

Graphical abstract
Keywords: Histone demethylation, Kabuki Syndrome, KDM6A, Molecular dynamics simulation, Computational biology
Abstract
Mutations in genes encoding for histone methylation proteins are associated with several developmental disorders. Among them, KDM6A is the disease causative gene of type 2 Kabuki Syndrome, a rare multisystem disease. While nonsense mutations and short insertions/deletions are known to trigger pathogenic mechanisms, the functional effects of missense mutations are still uncharacterized. In this study, we demonstrate that a selected set of missense mutations significantly hamper the interaction between KDM6A and the histone H3, by modifying the dynamics of the linker domain, and then causing a loss of function effect.
1. Introduction
Histone modifications determine the accessibility of promoter and enhancers to transcription factors with the effect of enabling the gene expression. Different types of histone modifications occur, i.e., acetylation, ubiquitinylation, methylation of lysine, arginine, and histidine, and serine phosphorylation. In particular, histone methylation is the most relevant one, since it is involved in critical biological processes, such as genomic imprinting, pluripotency, and cell differentiation programs. Lysine can be mono-, di- or tri-methylated by specific lysine methyltransferases (KMTs), whereas lysine demethylases (KDMs) catalyze the inverse process. Those enzymes coordinate, spatially, and temporally, the expression of distinct gene networks in almost all stages of development.
Mutations in genes encoding for histone methylation regulators were often associated with developmental defects and genetic diseases [1], [2], [3], [4], [5]. Representative examples are the genes that encode for the histone-lysine N-methyltransferase 2D, KMT2D (also known as MLL2, ALR, and MLL4; MIM 602113) and the lysine-specific demethylase 6A, KDM6A (also known as UTX; MIM 300128), whose genetic alterations were first associated with congenital anomalies and intellectual disability [6]. To date, most pathogenic protein-truncating variants in KMT2D and KDM6A genes, intolerant to heterozygous loss-of-function variations, are associated with Kabuki Syndrome (KS).
KS is a rare, multisystem disorder, occurring in approximately 1 in 32,000 newborns, characterized by distinctive facial features, skeletal anomalies, dermatoglyphic abnormalities, varying degrees of intellectual disability, growth delay, and short stature. Two modes of inheritance are known: an autosomal-dominant pattern, when mutations in the KMT2D gene cause type 1 KS (KS1, KABUK1, MIM 147920), and an X-linked dominant model, when mutations in the KDM6A gene cause the syndrome (KS2, KABUK2, MIM 300867). Furthermore, a significant portion of missense mutations, around 16% in KMT2D, was also found in several human developmental disorders [1].
Most of the pathogenic mutations in these two genes are single-nucleotide variants and short insertions/deletions, causing missense, nonsense, frame-shifts, and splice-site mutations, which produce truncated or inactivated proteins. Their functional effects were studied in KMTs and KDMs [1], [2], [3], [4], [7] but, despite several efforts, their biological consequences and molecular mechanisms are still unknown [7]. This may be due to long-range interactions exhibiting potential pathogenic effects on critical protein regions, which cannot be easily grasped studying the sole localization of the variations in the protein structure, nor from energetic studies conducted on rigid in-silico models.
Molecular dynamics (MD) simulation represents a groundbreaking approach to evaluate the temporal motion of macromolecules over time. It owes its success to the impressive improvements in terms of computing capabilities of HPC hardware and, in particular, of GPU-enabled graphics cards [8]. Here, we applied techniques of MD to study a selected set of heterozygous and hemizygous pathogenic missense variants of KDM6A, found in individuals with features of X-linked dominant KS2. Using an enhanced sampling MD technique, the Gaussian accelerated Molecular Dynamics (GaMD), we described the pathogenic mechanisms caused by these variants on the physiological demethylation activity of KDM6A on the histone H3.
2. Materials and methods
2.1. The starting system
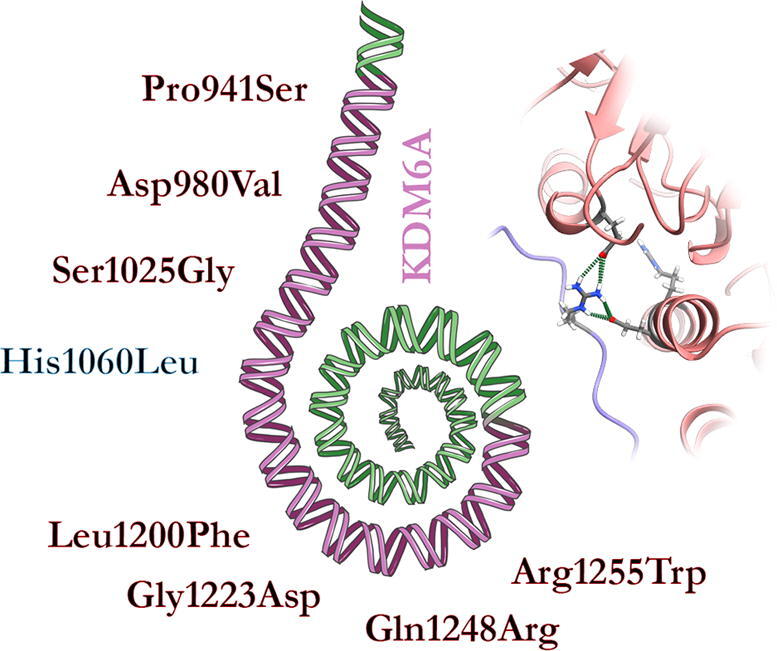
The KDM6A gene consists of 29 exons (NM_021140.3). It encodes for a protein of 1,401 amino acids (NP_066963.2) characterized by two main functional domains, an N-terminal containing eight tetratricopeptide repeat elements, with a more specific structural role, and a C-terminal, involved in the catalytic activity [9]. The former domain plays a role in chromatin remodeling by interacting with the SWI/SNF nucleosome remodeling complex [10]. The C-terminal region of KDM6A contains the catalytic Jumonji (JmjC) domain, with a histone demethylase activity, that is specific for the catalysis of mono, di, tri-demethylation of the lysine 27 of the histone H3 (H3K27) [11] (Fig. 1). At the time of this writing, fifteen missense pathogenic variants in KDM6A are known to cause KS, as reported in the Human Gene Mutation Database [12], with seven of them located in the catalytic domain and therefore suitable to be structurally analyzed. The remaining variants localize in the N-terminal TPR domain for which an experimentally solved structure is not yet available. In particular, three of the seven variants were also reported in large-scale studies and linked to specific phenotypes as intellectual disability (HP:0001249), microcephaly (HP:0000252), strabismus (HP:0000486), developmental delay (HP:0002194) and, thus not directly linked to KS (Tab. S1).
Fig. 1.

3D structure of the KDM6A-H3 complex. The linker (910–932), JmjC (933–1268), zinc (1315–1378), and helical (886–902/1269–1314/1379–1395) domains are colored in blue, green, magenta, and brown, respectively. Histone H3 is colored in gray.
2.2. System preparation
Atomic coordinates of KDM6A, in the H3-bound form, were obtained from the X-ray structure (3AVR) stored in the Protein Data Bank [13]. The residues 17–38 of the ligand-bound structure correspond to the histone H3K27me3 peptide (Fig. 1, gray), while the residues 880–1401 constitute the C-terminal catalytic fragment. Missing residues (from 1047 to 1078) of both structures were inferred using MODELLER v9.16 [14]. The wild-type structure was mutated in-silico to introduce p.Pro941Ser, p.Asp980Val, p.Ser1025Gly, p.His1060Leu, p.Leu1200Phe, p.Gly1223Asp, p.Gln1248Arg and p.Arg1255Trp variants (Fig. 1). The Predicted Side Chain Panel of the Schrodinger Suite was employed to check the orientation of the side chains after mutation [15]. The Amber ff14SB force field was applied to amino acids, where the Zinc AMBER force field (ZAFF) [16] was employed for the Zn(II) ion, covalently bonded to four cysteine residues in the Zinc domain, to preserve the tetrahedral coordination crucial for the overall stability of the protein. Using the tleap module of AmberTools18, each of the nine models was embedded in a simulation box, extending up to 12 Å, and solvated using the TIP3P water model. Furthermore, an appropriate number of Na+ and Cl- counter ions were added to neutralize the overall charge of the models. Each model was first subjected to energy minimization using steepest descent, followed by conjugate gradient methods. Then, they were gradually heated and then equilibrated for approximately 5 ns, by time steps of 1 fs. A 10 Å cutoff was used for non-bonded short-range interactions, while long-range electrostatics were treated with the particle-mesh Ewald method. Temperature and pressure were maintained at 300 K and 101.3 kPa, respectively, using the Langevin dynamics and Piston method.
2.3. Gaussian accelerated molecular dynamics simulation protocol
In this work, we used accelerated Molecular Dynamics (aMD) techniques to simulate processes that need to overcome large energy barriers to take place, and that occur on time scales of milliseconds or even larger. In these cases, classical MD techniques [17] are ineffective. In particular, we implemented a novel aMD technique, called Gaussian accelerated Molecular Dynamics (GaMD). It is an enhanced sampling method that works by providing a harmonic boost potential, which follows a Gaussian distribution, to smooth the system potential energy surface, thus causing the decrease of local energy barriers and accelerating the transitions between low-energy states [18]. The boost potential is applied in a dual-boost scheme; namely, two acceleration potentials are applied simultaneously to the system: (I) a dihedral potential boost and (ii) a total potential boost. A time step of 2 fs has been used in this study. Maximum, minimum, average, and standard deviation values of the system potential have been obtained from an initial ~two ns NPT simulation with no boost potential. Each GaMD simulation was performed starting with a ~50 ns run, in which the boost potential has been updated every 1.6 ns, thus reaching the equilibrium. Finally, 500 ns of GaMD simulations have been carried out in the NVT ensemble. Each system was simulated three times using the GPU version of AMBER 18 running on 1 NVIDIA Quadro P6000 and 3 NVIDIA RTX 2080Ti.
2.4. Simulation analysis
AmberTools18 was used to calculate the (i) Root-Mean-Square Deviation (RMSD) and (ii) Root-Mean-Square Fluctuation (RMSF), measuring respectively the average distance and the deviation over time between the positions of the Cα atomic coordinates of each residue and those of the reference X-ray structure. Finally, AmberTools18 was also employed to calculate the (iii) radius of gyration (Rg), which measures the root mean square distance from each atom of the protein to their centroids.
The PyReweighting script was used to reweight the GaMD simulations with the aim to calculate the potential of mean force (PMF) profiles and to examine the boost potential distributions. Based on the Gaussian distribution of the boost potential, cumulant expansion to second order was applied to reweight the aggregate GaMD trajectories. 2D PMF profiles were drawn using the RMSD and the radius of gyration (RMSD, Rg) as reaction coordinates to describe the rearrangement of the protein domains.
Principal Component Analysis (PCA) of atomic fluctuations was performed to infer large scale collective fluctuations of atoms and thus to predict a low-dimensional subspace in which essential protein motions are expected to take place. The fluctuations of particles in an MD simulation are, by definition, correlated due to interactions between the particles. Interactive particles give rise to structures, which are often directly related to protein functions or biophysical properties. PCA was thus used to construct the covariance matrix, and this was done using gmx_covar, as implemented in GROMACS v2018, which captures the degree of collinearity of atomic motions for each pair of atoms. The conformational changes of the systems, caused by the mutations under investigation, were explored using Dynamic Cross-Correlation Maps (DCCMs), generated using a custom Python script. The script took covariance matrices in input to generate the corresponding correlation matrices. DCCMs allowed us to study the long-range interactions between all pairs of atoms, whose correlated and anticorrelated motions were reported. The 3D movies of the temporal dynamics of the wild-type and mutated proteins were obtained using the g_anaeig GROMACS tool.
Finally, the GROMACS plugin g_hbond [19] was used to evaluate the per-residue hydrogen bonds, with an angle cutoff of 30° and a donor–acceptor distance of 3.5 Å. GetContacts (https://getcontacts.github.io) was used to quickly compute all the atomic interactions and contacts established in each frame of the trajectory. 3D figures and motions were generated using UCSF Chimera [20] and Visual Molecular Dynamics software (VMD) [21].
2.5. Network analysis with Pyntacle
An interesting approach to study the topology of complex systems, such as the three-dimensional structure of proteins, is the description of its inter-residue interactions as networks. This permits to exploit the theory of graphs to identify key-amino acids that could play essential roles in the stability of a protein as well as to identify fundamental cross-talks between amino acids. In a network representation of a protein, amino acids are nodes linked by edges, which can alternatively represent the spatial distances between amino acids, their physical interactions (Protein Contact Network, PCN), or their interaction energy (Residue Interaction Energy, RIN) [22]. A universal principle to these kinds of networks is that an external perturbation (e.g., an amino acid variant) can induce a dynamical cascade of fluctuations to their residues, whose effects can be studied locally or globally to the whole protein by ad-hoc centrality metrics.
Pyntacle (http://pyntacle.css-mendel.it/) was employed with this aim. It is an open-source Python suite of algorithms that allows the identification of groups of nodes that exhibit critical topological properties in a network. We studied how the betweenness [23] and the group-betweenness [24] topological metrics changed in our systems during the simulation of their dynamics. We relied on betweenness since it bases on the importance of links and of their distributions throughout a network.
A network has been built for every frame of the trajectories connecting residues through hydrogen bonds. The differences of the values of betweenness centrality calculated for a specified set of residues of the mutants and wild-type proteins were averaged across frames and then compared with the Mann-Whitney test (adjusted with the Benjamini-Hochberg procedure). Betweenness values were transformed into z-scores for easier comparisons. Additionally, we have computed and averaged the group-betweenness values of all node pairs involving Arg922 and each of the variants under consideration across all frames of all trajectories. Wild-type and mutant distributions of group-betweenness values were then compared with the Mann-Whitney test. Moreover, for each frame, we have made a background set of group-betweenness values by enumerating 100 random pairs of nodes, and thus computing their group-betweenness scores. The percentile of the group-betweenness of each Arg922-variant pair was then calculated over the background set of each frame and the average percentile was then calculated across frames. Wild-type and mutant percentile distributions were then compared with the Mann-Whitney test.
3. Results
In the following sections, we will present the results of an array of molecular dynamics analysis strategies performed on the seven considered KDM6A mutants, and we will compare them with the wild-type reference protein structure. To increase the confidence of these results, we will analyze a low frequency and benign variant and will comment on its dynamical properties.
3.1. Characterization and preliminary assessments of seven KDM6A pathogenic missense variants
It is well-established that the catalytic JmjC domain of the KDM6A protein strictly binds to the histone H3 residues 25–33 through a specific recognition sequence. The conserved zinc-binding domain stabilizes this interaction by both changing its conformation upon histone binding and recognizing the H3L20 side chain via a hydrophobic patch on its surface, which is inaccessible in the H3-free form [13]. Based on this consideration, we localized each of the disease-causing variants and visually inspected their local topology as a preliminary analysis step.
Ser1025Gly was the only substitution directly altering a residue located in the catalytic pocket, whose backbone amide group, in turn, forms a hydrogen bond with the H3T32 side chain in the wild-type structure. On the contrary, Leu1200Phe, Gly1223Asp, Gln1248Arg, and Arg1255Trp reside in the catalytic pocket but do not participate in any direct interaction with the H3. Pro941Ser and Asp980Val locate outside the catalytic pocket (Fig. 1). Conclusions on the potential pathogenic effects of these variants were not straightforward, but the observation that all the KS patients showed common phenotypic traits leaves room to the hypothesis that these mutations may lead to distinct alterations of common mechanisms. For this reason, their effect on protein structure and their potentially related pathogenic mechanisms were assessed by GaMD simulation.
3.2. RMSD and RMSF
Structural rearrangements during simulation were assessed by estimating the RMSD values of the alpha carbons (Cα) of the systems under investigation in comparison with those of the reference crystal structure. The wild-type protein was stable for about two-thirds of the simulation, with only small deviations from the initial reference structure. However, in the last 100 ns, a slightly unstable phase could be observed, with values increasing even up to 0.5 nm. On the other hand, all mutants exhibited more stable RMSD values, especially in the last 100 ns (Fig. 2A).
Fig. 2.

RMSD (A) and RMSF (B) profiles of the heavy atoms of KDM6A wild-type (black) and all mutants. The green box highlights the linker domain; the red box highlights the reconstructed region; dashed lines mark the positions of the mutated residues.
We then calculated per-residue RMSF (Fig. 2B). All simulated systems showed variable flexibility in two parts of the protein. The first corresponds to the linker domain (residues 910–932 highlighted in green in Fig. 2B, plus proximal residues), where the wild-type was, in general, less flexible than the mutants, except for Leu1200Phe, Arg1255Trp and Ser1025Gly that exhibited slightly higher RMSF values only in the second half of the domain. As expected, the reconstructed portion of the protein (residues 1047–1078 highlighted in red in Fig. 2B) showed increased mobility, and this was more evident in the wild-type protein than mutants, except for Leu1200Phe. The mean absolute difference of the RMSF values of the linker region between mutants and wild-type was compared with the mean absolute differences calculated for all other domains. The linker flexibility resulted significantly altered in all mutants, when compared with the JmjC and zinc domains (Mann-Whitney U, p < 0.05), except for Asp980Val where the linker domain was significantly more flexible only if compared with the zinc domain. Moreover, the linker domains of the Gln1248Arg and Ser1025Gly mutants were significantly more flexible than the JmjC and zinc domains, but not more flexible than the helical domain (Tab. S2). Finally, Ser1025Gly was characterized by a dramatic loss of the ligand flexibility (not visible in Fig. 2B).
It is worth noticing that the standard deviation of the backbone RMSD (except for the reconstructed region) and RMSF values among the three repeated MD trajectories for the native and all mutants were < 0.1 Å, except for Asp980Val and Ser1025Gly, which exhibited SD values of about 0.2 Å and 0.6 Å, respectively. The small SD values (<1Å) indicate that these results are consistent through all replicas.
3.3. Potential of mean force
Potential of Mean Force (PMF) calculations were performed for each of the simulated trajectories, using RMSD and Radius of Gyration (Rg) as reaction coordinates. Fig. 3 reports the PMF plots obtained for all the H3-bound configurations, describing the conformational behavior of KDM6A in the presence of the histone H3K27me3. The wild-type PMF profile highlights two distinct low-energy conformations that were reached by the system during the simulation. The protein starts, in fact, from a local energy minimum at (3.5 Å, 26 Å), from which it explores several intermediate configurations during a large part of the GaMD simulation, before reaching its final local energy minimum at (5 Å, 25.5 Å). On the contrary, all the other mutant PMF profiles showed one clear and sharp energy minimum around (4–5 Å, 26 Å), owed to the presence of fixed configurations during all frames of simulation. Only Pro941Ser explored two low energy-conformations at (4 Å, 25.5 Å) and (5 Å, 26 Å), which however were closer than the local minima of the wild-type protein.
Fig. 3.

Potential of Mean Force (PMF) profiles of KDM6A wild-type (top-left) and KDM6A mutants. RMSD values are reported in the X-axis and the radius of gyration in the Y-axis. Colors represent PMF levels, from the absolute minimum (blue) to the absolute maximum (firebrick red).
3.4. Dynamic Cross-Correlation maps
DCCMs were computed for the wild-type and mutant proteins to track and characterize the conformational changes during simulation of each protein domain (Fig. 4). The Asp980Val and Pro941Ser correlation matrices showed a dramatic decrease in all motions. This is suggestive of overall blocked dynamics. In the wild-type model, the first part of the linker domain (residues 910–917) (leftmost blue box) moved in a highly anticorrelated way with the C-terminal region of the JmjC domain (residues 1078–1268), while the movement of the second part of the linker domain (residues 918–932) correlated positively with the C-terminal region of the JmjC domain (rightmost blue box). This movement was partially or totally lost in all mutants, but Leu1200Phe. DCCMs of Ser1025Gly, Gln1248Arg, and Arg1255Trp describe a clear decline of correlated motions in the 1078–1268 JmjC region (red and green boxes in Fig. 4). The reconstructed region nearby the JmjC domain (residues 1048–1077) moved in an anticorrelated way with the whole JmjC domain. Even this movement was totally or partially lost in mutants (violet box). Finally, Leu1200Phe showed a correlation matrix comparable with that of the wild-type, where movements of residues were slightly decreased in all regions.
Fig. 4.

DCCMs of wild-type (top-left) and KDM6A mutants. Perfect correlations are highlighted in red (direct) or violet (inverse). Boxes in the wild-type DCCM highlight: (blue) the JmjC region spanning residues from 1078 to 1268, describing correlated and anticorrelated motions with the linker domain region (residues 910–933); (red) the N-terminal region of JmjC (935–985) correlates with the C-terminal region (1078–1268); (violet) the region spanning residues 1048 to 1077 correlates with the whole JmjC domain; (green) correlated internal subregions of the JmjC domain.
The Movie S1, obtained by interpolating the intermediate frames between the two extreme projections of each trajectory, accounted for >80% of the overall motions, thereby revealing a notable global rearrangement of the wild-type structure compared to mutants. In particular, the wild-type protein showed a precise translational motion along its central axis that led, in turn, to a rotational movement of the histone. This partial reshuffling of the various domains allowed the consequent rearrangement of the ligand and its active interaction with the binding pocket. On the other hand, the linker domain revealed significantly higher mobility in all mutants but the wild-type protein. This increased flexibility is due to the loss of an essential interaction between the linker domain and the rest of the protein, later described. As a final note, during the simulations, the observed wild-type structural transition, lost in all mutants, resulted in mainly involving the JmjC domain with a modest shift of the Zinc-domain region.
3.5. Hydrogen bonds analysis
We have monitored the establishment of the hydrogen bonds (h-bonds) in all simulations of all systems. The most striking result regarded Ser1025Gly that missed the h-bond between the residue 1149 of KDM6A and 29 of H3. This bond involves the residue adjacent to K27me3 and is crucial for the correct orientation of K27me3 inside the binding pocket. The h-bonds of all the other mutants displayed similar residence times to those of the wild-type system, except for those involving the linker domain and the surrounding regions of the JmjC domain. In Table 1, we report their identity and residence times during the simulations.
Table 1.
H-bonds established between residues in all simulated systems, thus not including the H3 histone, and relative residence times (>50% in at least in one simulated system). The underlined values refer to the residence times that differ by at least 20% between mutants and wild-type proteins.
| Acceptor residue number | Acceptor atom | Donor residue number | Donor atom | Wild-type | Asp980Val | Ser1025Gly | Gln1248Arg | Arg1255Trp | Leu1200Phe | Gly1223Asp | Pro941Ser |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1255 | H11 | 1147 | OE1 | 0 | 67.0 | 83.5 | 2.5 | 0 | 64.4 | 80.2 | 38.7 |
| 1255 | H21 | 1147 | OE1 | 0 | 62.6 | 74.2 | 2.3 | 0 | 61.4 | 52.5 | 23.2 |
| 1255 | H21 | 1171 | OE2 | 0 | 0 | 0 | 53.6 | 0 | 0 | 0 | 0 |
| 1255 | H | 1251 | O | 82.5 | 77.5 | 54.9 | 83.3 | 64.0 | 79.9 | 84.4 | 75.8 |
| 1255 | H21 | 1254 | OE1 | 80.8 | 1.1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1255 | HE | 1254 | OE2 | 69.6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1255 | H21 | 922 | O | 54.7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1256 | H | 1252 | O | 80.6 | 88.0 | 88.9 | 83.8 | 89.3 | 89.0 | 82.6 | 86.7 |
| 1256 | HH | 1337 | OE1 | 47.5 | 55.7 | 59.1 | 44.9 | 42.9 | 43.8 | 53.5 | 37.6 |
| 1256 | HH | 1337 | OE2 | 44.8 | 55.1 | 31.9 | 52.2 | 48.0 | 51.5 | 53.9 | 60.6 |
| 1257 | H | 1253 | O | 88.5 | 94.7 | 93.7 | 96.3 | 87.0 | 92.1 | 90.0 | 96.7 |
| 922 | H11 | 1171 | OE1 | 65.0 | 18.1 | 18.0 | 0 | 0 | 17.2 | 0 | 0 |
| 922 | H11 | 1171 | OE2 | 61.6 | 22.2 | 17.6 | 0 | 0 | 23.5 | 0 | 0 |
| 922 | H21 | 1171 | OE1 | 51.2 | 50.0 | 53.3 | 0 | 0 | 34.4 | 0 | 0 |
| 922 | H21 | 1171 | OE2 | 45.9 | 50.7 | 48.2 | 0 | 0 | 31.4 | 0 | 0 |
One of the disrupted h-bonds is the one established between Arg922, in the linker domain, and Arg1255, in the JmjC domain, which is lost or severely perturbed in all mutants. In particular, Arg922 establishes an h-bond with Glu1171 in the wild-type system (Fig. 5), which is lost in Gln1248Arg, Arg1255Trp, Gly1223Asp, and Pro941Ser, and partially lost in Leu1200Phe and Asp980Val, for which variable residence times can be observed. Finally, Arg1255 establishes h-bonds with Gln1147 in Asp980Val, Ser1025Gly, Leu1200Phe, Gly1223Asp, and Pro941Ser and with Glu1171 in Gln1248Arg, which are never created in the wild-type.
Fig. 5.

Residues at the interface between the linker and JmjC domains for wild-type and mutants (last frame of the simulation). H-bonds are represented as green dashed lines in the wild-type protein.
3.6. Dynamic residue interaction network analysis
While the basic motions of KDM6A were assessed, globally, by the analysis of h-bonds, the dynamic fluctuations of residues were determined using Pyntacle. In particular, we built a network for each simulated frame, where nodes were amino acids and edges represented h-bonds between them. Each of these networks was analyzed topologically, calculating the betweenness and group-betweenness local metrics for nodes and groups of nodes. The nodes with the highest betweenness centrality values fell into the JmjC and the linker domains. We recall here that the betweenness centrality metric assesses the extent to which a node or a group of nodes stand between each other. It is thus a measure of centrality, which is tightly related to the concept of connectivity of networks.
Residue betweenness decreased clearly in the 1240–1260 region (JmjC region mainly involved in the interaction with the linker domain) for all the mutants and for the residue Arg922 belonging to the linker domain (Tab. S3), thereby indicating a generally reduced connectivity involving this region. This region resulted in being significantly less connected than 100 random sets of residues of equal size (Mann-Whitney adjusted p < 0.05) in 3 mutants: Gln1248Arg, Leu1200Phe, and Gly1223Asp. Arg922 showed remarkably low betweenness in all mutants, especially in these three mutants. The z-score of the difference of betweenness centrality (-3.11, -2.69 and -3.76, respectively) revealed a remarkable difference of betweenness values compared to all the other residues in the complex.
Hence, having determined the individual importance of the residue Arg922, we calculated the betweenness centrality of the group made by Arg922 and each other mutant, for each frame of the trajectories when these residues were not isolated nodes. The group-betweenness scores were significantly lower when comparing the Arg922-wild-type-residue pair and the corresponding Arg922-variant-residue pair across the frames (Mann-Whitney p ≪ 0.01). Moreover, we built a background distribution of group-betweenness values for each frame by selecting 100 random pairs of residues in the network. We then calculated the percentile associated with the actual pair of residues (e.g., Arg922 plus a variant) in this background distribution and calculated the average percentile across all the simulation frames. Arg922, paired with the variants achieved an average percentile of 58.1 across the variants, while Arg922 paired with the corresponding wild-type sites averaged 77.5 in the wild-type structure. We observed a statistically significant difference between mutants and wild-type (Mann-Whitney p ≪ 0.01, except for Gly1223Asp where p < 0.05). From this analysis, we can conclude that Arg922 paired with the variant sites, occupies a key position in the network.
3.7. Study of a putatively benign missense variant
To increase the reliability of this study, we have applied the same analytical approach so far described to another variant, His1060Leu. It was selected from the Genome Aggregation Database (gnomAD v2.2.1, rs141303384) [25] because it was (i) the only annotated missense variant located in the catalytic domain of KDM6A, (ii) extremely rare (MAF 0.03%), (iii) and classified as benign in ClinVar [26]. This variant is located in a structurally disordered region, which was successfully reconstructed as described in section 2.2 of the Materials and Methods section. It is, however, important to notice that these kinds of regions usually exhibit extreme flexibility and accurate characterization of their conformational ensembles is not always a straightforward task.
His1060Leu exhibited stable RMSD values along most of its trajectory (Fig. 6A, left). The reconstructed portion (red band in Fig. 6A, right) exhibited high differential mobility, as expected, compared to the wild-type, while the linker domain (green band in Fig. 6A, right) revealed identical flexibility profiles. This behavior is confirmed in Fig. 6B, where the majority of the movements of the linker domain towards the JmjC domain (blue square) were conserved. Enumerating the h-bonds formed during simulation, we did not observe the drastic disruption of the interactions between the linker and the JmjC regions, which characterize most of the mutants, with the Arg922-Glu1171 connection partially conserved. However, the critical h-bond between Arg922 and Arg1255, found in the wild-type protein, was not significantly present in this mutant. A more in-depth study revealed the presence of other, compensative, types of interactions occurring between the linker and the JmjC domains, such as van der Waals and hydrophobic bonds. In details, the Arg922 established a strong interaction with the JmjC domain during the entire molecular dynamics simulation of the wild-type protein, especially with the residues Arg1255 and Glu1171. The interaction with this latter residue was entirely conserved in His1060Leu, Asp980Val, and Ser1025Gly, and only partially in Leu1200Phe (Fig. 6C left). Remarkably, the connection with Arg1255, which in this case was mainly characterized by van der Waals interactions, was maintained only in the His1060Leu mutant (Fig. 6C right).
Fig. 6.

A. RMSD and RMSF profiles of the heavy atoms of KDM6A wild-type (black) and His1060Leu. B. DCCMs for wild-type (left) and His1060Leu mutant. The blue box demarks the JmjC region spanning residues from 1078 to 1268, whose motions correlate/anticorrelate with residues (910–933) belonging to the linker domain region. C. Distribution of the occurrences of contacts established between Arg922 and Glu1171 (left) and between Arg922 and Arg1255 (right) over time for all mutants. This last link is mainly characterized by van der Waals interactions and is maintained only in the His1060Leu mutant.
Finally, we performed the same network analysis procedure used with the other mutants and observed no significant differences between His1060Leu and the wild-type proteins. We did not observe any significant difference in the 1240–1260 region between His1060Leu and the wild-type structure (Mann Whitney p > 0.05), and Arg922 showed a positive difference of betweenness centrality value (z-score of + 1.86). Moreover, when Arg922 was paired with His1060Leu, we measured an average percentile of group-betweenness of 77.75 in the mutant and of 76.88 in the wild-type (therefore significantly higher, Mann-Whitney p < 0.05). From a network analysis perspective, His1060Leu appears to be in contrast with all the other mutants and more similar to the wild-type structure.
4. Conclusions
Our strategy of analysis provided the reader with a possible explanation of pathogenic mechanisms triggered by missense variants in KDM6A. Even though the analyzed variants determined specific local perturbations, we have verified that their indirect consequences impacted significantly on the interaction between the linker domain (and neighboring amino acids) and the JmjC-domain, thus causing a loss of function effect. The RMSD analysis showed that all mutants behave differently compared to the wild-type. Additionally, the RMSF profiles pointed out that the mainly altered mechanisms involve residues forming the linker domain, which in the wild-type system was less flexible than Gln1248Arg, Arg1255Trp, Leu1200Phe and Ser1025Gly, mostly involving the second half of the linker domain. The remaining mutants behaved similarly in the immediate neighboring regions of the linker domain. This evidence was confirmed by the analysis of the DCCMs that highlighted how correlated and anticorrelated movements of the linker domain were partially or totally lost in all mutants. In particular, a lack of correlation emerged between the motions of the subdomains of JmjC.
Although a certain degree of structural rigidity of the linker domain is necessary for the correct catalytic mechanism of KDM6A to occur, all mutants exhibited a disordered movement of the linker domain that caused an increased overall rigidity of the KDM6A-H3 complex, which finally interfered with the correct exposure and orientation of the trimethylated lysine in the catalytic site.
PMFs were calculated to profile the free energy landscape of the simulated systems. They confirmed the significant energetic differences between mutants and wild-type systems, this latter being characterized by two well-separated low-energy minima, through which KDM6A-H3 seems to physiologically transit. This mechanism appears to have been completely lost in the mutants.
Finally, the analysis of the h-bonds allowed us to explain how the physiological role of the linker domain was altered at a molecular level. In the wild-type protein, in particular, the network made by the h-bonds linking Arg1255, Glu1171, and Arg922 appeared to work, keeping a portion of the linker domain constrained. Conversely, in all mutants, these bonds were missing or organized differently, leaving the linker domain unconstrained and thus unable to coordinate the other domains of KDM6A. This evidence was confirmed when the dynamic residue interaction networks were analyzed with Pyntacle. Betweenness and group-betweenness topological metrics highlighted the critical roles of essential residues located in the JmjC and linker domains in sustaining the global conformational transitions of the wild-type protein. In addition to the considered variants, Arg922 resulted in being a possible partner variant that may exert a functional role in an epistatic fashion. Interestingly, two allelic variants are associated with this residue, Arg922Gly (MAF 0.0016%, rs1368359635, dbSNP153), and Arg922Lys, which were reported in a patient with chronic myelomonocytic leukemia [27].
In conclusion, this study disclosed putative mechanisms of the pathogenesis of KS2. A similar study is still not possible for KS1 because a complete and reliable model of the protein KMT2D is not available in the PDB. Hence, for the former and not for the latter type of the disease, specialized techniques of molecular dynamics simulation allowed us to shed light on specific properties of a selected set of missense mutations which proved to significantly hamper the interaction between KDM6A and the histone H3, by modifying the dynamics of the linker domain and finally causing a loss of function effect. Although the possibility that these variants might be compensated by the increase of the expression levels of the KDM6A isoforms or by leveling the methylation/demethylation balance of H3 may represent a limitation to this approach, these results underline the significance of developing and applying ad-hoc computational techniques to support the interpretation of the pathogenicity of variants resulting from sequencing studies with the aim of facilitating the evaluation of clinical effects, with a direct impact on diagnosis and genetic counseling.
Funding sources
We gratefully acknowledge NVIDIA Corporation, the Amber Team and the “5x1000” voluntary contribution for supporting this research.
CRediT authorship contribution statement
Francesco Petrizzelli: Conceptualization, Methodology, Writing - original draft. Tommaso Biagini: Conceptualization, Methodology, Writing - original draft. Alessandro Barbieri: Methodology. Luca Parca: Methodology. Noemi Panzironi: Methodology, Writing - review & editing. Stefano Castellana: Investigation. Viviana Caputo: Methodology, Writing - review & editing. Angelo Luigi Vescovi: Funding acquisition. Massimo Carella: Funding acquisition. Tommaso Mazza: Writing - review & editing, Conceptualization, Project administration.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.07.013.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Faundes V., Newman W.G., Bernardini L., Canham N., Clayton-Smith J., Dallapiccola B. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. 2018;102:175–187. doi: 10.1016/j.ajhg.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Micale L., Augello B., Maffeo C., Selicorni A., Zucchetti F., Fusco C. Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of Kabuki syndrome patients. Hum Mutat. 2014;35:841–850. doi: 10.1002/humu.22547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bögershausen N., Gatinois V., Riehmer V., Kayserili H., Becker J., Thoenes M. Mutation update for kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked kabuki syndrome subtype 2. Hum Mutat. 2016;37:847–864. doi: 10.1002/humu.23026. [DOI] [PubMed] [Google Scholar]
- 4.Fokstuen S., Makrythanasis P., Hammar E., Guipponi M., Ranza E., Varvagiannis K. Experience of a multidisciplinary task force with exome sequencing for Mendelian disorders. Hum Genomics. 2016;10:24. doi: 10.1186/s40246-016-0080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J.-H., Lee J.H., Lee I.-S., Lee S.B., Cho K.S. Histone Lysine methylation and neurodevelopmental disorders. Int J Mol Sci. 2017;18. doi: 10.3390/ijms18071404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bögershausen N., Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83:201–211. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- 7.Cocciadiferro D., Augello B., De Nittis P., Zhang J., Mandriani B., Malerba N. Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum Mol Genet. 2018;27:3651–3668. doi: 10.1093/hmg/ddy241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biagini T, Petrizzelli F, Truglio M, Cespa R, Barbieri A, Capocefalo D, et al. Are Gaming-Enabled Graphic Processing Unit Cards Convenient for Molecular Dynamics Simulation? Evol Bioinform Online 2019;15:1176934319850144. [DOI] [PMC free article] [PubMed]
- 9.Adam M. Insights into the molecular genetics of Kabuki syndrome. AGG. 2015;121 [Google Scholar]
- 10.Lan F., Bayliss P.E., Rinn J.L., Whetstine J.R., Wang J.K., Chen S. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 11.Hong S., Cho Y.-W., Yu L.-R., Yu H., Veenstra T.D., Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengoku T., Yokoyama S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011;25:2266–2277. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics 2014;47:5.6.1–32. [DOI] [PubMed]
- 15.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 16.Peters M.B., Yang Y., Wang B., Füsti-Molnár L., Weaver M.N., Merz K.M., Jr. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF) J Chem Theory Comput. 2010;6:2935–2947. doi: 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biagini T., Chillemi G., Mazzoccoli G., Grottesi A., Fusilli C., Capocefalo D. Molecular dynamics recipes for genome research. Brief Bioinform. 2018;19:853–862. doi: 10.1093/bib/bbx006. [DOI] [PubMed] [Google Scholar]
- 18.Miao Y., Feher V.A., McCammon J.A. Gaussian accelerated molecular dynamics: unconstrained enhanced sampling and free energy calculation. J Chem Theory Comput. 2015;11:3584–3595. doi: 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 20.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 21.Humphrey W., Dalke A., Schulten K.V.M.D. Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 22.Grewal R.K., Roy S. Modeling proteins as residue interaction networks. Protein Pept Lett. 2015;22:923–933. doi: 10.2174/0929866522666150728115552. [DOI] [PubMed] [Google Scholar]
- 23.Freeman L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry. 1977;40:35. doi: 10.2307/3033543. [DOI] [Google Scholar]
- 24.Borgatti S.P. Identifying sets of key players in a social network. Comput Math Org Theory. 2006;12:21–34. doi: 10.1007/s10588-006-7084-x. [DOI] [Google Scholar]
- 25.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jankowska A.M., Makishima H., Tiu R.V., Szpurka H., Huang Y., Traina F. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118 doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.