

Abstract
Manganese tricarbonyl complexes are promising catalysts for CO2 reduction, but complexes in this family are often photo-sensitive and decompose rapidly upon exposure to visible light. In this report, synthetic and photochemical studies probe the initial steps of light-driven speciation for Mn(CO)3(Rbpy)Br complexes bearing a range of 4,4′-disubstituted-2,2′-bipyridyl ligands (Rbpy, R = tBu, H, CF3, NO2). Transient absorption spectroscopy measurements for the Mn(CO)3(Rbpy)Br coordination compounds with R = tBu, H, and CF3 in acetonitrile reveal ultrafast loss of a CO ligand on the femtosecond timescale, followed by solvent coordination on the picosecond timescale. The Mn(CO)3(NO2bpy)Br complex is unique among the four compounds in having a longer-lived excited state that does not undergo CO release or the subsequent solvent coordination. The kinetics of photolysis and solvent coordination for the light-sensitive complexes depend on the electronic properties of the di-substituted bipyridyl ligand. The results implicate roles for both metal-to-ligand charge transfer (MLCT) and dissociative ligand field (dd) excited states in the ultrafast photochemistry. Taken together, the findings suggest that more robust catalysts could be prepared with appropriately designed complexes that avoid crossing between the excited states that drive photochemical CO loss.
Graphical Abstract
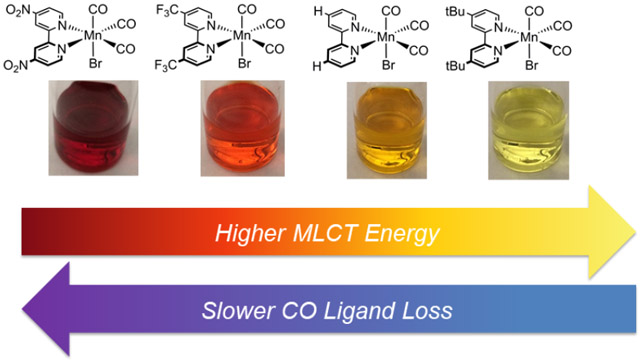
TOC Synopsis
In the presence of light, [Mn(CO)3] complexes bearing bipyridyl-type ligands decompose via CO loss, with subsequent solvent coordination in MeCN or other reactions in CHCl3. Ultrafast transient absorption studies reveal formation of both the five-coordinate and solvento intermediates involved in the photo-speciation process.
Introduction
Manganese tricarbonyl complexes of the form Mn(CO)3(Rbpy)Br (Rbpy = 4,4′-disubstituted–2,2′-bipyridyl) are promising catalysts for conversion of carbon dioxide (CO2) into carbon monoxide and/or formate, valuable precursors to chemical feedstocks and commodity chemicals.1,2 The synthetic chemistry for the Mn complexes was developed by Wilkinson and co-workers,3 and later extended by Wrighton, Meyer, and others.4 These complexes are remarkable for their ease of preparation and can be synthesized from commercially available or readily prepared ligands, Rbpy, and the synthon Mn(CO)5Br. Similar synthetic chemistry and catalytic properties have been demonstrated with analogous rhenium tricarbonyl complexes, Re(CO)3(Rbpy)Br, which were investigated for catalysis much earlier by Lehn5 and Meyer6, and popularized by Fujita7, Kubiak8, Ishitani9, and others.10 However, considering the significantly greater abundance of manganese, many research groups have continued to develop new [Mn(CO)3]-based catalysts. These efforts have led to a remarkable flourishing of reports showing that a variety of bidentate ligands can support and/or tune the electronic, photo-physical, and catalytic properties of [Mn(CO)3] complexes.11,12,13
A defining feature that distinguishes the rhenium complexes from their manganese analogues is markedly better stability upon irradiation with visible light.14 Importantly, many [Mn(CO)3] complexes are susceptible to speciation15 and/or degradation16 upon exposure to visible light. For example, our preliminary investigations in organic solvents revealed that Mn(CO)3(Rbpy)Br complexes begin to decompose within minutes under ambient fluorescent lighting, as evident from peak broadening in the 1H nuclear magnetic resonance (NMR) spectra. A review describing the photochemistry of a variety of metal carbonyl species provides context to the light sensitivity of Mn(CO)3(Rbpy)Br in solution.17 Specifically, the light sensitivity of many first-row transition metal complexes is often a consequence of the inherent excited-state electronic structure of the compounds. However, as the light sensitivity of the Mn(CO)3(Rbpy)Br complexes presents a potential challenge to their use in electrochemical or, especially, photoelectrochemical systems for CO2 conversion, we became interested in the underlying processes that contribute to speciation and/or degradation.
Separate from their role as catalysts, the light-induced reactivity of manganese complexes makes the [Mn(CO)3] moiety a promising motif for developing a relatively new class of putative therapeutics known as photo-induced carbon monoxide releasing molecules (photo-CORMs).18 Classic work on the photochemical properties of dimanganese decacarbonyl ([Mn(CO)5]2) revealed the susceptibility of low-valent manganese complexes to release CO under irradiation with visible light.19,20 However, [Mn(CO)5]2 absorbs only very weakly in the visible region, which is problematic from the standpoint of therapeutic development because UV light does not readily penetrate the skin.21 The need for efficient activation of photo-CORMs at longer wavelengths has motivated the development of novel [Mn(CO)3]-based compounds that absorb light in the visible region. Toward this goal, Schatzschneider and co-workers developed the first [Mn(CO)3]-based photo-CORMs supported by various tripodal ligands.22 Further work by several research groups has led to numerous platforms and ligand systems in this family that allow effective photo-induced CO release.23 Notably, Mascharak and co-workers highlighted the light sensitivity of Mn(CO)3(Rbpy)Br complexes, demonstrating that Mn(CO)3(Hbpy)Br is capable of visible light induced CO release at 420 nm.24
Despite the potential role of Mn(CO)3(Rbpy)Br complexes in diverse applications ranging from catalysis to phototherapeutics, the photophysical properties of these compounds have received surprisingly little attention. The analogous [Re(CO)3] complexes have been studied with nanosecond and femtosecond transient absorption spectroscopy, as well as time-resolved infrared absorption measurements.25,26 Vlček and co-workers in particular have examined various aspects of the ligand-dependent excited state behavior of [Re(CO)3] complexes bearing diimine-type ligands, providing important insights into electron transfer behavior and the resulting chemical reactivity.27,28 However, to the best of our knowledge, no femtosecond or picosecond pump-probe experiments have examined the fundamental photochemistry of [Mn(CO)3] complexes bearing diimine ligands. Only steady-state experiments involving spectroscopic characterization of the products following bulk photolysis,15 and a nanosecond pulse radiolysis study of the reduction-induced reactivity of Mn(CO)3(tBubpy)Br have been reported.29
In this contribution, we examine the fundamental photochemistry of Mn(CO)3(Rbpy)Br complexes with disubstituted ligands bearing tert-butyl (1), hydryl (2), trifluoromethyl (3), and nitro (4) groups (Chart 1) following exposure to visible light. Ultrafast transient absorption (TA) spectroscopy reveals the loss of a CO ligand on the femtosecond timescale, followed by solvent coordination on the picosecond timescale for 1, 2, and 3 in acetonitrile. The kinetics of these reactions depend on the identity of the substituents at the 4 and 4′ positions of the Rbpy ligand and correlate well with the Hammett parameters associated with the substituent groups. In contrast with the results for 1, 2, and 3, TA spectroscopy and gas chromatography (GC) measurements indicate that 4 does not undergo CO release. Taken together, these observations implicate metal-to-ligand charge transfer (MLCT) and dissociative ligand field (LF) states in driving the photoinduced reactivity, and suggest that the relative energies of these electronic states are tuned through the modification of the supporting ligand. Our results are discussed in the context of developing new design principles that could be used to selectively control light-driven CO release from [Mn(CO)3] complexes.
Chart 1:
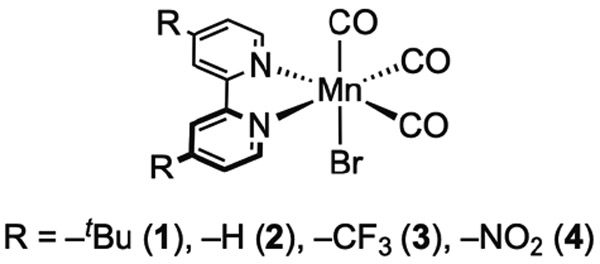
Mn(CO)3(Rbpy)Br complexes 1-4.
Results and Discussion
Synthesis and Characterization
In order to study [Mn(CO)3] complexes with uniformly tuned properties, we prepared compounds that differ only in the electron donating/withdrawing nature of the substituents in the 4 and 4′ positions of the supporting 2,2′-bipyridyl ligand. Complexes 1 and 2 were prepared according to prior reports1,2 and new derivatives 3 and 4 were synthesized and isolated using methods similar to those of Wrighton and co-workers (see Experimental Section for details).4 Overall, no unusual features were encountered for the synthesized complexes. All of the formally manganese(I) complexes were prepared from Mn(CO)5Br and isolated with inner-sphere bromide ligands. The compounds have Cs symmetry, and are soluble in common organic solvents (e.g., acetonitrile, chloroform) (see Figures S1-S5).
The Mn(CO)3(Rbpy)Br complexes have a variety of useful signatures that can be readily interrogated using NMR, IR, and electronic absorption spectroscopies. Trends in the spectra can be correlated with the Rbpy substituents using Hammett parameters.30 However, the traditional Hammett parameter (σ) does not consider stabilization of partial or full charges in reference to inductive and resonance effects of the conjugated ligand. These limitations are accounted for in Brown’s sigma plus parameter (σ+) and Kubota’s sigma minus parameter (σ−), which stabilize partial or full positive and negative charges, respectively.31,32 To determine which parameter is most appropriate for a given system or spectroscopic signature, a series of Hammett plots are constructed to compare goodness-of-fit with σ+, σ, and σ−. For example, the 1H-NMR chemical shifts for the ortho-pyridyl protons of each complex correlate best with Brown’s σ+ parameter (see Figures S6-S7), in agreement with prior literature.33 The correlation with σ+ indicates an inductive ground-state interaction between the ligand framework and the substituents.
Infrared absorption spectroscopy confirms the presence of the fac-tricarbonyl geometry for each of the complexes 1-4 in solution (see Figure S8), and demonstrates that the samples are free of Mn(CO)5Br starting material (associated with absorption bands at 2004 cm−1,2046 cm−1, and 2083 cm−1). Our results from IR spectroscopy are consistent with previous observations of heteroleptic carbonyl complexes, as we observe a systematic increase of the C─O stretching frequencies upon changing the character of the 4,4′-substituents of Rbpy from electron donating to electron withdrawing.34 Hammett plots of the A″-symmetry C─O stretching frequency35 as a function of ligand substituent are best fit by the σ parameter (R2 = 0.993), although the correlations with σ+ (R2 = 0.989) and σ− (R2 = 0.945) are not vastly different from the correlation with σ (see Figure S9). This observed correlation of C─O stretching frequencies is in agreement with previous work on para-substituted isocyanide complexes CpFe(CO)2(CNRPh) and CpMn(CO)2(CNRPh), where Cp is cyclopentadienyl.36,37 However, as C─O stretching modes are affected principally by π-bonding effects, the measured range of 15 cm−1 for the bipyridyl complexes under investigation here is consistent with the expected distal influence of the bipyridyl ligand substituents on the Mn metal center and the CO ligands.
Complexes 1-4 give vividly colored solutions when dissolved in common organic solvents like MeCN (see Figure S10). Figure 1 shows the electronic absorption spectra for all four complexes, and Table 1 lists the transition energies and molar absorptivities for each of the lowest-energy absorption bands. Plotting the energies of the first absorption band for 1-4 as a function of σ− parameter gives an excellent correlation (R2 = 0.998; see Figures S11-S15). On the basis of the molar absorptivity and comparison with related compounds, the lowest-energy absorption bands for 1-4 can all be confidently assigned as having significant MLCT character (See Table 1). These findings agree with prior work on Mn(CO)3(diimine)Br complexes showing that the highest occupied molecular orbital is primarily localized on the Mn center, while the lowest unoccupied molecular orbital has significantly more ligand character, suggesting that the visible transitions are primarily MLCT in nature.38 Plotting the molar absorptivity of the lowest-energy absorption bands of 1-4 as a function of the Hammett parameter also reveals a linear correlation with σ−, where the transition strength increases for more electron-donating substituents (see Figure S16). Thus, the trend in transition strength across all four compounds is roughly linear with σ−, consistent with previous studies of transition metal complexes featuring MLCT behavior.39 A likely explanation for the decreasing transition strength with increasing electron-withdrawing character of the ligand substituents is reduced overlap of the HOMO and LUMO due to increased localization of the LUMO on the electron-withdrawing substituents. The electron-donating groups, in contrast, localize the LUMO closer to the Mn metal center, leading to better overlap with the HOMO and therefore a stronger MLCT transition. Based on this reasoning, the relatively weak transition for 4 follows the trend in MLCT transition strengths set by the other three compounds, even though the lower absorption strength for 4 would typically be associated with a d-d transition. Although we cannot rule out the possibility of some d-d character in the lowest-energy transition for 4, the strong correlation of both the transition energies and strengths with σ− reinforces our assignment of the lowest-energy absorption bands as primarily MLCT in nature for all four compounds.
Figure 1:

Electronic absorption spectra of 1, 2, 3, and 4 in MeCN (left). Hammett plot of MLCT energy in eV as a function of the σ− parameter (right). The best fit line in the Hammett plot reveals a correlation with R2 = 0.998, and a slope of −0.41 ± 0.01.
Table 1:
Hammett parameters of the ligand substituents and selected spectral parameters for complexes 1-4.
| Compound | Hammett parameter (σ−) |
λ (nm, eV) | ε (M−1 cm−1) |
|---|---|---|---|
| 1 | −0.13 | 412, 3.00 | 2426 |
| 2 | 0.0 | 415, 2.99 | 2288 |
| 3 | 0.65 | 457, 2.71 | 1502 |
| 4 | 1.27 | 510, 2.43 | 228 |
Transient absorption spectroscopy
Ultrafast transient absorption (TA) spectroscopy probes the reaction dynamics following MLCT excitation of the [Mn(CO)3] complexes. Based on the shift of the MLCT bands with ligand substitution, we used excitation pulses at 420 nm for 1 and 2, 470 nm for 3, and 510 nm for 4. Figure 2 shows the evolution of the TA spectra for 1-4 in MeCN. The initial excited-state absorption bands for compounds 1-3 are double-peaked and partially decay in ≤1 ps, followed by the delayed appearance of a narrower absorption band at slightly longer wavelength within ~100 ps. The transient spectra are similar for all three compounds at longer time delays as well, except for a red-shift of the TA bands that is similar to the shift of the ground-state MLCT bands across the three compounds. Notably, both the initial excited-state absorption bands and the new features that appear within ~100 ps follow the same trend as the ground-state MLCT bands of 1-3, indicating that the Rbpy ligand remains bound to the Mn center for each of the transient species (see Figure S17).
Figure 2:

Transient absorption spectra of 1, 2, 3, and 4 (top to bottom) in MeCN. Left panel shows spectral evolution, right panel shows the absorption change at a single wavelength. For reference, the ground-state absorption spectra are scaled by 1/10 and shown as grey lines.
The evolution of the TA spectrum for the nitro disubstituted compound (4), also shown in Figure 2, is noticeably different from the other complexes. The initial excited-state absorption band is stronger, narrower, and decays more slowly than in the other compounds. Furthermore, there is not a secondary absorption feature that appears on the ~100 ps timescale in the transient spectrum of compound 4. Unlike 1-3, the shift in wavelength of the initial TA band of 4 also does not follow the same trend as the ground-state MLCT bands. The strongly electron withdrawing nitro groups likely localize the charge in the MLCT state, resulting in a more stable and longer-lived excited state for 4.
Based on the TA spectroscopy and kinetics, as well as the observation of CO in the headspace of irradiated samples using GC analysis (see Figures S18-S19), we propose a pathway for the early speciation process of complexes 1-3 that involves rapid CO loss followed by solvent coordination, as shown in Scheme 1. Optical excitation initially promotes an electron from a metal d-orbital to the π* orbital of the bpy ligand. The MLCT state is likely to be close in energy to a ligand field (LF) state with d-d excitation on the metal that has significant antibonding (σ*) character in the equatorial manganese-carbon bonds.40 The anti-bonding ligand field state is stabilized by increasing the Mn-CO bond length, which results in a curve crossing that allows adiabatic population transfer from the MLCT state to the directly dissociative LF state (Figure 3). Impulsive release of a CO ligand via the LF state generates an electron-deficient (16 e−), five-coordinate intermediate, Mn(CO)2(Rbpy)Br. In acetonitrile, such an intermediate species is likely to bind a solvent ligand in order to recover a more stable 18 e− configuration. Thus, we attribute the new signal that rises on the tens of picoseconds timescale to the formation of a solvent-coordinated complex. Reduced electron back-bonding by the newly associated MeCN ligand compared with the CO that it replaces explains the red-shift of the MLCT absorption bands of the solvent-coordinated complexes compared with the initial compounds.
Scheme 1:
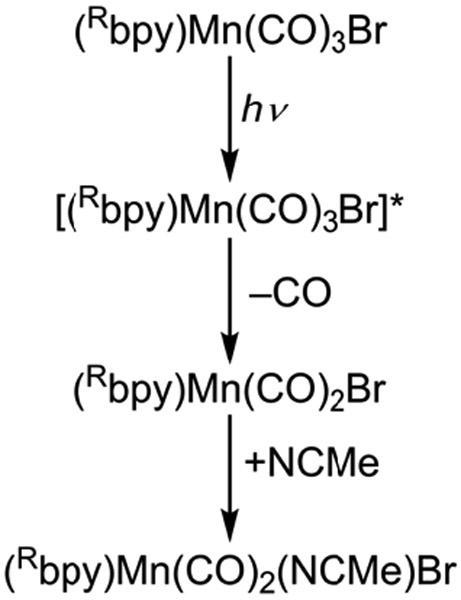
Proposed mechanism for the initial pathway leading to speciation for complexes 1-3.
Figure 3:

Schematic energy-level diagram illustrating how the change in Mn-CO bond length affects the curve crossing between MLCT and LF states. The barrier for adiabatic population transfer increases with decreasing MLCT energy across the series 1-4 (R = tBu, H, CF3, NO2).
A global fit to the TA spectrum of each compound using a bi-exponential function reveals the time constants for CO loss (τ1) and solvent coordination (τ2). The global fits give the time constants in Table 2 and the decay-associated spectra (DAS) in Figures S20-S22 of the SI.41 The time-constants indicate a slightly longer timescale for CO release and a shorter timescale for solvent coordination as the ligand becomes more electron withdrawing. The trend in τ2 is consistent with formation of a relatively more electron-poor 16 e− complex in the case of the more electron-withdrawing ligand for 3, compared with 2 and 1.
Table 2:
Time constants for CO release (τ1) and solvent coordination (τ2) a
| Compound | τ1 (ps) | τ2 (ps) |
|---|---|---|
| 1 | 0.50 ± 0.10 | 39 ± 4 |
| 2 | 0.46 ± 0.10 | 30 ± 4 |
| 3 | 0.68 ± 0.20 | 18 ± 3 |
From global fits to the TA spectra for ~2 mM solutions of 1-3 in MeCN.
In the proposed mechanism, the rate of CO loss is dependent on how quickly the MLCT state converts to the d-d dissociative LF state. Therefore, the observed rate of CO loss during photolysis likely depends on the initial relative energies of the MLCT and dissociative LF states. Changing the substituents on the 4 and 4′ positions of the bpy ligand should leave the metal d-orbitals relatively unaffected while tuning the ligand π* orbitals to a larger extent. The MLCT energy depends on π* of the ligand, but the LF energy should be relatively insensitive to substitution, as illustrated in Figure 3. This picture is consistent with the trend that we observe in the timescales for CO loss in Table 2. Changing the substituents on the bpy ligand from electron-donating to electron-withdrawing stabilizes the MLCT state and therefore increases the barrier to reach the LF state (Figure 3). Accordingly, the increasing electron-withdrawing character of Rbpy from 1 to 3 is associated with a corresponding decrease in the rate for CO loss (i.e. increase of τ1) due to stabilization of the MLCT state and, therefore, larger barrier to charge recombination by accessing the dissociative LF state.
As the substituents become even more electron withdrawing, the π* level of the bpy ligand is pushed substantially lower in energy compared with the occupied metal d-orbitals. In the limiting case of the NO2bpy complex (4), the MLCT band is > 0.5 eV below that of the prototypical Hbpy complex (2) due to charge stabilization and localization of electron density on the nitro groups. Thus the MLCT state of 4 is much lower than the dissociative LF state, significantly increasing the barrier for charge recombination and strongly disfavoring CO loss (see Figure S23 in SI). Instead, the MLCT state of 4 probably relaxes through a competing mechanism, possibly intersystem crossing (ISC) to a triplet state that is relatively long lived and persists beyond the time resolution of our experiment.
Measurement of CO release by infrared spectroscopy
To confirm that the [Mn(CO)3] complexes undergo CO loss followed by subsequent chemical reactivity in MeCN solvent, a series of photolysis experiments were carried out with infrared spectroscopic monitoring to interrogate the generation of new species by irradiation of 2 with visible light. Following 2 min. of irradiation with 415 nm light (at a total lamp power of 175 W), unique CO stretches appear in the IR spectrum, including stretches associated with Mn complexes between 2050–1825 cm−1 as well as the diagnostic stretch corresponding to free CO gas dissolved in MeCN near 2143 cm−1 (see Figure S24 for all spectra). Over time, the bands associated with 2 at 1924, 1934, and 2028 cm−1 decrease in intensity, and C─O stretches presumably associated with Mn-containing photoproducts grow in at 1883, 1961, and 1976 cm−1; two less intense features also appear at 1856 cm−1 and 2050 cm−1, but these features show minor variations in intensity over the 15 min. irradiation time, suggesting they may be associated with metastable intermediates. The presence of the [Mn(CO)3(bpy)]2 dimer can be inferred from related work examining electrochemical generation of the analogous [Mn(CO)3(tBubpy)]2 dimer by Kubiak and co-workers.42 As the speciation processes occurring at longer times are likely complex and multistep in nature, a portion of our future work will be devoted to further exploration by both time-resolved spectroscopies and more detailed steady-state photolysis experiments.
Involvement of MLCT vs. LF states
Although the changing energy of the MLCT state with ligand substituents is evident from the shifting of the ground-state absorption bands of 1-4, we have been unable to directly observe the relative energies of the LF states. The weak d-d transitions are not evident in the absorption spectra, due to the stronger overlapping MLCT bands. We also attempted to measure the d-d transition for the analogous bis-pyridine complex, Mn(CO)3Br(py)2 (5), in an effort to determine the relative energy of the LF state in [Mn(CO)3] complexes (see Figure S25).
Although the synthesis of 5 has been previously reported, the molecular structure of the complex was not reported. To obtain the structure, we grew single crystals of 5 suitable for X-ray diffraction (XRD) analysis by vapor diffusion of diethyl ether into a concentrated MeCN solution (see Figures S26-S27). Single crystals of the new complexes 3 and 4 suitable for XRD analysis were obtained with similar conditions (see Figures S28-S30). The expected atomic connectivity and fac-tricarbonyl geometry were confirmed in each case. While the donor strengths of py and bpy are similar (both molecules are conjugated imines with roughly the same electronic character), the average Mn─N bond distance in 5 (2.096(8) å) is significantly longer than that found in the bipyridyl complexes 3 or 4 (2.051(4) and 2.039(6) å, respectively), indicating a weaker interaction of the independent pyridyl rings in the (py)2 complex compared with bpy. Eliminating conjugation between the rings also pushes the π* orbital of py to higher energy and shifts the MLCT band of 5 to shorter wavelength. However, even with the simultaneous blue shift of the MLCT band and weaker donation by pyridine, we do not observe a distinct d-d transition in the absorption spectrum of 5. On the other hand, it is interesting to note the presence of a shoulder band near 390 nm in the absorption spectra of all four diimine complexes (Figure 1), as well as 5; this band could be consistent with the presence of an overlapping d-d feature at higher energy than the observed lowest-energy MLCT bands.
Considering all of these features, the ultrafast TA results show that we can selectively control the sensitivity of the initial CO release by tuning the orbital energy levels of the [Mn(CO)3Br(Rbpy)] complexes based on ligand substitution. Selectively controlling the loss of a CO ligand at the femtosecond timescale can lead to improved catalysts and more efficient photo-CORMs. On one hand, preventing CO release would give more stable catalysts, and on the other hand, complexes with tunable reactivity could serve as model compounds for regulating the therapeutic release of CO.
Secondary reactivity following CO loss
The CO release step is only the beginning of a more complex speciation process for these Mn complexes. After CO release in MeCN, the resulting electron deficient five-coordinate species [(Rbpy)Mn(CO)2Br] quickly binds a ligand to regain a full 18 e− valence. Table 2 shows that the time constants for solvent coordination (τ2) decrease markedly as the ligand becomes more electron withdrawing. Complexes with a more electron deficient Mn center due to the electron-withdrawing CF3bpy ligand (3) bind an acetonitrile more quickly than the complexes bearing electron-donating tBubpy (1) or Hbpy ligands (2).
In order to test the solvent dependence of the secondary reactivity occurring upon CO loss, we also measured the evolution of the TA spectrum of 2 in chloroform (CHCl3). Unlike MeCN, CHCl3 is not generally considered to be a coordinating solvent.43 Figure 4 shows that the evolution of the TA spectrum following MLCT excitation of 2 is very different in the two solvents. Specifically, the spectroscopic feature that appears on a picosecond timescale and is assigned as solvent-coordination in MeCN is completely attenuated in CHCl3, resulting in a broad and featureless spectrum that persists beyond the duration of our experiment.
Figure 4:

Transient absorption spectrum of 2 in CHCl3 (upper panel), and comparison of the kinetics at 575 nm for 2 in the coordinating solvent MeCN (blue markers) and in the noncoordinating solvent CHCl3 (red markers).
In both the cases of MeCN and CHCl3 as solvent, CO release exposes the coordinatively unsaturated, 16 e− metal center to the solvent. Although direct solvent coordination is suppressed in CHCl3, the electron-deficient, 5-coordinate complex is likely to be susceptible to other bimolecular reactions, possibly including oxidative addition of CHCl3 or hydrogen atom transfer from the solvent. The products from any of these reactions, or a combination of them, could be responsible for the broad, featureless TA spectrum that develops following excitation of 2 in CHCl3. Indeed, the reactivity of this complex in CHCl3 is consistent with recently reported transient absorption measurements for Mn tetracarbonyl complexes supported by anionic chelating ligands.44 However, regardless of the subsequent reactivity following solvent coordination, our results suggest that the feature that grows in at 100 ps in MeCN is likely a solvent coordinated species, and this event can be controlled based on the choice of solvent.
Conclusions
Mn(CO)3(Rbpy)Br complexes are stable in the absence of light, which allowed us to extensively characterize them using a variety of techniques. Transitions in the NMR, IR, and electronic absorption spectra are linearly correlated with the Brown (σ+), Hammett (σ), and Kubota (σ−) parameters, respectively, providing insight into how ligand substituents govern electronic properties at the metal center. In the presence of light, Mn(CO)3(Rbpy)Br complexes 1, 2, and 3 decompose via CO loss, with subsequent solvent coordination in MeCN or other reactions in CHCl3. Ultrafast TA spectroscopy revealed the previously unknown 5-coordinate and solvento intermediates in the photo-speciation process. Complex 4 follows a different reaction path that is likely the result of a long-lived triplet excited state that does not undergo decay to a dissociative LF (d-d) state. The different behavior of 1-4 illustrates how the reactivity and speciation of these complexes can be tuned by changing the electronic properties of the bidentate diimine ligand. Notably, as only limited computational modeling has examined the excited state electronic structure of Mn(CO)3Br(diimine) complexes,38,45 further experimental work combined with computational modeling would be useful in providing new insights into the speciation mechanisms operative during irradiation. Our ongoing experimental work aims to analyze the structure and electronics of the transient species generated upon irradiation using time resolved X-ray absorption and IR spectroscopies.
Experimental Section
Potential Hazards:
Working with carbonyl complexes poses a potential hazard of generating the colorless, odorless, tasteless, and acutely toxic gas carbon monoxide (CO). Work with carbonyl complexes should be carried out in a well ventilated fume hood and with use of a sensitive CO monitor. The synthesis of complexes 1-5 involves the displacement of two CO ligands, resulting in generation of a significant amount of CO during the reaction; this is especially true when working on larger scales, as has been done in the course of this work (hundreds of milligrams). Additionally, when complexes 1-5 are exposed to even low-intensity ambient visible light, they readily undergo photolysis to release CO. Caution should always be exercised when working with metal-carbonyl complexes.
General Considerations:
Manganese pentacarbonyl bromide (98%, Beantown Chemical Co.), 2,2′-bipyrdiyl (bpy) (98%; Alfa Aesar), 4,4′-bis(tert-butyl)-bipyridine (tBu-bpy) (98%, Sigma Aldrich), 2-chloro-4-trifluoromethyl-pyridine (98%; Oakwood Chemical), fuming nitric acid (90%, Alfa Aesar), and PCl3 (98%, Alfa Aesar) were used as received. 2,2′-bipyridyl-N,N′-dioxide46, 4,4′-dinitro-2,2′-bipyridyl-N,N′-dioxide46, 4,4′-dinitro-2,2′-bipyridyl46 (dnbpy), 4,4′-bis(trifluoromethyl)-2,2′-bipyridyl (CF3-bpy)47, Mn(CO)3Br(pyridine)248, Mn(CO)3Br(2,2′-bipyridyl)1, and Mn(CO)3Br(4,4′-bis(tert-butyl)-2,2′-bipyridyl)2 were prepared according to literature methods with minor modifications. If necessary, the ligands 2,2′-bipyridyl, 4,4′-bis(tert-butyl)-2,2′-bipyridyl, and 4,4′-bis(trifluoromethyl)-2,2′-bipyridyl can be sublimed (at ca. 80°C and 1 mTorr) if pre-purification is necessary.
Deuterated NMR solvents were purchased from Cambridge Isotope Laboratories; CD3CN was dried over molecular sieves. 1H, 13C, and 19F NMR spectra were collected with 400 or 500 MHz Bruker spectrometers. Spectra were referenced to the residual protio-solvent signal in the cases of 1H and 13C.49 Heteronuclear NMR spectra were referenced to the appropriate external standard following the recommended scale based on ratios of absolute frequencies (Ξ).50 19F NMR spectra are reported relative to CCl3F. Chemical shifts (δ) are reported in units of ppm, and coupling constants (J) are reported in Hz.
All manipulations were done in dry N2-filled gloveboxes (Vacuum Atmospheres Co. Hawthorne, CA) or under a N2 atmosphere using standard Schlenk techniques unless otherwise noted. All solvents were of commercial grade and dried over activated alumina using a Pure Process Technology (PPT; Nashua, NH) solvent purification system prior to use and were stored over molecular sieves. All chemicals were from major commercial suppliers and used as received after extensive drying.
IR spectra were collected on a PerkinElmer Spectrum 100 FTIR spectrometer. UV-visible spectra were collected with an Ocean Optics FLAME-S spectrometer equipped with a DH-Mini light source. Steady-state photolysis experiments were carried out using an Oriel HgXe arc lamp operating at 175 W and equipped with an Oriel Cornerstone 130 1/8 m monochromator accessory. For the experiments in which IR spectroscopic monitoring was used to examine the products of photolysis, an appropriate KBr-plate cell was charged with a 17 mM solution of 2 in MeCN under inert atmosphere, and an initial IR spectrum was collected prior to irradiation. Subsequent short periods of irradiation (totaling 2, 5, 10, and 15 min) with 415 nm light generated by the HgXe arc lamp operating at 175 W were followed in each case by collection of a new spectrum in order to examine evolution of the system over time. These spectra are given in Figure S24.
Elemental analyses were performed by Midwest Microlab, Inc. (Indianapolis, IN).
Single-crystal diffraction data were collected with a Bruker KAPPA APEX/II X-ray diffractometer. CCDC entries 1922040, 1922041, and 1922042 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
The transient absorption measurements used pump and probe pulses derived from the 800 nm output of a 1 kHz regeneratively amplified Ti:sapphire laser (Legend Elite, Coherent). A portion of the laser fundamental pumped an optical parametric amplifier with two stages of nonlinear frequency conversion (TOPAS) to generate the visible pump pulses. The beam diameter at the sample was 160 μm with an energy of 800 nJ per pulse. The relative polarization was set to magic angle by rotating the pump pulses with a zero-order λ/2 wave-plate. We used active background subtraction by passing the pump beam though a synchronized chopper wheel running at 500 Hz to block every other pump pulse. Broadband probe pulses with the desired wavelength range were generated by focusing a small fraction of the fundamental 800 nm laser light into a mechanically rotating CaF2 crystal. The sample is cycled through a flow cell with a path length of 0.5 mm. After passing through the sample the probe pulse is dispersed using a prism onto a 2069-element CCD array. Each TA spectrum is an average of 103 laser pulses per time delay.
Synthesis of Mn(CO)3(4,4′-bis(trifluoromethyl)-2,2′-bipyridine)Br (3):
To a 50 mL Schlenk flask equipped with a stir bar was added 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (0.0998 g, .342 mmol) in 50 mL of pentane. Then Mn(CO)5Br (0.0890 g, .324 mmol) was added and the reaction was brought to reflux under an argon atmosphere. The reaction was monitored by 1H NMR until consumption of the starting material was observed (~ 6 hours). Once the reaction had reached completion the Schlenk flask was placed into a refrigerator at −20°C for 30 minutes. The resulting solid is then filtered off with a fritted glass funnel and washed with cold pentane to afford the title compound as an orange-red solid. Yield: 0.0801 g (48%). 1H NMR (400 MHz, CD3CN): δ 9.48 (d, 2H, 3JH,H = 5.8 Hz), 8.77 (s, 2H), 7.91 (dd, 2H, 3JH,H = 5.8 Hz, 4JH,H = 1.8 Hz) ppm. 13C{1H} NMR (176 MHz, CD3CN): δ 185.5, 157.5, 156.4, 141.2 (q, 1JC,F = 35.3 Hz), 124.8, 123.9 (q, 4JC,F = 3.5 Hz), 122.6, 121.4 (q, 4JC,F = 3.5 Hz) ppm. 19F NMR (376 MHz, CD3CN) δ 65.4 ppm. Electronic absorption spectrum (MeCN): 208 (32000), 224 (33000), 293 (10000), 374 (1800), 454 nm (1500 M−1cm−1). IR (THF): νC═o 2027 (m) (A′), and νC═o 1944 (m) (A″), νC═o 1923 (m) (A′) cm−1. ESI-MS (positive) m/z: 471.9 (100%) (3-Br−+NCMe), 472.9 (23%), 474.0 (11%); 430.9 (87%) (3-Br−), 431.9 (16%), 432.9 (2%); 429.0 (23%) (3-Br−-3CO+2NCMe), 430.0 (4%); 402.9 (17%) (3-Br−-CO), 403.9 (3%); 388.0 (20%) (3-Br−-3CO+NCMe), 389.0 (4%); 374.9 (22%) (3-Br−−2CO), 375.9 (4%); 346.9 (11%) ((3-Br−−3CO), (2%) (see Figure S15). Anal. Calcd. for MnC15H6BrF6N2O3: C, 35.25; H, 1.18; N, 5.48. Found: C, 35.20; H, 1.24; N, 5.46.
Synthesis of Mn(CO)3(4,4′-dinitro-2,2′-bipyridyl)Br (4):
To a Schlenk flask equipped with a stir bar was added 4,4′-dinitro-2,2′-bipyridine (0.3761 g, 1.53 mmol) and 50 mL of Et2O. Then Mn(CO)5Br (0.4005 g, 1.46 mmol) was added and the reaction was brought to reflux under an argon atmosphere. The reaction was monitored by 1H NMR until consumption of the starting material was observed (~ 12 hours). Once the reaction had reached completion the Schlenk flask was placed into a −20°C refrigerator for 30 minutes. The resulting solid is then filtered off with a fritted glass funnel and washed with cold Et2O to afford the title compound as a purple solid. Yield: 0.6409 g (95 %). 1H NMR (400 MHz, CD3CN): δ 9.58 (d, 2H, 3JH,H = 6.0 Hz), 9.21 (s, 2H), 8.29 (dd, 2H, 4JH,H = 6.0 Hz) ppm. 13C{1H} NMR (176 MHz, CD3CN): δ 158.1, 157.1, 156.3, 120.5, 118.2 ppm. Electronic absorption spectrum (MeCN): 223 (1800), 245 (1400), 325 (800), 393 (200), 510 nm (230 M−1 cm−1). IR (THF): νC═o 2027 (m) (A′), νC═o 1945 (m) (A″), vC═o 1927 (m) (A′) cm−1. ESI-MS (positive) m/z: 425.9 (100%) (4-Br−+NCMe), 426.9 (17%), (3%); 411.0 (31%) (4-Br−−2CO+2NCMe), 412.0 (6%) 413.0 (1%); 384.9 (9%) (4-Br−), 385.9 (1%), 386.9 (1%); 383.0 (51%) (4-Br−−3CO+2NCMe), 384.0 (9%); 369.9 (7%) (4-Br−−2CO+NCMe), 370.9 (1%), 372.0 (1%); 368.0 (20%) (4-Br−-CO-2O+2H+NCMe), 369.0 (3%) (see Figure S16). Anal. Calcd. for MnC13H6BrN4O7 (+1H2O): C, 32.32; H, 1.67; N, 11.60. Found: C, 32.52; H, 1.41; N, 11.67.
Supplementary Material
Acknowledgements
The authors thank Dr. Davide Lionetti for numerous helpful discussions, Dr. Victor Day for assistance with X-ray crystallography, Keaton Prather for assistance with preparation of 4,4′-dinitro-2,2′-bipyridine, and Prof. Misha Barybin for generous access to a benchtop infrared spectrometer. This work was supported by the Hall Chemical Research Fund at the University of Kansas (to J.D.B. and C.G.E.) and a grant from the U.S. National Science Foundation (CHE-1151555; to C.G.E.). WCH was supported by the U.S. National Institutes of Health Graduate Training Program in the Dynamic Aspects of Chemical Biology (T32 GM008545-25).
Footnotes
Supporting Information. Supporting Information is available free of charge on the ACS Publications website: NMR spectra; IR spectra, crystallographic details; electronic absorption spectra; Cartesian coordinates (XYZ).
The authors declare no competing financial interest.
References
- 1.Bourrez M; Molton F; Chardon-Noblat S; Deronzier A, [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed 2011, 50, 9903–9906. [DOI] [PubMed] [Google Scholar]
- 2.Smieja JM; Sampson MD; Grice KA; Benson EE; Froehlich JD; Kubiak CP, Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem 2013, 52, 2484–2491. [DOI] [PubMed] [Google Scholar]
- 3.Abel EW; Bennett MA; Wilkinson G, Substituted carbonyl compounds of chromium, molybdenum, tungsten, and manganese. J. Chem. Soc 1959, 2323–2327. [Google Scholar]
- 4(a).Luong JC; Faltynek RA; Wrighton MS, Ground- and excited-state oxidation-reduction chemistry of (triphenyltin)- and (triphenylgermanium)tricarbonyl(1,10-phenanthroline)rhenium and related compounds. J. Am. Chem. Soc 1980, 102, 7892–7900. [Google Scholar]; (b) Caspar JV; Meyer TJ, Application of the energy gap law to nonradiative, excited-state decay. J. Phys. Chem 1983, 87, 952–957. [Google Scholar]; (c) Miguel D; Riera V, Synthesis of manganese(I) carbonyls with σ-bonded alkynyl ligands. J. Organomet. Chem 1985, 293, 379–390. [Google Scholar]
- 5(a).Hawecker J; Lehn JM; Ziessel R, Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–Co2+ combinations as homogeneous catalysts. J. Chem. Soc., Chem. Commun 1983, 536–538. [Google Scholar]; (b) Hawecker J; Lehn JM; Ziessel R, Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun 1984, 328–330. [Google Scholar]
- 6(a).Sullivan BP; Bolinger CM; Conrad D; Vining WJ; Meyer TJ, One- and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun 1985, 1414–1416. [Google Scholar]; (b) O'Toole TR; Margerum LD; Westmoreland TD; Vining WJ; Murray RW; Meyer TJ, Electrocatalytic reduction of CO2 at a chemically modified electrode. J. Chem. Soc., Chem. Commun 1985, 1416–1417. [Google Scholar]
- 7.Fujita E; Hayashi Y; Kita S; Brunschwig BS, Spectroscopic characterization of intermediates in CO2 reduction with rhenium photocatalysts. Vol. 153, pp 271–276, Elsevier, Amsterdam, 2004. [Google Scholar]
- 8(a).Smieja JM; Kubiak CP, Re(bipy-tBu)(CO)3Cl-improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem 2010, 49, 9283–9289. [DOI] [PubMed] [Google Scholar]; (b) Clark ML; Cheung PL; Lessio M; Carter EA; Kubiak CP, Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 5, 2021–2029. [Google Scholar]
- 9.Takeda H; Koike K; Morimoto T; Inumaru H; Ishitani O, Photochemistry and photocatalysis of rhenium(I) diimine complexes. Adv. Inorg. Chem, 2011; 63, 137–186. [Google Scholar]
- 10.El Nahhas A; van der Veen RM; Penfold TJ; Pham VT; Lima FA; Abela R; Blanco-Rodriguez AM; Zalis S; Vlcek A; Tavernelli I; Rothlisberger U; Milne CJ; Chergui M, X-ray Absorption Spectroscopy of Ground and Excited Rhenium–Carbonyl–Diimine Complexes: Evidence for a Two-Center Electron Transfer. J. Phys. Chem. A, 2013, 777, 361–369. [DOI] [PubMed] [Google Scholar]
- 11(a).Blakemore JD; Gupta A; Warren JJ; Brunschwig BS; Gray HB, Noncovalent Immobilization of Electrocatalysts on Carbon Electrodes for Fuel Production. J. Am. Chem. Soc 2013, 135, 18288–18291. [DOI] [PubMed] [Google Scholar]; (b) Agarwal J; Shaw TW; Stanton CJ; Majetich GF; Bocarsly AB; Schaefer HF, NHC-containing manganese(I) electrocatalysts for the two-electron reduction of CO2. Angew. Chem., Int. Ed 2014, 53, 5152–5155. [DOI] [PubMed] [Google Scholar]; (c) Grice KA; Kubiak CP, Recent studies of rhenium and manganese bipyridine carbonyl catalysts for the electrochemical reduction of CO2. Adv. Inorg. Chem 2014, 66, 163–188. [Google Scholar]; (d) Grills DC; Farrington JA; Layne BH; Lymar SV; Mello BA; Preses JM; Wishart JF, Mechanism of the Formation of a Mn-Based CO2 Reduction Catalyst Revealed by Pulse Radiolysis with Time-Resolved Infrared Detection. J. Am. Chem. Soc 2014, 136, 5563–5566. [DOI] [PubMed] [Google Scholar]; (e) Sampson MD; Nguyen AD; Grice KA; Moore CE; Rheingold AL; Kubiak CP, Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: Eliminating dimerization and altering catalysis. J. Am. Chem. Soc 2014, 136, 5460–5471. [DOI] [PubMed] [Google Scholar]; (f) Walsh JJ; Neri G; Smith CL; Cowan AJ, Electrocatalytic CO2 reduction with a membrane supported manganese catalyst in aqueous solution. Chem. Commun 2014, 50, 12698–12701. [DOI] [PubMed] [Google Scholar]; (g) Zeng Q; Tory J; Hartl F, Electrocatalytic Reduction of Carbon Dioxide with a Manganese(I) Tricarbonyl Complex Containing a Nonaromatic α-Diimine Ligand. Organometallics 2014, 33, 5002–5008. [Google Scholar]
- 12(a).Agarwal J; Shaw TW; Schaefer HF III; Bocarsly AB, Design of a Catalytic Active Site for Electrochemical CO2 Reduction with Mn(I)-Tricarbonyl Species. Inorg. Chem. 2015, 54, 5285–5294. [DOI] [PubMed] [Google Scholar]; (b) Fei H; Sampson MD; Lee Y; Kubiak CP; Cohen SM, Photocatalytic CO2 Reduction to Formate Using a Mn(I) Molecular Catalyst in a Robust Metal- Organic Framework. Inorg. Chem 2015, 54, 6821–6828. [DOI] [PubMed] [Google Scholar]; (c) Machan CW; Stanton CJ III; Vandezande JE; Majetich GF; Schaefer HF III; Kubiak CP; Agarwal J, Electrocatalytic Reduction of Carbon Dioxide by Mn(CN)(2,2'-bipyridine)(CO)3: CN Coordination Alters Mechanism. Inorg. Chem 2015, 54, 8849–8856. [DOI] [PubMed] [Google Scholar]; (d) Riplinger C; Carter EA, Influence of Weak Bronsted Acids on Electrocatalytic CO2 Reduction by Manganese and Rhenium Bipyridine Catalysts. ACS Catal. 2015, 5, 900–908. [Google Scholar]; (e) Walsh JJ; Smith CL; Neri G; Whitehead GFS; Robertson CM; Cowan AJ, Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 2015, 183, 147–160. [DOI] [PubMed] [Google Scholar]; (f) Agnew DW; Sampson MD; Moore CE; Rheingold AL; Kubiak CP; Figueroa JS, Electrochemical Properties and CO2-Reduction Ability of m-Terphenyl Isocyanide Supported Manganese Tricarbonyl Complexes. Inorg. Chem 2016, 55, 12400–12408. [DOI] [PubMed] [Google Scholar]; (g) Rawat KS; Mahata A; Choudhuri I; Pathak B, N-Heterocylic Carbene-Based Mn Electrocatalyst for Two-Electron CO2 Reduction over Proton Reduction. J. Phys. Chem. C 2016, 120, 8821–8831. [Google Scholar]
- 13(a).Sampson MD; Kubiak CP, Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids To Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc 2016, 138, 1386–1393. [DOI] [PubMed] [Google Scholar]; (b) Spall SJP; Keane T; Tory J; Cocker DC; Adams H; Fowler H; Meijer AJHM; Hartl F; Weinstein JA, Manganese Tricarbonyl Complexes with Asymmetric 2-Iminopyridine Ligands: Toward Decoupling Steric and Electronic Factors in Electrocatalytic CO2 Reduction. Inorg. Chem. 2016, 55, 12568–12582. [DOI] [PubMed] [Google Scholar]; (c) Stanton CJ; Vandezande JE; Majetich GF; Schaefer HF; Agarwal J, Mn-NHC Electrocatalysts: Increasing π Acidity Lowers the Reduction Potential and Increases the Turnover Frequency for CO2 Reduction. Inorg. Chem 2016, 55, 9509–9512. [DOI] [PubMed] [Google Scholar]; (d) Ngo KT; McKinnon M; Mahanti B; Narayanan R; Grills DC; Ertem MZ; Rochford J, Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes. J. Am. Chem. Soc 2017, 139, 2604–2618. [DOI] [PubMed] [Google Scholar]; (e) Reuillard B; Ly KH; Rosser TE; Kuehnel MF; Zebger I; Reisner E, Tuning Product Selectivity for Aqueous CO2 Reduction with a Mn(bipyridine)-pyrene Catalyst Immobilized on a Carbon Nanotube Electrode. J. Am. Chem. Soc 2017, 139, 14425–14435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Franco F; Pinto MF; Royo B; Lloret-Fillol J, A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem., Int. Ed 2018, 57, 4603–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Myren THT; Lilio AM; Huntzinger CG; Horstman JW; Stinson TA; Donadt TB; Moore C; Lama B; Funke HH; Luca OR, Manganese N-Heterocyclic Carbene Pincers for the Electrocatalytic Reduction of Carbon Dioxide. Organometallics 2019, 38, 1248–1253. [Google Scholar]; (h) Takeda H; Kamiyama H; Okamoto K; Irimajiri M; Mizutani T; Koike K; Sekine A; Ishitani O, Highly Efficient and Robust Photocatalytic Systems for CO2 Reduction Consisting of a Cu(I) Photosensitizer and Mn(I) Catalysts. J. Am. Chem. Soc 2018, 140, 17241–17254. [DOI] [PubMed] [Google Scholar]; (i) Tignor SE; Kuo H-Y; Lee TS; Scholes GD; Bocarsly AB, Manganese-Based Catalysts with Varying Ligand Substituents for the Electrochemical Reduction of CO2 to CO. Organometallics 2019, 38, 1292–1299, [Google Scholar]; (j) Walsh JJ; Neri G; Smith CL; Cowan AJ, Water-Soluble Manganese Complex for Selective Electrocatalytic CO2 Reduction to CO. Organometallics 2019, 38, 1224–1229. [Google Scholar]
- 14.Vollmer MV; Machan CW; Clark ML; Antholine WE; Agarwal J; Schaefer HF; Kubiak CP; Walensky JR , Synthesis, Spectroscopy, and Electrochemistry of (α-Diimine)M(CO)3Br, M = Mn, Re, Complexes: Ligands Isoelectronic to Bipyridyl Show Differences in CO2 Reduction. Organometallics 2015, 34, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda H; Koizumi H; Okamoto K; Ishitani O, Photocatalytic CO2 reduction using a Mn complex as a catalyst. Chem. Commun 2014, 50, 1491–1493. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal J; Stanton Iii CJ; Shaw TW; Vandezande JE; Majetich GF; Bocarsly AB; Schaefer III HF, Exploring the effect of axial ligand substitution (X = Br, NCS, CN) on the photodecomposition and electrochemical activity of [MnX(N─C)(CO)3] complexes. Dalton Trans. 2015, 44, 2122–2131. [DOI] [PubMed] [Google Scholar]
- 17.Perutz RN; Torres O; Vlcek A, “Photochemistry of Metal Carbonyls,” in Comprehensive Inorganic Chemistry II, Reedijk J, Poeppelmeier K, Eds., 2nd Ed., 2013, Vol. 8. [Google Scholar]
- 18(a).Schatzschneider U, PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications. Inorg. Chim. Acta 2011, 374, 19–23. [Google Scholar]; (b) Kottelat E, Fabio Z, Visible Light Activated PhotoCORMs. Inorganics 2017, 5, 24. [Google Scholar]; (c) Ling K; Men F; Wang W-C; Zhou Y-Q; Zhang H-W; Ye D-W, Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [DOI] [PubMed] [Google Scholar]
- 19(a).Hughey JL; Anderson CP; Meyer TJ, Photochemistry of Mn2(CO)10. J. Organomet. Chem 1977, 125, C49–C52. [Google Scholar]; (b) Meyer TJ; Caspar JV, Photochemistry of metal-metal bonds. Chem. Rev 1985, 85, 187–218. [Google Scholar]
- 20.Motterlini RC, James E; Foresti, Roberta; Sarathchandra, Padmini; Mann, Brian E.; Green, Colin J., Carbon Monoxide-Releasing Molecules: Characterization of Biochemical and Vascular Activities. J. Am. Heart Assoc 2002, 90, e17–e24. [DOI] [PubMed] [Google Scholar]
- 21.Bolze F; Jenni S; Sour A; Heitz V Molecular photosensitisers for two-photon photodynamic therapy. Chem. Commun 2017, 53, 12857–12877. [DOI] [PubMed] [Google Scholar]
- 22.Niesel J; Pinto A; N'Dongo HWP; Merz K; Ott I; Gust R; Schatzschneider U, Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex. Chem. Commun 2008, 1798–1800. [DOI] [PubMed] [Google Scholar]
- 23(a).He Q; Kiesewetter DO; Qu Y; Fu X; Fan J; Huang P; Liu Y; Zhu G; Liu Y; Qian Z; Chen X, NIR-Responsive On-Demand Release of CO from Metal Carbonyl-Caged Graphene Oxide Nanomedicine. Adv. Mater 2015, 27, 6741–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ruggi A; Zobi F, Quantum-CORMs: quantum dot sensitized CO releasing molecules. Dalton Trans. 2015, 44, 10928–31. [DOI] [PubMed] [Google Scholar]; (c) Aucott BJ; Ward JS; Andrew SG; Milani J; Whitwood AC; Lynam JM; Parkin A; Fairlamb IJS, Redox-tagged carbon monoxide-releasing molecules (CORMs): ferrocene-containing [Mn(C-N)(CO)4] complexes as a promising new CORM class. Inorg. Chem 2017, 56, 5431–5440. [DOI] [PubMed] [Google Scholar]; (d) Carmona FJ; Jimenez-Amezcua I; Rojas S; Romao CC; Navarro JAR; Maldonado CR; Barea E, Aluminum Doped MCM-41 Nanoparticles as Platforms for the Dual Encapsulation of a CO-Releasing Molecule and Cisplatin. Inorg. Chem 2017, 56, 10474–10480. [DOI] [PubMed] [Google Scholar]; (e) Li Z; Pierri AE; Huang P-J; Wu G; Iretskii AV; Ford PC, Dinuclear PhotoCORMs: Dioxygen-Assisted Carbon Monoxide Uncaging from Long-Wavelength-Absorbing Metal-Metal-Bonded Carbonyl Complexes. Inorg. Chem 2017, 56, 6094–6104. [DOI] [PubMed] [Google Scholar]; (f) Mansour AM, Rapid green and blue light-induced CO release from bromazepam Mn(I) and Ru(II) carbonyls: synthesis, density functional theory and biological activity evaluation. Appl. Organomet. Chem 2017, 31, e3564. [Google Scholar]; (g) Mede R; Hoffmann P; Klein M; Goerls H; Schmitt M; Neugebauer U; Gessner G; Heinemann SH; Popp J; Westerhausen M, Water-Soluble Mn(CO)3-Based and Non-Toxic PhotoCORM for Administration of Carbon Monoxide Inside of Cells. Anorg. Allg. Chem 2017, 643, 2057–2062. [Google Scholar]; (h) Reddy GU; Liu J; Gorls H; Askes SHC; Schiller A; Hoffmann P; Neugebauer U; Hoffmann P; Neugebauer U; Steinmetzer J; Kupfer S; Grafe S, Light-responsive paper strips as CO-releasing material with a colourimetric response. Chem. Sci 2017, 8, 6555–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Mansour AM; Shehab OR, Reactivity of visible-light induced CO releasing thiourea-based Mn(I) tricarbonyl bromide (CORM-NS1) towards lysozyme. Inorg. Chim. Acta 2018, 480, 159–165. [Google Scholar]
- 24.Carrington SJ; Chakraborty I; Mascharak PK, Exceptionally rapid CO release from a manganese(I) tricarbonyl complex derived from bis(4-chloro-phenylimino)acenaphthene upon exposure to visible light. Dalton Trans. 2015, 44, 13828–13834. [DOI] [PubMed] [Google Scholar]
- 25.Glyn P; George MW; Hodges PM; Turner JJ, Fast time-resolved IR studies of the excited states of co-ordination compound: direct observation of intramolecular charge transfer. Chem. Commun 1989, 1655–1657. [Google Scholar]
- 26.Sato S; Matubara Y; Koike K; Falkenstrom M; Katayama T; Ishibashi Y; Miyasaka H; Taniguchi S; Chosrowjan H; Mataga N; Fukazawa N; Koshihara S; Onda K; Ishitani O, Photochemistry of fac-[Re(bpy)(CO)3Cl]. Chem.: Eur. J 2012, 18, 15722–15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stufkens DJ; Vlcek A Ligand-dependent excited state behaviour of Re(I) and Ru(II) carbonyl–diimine complexes. Coord. Chem. Rev 1998, 177, 127–179. [Google Scholar]
- 28(a).Cannizzo A; Blanco-Rodriguez AM; El Nahhas A; Sebera J; Zalis S; Vlcek A; Chergui M Femtosecond Fluorescence and Intersystem Crossing in Rhenium(I) Carbonyl–Bipyridine Complexes. J. Am. Chem. Soc 2008, 130, 8967–8974. [DOI] [PubMed] [Google Scholar]; (b) El Nahhas A; Consani C; Blanco-Rodriguez AM; Lancaster KM; Braem O; Cannizzo A; Towrie M; Clark IP; Zalis S; Chergui M; Vlcek A Ultrafast Excited-State Dynamics of Rhenium(I) Photosensitizers [Re(Cl)(CO)3(N,N)] and [Re(imidazole)(CO)3(N,N)]+: Diimine Effects. Inorg. Chem 2011, 50, 2932–2943. [DOI] [PubMed] [Google Scholar]
- 29.Grills DC; Farrington JA; Layne BH; Lymar SV; Mello BA; Preses JM; Wishart JF, Mechanism of the Formation of a Mn-Based CO2 Reduction Catalyst Revealed by Pulse Radiolysis with Time-Resolved Infrared Detection. J. Am. Chem. Soc 2014, 136, 5563–5566. [DOI] [PubMed] [Google Scholar]
- 30.Hammett LP, The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc 1937, 59, 96–103. [Google Scholar]
- 31.Brown HC; Okamoto Y, Electrophilic Substituent Constants. J. Am. Chem. Soc 1958, 80, 4979–4987. [Google Scholar]
- 32.Yoshioka M; K. H, and Kubota T; Relationship between Acid and Dissociation Constants of N1-Aryl-sulfanilamides and the Hammett Equation. Bull. Chem. Soc. Japan 1962, 35, 1723–1728. [Google Scholar]
- 33.von Philipsborn W, Probing organometallic structure and reactivity by transition metal NMR spectroscopy. Chem. Soc. Rev 1999, 28, 95–105. [Google Scholar]
- 34.Hoye TR; Rehberg GM, Reactions of (CO)5Cr:C(Me)N(CH2CH2)2 with enynes: mechanistic insight and synthetic value of changing a carbene donor group from alkoxy to dialkylamino. Organometallics 1989, 8, 2070–2071. [Google Scholar]
- 35.Stor GJ; Morrison SL; Stufkens DJ; Oskam A, The Remarkable Photochemistry of fac-XMn(CO)3(α-diimine) (X = Halide): Formation of Mn2(CO)6(α-diimine)2 via the mer Isomer and Photocatalytic Substitution of X- in the Presence of PR3. Organometallics 1994, 13, 2641–2650. [Google Scholar]
- 36.Zimmer P; Sun Y; Thiel WR, Cationic isonitrile complexes of the CpFe(CO)2 fragment. J. Organomet. Chem 2014, 774, 12–18. [Google Scholar]
- 37.Herberhold M; Brabetz H, Übergangsmetallkomplexe mit N-haltigen Liganden, III. Donator-Akzeptor-Eigenschaften organischer Nitrile im System C5H5Mn(CO)2L. Chem. Ber 1970, 103, 3909–3917, [Google Scholar]
- 38.Jiang Q; Xia Y; Barrett J; Mikhailovsky A; Wu G; Wang D; Shi P; Ford PC, Near-Infrared and Visible Photoactivation to Uncage Carbon Monoxide from an Aqueous-Soluble PhotoCORM. Inorg. Chem 2019, 58, 11066–11075, [DOI] [PubMed] [Google Scholar]
- 39.Papadakis R; Tsolomitis A, Study of the correlations of the MLCT Vis absorption maxima of 4-pentacyanoferrate- 4′-arylsubstituted bispyridinium complexes with the Hammett substituent parameters and the solvent polarity parameters E and AN. J. Phys. Org. Chem 2009, 22, 515–521. [Google Scholar]
- 40.Hummel P; Oxgaard J; Goddard WA; Gray HB Ligand-Field Excited States of Metal Hexacarbonyls. Inorg. Chem 2005, 44, 2454–2458. [DOI] [PubMed] [Google Scholar]
- 41.van Stokkum IHM; Larsen DS; van Grondelle R Global and target analysis of time-resolved spectra. Biochem. Biophys. Acta – Bioenerg 2004, 1657, 82–104. [DOI] [PubMed] [Google Scholar]
- 42.Machan CW; Sampson MD; Chabolla SA; Dang T; Kubiak CP, developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 2014, 33, 4550–4559. [Google Scholar]
- 43.Díaz-Torres R; Alvarez S Coordinating ability of anions and solvents towards transition metals and lanthanides. Dalton Trans. 2011, 40, 10742–10750. [DOI] [PubMed] [Google Scholar]
- 44.Hammarback LA; Clark IP; Sazanovich IV; Towrie M; Robinson A; Clarke F; Meyer S; Fairlamb IJS; Lynam JM Mapping out the key carbon–carbon bond-forming steps in Mn-catalysed C─H functionalization. Nature Catalysis 2018, 1, 830–840. [Google Scholar]
- 45.Mansour AM; Ragab MS, Spectroscopic and DFT studies of photoactivatable Mn(I) tricarbonyl complexes. Appl. Organomet. Chem 2019, 33, e4944. [Google Scholar]
- 46.Maerker G; Case FH, The Synthesis of Some 4,4'-Disubstituted 2,2'-Bipyridines. J. Am. Chem. Soc 1958, 80, 2745–2748. [Google Scholar]
- 47.Li H; Oppenheimer J; Smith III MR; Maleczka RE Jr, Improved synthesis of electron deficient bipyridines. Tet. Lett 2016, 57, 2231–2232. [Google Scholar]
- 48.Pons M; Herberich G, Bromotricarbonyldi(pyridine)manganese(I). Inorg. Synth 2014, 36, 148–149. [Google Scholar]
- 49.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar]
- 50(a).Harris RK, Becker ED, Cabral De Menezes SM, Goodfellow R, Granger P, NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem 2001, 73, 1795–1818. [DOI] [PubMed] [Google Scholar]; (b) Harris RK, Becker ED, Cabral De Menezes SM, Granger P, Hoffman RE, Zilm KW, Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008). Pure Appl. Chem 2008, 80, 59–84. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.