

Abstract
Objective:
Hepcidin, a mediator of innate immunity, binds the iron exporter ferroportin, leading to functional hypoferremia through intracellular iron sequestration. We explored hepcidin-ferroportin interactions in neonates clinically diagnosed with early-onset neonatal sepsis (EONS).
Study Design:
Hepcidin and IL-6 were quantified by ELISA in 92 paired cord blood-maternal blood samples in the following groups: “Yes” EONS (n=41, GA 29±1weeks) and “No” EONS (n=51, GA 26±1 weeks). Placental hepcidin and ferroportin expression were evaluated by immunohistochemistry and RT-PCR. Liver hepcidin and ferroportin expression patterns were ascertained in autopsy specimens of neonates (n=8) who died secondary to culture-proven sepsis.
Results:
Cord blood hepcidin (GA corrected p=0.018) was positively correlated with IL-6 (r=0.379, p=0.001) in EONS. Hepcidin localized at syncytiotrophoblast and fetal vascular endothelium. Placental ferroportin, but not hepcidin mRNA correlated with cord blood hepcidin levels (r=0.46, p=0.039) and funisitis severity (r=0.50, p=0.018). Newborns who died from sepsis (n=4) had higher hepatic hepcidin and iron sequestration, but lower ferroportin staining than those who died of non-sepsis causes (n=4).
Conclusion:
Premature fetuses with EONS have elevated circulating hepcidin, likely related to lower placenta and liver ferroportin expression. Fetal hepcidin-ferroportin interaction appears to play a role in EONS pathophysiology independent of maternal response to intrauterine inflammation.
Keywords: Chorioamnionitis, Funisitis, Placenta, Cord blood
Introduction
The essential trace element, iron, is critical for development and survivability of living organisms.1 Iron is an indispensable cofactor for many proteins responsible for fine tuning many biologic processes including oxygen transport, erythropoiesis, mitochondrial energy generating processes, and DNA/neurotransmitter synthesis.2,3 Although many transport proteins and enzymes have been implicated, there is agreement that iron’s metabolic functions are tightly controlled by hepcidin, a regulatory cytokine produced primarily by hepatocytes.4 As shown in Figure 1, hepcidin achieves its function by binding to and inactivating the only known iron cell exporter, ferroportin, a transmembrane protein that extrudes iron from inside a cell.4 High levels of ferroportin are known to be present in enterocytes and tissue macrophages of the spleen and the liver. Hepcidin binding of ferroportin results in the internalization and degradation of ferroportin, which increases intracellular iron content and decreases its extra-cellular serum concentration. In normal physiological conditions, the result is maintenance of constant serum iron levels.
Fig. 1.
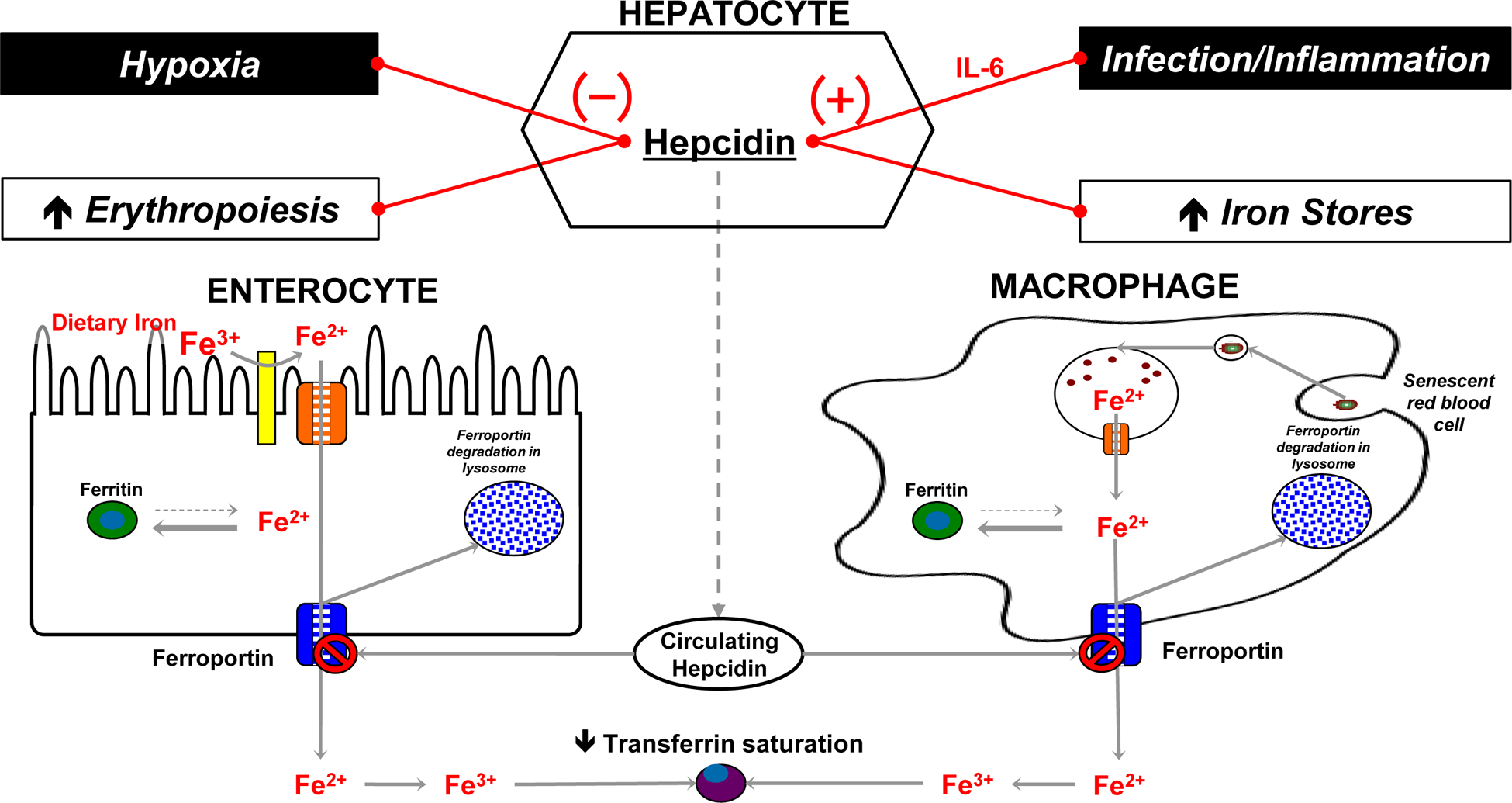
Schematic representation of the involvement of hepcidin in iron homeostasis.
For most living organisms, including pathogenic bacteria, iron is essential and its absence potentially damaging.5 Regulating iron sequestration and microbial access to iron represents an important host defense mechanism of innate immunity.6, 7 Along with transferrin and lactoferrin, which bind to free iron and render it inaccessible to most bacteria, hepcidin participates as an acute phase reactant to prompt hypoferremia in response to microbial challenge.5 In this role, hepcidin has been demonstrated to be paramount to the replication and metabolism of microbial pathogens.8 In addition to being a key regulator of iron availability, hepcidin, was shown to have defined antimicrobial properties.9 Inflammation, iron stores, hypoxia and anemia are major regulatory factors of hepcidin expression.10, 11 Persistent elevation of hepcidin through transcriptional regulation by interleukin (IL)-6 has been implicated as a pathogenic mechanism for anemia of inflammation and chronic disease (AICD), a common form of anemia with normal iron levels that accompanies chronic autoimmune conditions, infections, malignancies and aging.12, 13
The placental syncytiotrophoblast expresses ferroportin, which participates in maternal-fetal iron transfer across the placenta.14 Despite well-established relationships among intra-uterine infection, preterm birth, and early-onset neonatal sepsis (EONS)15, little is known about their impact on the interplay between hepcidin and ferroportin across the maternal-fetal interface. Recent evidence has demonstrated increased hepcidin levels in cord or peripheral blood of newborns with suspected early- and late-onset neonatal sepsis.16, 17 However, these studies included a low number of EONS neonates. In addition, questions about the relative differences of hepcidin expression between the maternal and fetal compartments, the potential source of hepcidin in the setting of intra-amniotic infection (IAI), and hepcidin expression in the liver of neonates with EONS remain unexplored. Therefore, we sought to examine systematically the hepcidin-ferroportin interplay in the mother, fetus and placenta in the setting of IAI and EONS. We hypothesized that premature neonates with EONS demonstrate a significant increase in circulating hepcidin resulting in altered ferroportin expression at the maternal-fetal interface.
Materials and Methods
Study population, definitions and research design.
We studied 92 mother-newborn dyads affected by preterm birth. Women were recruited following admission to Labor and Delivery ward or to Maternal Special Care antepartum unit at Yale New Haven Hospital. All newborns were admitted to the Yale Newborn Special Care Unit (NBSCU). Enrollment was based on the decision to perform an amniocentesis based on clinical suspicion of IAI in women presenting with symptoms of preterm labor, preterm premature rupture of membranes (PPROM), advanced cervical dilatation (≥3 cm), and/or uterine contractions intractable to tocolysis. The decision to perform amniocentesis was made by clinicians responsible for the care of the patient, independent of our study protocol. The Human Investigation Committees of Yale University, The Ohio State University, and Nationwide Children’s Hospital approved the study and all women signed informed consent. Inclusion criteria were: consecutive singleton pregnancies, gestational age (GA) at enrollment ≥ 23 weeks, no structural fetal anomalies, symptoms of preterm labor ≤ 34 weeks GA, who had a clinical indication for amniocentesis to rule-out IAI. Exclusion criteria consisted of GA < 23 weeks, anhydramnios, human immunodeficiency or hepatitis viral infections. GA was determined based on last menstrual period and ultrasound information obtained prior to 20 weeks GA.18 Preterm labor was defined as the presence of regular uterine contractions and documented cervical effacement and/or dilatation in patients <37 weeks GA. The diagnosis of PPROM was confirmed by vaginal “pooling”, “nitrazine”, “ferning” or by an amniocentesis-dye positive test. No digital exams were permitted, and in the absence of infection, PPROM was managed expectantly. Patients received corticosteroids for fetal lung maturity and antibiotic therapy in accordance to the American Congress of Obstetrics and Gynecology (ACOG) recommendations.6 Delivery was performed for routine obstetrical indications including amniocentesis results suggestive of IAI. The decision to deliver was made by the clinical team responsible for the care of the patient. The neonatology resuscitation team was present at the time of delivery for all cases.
For clinical purposes, neonatal blood specimens and microbial cultures were obtained within 2 h from the time of birth. A diagnosis of EONS was based on clinical symptoms corroborated with hematological and microbiological laboratory results.19, 20 EONS was defined as either confirmed or suspected sepsis at <72 h after birth and dichotomized into present or absent. Any two or more of the following hematological criteria were used as indicators of suspected EONS: 1) absolute neutrophil count of <7,500 or >14,500 cells /mm3; 2) absolute band count >1,500 cells/mm3; 3) immature/total neutrophil (I:T) ratio >0.16; 4) platelet count <150,000 cells/ mm3. EONS was considered confirmed in the presence of positive neonatal blood culture results. Per institutional protocol, all neonates with EONS received antibiotic therapy.
Biological samples.
Amniotic fluid (AF) was collected by ultrasound-guided amniocentesis for all the participants. After retrieval, AF was cultured for aerobic and anaerobic bacteria, Ureaplasma urealyticum, and Mycoplasma hominis. Additional clinical laboratory tests performed for the purpose of diagnosing (IAI) and/or inflammation included glucose concentration, lactate dehydrogenase (LDH), Gram stain, and white blood cell (WBC) count. For clinical management, an AF glucose cut-off of ≤15 mg/dL and LDH levels ≥419 U/L were considered suggestive of IAI.21, 22 In the present study, IAI was defined by positive AF culture or positive Gram stain.
Maternal blood samples were retrieved by venipuncture within 1 h prior to amniocentesis. Plasma was collected on sodium citrate. Serum and plasma samples were spun at 3,000×g at 4°C for 20 min. The supernatant was aliquoted in sterile polypropylene tubes and immediately stored at –80°C until hepcidin and IL-6 levels were measured.
Umbilical cord blood was obtained by aseptic puncture of the clamped umbilical vein at the time of delivery. Acid-base status was determined within 10 min of delivery. Immediately following collection, cord blood was centrifuged at 1,000×g for 15 min. Serum was aliquoted in sterile polypropylene tubes and stored at –80°C until hepcidin and IL-6 levels were measured.
Immediately following delivery of the placenta, a central cotyledon was dissected and a villous biopsy free of decidua was snap frozen in liquid nitrogen and maintained at –80°C for use in quantitative real-time PCR studies. A full thickness adjacent placental tissue specimen was fixed in formalin for immunohistochemical analysis.
Pathologic examination of the placenta.
For clinical purposes, histological examination of the placentas was performed by a perinatal pathologist unaware of the clinical presentation or outcome. From each placenta, sections of chorionic plate, fetal membranes, and umbilical cord were examined systematically for the presence or absence of chorioamnionitis and funisitis. Three histological stages of chorioamnionitis were complemented by the histological grading system devised by Salafia et al., which includes four grades of inflammation of the amnion, chorion-decidua and umbilical cord.23, 24
Neonatal liver specimens.
Neonatal liver sections fixed in formalin were obtained from autopsies performed in the Nationwide Children’s Hospital Department of Pathology and Laboratory Medicine. Specimens were divided into culture proven sepsis related (n = 4) and non-sepsis related (n = 4) causes of death (alveolar capillary dysplasia, n = 1; congenital heart defect, n = 3). Immunohistochemical analyses for hepcidin and ferroportin expression were performed as described below.
Immunoassays for hepcidin and IL-6.
Enzyme-linked immunosorbent assays for hepcidin-25 (DRG Diagnostics, Springfield, NJ) and IL-6 (Pierce-Endogen, Rockford, IL) were performed in duplicate according to manufacturers’ instructions by investigators unaware of the maternal blood and umbilical cord blood sample origin. The minimal detectable concentration for maternal and umbilical cord plasma hepcidin was 1 ng/mL and 3 ng/mL, respectively. The minimal detectable concentration for IL-6 in both compartments was 1 pg/mL. Inter- and intra- assay coefficients of variation were <10%.
Histology and immunohistochemistry.
Immunohistochemistry was used to localize placental expression of hepcidin and ferroportin in a subset of cases in the study: No IAI & No EONS (idiopathic preterm birth [iPTB], n = 12), Yes IAI & No EONS (n = 6), and Yes IAI & Yes EONS (n = 6 of which culture confirmed EONS, n = 3). Paraffin sections (5 μm) of placental villous tissue were deparaffinized in xylene and rehydrated with graded ethanol to potassium-phosphate-buffered saline solution, pH 7.2. Following antigen retrieval with citrate buffer, the sections were pretreated with 1% hydrogen peroxide for 15 min and then incubated overnight at 4°C with hepcidin primary antibody (1:50, rabbit anti-human, Cat. no. ab30760, Abcam, Cambridge, MA). Matched sections were incubated with a primary antibody to ferroportin (1:250, rabbit anti-human ferroportin antibody, Cat. no. ab85370, Abcam). Negative control sections were incubated with non-immune rabbit IgG at an equivalent dilution (Cat. no. I5256, Sigma-Aldrich, St. Louis, MO). The slides were then incubated for 1 h at room temperature with biotinylated donkey anti-rabbit IgG (1:600, Jackson Immunochemicals, West Grove, PA). Immunohistochemical staining was performed with avidin-biotin staining (VECTASTAIN® Elite ABC, Vector Laboratories, Burlingame, CA) with NovaRED as the chromogen solution. The tissue sections were dehydrated in graded ethanols, cleared, and mounted. Histological scores were assigned by two independent investigators, visualizing three different areas from each slide. Staining intensity of villous stroma versus the outer trophoblast layer was graded using a semi quantitative 0–5 scale. Non-heme iron was visualized in the same sections using the DAB-enhanced Turnbull method.25
Quantitative real-time PCR procedures and primer sequences.
RNA was extracted from placental villous tissue biopsies of a subset of 11 cases in the study: No IAI & No EONS (iPTB, n = 7) and Yes IAI & Yes EONS (n = 4). Total RNA was isolated using TRIzol® Reagent (Invitrogen Life Technologies, Carlsbad, CA) with homogenization, followed by chloroform separation, isopropanol precipitation and rehydration with nuclease free water. Reverse transcription was carried out with Superscript® II Reverse Transcriptase (Invitrogen Life Technologies) using oligo(deoxythymidine) primers to synthesize first strand complementary DNA (cDNA). The following TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) were used for qRT-PCR: hepcidin: Hs00221783_m1, ferroportin: Hs00205888_m1, Hs00265497_m1 (RPL30); and Hs99999907_m1 (B2M). Each 20 μl reaction consisted of 1 μl cDNA (500 ng), 1 μl of TaqMan Gene Expression Assay, 10 μl TaqMan Fast Advanced Master Mix (Applied Biosystems), and 8 μl of nuclease free water. Amplifications were performed on the StepOnePlus Real-Time PCR System (Applied Biosystems).
Statistical analysis.
Data normality was tested using the Shapiro-Wilk method, and reported as median and interquartile range (IQR). Data were analyzed using Mann-Whitney Rank Sum test, Student-t test or Chi-square tests as appropriate. Spearman correlations were used to measure co-linearity between the selected independent variables. ELISA data was log-transformed before statistical testing. Multivariable linear regression was used to correct for influences of GA and other variables. Data analysis was performed with Sigma Stat, version 12.5 (RockWare, Golden, CO). P < 0.05 was considered significant in all studies.
Results
The prevalence of IAI in our study population was 76% (70/92) while that of EONS was 45% (41/92). The demographics and clinical characteristics of the maternal and neonatal study population grouped by presence or absence of EONS are presented in Table 1. Infants diagnosed with EONS were more likely to have a lower GA at birth (p < 0.001), and positive AF culture and IAI (40/41, p < 0.001 for both). Newborns with EONS had lower birth weight and lower Apgar scores. Neonatal sepsis work-up showed hematological indices of babies with EONS were consistent with the definition. Although the majority of EONS remained unconfirmed by blood cultures, babies with EONS had a higher degree of anemia, shown by a lower hemoglobin and hematocrit (p < 0.001 for both).
Table 1.
Demographic and clinical characteristics
| Variable | Yes EONS (n = 41) |
No EONS (n = 51) |
p-Value |
|---|---|---|---|
| Demographic and clinical characteristics | |||
| Maternal age (y)a | 26 [21–34] | 30 [22–35] | 0.637 |
| African-American raceb | 20 (49) | 25 (49) | 0.982 |
| PPROMb | 20 (49) | 30 (59) | 0.402 |
| Clinical chorioamnionitisb | 4 (10) | 8 (16) | 0.401 |
| Positive amniotic fluid Gram stainb | 36 (88) | 21 (41) | 0.003 |
| Positive amniotic fluid cultureb | 40 (98) | 24 (47) | <0.001 |
| Intra-amniotic infection (IAI)b | 40 (98) | 30 (59) | <0.001 |
| Gestational age at delivery (wks)a | 25 [24–27] | 29 [28–32] | <0.001 |
| Birthweight (g)a | 780 [675–968] | 1,370 [1,020–1,930] | <0.001 |
| Cesarean deliveryb | 12 (29) | 24 (47) | 0.091 |
| 1 min Apgar <7b | 30 (73) | 21 (41) | 0.003 |
| 5 min Apgar <7b | 17 (41) | 7 (14) | 0.004 |
| Arterial pHa | 7.32 [7.25–7.35] | 7.31 [7.28–7.35] | 1.000 |
| Funisitisbc | 0.003 | ||
| Absent | 10 (25) | 31 (61) | |
| Mild (grade 1 or 2) | 7 (18) | 4 (8) | |
| Severe (grade 3 or 4) | 23 (57) | 16 (31) | |
| Neonatal sepsis workup | |||
| Neonatal ANC (cells/mm3)a | 4,050 [1,948–7,165] | 3,861 [2,185–6,630] | 0.594 |
| Neonatal ABC (cells/mm3)a | 2,550 [1,602–3,808] | 270 [0–672] | <0.001 |
| I:T ratio (%)a | 19 [13–25] | 3 [0–7] | <0.001 |
| Hemoglobin (mg/dL)a | 13.1 [12.1–14.2] | 14.5 [13.3–16.1] | <0.001 |
| Hematocrit (%)a | 40.9 [37.3–43.8] | 44.5 [41.2–50.7] | <0.001 |
| Platelets (cells × 1000/mm3)a | 233 [157–329] | 244 [213–286] | 0.396 |
| Culture confirmed EONSb | 4 (11) | 0 (0) | 0.028 |
Data presented as median [interquartile range] and analyzed by Mann Whitney tests.
Data presented as n (%) and analyzed by Fisher Exact test.
Pathology results missing in one case
Abbreviations: ANC, absolute neutrophil count; ABC, absolute band count; I:T ratio, immature:total neutrophil ratio.
Newborns with EONS had higher cord blood hepcidin concentrations compared to those without EONS (p = 0.004; Fig. 2A). The difference remained significant after correction for GA and IAI status (p = 0.018). In contrast, no EONS-related hepcidin difference was observed in maternal blood (Fig. 2B). Overall, cord blood hepcidin levels did not correlate with GA at birth (r = −0.068, p = 0.522), birth weight (r = −0.109, p = 0.302), or maternal blood hepcidin concentration (r = 0.213, p = 0.088). However, cord blood hepcidin correlated significantly with cord blood IL-6 concentration (r = 0.379, p < 0.001; Fig. 2C). Moreover, newborns with severe funisitis (grades 3–4) had higher cord blood hepcidin compared to newborns with mild or absent funisitis (grades 0–2) (p = 0.027; Fig. 2D). A significant relationship with cord blood IL-6 and severity of funisitis was maintained after correction for GA and EONS status (p < 0.05).
Fig. 2.
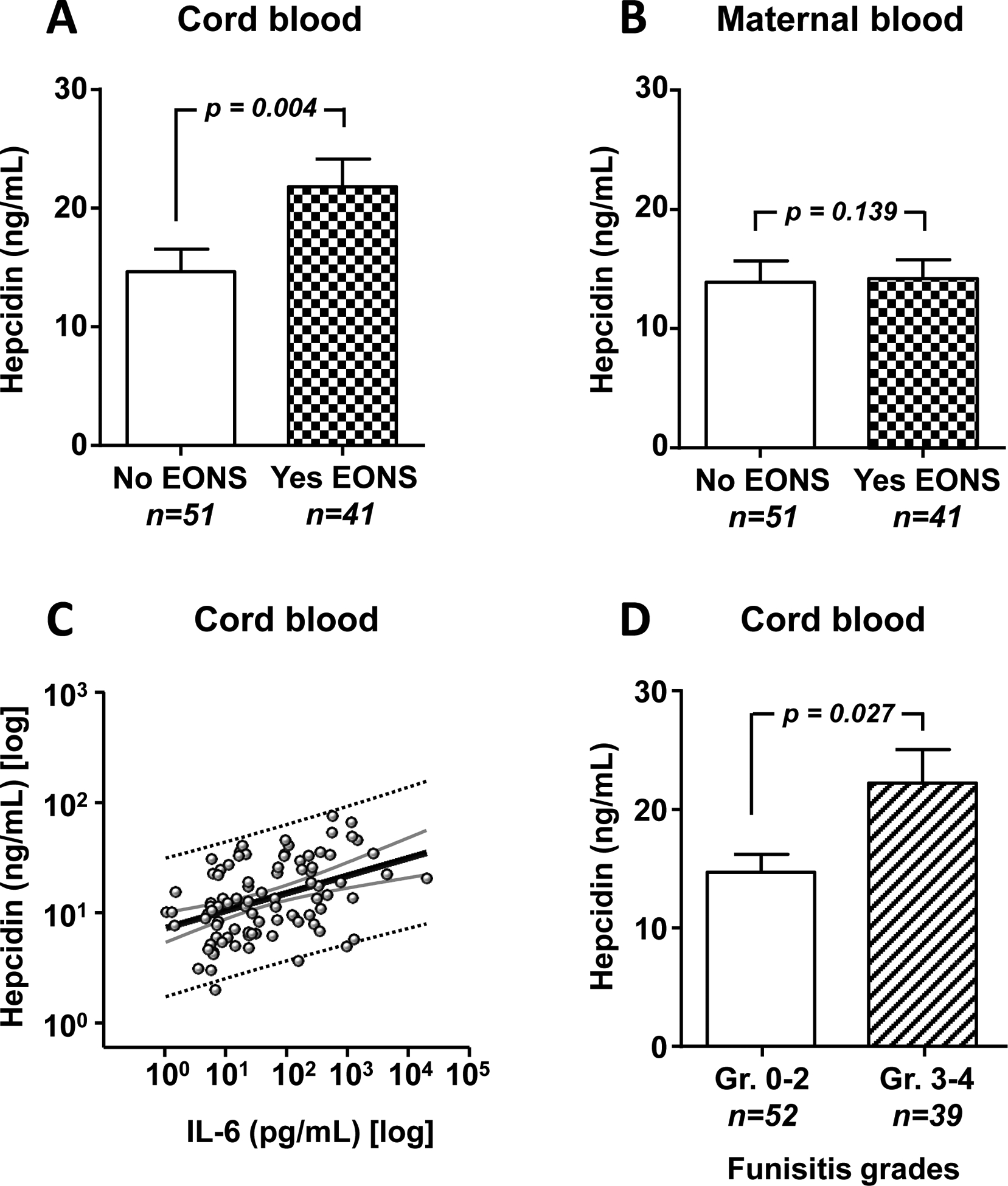
Relationships between the umbilical cord blood hepcidin levels and EONS, IL-6 levels, and funisitis, and between maternal blood hepcidin and EONS. (A) Hepcidin levels in cord blood of newborns with (Yes EONS) and without (No EONS) a clinical diagnosis of EONS. (B) Hepcidin levels in corresponding maternal blood samples. (C) Distribution of cord blood hepcidin levels (Y axis) in relation to IL-6 levels (X axis). (D) Cord blood hepcidin levels by severity of funisitis (Gr. 0–2, absent or mild vs. Gr. 3–4, severe). Data presented as mean and standard error of the mean (SEM) and compared using Mann-Whitney Rank sum tests after logarithmic transformation.
Next, we asked the question whether the elevated hepcidin concentration observed in fetal circulation in newborns with EONS is associated with altered expression of hepcidin and ferroportin in the placenta, which would impair placental transport of iron. In iPTB placenta, hepcidin immunostaining was weak to near absent in both villous stroma and trophoblast (Fig. 3A, arrow). Ferroportin staining was largely confined to cytotrophoblasts, syncytiotrophoblast and fetal endothelium, which sharply delineated the villous stroma from maternal and fetal vascular spaces (Fig. 3B, arrow). In IAI, placental hepcidin staining appeared overall diffuse, yet more conspicuous than in iPTB placenta. At higher magnification, the most intense hepcidin staining was observed at the apical edge of the villi (Fig. 3C, arrow) and along fetal capillaries (Fig. 3C, arrowhead). A correspondingly diffuse stromal staining pattern was observed for ferroportin with contiguous patches of intensely stained syncytiotrophoblast discernible at higher magnification in IAI placentas (Fig. 3D, arrow). Semi quantitative scoring reflected the change in ferroportin staining pattern with a decrease in staining intensity of the villous trophoblast and syncytiotrophoblast (Fig. 3E, 2-way ANOVA p < 0.001) and increase in stromal staining (Fig. 3E, 2-way ANOVA p = 0.03). The shift in staining from the outer villous trophoblast to the villous stroma (VT-VS ratio) was significantly correlated (multivariable analysis R = 0.683, p = 0.001) with the severity of chorionic plate inflammation (p < 0.001) and severity of funisitis (p = 0.009), independent of cord blood hepcidin, GA at birth, or postnatal EONS status (suspected or confirmed). Together, the placental staining pattern described above supports the notion that fetal exposure to IAI results in degradation of ferroportin at the maternal-fetal interface. No significant non-heme iron deposits were observed in the placenta.
Fig. 3.
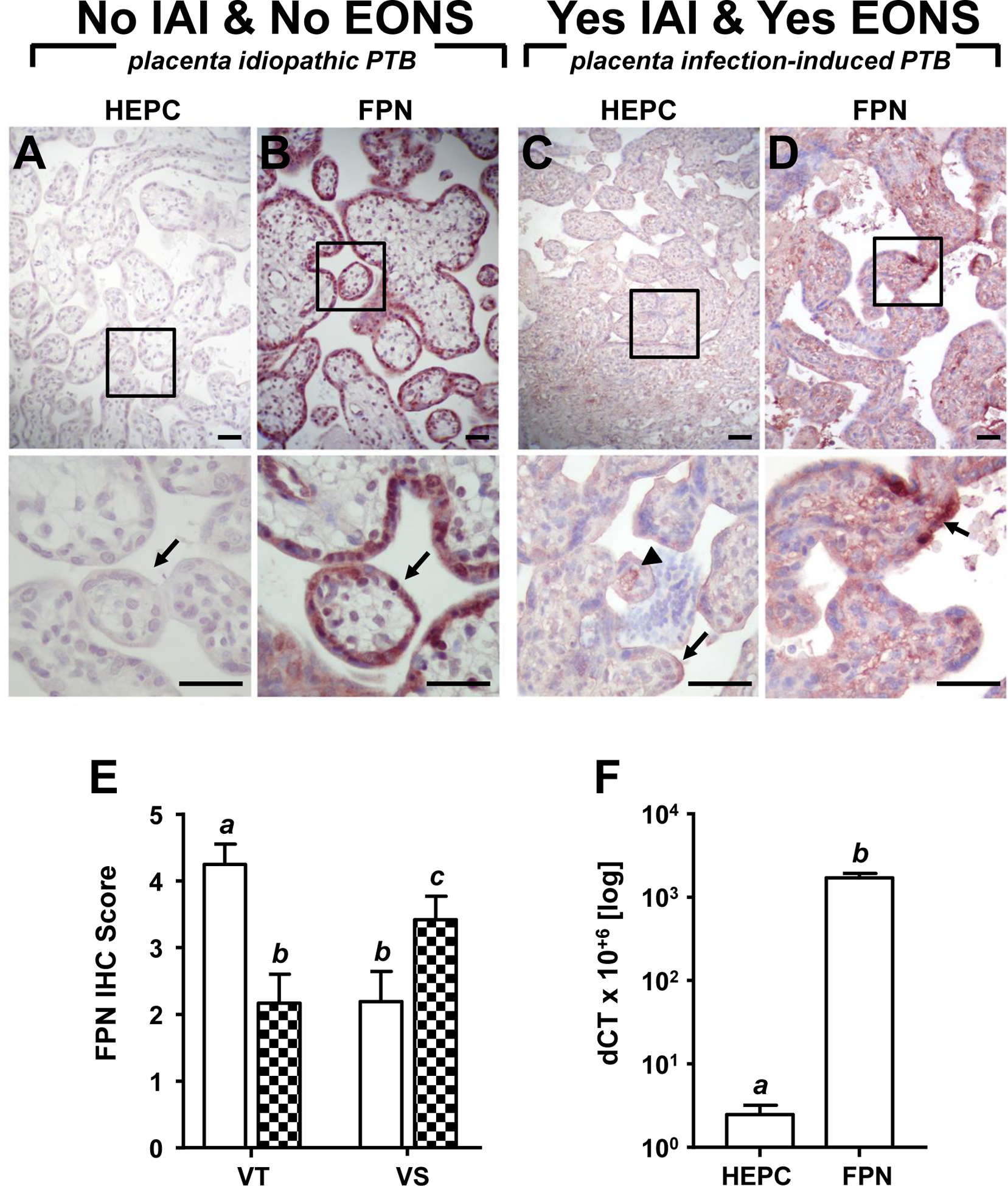
Representative placental immunohistochemical staining of hepcidin (HEPC) and ferroportin (FPN) in idiopathic preterm birth (iPTB, 29 weeks GA at birth) and preterm birth in the context of infection-induced preterm birth (IAI)-determined early-onset sepsis (EONS, 30 weeks GA at birth). (A) Minimal HEPC staining in villous stroma and trophoblast (arrow), and (B) intense FPN restricted to cytotrophoblasts, syncytiotrophoblast and fetal endothelium, delineating villus stroma from maternal and fetal vascular spaces (arrow) observed in iPTB placenta. (C) Intense HEPC staining at apical edge of villi (arrow) and along fetal capillary (arrowhead), and (D) diffuse FPN stained stroma with contiguous patches of intensely stained syncytiotrophoblast (inset, arrow) in IAI placenta. Upper panels, 200× magnification; bottom panels represent boxed region in upper panels, 600× magnification; scale bar = 50 μm (all panels). (E) Semi quantitative score of placental ferroportin staining intensity in villous trophoblast (VT) and villous stroma (VS) in cases of iPTB (open bars, n = 12) and preterm birth in the context of IAI (n = 12, checkered bars). (F) Relative hepcidin and ferroportin mRNA abundance in preterm placenta. Means with different letters are statistically different at a value of p < 0.05.
Using real-time PCR, we determined that placental expression of hepcidin at the mRNA level was at least three orders of magnitude lower than that of ferroportin suggesting that the placenta is not a major site of hepcidin synthesis (Fig. 3F). Placental ferroportin (but not hepcidin) mRNA correlated with cord blood hepcidin (r = 0.46, p = 0.039) and severity of funisitis (r = 0.500, p = 0.018).
As the liver is the primary site of hepcidin synthesis26, we extended the immunohistochemical staining to hepatic sections of newborns that died shortly after birth. In those that died of non-septic causes, hepcidin signal was localized in hepatocytes in a granular and dispersed intracellular pattern (Fig. 4A, arrow). Hepatic ferroportin staining in non-septic newborns was more prominent around the portal veins (zone 1) (Fig. 4B, arrow) while lower levels were seen around the central vein (zone 3) (Fig. 4B). In contrast, in livers of newborns that died in the context of EONS, intense hepcidin staining was noted lining the sinusoids (Fig. 4C). Concurrently, hepatocyte ferroportin signal shifted to a diffuse pattern that became conspicuous around the central vein (Fig. 4D). Compared to newborns who died of non-septic causes (Fig. 4E), those that died from sepsis (Fig. 4F) had increased liver iron stores suggesting that the changes in hepatic hepcidin and ferroportin were associated with hepatic sequestration of iron.
Fig. 4.
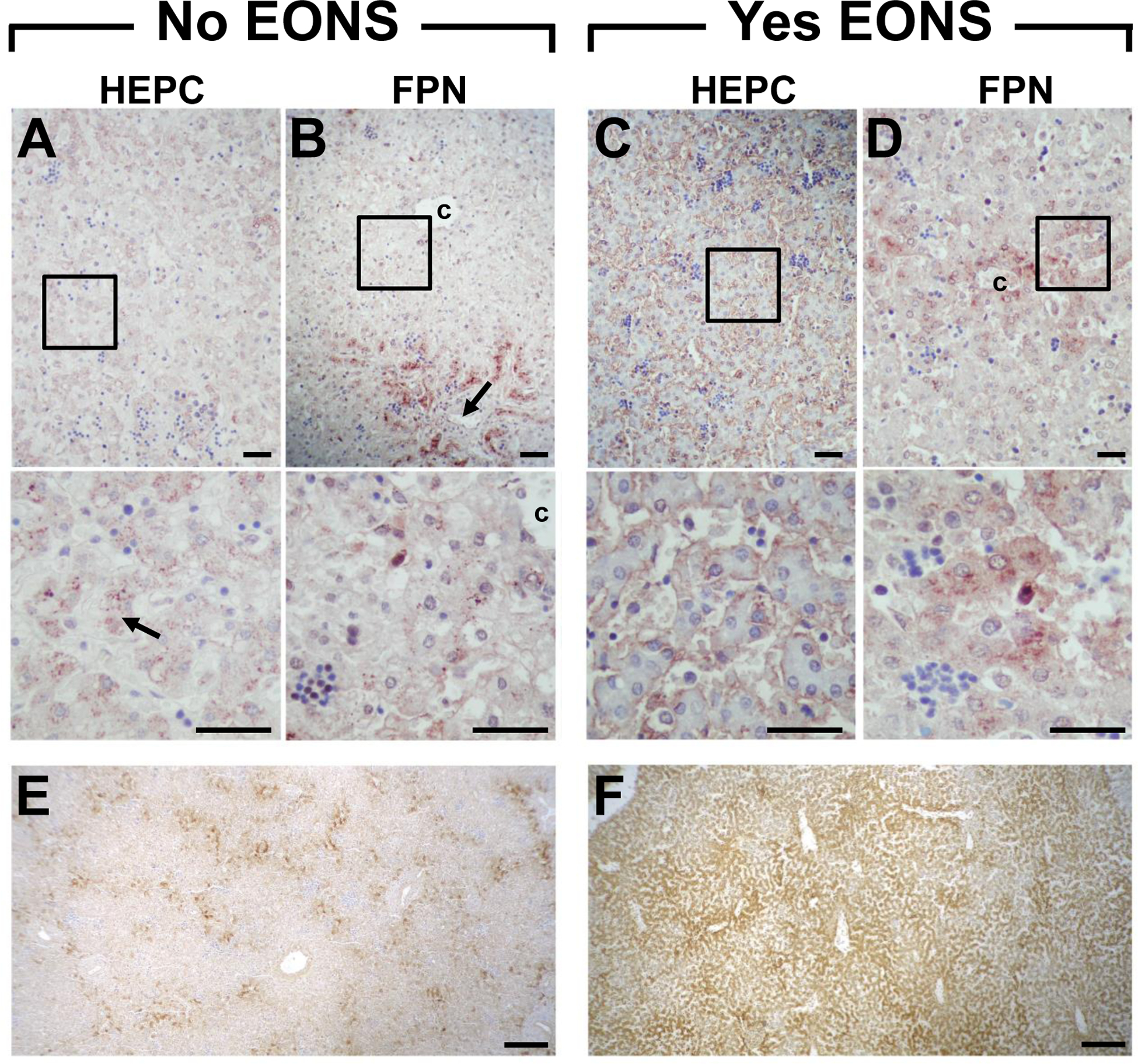
Representative immunohistochemical staining of liver hepcidin (HEPC, A&C) and ferroportin (FPN, B&D) of term newborns who died ~1 h postnatal. Liver of newborn who died of a non-septic cause (congenital diaphragmatic hernia, No EONS) shows (A) HEPC localized in hepatocytes in a granular and dispersed intracellular pattern (arrow), and (B) FPN staining more prominent around portal veins (arrow) and central vein. Liver of newborn who died of culture confirmed early-onset sepsis (Yes EONS) shows (C) intense HEPC staining lining sinusoids, and (D) diffuse FPN staining more conspicuous near central vein. Magnified insets of boxed regions are shown below the respective panel (A-D). Histochemical staining for liver iron stores for No EONS (E) and Yes EONS (F) sections shown in A-D. Original magnification: upper panels 200×; middle panels 600×; bottom panels 40×. Scale bar = 50 μm (A-D); scale bar = 200 μm (E&F); c = central vein.
Discussion
The results of this study support a hepcidin-ferroportin interaction across the maternal-fetal interface that is modified in the context of IAI-determined EONS. Our results add to the previously published reports of elevated cord and peripheral blood hepcidin in the setting of EONS and late onset sepsis.16, 17 As the hepcidin-ferroportin interaction is a regulator of iron transport across the placenta and of utilization of iron stores in both the antenatal and postnatal period, our findings support the premise that intra-uterine exposure to IAI likely induces an iron deficient state of the fetus. The observed significantly lower hematocrit in newborns with EONS is supportive of a relatively iron deficient state in this setting. Premature neonates are considered iron deficient even in the absence of EONS as they have been deprived of the ongoing transplacental transfer of iron, which reaches its peak only in the third trimester.27 Accelerated body growth velocities, low erythropoietin, frequent blood draws and suboptimal iron replacement therapies further deplete postnatal iron stores of premature babies.28 This is problematic because a strong association exists between iron deficiency in infancy and long-term neurodevelopmental impairment.29 Although it appears that early enteral iron supplementation may prevent some neurocognitive and psychomotor deficits30, iron excess is postulated to generate free radical injury that can also adversely impact the development of premature infants.28 Our data supports the premise that premature newborns with EONS have a more profound state of iron deficiency compared to those without EONS. Although up-regulation of hepcidin in cord blood of neonates with EONS was correlated with IL-6 levels, and it may be an effective innate defense mechanism to thwart ongoing bacterial replication, the relative lack of iron at critical developmental stages may be a contributor to adverse composite morbidity. Because hepcidin was not up regulated in the maternal compartment in relation to either IAI or EONS, it cannot be used as an antenatal diagnostic tool for fetal infection. This further supports previous reports of compartmentalization of the maternal-fetal inflammatory response pathways.31
Based on our results, several mechanisms can be responsible for the relatively iron deficient state on neonates with EONS: 1) degradation of placental ferroportin that is intended to limit the transfer of iron from the mother to the fetus through the placenta, 2) degradation of ferroportin in the periportal space to limit iron that is anticipated to reach the liver, and 3) sequestration of iron in the liver. Our histological data indicated a change in the ferroportin staining pattern in the placenta of EONS neonates. Specifically, decreases in ferroportin staining intensity of the villous trophoblast and syncytiotrophoblast, and an increase in stromal immunoreactivity in EONS. This change in pattern possibly contributes to limiting iron transfer to the fetus through the placenta because, in EONS, the cell exporter, ferroportin, is primarily located in the stroma and not at the maternal-fetal interface where the exchange usually takes place. Changes in placental hepcidin and ferroportin were observed to correlate with histological evidence of fetal inflammation (funisitis, chorionic plate inflammation) even in the absence of culture-confirmed EONS. We are aware of the imprecision in the clinical diagnosis of EONS; therefore, a strength of our study compared to previous reports of elevated hepcidin associated with EONS is the precise knowledge of the infection status and inflammation severity of AF and of the placenta.
To our knowledge this is the first study to investigate the expression pattern of hepcidin and ferroportin in the liver of neonates who died from sepsis. We observed significant differences in the expression pattern for both hepcidin and ferroportin in EONS. Hepatic ferroportin staining in newborns that died of causes unrelated to sepsis was more prominent around the portal veins implying increased role in iron metabolism once this element is absorbed from the gut. Conversely, intense hepcidin staining was noted lining the sinusoids in newborns that died in the context of EONS. Because the Kupffer cells are located inside the sinusoids and can take up and destroy bacteria, the observed histologic pattern is biologically plausible.
In conclusion, premature fetuses with EONS have elevated circulating hepcidin levels, which likely relates to the altered expression of ferroportin in the liver and placenta. Activation of the fetal hepcidin-ferroportin axis appears to play a role in the pathophysiology of EONS independent of the maternal response to intrauterine infection and may impact the postnatal hematologic adaptation of affected newborns.
Acknowledgements:
The authors would like to thank Dr. Stephen Thung for his assistance in patient recruitment and input on study design. We would also like to thank Lisa Zhao and Taryn Summerfield for their assistance with sample preparation and for providing valuable technical expertise. We are grateful to the patients who consented for their participation as part of this research.
Funding: This work was supported by grants from National Institutes of Health (NIH) Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) R01HD062007 (CSB&IAB) and R01HD047321 (IAB). In addition, the Division of Maternal Fetal Medicine of The Ohio State University College of Medicine and The Center for Perinatal Research of The Research Institute at Nationwide Children’s Hospital participated with funds allocated for MFM fellow research projects. The funding sources had no involvement in study design, interpretation of data, writing of the report or decision to submit the paper for publication.
References
- 1.Lasocki S, Gaillard T, Rineau E. Iron is essential for living! Crit Care 2014;18:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang C Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell 2014; 5:750–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unger EL, Bianco LE, Jones BC, Allen RP, Earley CJ. Low brain iron effects and reversibility on striatal dopamine dynamics. Exp Neurol 2014;261:462–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo S, Frazer DM, Anderson GJ. Iron homeostasis: transport, metabolism, and regulation. Curr Opin Clin Nutr Metab Care 2016;19:276–281 [DOI] [PubMed] [Google Scholar]
- 5.Andrews SC, Robinson AK, Rodríguez-Quiñones F. Bacterial iron homeostasis. FEMS Microbiol Rev 2003;27:215–237 [DOI] [PubMed] [Google Scholar]
- 6.Parrow NL, Fleming RE, Minnick MF. Sequestration and scavenging of iron in infection. Infect Immun 2013; 81:3503–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Rovin BH. Beyond anemia.: hepcidin, monocytes and inflammation. Biol Chem 2013; 394:231–238 [DOI] [PubMed] [Google Scholar]
- 8.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science 2012;338:768–772 [DOI] [PubMed] [Google Scholar]
- 9.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276:7806–7810 [DOI] [PubMed] [Google Scholar]
- 10.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001;276:7811–7819 [DOI] [PubMed] [Google Scholar]
- 11.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002;110:1037–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med 2005;352:1011–1023 [DOI] [PubMed] [Google Scholar]
- 14.Gambling L, Lang C, McArdle HJ. Fetal regulation of iron transport during pregnancy. Am J Clin Nutr 2011;94:1903S–1907S [DOI] [PubMed] [Google Scholar]
- 15.Buhimschi IA, Buhimschi CS. Proteomics/diagnosis of chorioamnionitis and relationships with the fetal exposome. Semin Fetal Neonatal Med 2012;17:36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cizmeci MN, Kara S, Kanburoglu MK, Simavli S, Duvan CI, Tatli MM. Detection of cord blood hepcidin levels as a biomarker for early-onset neonatal sepsis. Med Hypotheses 2014; 82:310–312 [DOI] [PubMed] [Google Scholar]
- 17.Wu TW, Tabangin M, Kusano R, Ma Y, Ridsdale R, Akinbi H. The utility of serum hepcidin as a biomarker for late-onset neonatal sepsis. J Pediatr 2013;162:67–71 [DOI] [PubMed] [Google Scholar]
- 18.Hadlock FP, Deter RL, Harrist RB, Park SK. Computer assisted analysis of fetal age in the third trimester using multiple fetal growth parameters. J Clin Ultrasound 1983;11:313–316 [DOI] [PubMed] [Google Scholar]
- 19.Bhandari V, Wang C, Rinder C, Rinder H. Hematologic profile of sepsis in neonates: neutrophil CD64 as a diagnostic marker. Pediatrics 2008;121:129–134 [DOI] [PubMed] [Google Scholar]
- 20.Buhimschi CS, Buhimschi IA, Abdel-Razeq S, et al. Proteomic biomarkers of intra-amniotic inflammation: relationship with funisitis and early-onset sepsis in the premature neonate. Pediatr Res 2007;61:318–324 [DOI] [PubMed] [Google Scholar]
- 21.Garry D, Figueroa R, Aguero-Rosenfeld M, Martinez E, Visintainer P, Tejani N. A comparison of rapid amniotic fluid markers in the prediction of microbial invasion of the uterine cavity and preterm delivery. Am J Obstet Gynecol 1996;175;1336–1341 [DOI] [PubMed] [Google Scholar]
- 22.Edwards RK, Clark P, Locksmith Gregory J, Duff P. Performance characteristics of putative tests for subclinical chorioamnionitis. Infect Dis Obstet Gynecol 2001;9:209–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naeye RL. Disorders of the placenta and decidua In: Naeye RL, ed. Disorder of the Placenta, Fetus and Neonate: Diagnosis and Clinical Significance. St. Louis: Mosby; 1992:118–247 [Google Scholar]
- 24.Salafia CM, Weigl C, Silberman L. The prevalence and distribution of acute placental inflammation in uncomplicated term pregnancies. Obstet Gynecol 1989;73:383–389 [PubMed] [Google Scholar]
- 25.Meguro R, Asano Y, Iwatsuki H, Shoumura K. Perfusion-Perls and -Turnbull methods supplemented by DAB intensification for nonheme iron histochemistry: demonstration of the superior sensitivity of the methods in the liver, spleen, and stomach of the rat. Histochem Cell Biol 2003;120:73–82 [DOI] [PubMed] [Google Scholar]
- 26.Zhang AS, Xiong S, Tsukamoto H, Enns CA. Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood 2004;103:1509–1514 [DOI] [PubMed] [Google Scholar]
- 27.Strauss RG. Anaemia of prematurity: pathophysiology and treatment. Blood Rev 2010;24:221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller KF, Lorenz L, Poets C, Westerman M, Franz A. Hepcidin Concentrations in serum and urine correlate with iron homeostasis in preterm infants. J Pediatr 2012;160:949–953 [DOI] [PubMed] [Google Scholar]
- 29.Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev 2006;64:S34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinmacher J, Pohlandt F, Bode H, Sander S, Kron M, Franz AR. Randomized trial of early versus late enteral iron supplementation in infants with a birth weight of less than 1301 grams: neurocognitive development at 5.3 years’ corrected age. Pediatrics 2007;120:538–546 [DOI] [PubMed] [Google Scholar]
- 31.Dulay AT, Buhimschi IA, Zhao G, et al. Compartmentalization of acute phase reactants interleukin-6, C-reactive protein and procalcitonin as biomarkers of intra-amniotic infection and chorioamnionitis. Cytokine 2015;76:236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]