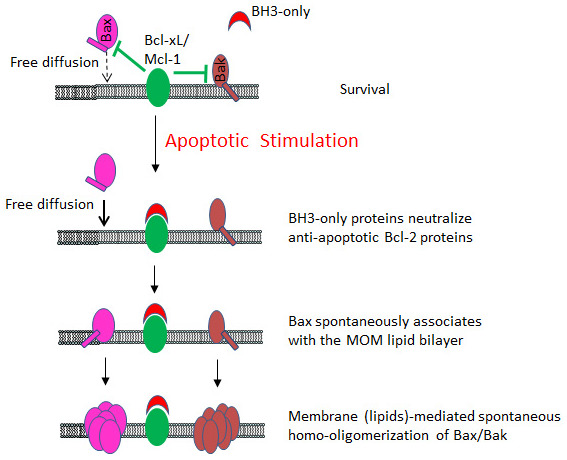
In this model, upon apoptotic stimulation, the BH3-only proteins neutralize/inactivate the anti-apoptotic Bcl-2 proteins Bcl-xL/Mcl-1. Such inactivation allows Bax to freely migrate to the mitochondrial outer membrane (MOM) through the interaction between helix 9 and the lipid bilayer. Anchored to the MOM through helix 9, both Bax and Bak spontaneously homo-oligomerize and form apoptotic pores within the lipid millieu. This figure is adapted from Figure 7 of our previous paper in Genes & Development 66.
What are the targets of BH3-only proteins: anti-apoptotic Bcl-2 proteins, Bax/Bak, or both?
Our understanding of the functions of the BH3-only proteins was significantly enhanced when Chen et al. defined the binding specificity of various BH3-only proteins toward different anti-apoptotic Bcl-2 proteins 69. Using peptides of the BH3 domains of the BH3-only proteins, Chen et al. found that while Bid, Puma, and Bim are promiscuous in binding all five anti-apoptotic Bcl-2 proteins, Bad only selectively binds to Bcl-w, Bcl-xL, and Bcl-2, and Noxa binds only Mcl-1 and A1. The combination of Bad and Noxa potently induced apoptosis 69. Using derivatives of the OctaKO cells that also lack Bcl-xL or Mcl-1, the Huang et al. study confirmed and refined these findings by demonstrating that while Bad and Noxa preferentially bind Bcl-xL and Mcl-1, respectively, tBid, Bim, Puma, Bmf, Bik, and Hrk are promiscuous binders toward both 59 ( Figure 4A).
Figure 4. Apoptotic signaling through membrane (lipids)-mediated spontaneous activation of Bax/Bak.
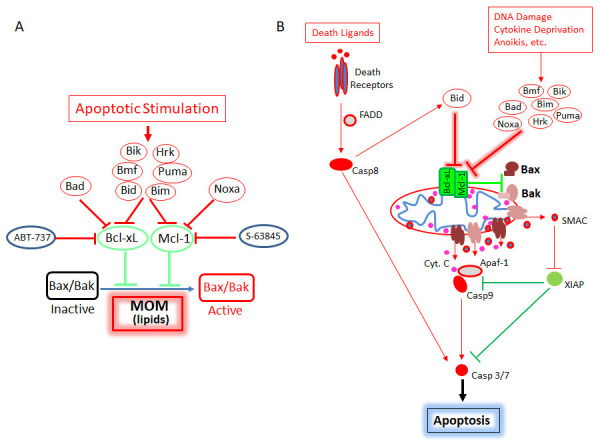
A) The intrinsic (mitochondrial-dependent) apoptotic pathway based on the membrane (lipids)-mediated spontaneous (MMS) Bax/Bak activation model. B) The intrinsic (mitochondrial-dependent) and extrinsic (death receptor-mediated) apoptotic pathways based on the MMS activation model. Apaf-1, apoptotic protease activating factor-1; Casp, caspase; Cyt c, cytochrome c; FADD, Fas associated death domain protein; MOM, mitochondrial outer membrane; SMAC, second mitochondria-derived activator of caspase; XIAP, X-linked inhibitor of apoptosis protein.
The critical role of Bcl-xL and Mcl-1 in cell survival was first demonstrated in HeLa cells in which siRNA knockdown of both Bcl-xL and Mcl-1 resulted in robust apoptosis 70, 71. This assay was subsequently carried out in multiple cell lines with deficiencies in several BH3-only proteins and ultimately the OctaKO cells. Without exception, the double siRNA knockdown induced robust apoptosis, strongly suggesting that Bcl-xL and Mcl-1 are the major targets of the BH3-only proteins during apoptosis 36, 64, 65, 70, 71. In support of Bcl-xL and Mcl-1’s critical role as gatekeepers against Bax/Bak activation, an elegant mouse genetic study found that while Mcl-1 Bcl-2 +/– and Bcl-xL +/–Bcl-2 +/– mice are almost normal, Mcl-1 +/–Bcl-xL +/– mice displayed craniofacial defects 72.
Strikingly, Huang et al. found that Bad, the sensitizer protein, induced apoptosis as efficiently as the activator proteins tBid and Bim in the OctaKO derivative cells 59. Additionally, mutant activator BH3-only proteins with their BH3 domain replaced by that of Bad showed no difference from the wild-type activators in the induction of apoptosis in the OctaKO cells. Furthermore, tBid and Bim (both activators) as well as Bad (sensitizer) were found to induce apoptosis without distinguishing Bax from Bak, consistent with the conclusions of a recent in vitro study 35 but in contrast to an earlier study 73. Lastly, tBid and Bim are unable to accelerate the spontaneous activation of Bax in the Bcl-2 allKO cells, suggesting that there is no functional/physical interaction between the activator BH3-only proteins and Bax/Bak after the neutralization of the anti-apoptotic Bcl-2 proteins 59. These results are incompatible with the direct activation model and argue strongly that there is no distinction between direct activators and sensitizers and that the only target of the BH3-only proteins is the anti-apoptotic Bcl-2 proteins. These findings unify the functions of BH3-only proteins and provide an elegantly simple model for apoptotic signaling ( Figure 4B).
Of note, loss-of-function studies in mice have clearly demonstrated differential requirements for various BH3-only proteins in vivo 74– 84. These observations are consistent with the notion that the BH3-only proteins’ roles are determined by their expression level, location, various regulations, and affinity with different anti-apoptotic Bcl-2 proteins, etc.
A bigger role for MOM in Bax and Bak activation
MOM has traditionally been viewed as a passive victim of activated Bax/Bak. However, some in vitro studies have suggested that MOM can serve as a platform for multiple interactions among the Bcl-2 family proteins, including tBid–Bax, tBid–Bcl-xL interactions, etc. 31, 85, 86. Results from the new series of studies, especially the finding that the TD (helix 9) of Bax/Bak was required and sufficient for Bax/Bak activation in the absence of all BH3-only and anti-apoptotic Bcl-2 proteins, suggested a much bigger role for the MOM in the regulation of Bax/Bak: the direct activator 59, 65, 66. In other words, the MOM itself functions as the instigator of Bax/Bak’s attack by directly recruiting and activating Bax/Bak. These results also suggested a much bigger role for helix 9: the trigger site. Importantly, the interaction between helix 9 and the MOM is spontaneous, stable, and easily detectable in the cell. On the other hand, the transient activator BH3-only–Bax/Bak interaction observed in vitro has been shown to be non-essential in cellular assays.
How does the MOM activate Bax? First, as Bax has been shown to exist in a dynamic equilibrium between the cytosol and mitochondria, and that Bcl-xL constantly retro-translocates Bax/Bak into the cytoplasm 67, 68, 87, it is essential that the anti-apoptotic Bcl-2 proteins are inactivated by the BH3-only proteins or other inhibitors before the MOM becomes a direct activator 59, 66. Second, a systematic mapping of the helices of Bax for their capacity to associate with the MOM indicated that helices 4, 5, 6, and 9 contain mitochondrial targeting signals (MTS) 88. It is not difficult to imagine that the MOM has the capacity to reconfigure the whole molecule through these multiple lipid–protein interactions to promote Bax/Bak homo-oligomerization upon the initial contact with helix 9. Third, as helix 9 of Bax was recently shown to form dimers in the MOM, such dimerization through helix 9 should provide further impetus for homo-oligomerization and activation of Bax/Bak molecules 52, 89– 91.
Thus, lipids are most likely the direct activator of Bax/Bak. Consistent with this suggestion, cardiolipin was found to be required for Bax activation 29, 92. Interestingly, it has been shown that non-ionic detergents, which somewhat mimic the lipid bilayer environment, are excellent activators of Bax/Bak 12, 93. It is therefore of great interest to test if certain lipid compositions can support Bax activation without the activator BH3-only proteins, even though the commonly used preparations prohibit such activity. A liposome composition that supports the constitutive activation of Bax in the absence of tBid should recapitulate what is observed in the cell.
MOM proteins may help recruit Bax to the MOM. VDAC2, despite earlier observations that it sequesters and suppresses Bak 94, 95, has recently been shown to be required for the mitochondrial translocation of Bax 96, 97. In addition, the mitochondrial fission regulator Drp1 has been shown to play a positive role in the regulation of Bax/Bak activation 98– 100. As both VDAC2 and Drp1 are present in the Bcl-2 allKO cells, it is possible that these proteins play a positive role in recruiting Bax/Bak along with the lipids to allow spontaneous localization 59, 66.
De novo activation versus auto-activation of Bax/Bak
Based on results from a liposome study with an extended BH3 peptide and Bax, it has been hypothesized that active Bax uses its BH3 domain to directly bind and activate an inactive Bax to accelerate Bax activation. This putative feed-forward mechanism is termed “auto-activation” 101. In support of this hypothesis, during apoptosis, Bax/Bak undergo conformational changes and expose the BH3 domain 17, 40. Auto-activation has recently been suggested to be a critical feed-forward mechanism for Bax/Bak activation in response to DNA damage and other death stimuli 36, 102. However, the origin of the first active Bax/Bak molecule and the physiological significance of auto-activation are unclear. Furthermore, there are no experimental data to support the hypothesis that active Bax can recruit cytosolic Bax to the mitochondria.
Both the direct activation and MMS model probe the mechanism of Bax/Bak’s de novo activation. Although there is potential involvement of auto-activation as a feed-forward mechanism to sustain Bax/Bak activation through homo-oligomerization, it is not the triggering mechanism that initiates the activation of Bax/Bak per se. For example, although the “interconnected” model contains an element of auto-activation, the origin of the first active Bax/Bak molecules was attributed to direct activation 36.
How do the BH3 mimetics work?
Thanks to a close collaboration between basic researchers and the pharmaceutical industry, numerous small molecules that mimic the structures of the BH3 domains of the BH3-only proteins have been developed to kill cancer cells and overcome resistance 103. For example, through NMR-based fragment screen and structure-guided design, ABT-737 was developed to mimic BadBH3, and it is highly selective against Bcl-xL, Bcl-2, and Bcl-w 104. Using similar strategies, Kotschy et al. designed S63845, a highly selective inhibitor of Mcl-1 105. Remarkably, venetoclax, a BH3 mimetic and the first small-molecule drug that disrupts a protein–protein interaction, was FDA approved for the treatment of a subtype of chronic lymphocytic lymphoma 103, 106– 109. However, the obvious question is this: which interaction does it disrupt in the cell? According to the direct activation model, the Bcl-2 mimetic disrupts two interactions: activator BH3-only–Bcl-2 and Bax/Bak–Bcl-2. The liberated activator BH3-only proteins then directly activate the liberated Bax or Bak 106. However, this scenario is difficult to prove experimentally and is not consistent with the observation that combinations of BH3 mimetics against Bcl-xL and Mcl-1 are sufficient to kill OctaKO HCT116 cells 59, 110. Such an observation is explained by the MMS model, in which the BH3 mimetics neutralize Bcl-xL/Mcl-1 and allow the MOM to directly activate Bax/Bak. It is of interest, however, that over-expression of Bcl-xL confers resistance to the apoptotic effects of Bcl-xL-targeting BH3 mimetics, suggesting limitations of these small-molecule inhibitors 111, 112.
How do the anti-apoptotic Bcl-2 suppress Bax/Bak activation?
It is well established that Bax exists mostly as an inactive and monomeric protein in cytosol 14. However, the spontaneous activation of Bax in the Bcl-2 allKO strongly suggests that Bax is constantly trying to land on the mitochondria through its C-terminal tail (α9) 59, 66, consistent with the observation of a natural tendency of Bax to move to the mitochondria 67, 68. It is also clear that Bcl-xL and Mcl-1 play a critical role in keeping Bax in the cytosol. How do Bcl-xL and Mcl-1 achieve this feat? Edlich et al. demonstrated that Bcl-xL has the ability to push the mitochondrially localized Bax molecules into the cytosol 67. Interestingly, Todt et al. demonstrated that Bak can also be retro-translocated by Bcl-xL, albeit at a much slower rate 87. However, the molecular mechanisms of such a retro-translocation activity remain unclear. Nevertheless, helix 9 of Bcl-xL may play a role in mediating this mysterious activity 113, through which the Bax molecule may be re-packed into a globular shape so that helix 9 can fit into the hydrophobic groove. Once this re-packing is finished, Bax is expected to leave the mitochondria and become cytosolic. The molecular details of such extraordinary activity will be a focus of future studies.
The membrane (lipids)-mediated spontaneous model versus indirect and direct activation models
The MMS model combines some important elements from both the direct and the indirect activation models yet for the first time introduced a new interaction as the trigger for Bax/Bak activation ( Figure 2 and Figure 3, Table 1). First, in agreement with indirect activation, the MMS model proposes that there is no hierarchy among the BH3-only proteins and the only target of the BH3-only proteins are the anti-apoptotic Bcl-2 proteins. Second, similar to the direct activation model, the MMS model proposes that a de novo direct activation, instead of de-repression, is the mechanism of Bax/Bak activation. However, the MOM, instead of the activator BH3-only proteins, is the direct activator. Third, the MMS model proposes that helix 9 is the trigger site for Bax/Bak activation. Instead of a transient, “hit-and-run” interaction as proposed in the direct activation model, the interaction between Bax/Bak and the MOM is visible and stable and consists of multiple interfaces once homo-oligomerization initiates.
Table 1. Comparison of the three models of BH3-only-mediated Bax/Bak activation.
| Model of Bax/Bak
Activation |
Targets of the
BH3-only proteins |
BH3-only–
Bax/Bak Interaction |
Activation
Mechanism |
Direct Activator | Trigger Site on
Bax/Bak |
Driving Force |
|---|---|---|---|---|---|---|
|
Indirect Activation
(Displacement) |
Anti-apoptotic Bcl-
2 proteins |
None | Displacement
and De- repression |
None | None | Stable BH3-
only–Bcl-xL/Mcl-1 interaction |
| Direct Activation | Bax/Bak and anti-
apoptotic Bcl-2 proteins |
Yes |
De novo and
direct activation |
Activator BH3-
only proteins |
Helices 1/6 or
the hydrophobic groove |
Transient BH3
(BH3-only)–groove (Bax/Bak) interaction |
|
Membrane
(lipids)-mediated Spontaneous Activation |
Anti-apoptotic Bcl-
2 proteins |
None |
De novo and
direct activation |
Mitochondrial
outer membrane (lipids) |
C-terminal
tail (helix 9) |
Stable BH3-
only–Bcl-xL/Mcl-1 interaction, free diffusion, and stable Bax/Bak α9-membrane interaction |
Some questions remain to be answered: what are the molecular details of MOM-mediated spontaneous Bax/Bak activation? What are the dynamics of the helix 9–hydrophobic groove interaction in the cytosol? Can in vitro liposome assays recapitulate the MMS model? What are the mechanisms of retro-translocation?
Conclusion
The balance among the Bcl-2 family members, namely the relative amounts of the BH3-only proteins and the anti-apoptotic Bcl-2 proteins, determines the fate of the cell through the activation of Bax and Bak 59, 66. According to the MMS model, during apoptotic signaling, once all the Bcl-xL/Mcl-1 molecules are neutralized by the BH3-only proteins, Bax/Bak spontaneously (through free diffusion) associate with the MOM, which acts as a direct activator to trigger Bax/Bak’s self-aggregation and pore formation 59, 66. This remarkably efficient and simple mechanism might provide new thinking on pharmacological intervention. For example, is it possible to manipulate the lipid composition or other properties of the MOM to either accelerate or slow down the attack by the Bax/Bak molecules? Also, since helix 9’s exposure and access to the MOM is essential to this process, is it possible to design small molecules to promote or suppress the dislodging of helix 9 from the hydrophobic groove of Bax/Bak, therefore modulating the targeting of these molecules to the mitochondria 114, 115?
Acknowledgements
We are grateful to Dr. Keith Johnson (Department of Oral Biology and Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, USA) for his suggestions and critical reading of the manuscript. Xu Luo dedicates this work to the loving memory of his father, Dr. Shiqi Luo, one of the finest neurosurgeons ever, whose encouragement, support, and advice made the work possible.
Editorial Note on the Review Process
F1000 Faculty Reviews are commissioned from members of the prestigious F1000 Faculty and are edited as a service to readers. In order to make these reviews as comprehensive and accessible as possible, the referees provide input before publication and only the final, revised version is published. The referees who approved the final version are listed with their names and affiliations but without their reports on earlier versions (any comments will already have been addressed in the published version).
The referees who approved this article are:
Vishva Dixit, Department of Physiological Chemistry, Genentech, South San Francisco, CA, USA
Andrew Gilmore, Wellcome Trust Centre for Cell/Matrix Research, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, UK
Andreas Strasser, Molecular Genetics of Cancer Division, The Walter and Eliza Hall Institute, Melbourne, VIC, 3050, Australia
Funding Statement
This work was supported by a pilot grant from the Nebraska Center for Cellular Signaling (P30GM106397) and National Institutes of Health (NIH) Grants R03CA205496 and R01GM118437 to Xu Luo.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 1; peer review: 3 approved]
References
- 1. Danial NN, Korsmeyer SJ: Cell death: Critical control points. Cell. 2004;116(2):205–19. 10.1016/s0092-8674(04)00046-7 [DOI] [PubMed] [Google Scholar]
- 2. Jiang X, Wang X: Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. 10.1146/annurev.biochem.73.011303.073706 [DOI] [PubMed] [Google Scholar]
- 3. Kalkavan H, Green DR: MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. 10.1038/cdd.2017.179 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 4. Cleary ML, Smith SD, Sklar J: Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47(1):19–28. 10.1016/0092-8674(86)90362-4 [DOI] [PubMed] [Google Scholar]
- 5. Tsujimoto Y, Cossman J, Jaffe E, et al. : Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–3. 10.1126/science.3874430 [DOI] [PubMed] [Google Scholar]
- 6. Vaux DL, Cory S, Adams JM: Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–2. 10.1038/335440a0 [DOI] [PubMed] [Google Scholar]
- 7. Cory S, Huang DCS, Adams JM: The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene. 2003;22(53):8590–607. 10.1038/sj.onc.1207102 [DOI] [PubMed] [Google Scholar]
- 8. Puthalakath H, Villunger A, O'Reilly LA, et al. : Bmf: A proapoptotic BH3 only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293(5536):1829–32. 10.1126/science.1062257 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 9. Chipuk JE, Moldoveanu T, Llambi F, et al. : The BCL-2 family reunion. Mol Cell. 2010;37(3):299–310. 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moldoveanu T, Follis AV, Kriwacki RW, et al. : Many players in BCL-2 family affairs. Trends Biochem Sci. 2014;39(3):101–11. 10.1016/j.tibs.2013.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kale J, Osterlund EJ, Andrews DW: BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018;25(1):65–80. 10.1038/cdd.2017.186 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 12. Suzuki M, Youle RJ, Tjandra N: Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell. 2000;103(4):645–54. 10.1016/s0092-8674(00)00167-7 [DOI] [PubMed] [Google Scholar]
- 13. Nechushtan A, Smith CL, Hsu YT, et al. : Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999;18(9):2330–41. 10.1093/emboj/18.9.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolter KG, Hsu YT, Smith CL, et al. : Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139(5):1281–92. 10.1083/jcb.139.5.1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moldoveanu T, Liu Q, Tocilj A, et al. : The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24(5):677–88. 10.1016/j.molcel.2006.10.014 [DOI] [PubMed] [Google Scholar]
- 16. Youle RJ, Strasser A: The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 17. Dewson G, Kratina T, Sim HW, et al. : To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell. 2008;30(3):369–80. 10.1016/j.molcel.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 18. George NM, Evans JJD, Luo X: A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 2007;21(15):1937–48. 10.1101/gad.1553607 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 19. Strasser A, Cory S, Adams JM: Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–83. 10.1038/emboj.2011.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ke FFS, Vanyai HK, Cowan AD, et al. : Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell. 2018;173(5):1217–1230.e17. 10.1016/j.cell.2018.04.036 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 21. Llambi F, Wang YM, Victor B, et al. : BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell. 2016;165(2):421–33. 10.1016/j.cell.2016.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei MC, Zong WX, Cheng EH, et al. : Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–30. 10.1126/science.1059108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chipuk JE, Green DR: How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–64. 10.1016/j.tcb.2008.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang K, Yin XM, Chao DT, et al. : BID: A novel BH3 domain-only death agonist. Genes Dev. 1996;10(22):2859–69. 10.1101/gad.10.22.2859 [DOI] [PubMed] [Google Scholar]
- 25. Letai A, Bassik MC, Walensky LD, et al. : Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–92. 10.1016/s1535-6108(02)00127-7 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 26. Kim H, Rafiuddin-Shah M, Tu HC, et al. : Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–58. 10.1038/ncb1499 [DOI] [PubMed] [Google Scholar]
- 27. Kim H, Tu HC, Ren D, et al. : Stepwise Activation of BAX and BAK by tBID, BIM and PUMA Initiates Mitochondrial Apoptosis. Mol Cell. 2009;36:487–99. 10.1016/j.molcel.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 28. Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. : BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525–35. 10.1016/j.molcel.2005.02.003 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 29. Kuwana T, Mackey MR, Perkins G, et al. : Bid, Bax, and Lipids Cooperate to Form Supramolecular Openings in the Outer Mitochondrial Membrane. Cell. 2002;111(3):331–42. 10.1016/s0092-8674(02)01036-x [DOI] [PubMed] [Google Scholar]
- 30. Llambi F, Moldoveanu T, Tait SWG, et al. : A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44(4):517–31. 10.1016/j.molcel.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 31. Lovell JF, Billen LP, Bindner S, et al. : Membrane Binding by tBid Initiates an Ordered Series of Events Culminating in Membrane Permeabilization by Bax. Cell. 2008;135(6):1074–84. 10.1016/j.cell.2008.11.010 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 32. Bogner C, Kale J, Pogmore J, et al. : Allosteric Regulation of BH3 Proteins in Bcl-xL Complexes Enables Switch-like Activation of Bax. Mol Cell. 2020;77(4):901–912.e9. 10.1016/j.molcel.2019.12.025 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 33. Dai H, Smith A, Meng XW, et al. : Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011;194(1):39–48. 10.1083/jcb.201102027 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 34. Du H, Wolf J, Schafer B, et al. : BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286(1):491–501. 10.1074/jbc.M110.167148 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 35. Hockings C, Anwari K, Ninnis RL, et al. : Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax and show no preference for Bak versus Bax. Cell Death Dis. 2015;6(4):e1735. 10.1038/cddis.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen HC, Kanai M, Inoue-Yamauchi A, et al. : An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17(10):1270–81. 10.1038/ncb3236 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 37. Edwards AL, Gavathiotis E, LaBelle JL, et al. : Multimodal Interaction with BCL-2 Family Proteins Underlies the Proapoptotic Activity of PUMA BH3. Chem Biol. 2013;20(7):888–902. 10.1016/j.chembiol.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cartron PF, Gallenne T, Bougras G, et al. : The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16(5):807–18. 10.1016/j.molcel.2004.10.028 [DOI] [PubMed] [Google Scholar]
- 39. Gallenne T, Gautier F, Oliver L, et al. : Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol. 2009;185(2):279–90. 10.1083/jcb.200809153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gavathiotis E, Reyna DE, Davis ML, et al. : BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40(3):481–92. 10.1016/j.molcel.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 41. Gavathiotis E, Suzuki M, Davis ML, et al. : BAX activation is initiated at a novel interaction site. Nature. 2008;455(7216):1076–81. 10.1038/nature07396 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 42. Czabotar PE, Westphal D, Dewson G, et al. : Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152(3):519–31. 10.1016/j.cell.2012.12.031 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 43. Moldoveanu T, Grace CR, Llambi F, et al. : BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20(5):589–97. 10.1038/nsmb.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 44. Brouwer JM, Lan P, Cowan AD, et al. : Conversion of Bim-BH3 from Activator to Inhibitor of Bak through Structure-Based Design. Mol Cell. 2017;68(4):659–672.e9. 10.1016/j.molcel.2017.11.001 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 45. Robin AY, Krishna Kumar K, Westphal D, et al. : Crystal structure of Bax bound to the BH3 peptide of Bim identifies important contacts for interaction. Cell Death Dis. 2015;6(7):e1809–e1809. 10.1038/cddis.2015.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brouwer JM, Westphal D, Dewson G, et al. : Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55(6):938–46. 10.1016/j.molcel.2014.07.016 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 47. Leshchiner ES, Braun CR, Bird GH, et al. : Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110(11):E986–95. 10.1073/pnas.1214313110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dengler MA, Robin AY, Gibson L, et al. : BAX Activation: Mutations Near Its Proposed Non-canonical BH3 Binding Site Reveal Allosteric Changes Controlling Mitochondrial Association. Cell Rep. 2019;27(2):359–373.e6. 10.1016/j.celrep.2019.03.040 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 49. Peng R, Tong JS, Li H, et al. : Targeting Bax interaction sites reveals that only homo-oligomerization sites are essential for its activation. Cell Death Differ. 2013;20(5):744–54. 10.1038/cdd.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 50. Leber B, Lin J, Andrews DW: Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12(5):897–911. 10.1007/s10495-007-0746-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leber B, Lin J, Andrews DW: Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene. 2010;29(38):5221–30. 10.1038/onc.2010.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bleicken S, Jeschke G, Stegmueller C, et al. : Structural model of active Bax at the membrane. Mol Cell. 2014;56(4):496–505. 10.1016/j.molcel.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 53. Czabotar PE, Lessene G, Strasser A, et al. : Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. 10.1038/nrm3722 [DOI] [PubMed] [Google Scholar]
- 54. Westphal D, Kluck RM, Dewson G: Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21(2):196–205. 10.1038/cdd.2013.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Willis SN, Chen L, Dewson G, et al. : Proapoptotic Bak is sequestered by Mcl-1 and Bcl-x L, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–305. 10.1101/gad.1304105 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 56. Willis SN, Fletcher JI, Kaufmann T, et al. : Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–9. 10.1126/science.1133289 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 57. Willis SN, Adams JM: Life in the balance: How BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17(6):617–25. 10.1016/j.ceb.2005.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fletcher JI, Meusburger S, Hawkins CJ, et al. : Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci U S A. 2008;105(47):18081–7. 10.1073/pnas.0808691105 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 59. Huang K, O’Neill KL, Li J, et al. : BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Res. 2019;29(11):942–52. 10.1038/s41422-019-0231-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mérino D, Giam M, Hughes PD, et al. : The role of BH3-only protein Bim extends beyond inhibiting Bcl-2–like prosurvival proteins. J Cell Biol. 2009;186(3):355–62. 10.1083/jcb.200905153 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 61. Borner C, Andrews DW: The apoptotic pore on mitochondria: Are we breaking through or still stuck? Cell Death Differ. 2014;21(2):187–91. 10.1038/cdd.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. García-Sáez AJ: The secrets of the Bcl-2 family. Cell Death Differ. 2012;19(11):1733–40. 10.1038/cdd.2012.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ren D, Tu HC, Kim H, et al. : BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330(6009):1390–3. 10.1126/science.1190217 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 64. Senft D, Weber A, Saathoff F, et al. : In non-transformed cells Bak activates upon loss of anti-apoptotic Bcl-XL and Mcl-1 but in the absence of active BH3-only proteins. Cell Death Dis. 2015;6(11):e1996–e1996. 10.1038/cddis.2015.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang J, Huang K, O'Neill KL, et al. : Bax/Bak activation in the absence of Bid Bim, Puma, and p53. Cell Death Dis. 2016;7(6):e2266. 10.1038/cddis.2016.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Neill KL, Huang K, Zhang J, et al. : Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30(8):973–88. 10.1101/gad.276725.115 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 67. Edlich F, Banerjee S, Suzuki M, et al. : Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell. 2011;145(1):104–16. 10.1016/j.cell.2011.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 68. Schellenberg B, Wang P, Keeble JA, et al. : Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell. 2013;49(5):959–71. 10.1016/j.molcel.2012.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 68. Chen L, Willis SN, Wei A, et al. : Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol Cell. 2005;17(3):393–403. 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 70. Lopez H, Zhang L, George NM, et al. : Perturbation of the Bcl-2 network and an induced Noxa/Bcl-xL interaction trigger mitochondrial dysfunction after DNA damage. J Biol Chem. 2010;285(20):15016–26. 10.1074/jbc.M109.086231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang L, Lopez H, George NM, et al. : Selective involvement of BH3-only proteins and differential targets of Noxa in diverse apoptotic pathways. Cell Death Differ. 2011;18(5):864–73. 10.1038/cdd.2010.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Grabow S, Kueh AJ, Ke F, et al. : Subtle Changes in the Levels of BCL-2 Proteins Cause Severe Craniofacial Abnormalities. Cell Rep. 2018;24(12):3285–3295.e4. 10.1016/j.celrep.2018.08.048 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 73. Sarosiek KA, Chi X, Bachman JA, et al. : BID Preferentially Activates BAK while BIM Preferentially Activates BAX, Affecting Chemotherapy Response. Mol Cell. 2013;51(6):751–65. 10.1016/j.molcel.2013.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Coultas L, Bouillet P, Loveland KL, et al. : Concomitant loss of proapoptotic BH3-only Bcl-2 antagonists Bik and Bim arrests spermatogenesis. EMBO J. 2005;24(22):3963–73. 10.1038/sj.emboj.7600857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Coultas L, Bouillet P, Stanley EG, et al. : Proapoptotic BH3-Only Bcl-2 Family Member Bik/Blk/Nbk Is Expressed in Hemopoietic and Endothelial Cells but Is Redundant for Their Programmed Death. Mol Cell Biol. 2004;24(4):1570–81. 10.1128/mcb.24.4.1570-1581.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bouillet P, Metcalf D, Huang DC, et al. : Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–8. 10.1126/science.286.5445.1735 [DOI] [PubMed] [Google Scholar]
- 77. Yin XM, Wang K, Gross A, et al. : Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400(6747):886–91. 10.1038/23730 [DOI] [PubMed] [Google Scholar]
- 78. Shibue T, Takeda K, Oda E, et al. : Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17(18):2233–8. 10.1101/gad.1103603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jeffers JR, Parganas E, Lee Y, et al. : Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–8. 10.1016/s1535-6108(03)00244-7 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 80. Villunger A, Michalak EM, Coultas L, et al. : p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–8. 10.1126/science.1090072 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 81. Hübner A, Cavanagh-Kyros J, Rincon M, et al. : Functional Cooperation of the Proapoptotic Bcl2 Family Proteins Bmf and Bim In Vivo. Mol Cell Biol. 2010;30(1):98–105. 10.1128/MCB.01155-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hausmann M, Leucht K, Ploner C, et al. : BCL-2 modifying factor (BMF) is a central regulator of anoikis in human intestinal epithelial cells. J Biol Chem. 2011;286(30):26533–40. 10.1074/jbc.M111.265322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Coultas L, Terzano S, Thomas T, et al. : Hrk/DP5 contributes to the apoptosis of select neuronal populations but is dispensable for haematopoietic cell apoptosis. J Cell Sci. 2007;120(Pt 12):2044–52. 10.1242/jcs.002063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ranger AM, Zha J, Harada H, et al. : Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100(16):9324–9. 10.1073/pnas.1533446100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Huang K, Zhang J, O'Neill KL, et al. : Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J Biol Chem. 2016;291(22):11843–51. 10.1074/jbc.M115.711051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. García-Sáez AJ, Ries J, Orzáez M, et al. : Membrane promotes tBID interaction with BCL(XL). Nat Struct Mol Biol. 2009;16(11):1178–85. 10.1038/nsmb.1671 [DOI] [PubMed] [Google Scholar]
- 87. Todt F, Cakir Z, Reichenbach F, et al. : Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2014;34(1):67–80. 10.15252/embj.201488806 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 88. George NM, Targy N, Evans JJD, et al. : Bax contains two functional mitochondrial targeting sequences and translocates to mitochondria in a conformational change- and homo-oligomerization-driven process. J Biol Chem. 2010;285(2):1384–92. 10.1074/jbc.M109.049924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gahl RF, He Y, Yu S, et al. : Conformational Rearrangements in the Pro-Apoptotic Protein, Bax, as It Inserts Into Mitochondria. J Biol Chem. 2014;289(47):32871–82. 10.1074/jbc.M114.593897 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 90. Zhang Z, Subramaniam S, Kale J, et al. : BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35(2):208–36. 10.15252/embj.201591552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Iyer S, Bell F, Westphal D, et al. : Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015;22(10):1665–75. 10.1038/cdd.2015.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Terrones O, Antonsson B, Yamaguchi H, et al. : Lipidic Pore Formation by the Concerted Action of Proapoptotic BAX and tBID. J Biol Chem. 2004;279(29):30081–91. 10.1074/jbc.M313420200 [DOI] [PubMed] [Google Scholar]
- 93. Hsu YT, Youle RJ: Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272(21):13829–34. 10.1074/jbc.272.21.13829 [DOI] [PubMed] [Google Scholar]
- 94. Cheng EHY, Sheiko TV, Fisher JK, et al. : VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301(5632):513–7. 10.1126/science.1083995 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 95. Ren D, Kim H, Tu HC, et al. : The VDAC2-BAK rheostat controls thymocyte survival. Sci Signal. 2009;2(85):ra48. 10.1126/scisignal.2000274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chin HS, Li MX, Tan IKL, et al. : VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat Commun. 2018;9(1):4976. 10.1038/s41467-018-07309-4 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 97. Lauterwasser J, Todt F, Zerbes RM, et al. : The porin VDAC2 is the mitochondrial platform for Bax retrotranslocation. Sci Rep. 2016;6:32994. 10.1038/srep32994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Karbowski M, Lee YJ, Gaume B, et al. : Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159(6):931–8. 10.1083/jcb.200209124 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 99. Milani M, Beckett AJ, Al-Zebeeby A, et al. : DRP-1 functions independently of mitochondrial structural perturbations to facilitate BH3 mimetic-mediated apoptosis. Cell Death Discov. 2019;5:117. 10.1038/s41420-019-0199-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Montessuit S, Somasekharan SP, Terrones O, et al. : Membrane Remodeling Induced by the Dynamin-Related Protein Drp1 Stimulates Bax Oligomerization. Cell. 2010;142(6):889–901. 10.1016/j.cell.2010.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 101. Tan C, Dlugosz PJ, Peng J, et al. : Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J Biol Chem. 2006;281(21):14764–75. 10.1074/jbc.M602374200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dai H, Ding H, Meng XW, et al. : Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells. Genes Dev. 2015;29(20):2140–52. 10.1101/gad.267997.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ashkenazi A, Fairbrother WJ, Leverson JD, et al. : From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–84. 10.1038/nrd.2016.253 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 104. Oltersdorf T, Elmore SW, Shoemaker AR, et al. : An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–81. 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 105. Kotschy A, Szlavik Z, Murray J, et al. : The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–82. 10.1038/nature19830 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 106. Green DR: A BH3 Mimetic for Killing Cancer Cells. Cell. 2016;165(7):1560. 10.1016/j.cell.2016.05.080 [DOI] [PubMed] [Google Scholar]
- 107. Cory S, Roberts AW, Colman PM, et al. : Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer. 2016;2(8):443–60. 10.1016/j.trecan.2016.07.001 [DOI] [PubMed] [Google Scholar]
- 108. Merino D, Kelly GL, Lessene G, et al. : BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell. 2018;34(6):879–91. 10.1016/j.ccell.2018.11.004 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 109. Souers AJ, Leverson JD, Boghaert ER, et al. : ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–8. 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 110. Greaves G, Milani M, Butterworth M, et al. : BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-X L. Cell Death Differ. 2019;26(6):1037–47. 10.1038/s41418-018-0183-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pécot J, Maillet L, Le Pen J, et al. : Tight Sequestration of BH3 Proteins by BCL-xL at Subcellular Membranes Contributes to Apoptotic Resistance. Cell Rep. 2016;17(12):3347–58. 10.1016/j.celrep.2016.11.064 [DOI] [PubMed] [Google Scholar]
- 112. Inoue-Yamauchi A, Jeng PS, Kim K, et al. : Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun. 2017;8:16078. 10.1038/ncomms16078 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 113. Todt F, Cakir Z, Reichenbach F, et al. : The C-terminal helix of Bcl-x( L) mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2013;20(2):333–42. 10.1038/cdd.2012.131 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 114. Reyna DE, Garner TP, Lopez A, et al. : Direct Activation of BAX by BTSA1 Overcomes Apoptosis Resistance in Acute Myeloid Leukemia. Cancer Cell. 2017;32(4):490–505.e10. 10.1016/j.ccell.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 115. Garner TP, Amgalan D, Reyna DE, et al. : Small-molecule allosteric inhibitors of BAX. Nat Chem Biol. 2019;15(4):322–30. 10.1038/s41589-018-0223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation