

Abstract
Background
The focus of this study was to explore potential differences in colonic mucosal microbiota in irritable bowel syndrome (IBS) patients compared to a control group utilizing a metagenomic study.
Methods
Mucosal microbiota samples were collected from each IBS patient utilizing jet‐flushing colonic mucosa in unified segments of the colon with distilled water, followed by aspiration, during colonoscopy. All the purified dsDNA was extracted and quantified before metagenomic sequencing using an Illumina platform. An equal number of healthy age‐matched controls were also examined for colonic mucosal microbiota, which were obtained during screening colonoscopies.
Results
The microbiota data on 50 IBS patients (31 females), with a mean age 43.94 ± 14.50 (range19–65), were analyzed in comparison to 50 controls. Satisfactory DNA samples were subjected to metagenomics study, followed by comprehensive comparative phylogenetic analysis. Metagenomics analysis was carried out, and 3.58G reads were sequenced. Community richness (Chao) and microbial structure in IBS patients were shown to be significantly different from those in the control group. Enrichment of Oxalobacter formigenes, Sutterella wadsworthensis, and Bacteroides pectinophilus was significantly observed in controls, whereas enrichment of Collinsella aerofaciens, Gemella morbillorum, and Veillonella parvula Actinobacteria was observed significantly in the IBS cohort.
Conclusion
The current study has demonstrated significant differences in the microbiota of IBS patients compared to controls.
Keywords: collinsella aerofaciens, gemella morbillorum, irritable bowel syndrome, metagenomic study, veillonella parvula Actinobacteria

The current study gives an insight into the microbiota of irritable bowel syndrome (IBS) patients in Saudi Arabia and has identified various significantly present genera in these patients.
Introduction
Irritable bowel syndrome (IBS), characterized by recurrent abdominal pain and disordered bowel habits, is one of the most common functional bowel disorders in clinical practice. The pathogenesis of IBS is not entirely understood, and several pathophysiologic mechanisms have been postulated. While altered gastrointestinal motility, visceral hypersensitivity, changes in gut permeability, and immune activation have been proposed from time to time, gut microbiota alterations are emerging as one of the important hypotheses in the pathogenesis of IBS. The gastrointestinal tract is the natural reservoir of microbiota, which begins harboring it soon after birth. The dynamic interactions between the gut and microbiota are known to play a physiological role in the metabolic and structural functions of the alimentary canal. Conceivably, dysbiosis contributes to several diseases of the gut, including IBS. Various clues to this hypothesis come from the fact that a bout of gastroenteritis triggers IBS. Both qualitative and quantitative differences have been observed in the gut microbiota of IBS patients compared to healthy controls. Furthermore, a positive effect of treatment with nonabsorbable antibiotics and probiotics in IBS provides another clue to the alteration of gut microbiota in IBS.1
Nowadays, metagenomics, which is a molecular method of culture‐independent microbiology, has been used as one of the most potent sequence‐driven approaches to study the composition and the genetic potential of the mucosal gut microbiota. The genetic material is recovered directly from environmental samples and is investigated utilizing this technology. It is believed that abnormal microbiota activate innate mucosal responses, resulting in a change in the intestinal permeability. This phenomenon activates nociceptive sensory pathways, causing dysregulation of the enteric nervous system. While this conceptualization continues to grow, various dietary manipulations, prebiotics, probiotics, symbiotics, and nonabsorbable antibiotics have already been in use in clinical practice as potential therapeutic agents in the management of IBS.2
Various aspects of IBS in Saudi Arabia have been addressed by different investigators from time to time.3, 4, 5, 6 To our knowledge, from a Medline search, there appears to be no existing data regarding microbiota in Saudi Arabian IBS patients, and the current study is the first of its kind in this region. The microbiota in IBS patients were compared with that of age‐ and gender‐matched controls. Many studies on gut microbiota and their relation to IBS have been based on the analyses of fecal samples.7, 8 However, although tedious, we preferred to estimate mucosal microbiota in IBS patients rather than perform an analysis on fecal samples based on the assumption that the mucosal microbiota live closer to the intestinal epithelium. Thus, their interaction with the immune and neuroendocrine systems must be more significant than their luminal counterparts. Substantial differences in mucosal and fecal microbiota composition have already been demonstrated by researchers in various gastrointestinal disorders.9, 10
Methods
This study was conducted in full compliance with the research guidelines of the King Fahad Medical City (KFMC), Riyadh. The study was approved by the Ethics Committee of King Abdulaziz City for Science and Technology and KFMC, Riyadh.
Inclusion criteria for study participants
During the study period (July 2010 to November 2012), all IBS and screening colonoscopy patients at King Fahad Medical City (KFMC), Riyadh, were asked to provide specific written consent for possible inclusion in this study and were included after they met the enrolment criteria.
- Cases:
- Adult patients with diagnosed IBS, the diagnoses of whom were based on the exclusion of all organic causes following Rome III criteria.
- Controls:
- Patients with normal colonoscopy who underwent screening colonoscopic examination for anemia and colorectal cancer surveillance.
Exclusion criteria
IBS patients who had used antibiotics 4 weeks before colonoscopy were excluded.
Data collection
Demographic and clinical data were collected from all IBS participants, including age, gender, and body mass index (BMI), and the data were entered in a Microsoft Excel file.
Bowel preparation and sample collection
After standard bowel preparation during the colonoscopy procedure, distilled water was pushed through the biopsy channel of the scope and collected back by aspiration. The mucosal jet wash from cecum, transverse, left, and rectal colon segments was collected from all study participants. Finally, 50 ml of the washes, obtained from each colonoscopy, were collected in four (15 ml) test tubes and immediately stored at −80°C for further processing as discussed in our previous metagenomic studies.11, 12
DNA extraction for Total metagenomic sequencing
DNA samples were centrifuged at 5000 × g for 15 min, and the supernatants were discarded. The pellet was resuspended in 10‐ml lysis buffer (0.5 M Tris–HCl; 20 mm EDTA; 10 mm NaCl; 0.1% SDS; pH 9.0), and the mixtures were homogenized by centrifuging and shaking for 5–10 min as discussed in our previous metagenomic studies.11, 12
Samples were then diluted (1: 2) with a 10‐ml lysis buffer and homogenized for another 5 min. Genomic DNA (gDNA) was precipitated by adding 5 ml of 7.5 M ammonium acetate and 25 mL of ice‐cold ethanol (95–100%). The samples were subsequently incubated at −20°C for 20–30 min, and gDNA was collected following centrifugation at 4500 × g for 15 min at room temperature. DNA pellets were resuspended in 600 μL of TE buffer (Tris‐EDTA, pH 8.0) and incubated at 65°C for 15 min. An equal volume of phenol–chloroform: isoamyl alcohol solution was briefly mixed with the DNA, and the mixtures were centrifuged at 12 000g for 5 min at room temperature. The supernatant aqueous phase was then transferred to a new tube, while the interface and the organic phase were discarded. This step was repeated until no protein was visible at the interface. The final supernatant aqueous phase was then transferred to a new tube; twice the amount of ethanol was added to the aqueous phase, and the solution was stored overnight. The following day, the DNA was precipitated in ethanol as described above. The resulting DNA pellet was resuspended in 50 μL of TE buffer. The quality and concentration of the extracted gDNA were verified using 1% agarose gel electrophoresis and a Qubit fluorometer 3.0 (Life Technologies, Carlsbad, CA).
Metagenomics DNA library construction and sequencing
Paired‐end (PE) metagenomics DNA library construction was performed based on the manufacturer's instructions (Sequencing Kits and Reagents, Illumina, San Diego, CA, USA). High‐quality reads were separated from low‐quality reads with “N” bases, adapter contamination, or human DNA contamination from the Illumina raw data using the BWA‐SW algorithm.13 On average, the proportion of high‐quality reads in all samples was approximately 98.1%, and the insert sizes of our PE clones ranged from 313 to 381 bp. Sequencing and data processing were performed at the Beijing Genomics Institute, where Illumina GAIIx and HiSeq 2000 platforms were utilized to sequence the samples.
Gene catalog construction
The first 72 randomly chosen samples were combined to establish the nonredundant gene set. Predicted open reading frames (ORFs) of the 72 samples were aligned with each other, and gene pairs with an identity higher than 95% were grouped. Groups with similar genes were merged, and the longest ORF in each group was used to represent that group. We, therefore, organized the nonredundant gene set from all predicted genes by excluding redundant ORFs. For 159 high‐quality reads in stages I, II, and III, we performed de novo assembly and gene predictions using SOAPdenovo v1.0614 and GeneMark v2.7,15 respectively. All predicted genes were aligned pairwise using BLAT, and genes that could be aligned (>90% of gene length) with another gene with more than 95% identity (no gaps allowed) were removed as redundancies, resulting in a nonredundant gene catalog. This catalog of colonic samples was further combined with the previously constructed Meta HIT gene catalog by eliminating redundancies in the same manner.
Bioinformatics analysis‐pipeline
We subjected multiple samples from the mucosal microbiota metagenome to comparative phylogenetic analyses to understand the ecology of cultivation‐independent gut microbiota and the phylogenetic differences between samples. We aligned all high‐quality reads with known bacterial, fungal, protozoal, or human gut gene databases from NCBI, RDP, or MetaHIT. For each sample, we compared paired alignment and single alignments to the databases.
Phylogenetic classification of ORFs
Taxonomic assignments were performed with the BLASTp alignment tool against the NR90 database. Alignment hits with values greater than 1E‐5 were removed, and significant matches with values in the same order of the top hitters were used to determine taxonomic groups. We assessed the taxonomic association of each gene by a lowest common ancestor (LCA)‐based algorithm implemented in MEGAN43.
Statistical analysis
The module extracted from the i path (reference) test was assessed utilizing the Wilcoxon test between the control and the IBS groups. The Chao1 Richness Index, Shannon Index, and the Simpson Diversity Index were used to describe the α diversity features of our bacterial community. A value of <0.05 was considered significant.
Beta diversity was used to assess diversity between cases and controls. Principal coordinate analysis (PCoA) based on the unweighted UniFrac distance metrics was used to demonstrate that there was a difference in the mucosal bacterial communities between the cases and controls, which in turn was confirmed by permutational multivariate analysis of variance (PERMANOVA) PCoA in the IBS patients.
Results
The microbiota data of 50 IBS patients (31 females), with a mean age 43.94 ± 14.50 (range 19–65), were analyzed in comparison to age‐ and gender‐matched 50 controls. The patients were diagnosed as per Rome III classification. None of our IBS cohorts had alarm symptoms, and the mean duration of their symptoms was 33.24 ± 14.50 months. The majority of our study cohort were obese. (BMI = 26.5–46.43 kg/M2). All patients had normal baseline investigations.
A metagenomics analysis was carried out, and a total of 3.58G reads were sequenced. About 321.91G of data were used in this study. The useful reads in each sample were aligned with the 4.3 M gene set1 by soap2. On average, 70.33% reads could be mapped to the gene set.
The Chao1 Richness Index, Shannon Index, and the Simpson Diversity Index, used to describe the α diversity features of our bacterial community, showed the following results after the Wilcox test. The results of the PERMANOVA showed that the disease has the most significant impact on gut bacterial composition compared to age and gender.
The module extracted from the ipath (reference) test by the Wilcoxon test between the control and case groups was 0.23. All the results were mapped to the 9.9 M gene set. The difference between the groups in rarefaction was quite significant. Beta diversity showed significant diversity between cases and controls. PCoA based on the unweighted UniFrac distance metrics demonstrated that there was a separation in the mucosal bacterial communities between the cases and controls, which was confirmed by PERMANOVA PCoA.
It was further observed that IBS patients had statistically significant enrichment of four genera Actinomyces, Collinsella, Gordonibacter, and Veillonella compared to controls. There was a selective enrichment of three genera Brachyspira, Desulfitobacterium, and Nitrosococcus in the control group as shown in Table 1 and Figure 1.
Table 1.
Significant enrichment in case (1) and controls (0)
| Genus, Phylum, class, order, family | Control vs. IBS | |||
|---|---|---|---|---|
| P‐value | Mean rank sum in 1 | Mean rank sum in 0 | Enrichment | |
| Actinomyces | 0.0009 | 50 | 33 | 1 |
| Actinobacteria, Actinobacteria, Actinomycetales, Actinomycetaceae, Actinomyces | ||||
| Brachyspira | 0.0079 | 35 | 47 | 0 |
| Spirochaetes,Spirochaetes, Brachyspirales, Brachyspiraceae, Brachyspira | ||||
| Collinsella | 0.0062 | 48 | 34 | 1 |
| Actinobacteria, Actinobacteria, Coriobacteriales, Coriobacteriaceae, Collinsella | ||||
| Desulfitobacterium | 0.0015 | 33 | 49 | 0 |
| Firmicutes, Clostridia, Clostridiales, Peptococcaceae, Desulfitobacterium | ||||
| Gordonibacter | 0.0085 | 48 | 34 | 1 |
| Actinobacteria; Coriobacteriia; Eggerthellales; Eggerthellaceae; Gordonibacter | ||||
| Nitrosococcus | 0.0036 | 37 | 45 | 0 |
| Proteobacteria; Gammaproteobacteria; Chromatiales; Chromatiaceae; Nitrosococcus | ||||
| Veillonella | 0.0093 | 48 | 34 | 1 |
| Firmicutes; Negativicutes; Veillonellales; Veillonellaceae; Veillonella | ||||
IBS, irritable bowel syndrome.
Figure 1.
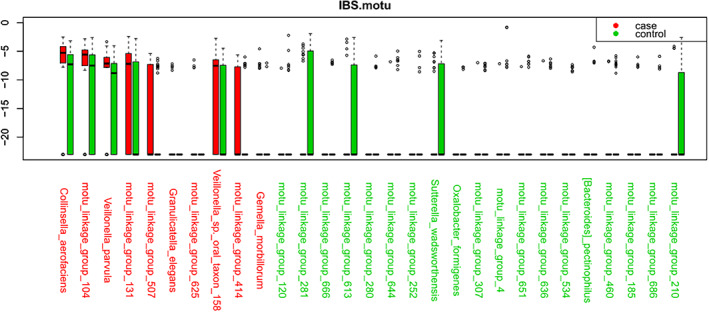
Metagenomic analysis of irritable bowel syndrome (IBS) and controls.
Further subanalysis of Enterobacteriaceae showed that enterotype one was observed in 27 IBS patients, while enterotype two was not observed in any of IBS patients. There were 13 IBS patients with enterotype three positivity (Figure 2).
Figure 2.
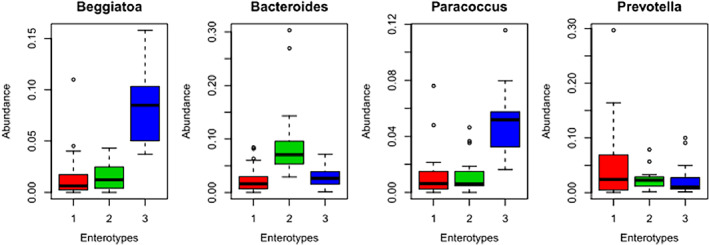
Enterotypes in irritable bowel syndrome (IBS) and controls.
Discussion
The results of the systemic review and meta‐analysis by Zhuang et al. demonstrated an alteration of gut microbiota in IBS patients and pointed to the role of microbiota in the pathogenesis of IBS.16 We observed a distinct difference in microbiota between IBS patients and the control group. Our results are in line with various other studies across different ethnicities of the world as summarized in Table 2.17, 18, 19, 20, 21, 22
Table 2.
Summary of molecular studies of intestinal microbiota in irritable bowel syndrome across various ethnicities and comparison with current study
| Reference | Ethnicity | IBS | Controls | Method | Sample | Changes in intestinal microbiota composition |
|---|---|---|---|---|---|---|
| Current study | Saudi Arabia | 50 | 50 | Metagenomic study | Mucosal washes |
Controls: Bacteroides pectinophilus↑ Oxalobacter formigenes↑ Oxalobacter formigenes↑ Bacteroides pectinophilus↑ Sutterella wadsworthensis↑ IBS: Collinsella aerofaciens↑ Gemella morbillorum, ↑ Veillonella parvula↑ Actinobacteria ↑ |
| Malinen et al.17 | Finland | 27 | 22 | qPCR covering bacteria 300 bacterial species | Feces |
↓ lactobacillus ↑ veillonella spp. ↓ clostridium coccoides subgroup, Bifidobacterium catenulatum group |
| Kerckhoffs et al.18 | The Netherlands | 41 | 26 | FISH, qPCR | Feces, duodenal mucosa | ↓ bifidobacterium catenulatum |
| Tana et al.19 | Japan | 26 | 26 | Culture, qPCR | Feces |
↑ veillonella spp. ↑ lactobacillus spp |
| Carroll et al.20 | USA | 16 | 21 | T‐RFLP fingerprinting of 16S rRNA‐PCR | Feces, Colonic biopsies | ↓ microbial biodiversity in D‐IBS fecal samples |
| Parkes et al.21 | UK | 53 | 26 | FISH, confocal microscopy | Colonic mucosa | Expansion of mucosa‐associated microbiota; mainly bacteroides and clostridia; association with IBS subgroups and symptoms |
| Jeffery et al.22 | Sweden | 37 | 20 | Pyrosequencing 16SrRNA | Feces | Increase of firmicutes‐associated taxa and a depletion of bacteroidetes‐related taxa) |
IBS, irritable bowel syndrome.
Bacteroides fragilis, Bacteroides stercoris, and Bifidobacterium bifidum were selectively enriched in our IBS cohort, but their levels did not reach significance possibly because our IBS cohort patients were in remission. A downregulation of bacterial colonization, including Lactobacillus, Bifidobacterium, and F. prausnitzii, was observed in IBS patients, particularly in diarrhea‐predominant IBS, by Liu et al. The authors were of the opinion that microbiota alteration occurs in the pathogenesis of IBS.23
Studies have shown that 1 in 10 patients with IBS believe that their symptoms begin with an infectious illness, and 3–36% of enteric infections lead to persistent new IBS symptoms.24 There was a significant enrichment of enterococcus type 1 and type 3 in our IBS cohort. It is entirely possible that residual inflammation or persistent changes in mucosal immunocytes, enterochromaffin, may be causative factors in the etiology of postinfectious IBS, but such a correlation continues to be under investigation. The prognosis is, fortunately, better with postinfectious IBS even though it may take several years to resolve25 There was a female predominance in our IBS cohort, which has been described to be one of the risk factors of the development of IBS. While we did not study the risk factors of IBS, studies have shown that adverse events in the preceding 3 months, such as depression and hypochondriasis, affect IBS. We demonstrated enrichment of Bacteroides (type I) and Prevotella (type II), in the IBS cohort compared to controls, but their enrichment did not reach statistically significant levels in this study. Liu et al. studied enrichment of these bacteria. It was observed by the authors that the fecal microbiota signatures were similar between patients with diarrhea‐predominant IBS and depression in that they were less diverse than samples from controls highlighting the disturbance of the brain–gut axis in IBS patients. The authors concluded that these microbial profiles in patients could indicate different subtypes of IBS and depression.23
Collinsella aerofaciens was significantly enriched in our IBS cohort as shown in Table 1 Contrary to our results, Kassinen et al.7 in their Finnish IBS cohort observed a significant reduction of C. aerofaciens. It is entirely possible that some genetic factors might be playing a role in the selective enrichment of this microbe in IBS. In addition, the enrichment of C. aerofaciens has been linked to the BMI of the person as Turnbaugh et al.26 observed C. aerofaciens to be more prominent in obese than lean twins and their mothers. The majority of our IBS cohort were obese (BMI of >30 kg/M2), which could explain selective enrichment in our study cohort. Although obesity has been associated with a low‐grade systemic inflammation in which the GI microbiota may be involved, it may be difficult to conclude whether C. aerofaciens could have any role in IBS.
Gemella morbillorum was significantly enriched in our IBS cohort. The association of this gram‐negative coccus has ranged from endocarditis27 to various malignancies,28 including colonic malignancy.29 In fact, we demonstrated significant enrichment of this gram‐negative coccus in our colorectal cancer metagenomic study.11 Thus, Gemella morbillorum may not be a future biomarker in IBS despite its significant enrichment observed in this study. Contrary to this, Oxalobacter formigenes enrichment was significant in the control group. This commensal has been shown to play a role in the regulation of host oxalate homeostasis. Many factors may be responsible for the variable decline of this bacterium in IBS cohorts as was observed in this study. These factors include the amount of oxalate in the diet, unfavorable environmental conditions including pH, or other growth inhibitory intestinal components from other organisms.30, 31 All these factors connote dysbiosis among IBS patients and point toward the role of various interventions aimed to regulate the microbiota in IBS.
Veillonella parvula species were significantly enriched in our IBS cohort compared to control. Malinen et al. observed similar results in their Finish IBS cohort17 and Tana et al. in their Japanese IBS cohorts.19 This highlights uniform enrichment of Veillonella parvula across various ethnicities among IBS populations as summarized in Table 2.
We demonstrated enrichment of Methanobrevibacter smithii in our controls, but it did not reach significant levels. The reduced levels of methanogens in IBS patients have been found to correlate with the severity of IBS symptoms.17
The drawback of our study is that we did not subcategorize our study of colonic mucosal microbiota according to various subtypes of IBS, although we did include both IBS‐D (diarrhea predominant) and IBS‐C (constipation predominant) patients. Moreover, study sample inclusion of all our patients was conducted during their clinically inactive phase of the disease to avoid intolerance to the colonoscopic procedure during an active phase. We postulate that further colonic mucosal microbiota studies specifically addressing head‐to‐head comparison of active versus inactive phase of disease in all IBS subtypes might lead to a better understanding of the role of colonic microbiota in the pathogenesis of IBS. However, some studies have shown that, within 1 year, 75% of patients change subtypes, and in approximately 29% of patients, there is a switch between IBS‐D and IBS‐C, making the usefulness of these subtypes debatable.32
Conclusion
Based on the discussed results, it can be concluded that there is a significant difference in the microbiota of IBS and control groups. Our study identified enrichment of four genera in Saudi Arabian IBS patients compared to controls. Our study results were similar to various other studies across different ethnicities of the world.
Data availability statement
The [Metagenomic analysis] data used to support the findings of this study are available from the corresponding author upon request.
Acknowledgments
This research project was funded by KACST (Nu: AT‐29‐243). The funding was used for metagenomics analysis. There was no funding for writing the manuscript.
Declaration of conflict of interest: None.
REFERENCES
- 1. Ohman L, Simrén M. Intestinal microbiota and its role in irritable bowel syndrome (IBS). Curr. Gastroenterol. Rep. 2013. May; 15: 323. [DOI] [PubMed] [Google Scholar]
- 2. Xu X, Wu A, Zhang X, Su M, Jiang T, Yuan ZM. MetaDP: a comprehensive web server for disease prediction of 16S rRNA metagenomic datasets. Biophys. Rep. 2016; 2: 106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mosli M, Bamarhul M, Alharbi A et al Screening irritable bowel syndrome patients for symptoms predictive of Crohn's disease using the red flag score. Saudi J. Gastroenterol. 2017; 23: 229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. AlKhalifah MI, Al‐Aql AM, Al‐Mutairi MS et al Prevalence of irritable bowel syndrome among Qassim school teachers, and its impact on their performance and life duties. Saudi Med. J. 2016; 37: 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al‐Ajlan AS. Screening of coeliac disease in undetected adults and patients diagnosed with irritable bowel syndrome in Riyadh, Saudi Arabia. Saudi J. Biol. Sci. 2016; 23: 462–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Al‐Hazmi AH. Knowledge, attitudes, and practices of primary care physicians about irritable bowel syndrome in Northern Saudi Arabia. Saudi J. Gastroenterol. 2012; 18: 173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kassinen A, Krogius‐Kurikka L, Mäkivuokko H et al The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology. 2007; 133: 24–33. [DOI] [PubMed] [Google Scholar]
- 8. Rinttilä T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA‐targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real‐time PCR. J. Appl. Microbiol. 2004; 97: 1166–77. [DOI] [PubMed] [Google Scholar]
- 9. Bibiloni R, Mangold M, Madsen KL et al The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn's disease and ulcerative colitis patients. J. Med. Microbiol. 2006; 55(Pt 8): 1141–9. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi K, Nishida A, Fujimoto T et al Reduced abundance of butyrate‐producing bacteria species in the fecal microbial community in Crohn's disease. Digestion. 2016; 93: 59–65. [DOI] [PubMed] [Google Scholar]
- 11. Alomair AO, Masoodi I, Alyamani EJ et al Colonic mucosal microbiota in colorectal cancer: a single‐center metagenomic study in Saudi Arabia. Gastroenterol. Res. Pract. 2018; 16: 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masoodi I, Alshanqeeti AS, Ahmad S et al Microbial dysbiosis in inflammatory bowel diseases: results of a metagenomic study in Saudi Arabia. Minerva Gastroenterol. Dietol. 2019; 65: 177–86. [DOI] [PubMed] [Google Scholar]
- 13. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics. 2010; 26: 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo R, Liu B, Xie R et al SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. GigaScience. 2012; 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ter‐Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008; 18: 1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhuang X, Xiong L, Li L, Li M, Chen M. Alterations of gut microbiota in patients with irritable bowel syndrome: A systematic review and meta‐analysis. J. Gastroenterol. Hepatol. 2017; 32: 28–38. [DOI] [PubMed] [Google Scholar]
- 17. Malinen E, Rinttilä T, Kajander K et al Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real‐time PCR. Am. J. Gastroenterol. 2005; 100: 373–82. [DOI] [PubMed] [Google Scholar]
- 18. Kerckhoffs AP, Samsom M, van der Rest ME et al Lower Bifidobacteria counts in both duodenal mucosa‐associated and fecal microbiota in irritable bowel syndrome patients. World J. Gastroenterol. 2009; 15: 2887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tana C, Umesaki Y, Imaoka A et al Altered profiles of intestinal microbiota and organic acids may be the origin of symptoms in irritable bowel syndrome. Neurogastroenterol. Motil. 2010; 22: 512–19 e114–e115. [DOI] [PubMed] [Google Scholar]
- 20. Carroll IM, Ringel‐Kulka T, Keku TO et al Molecular analysis of the luminal‐ and mucosal‐associated intestinal microbiota in diarrhea‐predominant irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2011; 301: G799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parkes GC, Rayment NB, Hudspith BN et al Distinct microbial populations exist in the mucosa‐associated microbiota of sub‐groups of irritable bowel syndrome. Neurogastroenterol. Motil. 2012; 24: 31–9. [DOI] [PubMed] [Google Scholar]
- 22. Jeffery IB, O'Toole PW, Öhman L et al An irritable bowel syndrome subtype defined by species‐specific alterations in faecal microbiota. Gut. 2012; 61: 997–1006. [DOI] [PubMed] [Google Scholar]
- 23. Liu HN, Wu H, Chen YZ, Chen YJ, Shen XZ, Liu TT. Altered molecular signature of intestinal microbiota in irritable bowel syndrome patients compared with healthy controls: A systematic review and meta‐analysis. Dig. Liver Dis. 2017. Apr; 49: 331–7. [DOI] [PubMed] [Google Scholar]
- 24. Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009. May; 136: 1979–88. [DOI] [PubMed] [Google Scholar]
- 25. Neal KR, Barker L, Spiller RC. Prognosis in post‐infective irritable bowel syndrome: a six‐year follow up study. Gut. 2002; 51: 410–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Turnbaugh PJ, Hamady M, Yatsunenko T et al A core gut microbiome in obese and lean twins. Nature. 2009. Jan 22; 457: 480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shinha T. Endocarditis due to Gemella morbillorum. Intern. Med. 2017; 56: 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pushalkar S, Ji X, Li Y et al Comparison of oral microbiota in tumor and non‐tumor tissues of patients with oral squamous cell carcinoma. BMC Microbiol. 2012; 12: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. FitzGerald SF, Moloney AC, Maurer BJ, Hall WW. Gemella endocarditis: consider the colon. J. Heart Valve Dis. 2006; 15: 833–5. [PubMed] [Google Scholar]
- 30. Liu M, Koh H, Kurtz ZD et al Oxalobacter formigenes‐associated host features and microbial community structures examined using the American Gut Project. Microbiome. 2017; 5: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weese JS, Weese HE, Yuricek L, Rousseau J. Oxalate degradation by intestinal lactic acid bacteria in dogs and cats. Vet. Microbiol. 2004; 101: 161–6. [DOI] [PubMed] [Google Scholar]
- 32. Tap J, Derrien M, Törnblom H et al Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology. 2017; 152: 111–123.e8. 10.1053/j.gastro.2016.09.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The [Metagenomic analysis] data used to support the findings of this study are available from the corresponding author upon request.