

Abstract
Alzheimer’s disease (AD) is the main cause of dementia in the elderly. Although activation of brain insulin signaling protects neurons, preserves memory in AD models and appears beneficial in patients, the role of insulin-like growth factor 1 (IGF1) remains incompletely understood. We found reduced active/inactive IGF1 ratio and increased IGF1R expression in postmortem hippocampal tissue from AD patients, suggesting impaired brain IGF1 signaling in AD. Active/inactive IGF-1 ratio was also reduced in the brains of mouse models of AD. We next investigated the possible protective role of IGF1 in AD models. We used a recombinant adenoviral vector, RAd-IGF1, to drive the expression of IGF1 in primary hippocampal neuronal cultures prior to exposure to AβOs, toxins that accumulate in AD brains and have been implicated in early synapse dysfunction and memory impairment. Cultures transduced with RAd-IGF1 showed decreased binding of AβOs to neurons and were protected against AβO-induced neuronal oxidative stress and loss of dendritic spines. Interestingly, in vivo transduction with RAd-IGF1 blocked memory impairment caused by intracerebroventricular (i.c.v.) infusion of AβOs in mice. Our results demonstrate altered active IGF1 and IGF1R levels in AD hippocampi, and suggest that boosting brain expression of IGF1 may comprise an approach to prevent neuronal damage and memory loss in AD.
Keywords: dementia, human brain, hippocampus, insulin-like growth factor I, adenovirus, memory
Introduction
Alzheimer’s disease (AD), the main form of dementia in the elderly, is clinically defined by memory loss and progressive cognitive impairment. Despite extensive investigation and considerable insight gained during the past two decades [1–3], the mechanisms involved in AD pathogenesis remain to be fully elucidated.
The insulin/IGF1 signaling pathway, a major regulator of protein homoeostasis and longevity, has emerged as a critical pathway in AD [4–7]. Clinical and epidemiological studies have revealed that diabetic patients are at higher risk of developing dementia (e.g., [8–11]), suggesting a connection between peripheral and central insulin resistance in AD. Insulin has been investigated as a potential therapeutic agent to restore brain metabolism and cognition in AD, with results showing memory benefit in mild cognitive impairment and in early/moderate AD individuals [12–16]. On the other hand, IGF1 has not yet reached clinical trials in AD, likely in part because its role in AD remains controversial, with pre-clinical studies showing both beneficial and detrimental effects of IGF1 on disease pathogenesis and progression [17, 18].
Although IGF1 is known as a neuroprotective hormone [19], recent evidence suggests that IGF1 signaling could be detrimental in AD progression [20]. Moreover, clinical studies have found contrasting results regarding circulating levels of IGF1 in AD patients [21–23], and studies evaluating the relationship between plasma levels of IGF1 and AD risk have yielded controversial results [24–26]. Of note, brain IGF1 expression decreases with age, suggesting that peripheral IGF1 reaching the brain may play an important role in IGF1 signaling in the adult brain [17]. Steen et al. (2005) reported decreased immunoreactivities for IGF1 and IGF1 receptor (IGF1R), along with reduced expression of IGF1R and a trend of increase in IGF1 expression, in the hippocampi of AD patients. Further, IGF1 mRNA level was significantly reduced in the frontal cortex and hypothalamus of AD patients [27]. To the best of our knowledge, and despite the potential relevance to AD pathogenesis, no other studies have characterized protein levels of IGF1 or IGF1R in the AD hippocampus. Here, we show that IGF1 and IGF1R levels are altered in the hippocampi of AD patients and mouse models of AD, and investigate viral-mediated transduction of IGF1 as an approach to prevent AD-related neurotoxicity and memory impairment in vitro and in vivo.
Materials and Methods
Postmortem hippocampal tissue:
Human postmortem tissue samples were obtained from the Brain Bank of the Brazilian Aging Brain Study Group, School of Medicine of the University of Sao Paulo. Brains were obtained from the Sao Paulo Autopsy Service, after written informed consent. We studied 11 cases with a neuropathological diagnosis of AD confirmed for the presence of pathological hallmarks by an experienced neuropathologist, and 10 cases without neuropathological changes. Subject demographics are presented in Table 1. Clinical dementia rating (CDR) scores were determined by a validated interview conducted with the informant caregiver [28, 29]. The control (cognitively intact) group consisted of cases with CDR=0, whereas the AD group included cases with CDR ranging from 0.5 to 3.
Table 1.
Demographics of individual subjects in post-mortem analysis.
| Case number | Sex | Age | Postmortem Interval (h) | CDR | Braak Stage |
|---|---|---|---|---|---|
| 8203/05 | M | 65 | 10,85 | 0 | 1 |
| 9766/05 | M | 89 | 14,17 | 0 | 2 |
| 6947/05 | F | 82 | 14,83 | 0 | 1 |
| 10921/05 | M | 81 | 9 | 0 | 2 |
| 9828/05 | F | 61 | 14,66 | 0 | 1 |
| 7178/05 | F | 75 | 12,58 | 0 | 2 |
| 2275/05 | F | 60 | 20,42 | 0 | 1 |
| 1811/05 | F | 64 | 21,5 | 0 | 0 |
| 1372/05 | M | 69 | 16,5 | 0 | 0 |
| 2574/05 | M | 83 | 15,92 | 0 | 0 |
| 5734/05 | M | 71 | 8,67 | 0,5 | 4 |
| 6275/06 | F | 92 | 27,83 | 0,5 | 4 |
| 7240/05 | F | 84 | 8,33 | 0,5 | 3 |
| 5258/06 | M | 85 | 14,3 | 1 | 3 |
| 3980/05 | F | 81 | 13,88 | 2 | 5 |
| 7207/08 | F | 84 | 12,92 | 3 | 6 |
| 2682/08 | M | 82 | 15,33 | 3 | 6 |
| 2018/09 | F | 81 | 10,67 | 3 | 4 |
| 5248/07 | F | 86 | 17,08 | 3 | 5 |
| 691/07 | F | 87 | 17,72 | 3 | 5 |
| 2544/09 | M | 80 | 18,6 | 3 | 5 |
Animals:
All procedures were approved by the Federal University of Rio de Janeiro Institutional Animal Care and Use Committee (protocol number #IBqM 130/15) and were in full compliance with the NIH Guide for Care and Use of Laboratory Animals. Mice were obtained from the animal facility at Federal University of Rio de Janeiro and were housed (5 mice/cage) in a temperature controlled facility with 12-hour light/dark cycle (on at 7 am) and ad libitum access to food. AD mouse models used in this work consisted of (1) 16–19 month-old transgenic APPswe/PS1ΔE9 (APP/PS1) mice [30] and (2) 3 month-old Swiss mice that received a single intracerebroventricular (i.c.v.) infusion of amyloid-β oligomers (AβOs), a model that recapitulates AD-like brain pathology and memory impairment [31–35]. Numbers of animals used in each experiment are provided in the corresponding Figure Legends. Animals were euthanized by cervical dislocation followed by decapitation and dissection of hippocampi. Tissue was immediately frozen in liquid nitrogen and stored at −80 °C.
Preparation and characterization of AβOs:
AβOs were prepared from Aβ1–42 as previously described [36, 37]. Oligomer preparations were routinely characterized by size exclusion HPLC [31] and, occasionally, by Western blots using oligomer-sensitive NU4 monoclonal antibody [38]. Protein concentration was determined using the BCA assay (Thermo-Pierce).
Intracerebroventricular (i.c.v.) infusion of AβOs:
Swiss mice were anesthetized with 2.5% isoflurane (Cristália, São Paulo, Brazil) using a vaporizer system, and were gently restrained only during the injection procedure (< 7 min). A 2.5 mm-long needle was unilaterally inserted 1 mm to the right of the midline point equidistant from each eye and 1 mm posterior to a line drawn through the anterior base of the eyes [31, 32, 35]. AβOs (10 pmol) or vehicle were infused in a final volume of 3 μl and the needle was kept in place for an additional 30 s to prevent backflow. Mice that showed signs of misplaced injections or any sign of hemorrhage were excluded from further analysis.
Dot immunoblots:
Samples were thawed and homogenized in PBS buffer containing a phosphatase and protease inhibitor cocktail (Thermo Scientific Pierce). Samples were filtered using 10 kDa cutoff Amicon Ultra-0.5 mL Centrifugal Filters. Protein concentrations in the filtrate and retentate were determined using the BCA kit following manufacturer’s instructions. Samples (20 μg total protein in 200 μL) were spotted onto a nitrocellulose membrane using a vacuum-assisted dot blot apparatus (Bio-Dot Apparatus 1706545, Bio-Rad). Blots were blocked with 5% BSA in Tween-TBS at room temperature for 2 h and incubated at 4 °C overnight with anti-IGF1 antibody (1:500; Sigma Aldrich) in blocking buffer. Membranes were then incubated with anti-mouse secondary antibody conjugated to IRDye 800CW (Licor, Lincoln, NE; 1:10,000) at room temperature for 2 h, imaged on an Odyssey Imaging System (Licor) and analyzed using NIH Image J. The integrated density of each dot was normalized by protein concentration of the corresponding sample.
Western immunoblots:
Samples were thawed and homogenized in RIPA buffer containing a phosphatase and protease inhibitor cocktail (Thermo Scientific Pierce). Protein concentrations were determined using the BCA kit. Samples containing 30 μg protein were resolved in 15% polyacrylamide Tris-glycine gels (Invitrogen) and electrotransferred to nitrocellulose membranes at 350 mA for 1 h. Blots were blocked with 5% BSA in Tween-TBS at room temperature for 2 h and incubated at 4 °C overnight with polyclonal anti-IGF1Rα antibody (1:500; Santa Cruz) in blocking buffer. Membranes were then incubated with secondary antibody conjugated to IRDye (1:10,000) at room temperature for 2 h, imaged on an Odyssey Imaging System and analyzed using NIH Image J.
Recombinant adenoviral vector for IGF1:
We employed a RAd harboring the nucleotide sequence for rat IGF1 (kindly provided by Dr. Peter Rotwein, Oregon Health Sciences University) constructed and purified as previously described [39]. Briefly, the cDNA of IGF1 was placed under control of the mouse cytomegalovirus (mCMV) promoter of the shuttle pDC515 (AdMax® plasmid kit; Microbix, Canada). The shuttle plasmid so loaded was co-transfected with the Ad5 genomic plasmid pBHGfrt(del)E1,3FLP in Human Embryo Kidney (HEK) 293 cells, which led to the generation of the adenoviral vector RAd-IGF1. The adenovector was rescued from HEK293 cell lysates and plaque purified. It was further purified by ultracentrifugation in a CsCl gradient. Final adenoviral stock was titrated using a serial dilution plaque assay.
In vitro transduction:
Primary hippocampal neuronal cultures were prepared from Wistar rat embryos (E18) and maintained in Neurobasal medium supplemented with B27 (Invitrogen) and glutamine as described [40]. After 18 days in vitro (DIV), cultures were treated with RAd-IGF1 at a multiplicity of infection (MOI) of 300. Two days later, cell cultures were exposed to AβOs (500 nM) or vehicle (2% dimethyl sulfoxide/PBS) for different time intervals: 3 h for binding analysis, 4 h for determination of reactive oxygen species (ROS), and 24 h for quantification of dendritic spines, as previously described [34, 40, 41]. At least three experiments with independent neuronal cultures and AβO preparations were performed for each assay, each with triplicate culture wells per experimental condition.
AβO Binding analysis:
AβO binding to neurons was assessed by immunocytochemistry as previously described [37]. Briefly, cells were fixed, blocked, incubated with AβO-sensitive NU4 mouse monoclonal antibody (1 μg/mL; kindly provided by Dr. William L. Klein, Northwestern University [38]) overnight at 4 °C, and incubated for 3 h at room temperature with Alexa-conjugated secondary antibody. Coverslips mounted with Prolong containing DAPI were imaged on a Zeiss Axio Observer Z1 microscope.
Reactive oxygen species formation:
ROS formation was evaluated in live neurons using CM-H2DCFDA (Molecular Probes), a fluorescent probe that is sensitive to the formation of various types of ROS, including hydrogen peroxide, hydroxyl radical, peroxyl radicals, and peroxynitrite. ROS formation was assessed using 2 μM CM-H2DCFDA [40, 42], with 40 min of probe loading. After that, neurons were rinsed three times with warm PBS and two times with Neurobasal medium without phenol red. Cells were immediately visualized on a Nikon Eclipse TE300 microscope. Analysis of DCF fluorescence was carried out using Image J (Windows version; National Institutes of Health [43]) as described [40, 42, 44].
Dendritic spine density:
Dendritic spines were labeled with phalloidin as previously described [34, 35]. Briefly, cells were fixed, blocked, and incubated with 2U per coverslip of Alexa 488-conjugated phalloidin (which binds to spine-localized dense bundles of F-actin) for 20 min at room temperature, according to manufacturer’s instructions. Coverslips mounted with Prolong containing DAPI were imaged on a Zeiss Axio Observer Z1 microscope. Images were obtained from three coverslips per experimental condition. Five neurons were photographed per coverslip and the number of dendritic spines of three distal dendrite segments was quantified manually. Results are expressed as the mean number of spines per μm.
In vivo IGF1 transduction:
Three month-old female Swiss mice were used. Animals were randomly divided into four experimental groups (N for each experimental condition are provided in Figure Legend): Vehicle, AβOs, RAd-IGF1+vehicle, RAd-IGF1+AβOs. Mice in the RAd-IGF1+vehicle and RAd-IGF1+AβOs groups received an i.c.v. injection of 3 μl of a suspension containing 1.5 × 109 plaque forming units (pfu) of RAd-IGF1. Five days later, animals received an i.c.v. infusion of 10 pmol AβOs in 3 μl (or an equivalent volume of vehicle). I.c.v. injections were performed as described above. Memory was assessed using the Novel Object Recognition (NOR) test 24 hours and 7 days after AβO infusion, as described below.
Novel object recognition test:
The test was performed as previously described [31, 32, 35]. Briefly, animals were submitted to a 5 min habituation session, in which they were allowed to freely explore an empty open field arena. Training consisted of a 5 min session during which animals were placed at the center of the arena in the presence of two identical objects. Two hours after training, animals were again placed in the arena for the test session, when one of the two objects used in the training session was replaced by a new one. The amount of time spent exploring the objects was measured manually. Results were expressed as Recognition Index, calculated as (tnew object - tfamiliar object)/ttotal during the test session and were analyzed using a one-sample Student’s t-test comparing the discrimination index to chance level (0). By definition, animals that recognize the familiar object as such (i.e., learn the task) have a recognition index >0.
Statistical analysis:
All analyses were performed using GraphPad Prism (La Jolla, CA). Values are expressed as mean ± SEM. Statistical outliers identified by the Grubbs’ test in GraphPad Prism were excluded from further analysis. Statistical tests and post-test corrections are detailed in Figure Legends (* p < 0.05; ** p < 0.01).
Results
Active/inactive IGF1 and IGF1R levels in postmortem AD hippocampus and AD mouse models
To determine whether the IGF1 pathway is affected in AD brains, we examined human postmortem hippocampal tissue from AD and control individuals. Using 10-kDa cutoff filters, we separated mature, biologically active IGF1 (~7 kDa) from inactive IGF1 bound to IGF1-binding proteins (~25–45 kDa [45]) (Fig. 1a). Dot immunoblot analysis revealed reduced hippocampal immunoreactivity for active IGF1 (< 10 kDa; Fig. 1b) and increased levels of inactive forms of IGF1 (> 10 kDa; Fig. 1c) in AD hippocampi. As a consequence, the ratio between active and inactive IGF1 was significantly decreased in AD compared to control hippocampi (Fig. 1d). We further observed an increase in IGF1R immunoreactivity in AD hippocampal tissue (Fig. 1e), likely reflecting an adaptive response to lower levels of active IGF1 in AD brains. These results suggest that hippocampal IGF1 signaling is impaired in AD.
Fig. 1.
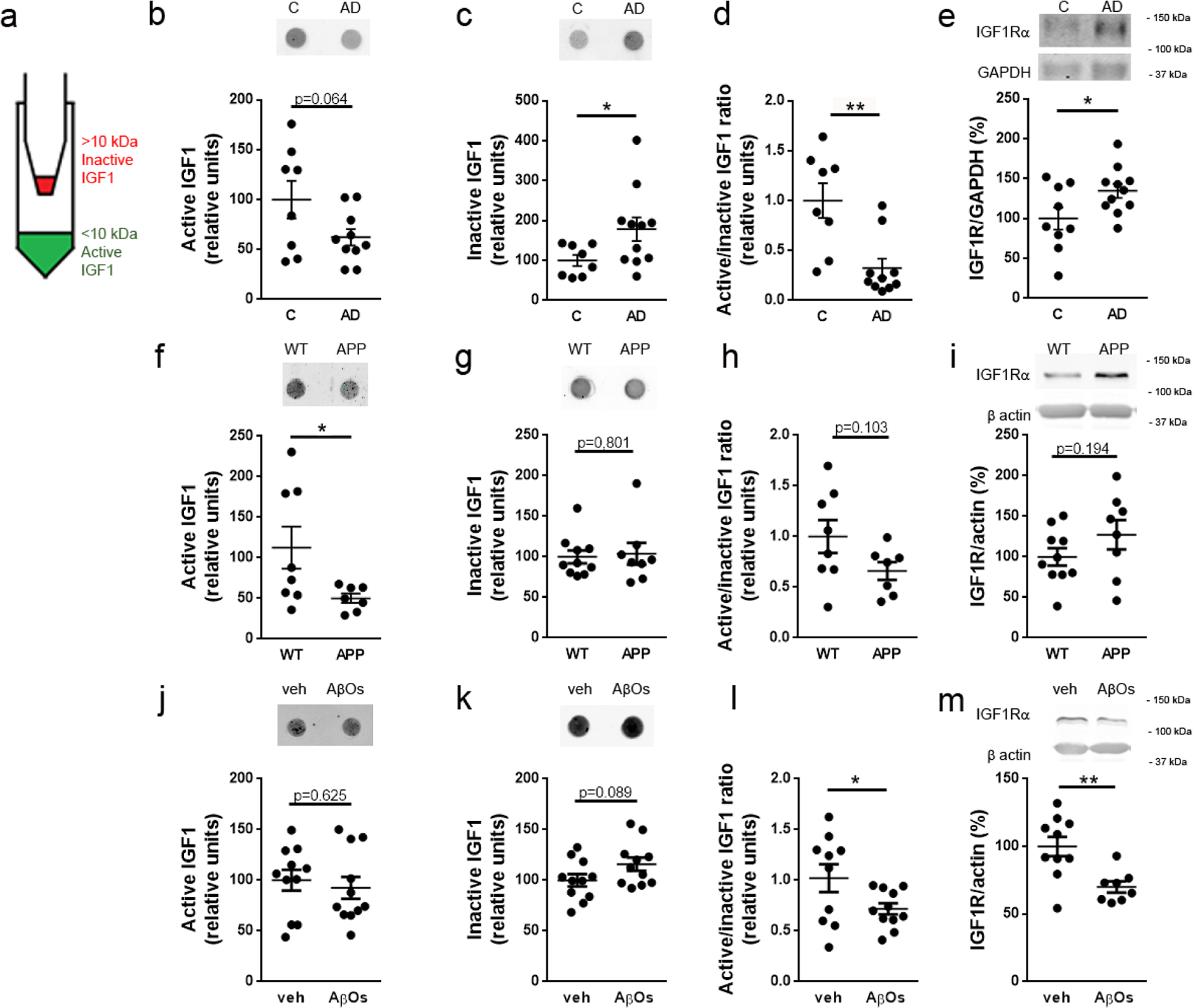
IGF1 and IGF1R immunoreactivities are altered in the hippocampi of AD subjects and AD mouse models. (a) Hippocampal homogenates were fractionated using a 10-kDa cutoff filter, which allowed separation of biologically active, mature IGF1 (< 10 kDa; filtrate) and inactive IGF1 bound to IGF1-binding proteins (> 10 kDa; retentate). (b-d) Dot immunoblot detection of active IGF1, inactive IGF1 and active/inactive IGF1 ratio, respectively, in AD versus control hippocampi; Intensities are normalized by protein mass. (e) IGF1R detected by western blotting in AD versus control hippocampi; IGF1R intensities are normalized by GAPDH. (f-h) Dot immunoblot detection of active IGF1, inactive IGF1 and active/inactive IGF1 ratio, respectively, in hippocampi from APP/PS1 versus WT mice; Intensities are normalized by protein mass. (i) IGF1R detected by western blotting in hippocampi from APP/PS1 versus WT mice; IGF1R intensities are normalized by actin. (j-l) Dot immunoblot detection of active IGF1, inactive IGF1 and active/inactive IGF1 ratio, respectively, in hippocampi from AβO- versus vehicle-infused mice; Intensities are normalized by protein mass. (m) IGF1R detected by western blotting in hippocampi from mice infused i.c.v. with AβOs (10 pmol) or vehicle; IGF1R intensities are normalized by actin. *p < 0.05, Student’s unpaired t-test.
To determine whether mouse models of AD recapitulated the findings in human brain tissue, we next analyzed IGF1/IGF1R contents in the hippocampi of two different AD mouse models. First, we detected decreased immunoreactivity for active IGF1 and unaltered immunoreactivity for inactive IGF1 in hippocampi from transgenic APP/PS1 mice (Fig. 1f,g), resulting in a trend in reduction in the ratio between active/inactive IGF1 in APP/PS1 mice (Fig. 1h). No statistically significant changes were detected in hippocampal IGF1R in APP/PS1 mice (Fig. 1i). Second, we analyzed the hippocampi of mice infused via i.c.v. with soluble oligomers of the Aβ peptide (AβOs). Considerable evidence accumulated during the past 20 years indicates that AβOs are key players in the development and progression of AD [1–3, 46, 47]. No change in immunoreactivity for active IGF1 was detected, but an increase in inactive forms of IGF1 was detected in AβO-infused mice (Fig. 1j,k). As a result, the active/inactive IGF1 ratio was significantly decreased in AβO-infused mice compared to vehicle-infused animals (Fig. 1l). IGF1R levels were decreased in the hippocampi of AβO-injected mice (Fig. 1m).
RAd-IGF1 protects hippocampal neurons against AβO-induced toxicity
We used a recombinant adenoviral vector (RAd-IGF1) to drive the expression of IGF1 in primary hippocampal neuronal cultures exposed to AβOs. Hippocampal cultures were treated with RAd-IGF1 at 18 DIV and were exposed to AβOs (500 nM) at 20 DIV. Because we have demonstrated that insulin signaling reduces the levels of AβOs bound to neurons [37], we first determined whether IGF1 would exert a similar effect. We found that transduction by RAd-IGF1 reduced AβO binding to neurons by about 20% (Fig. 2a,b), as indicated by a decrease in immunofluorescence of oligomer-sensitive monoclonal antibody NU4 [38].
Fig. 2.
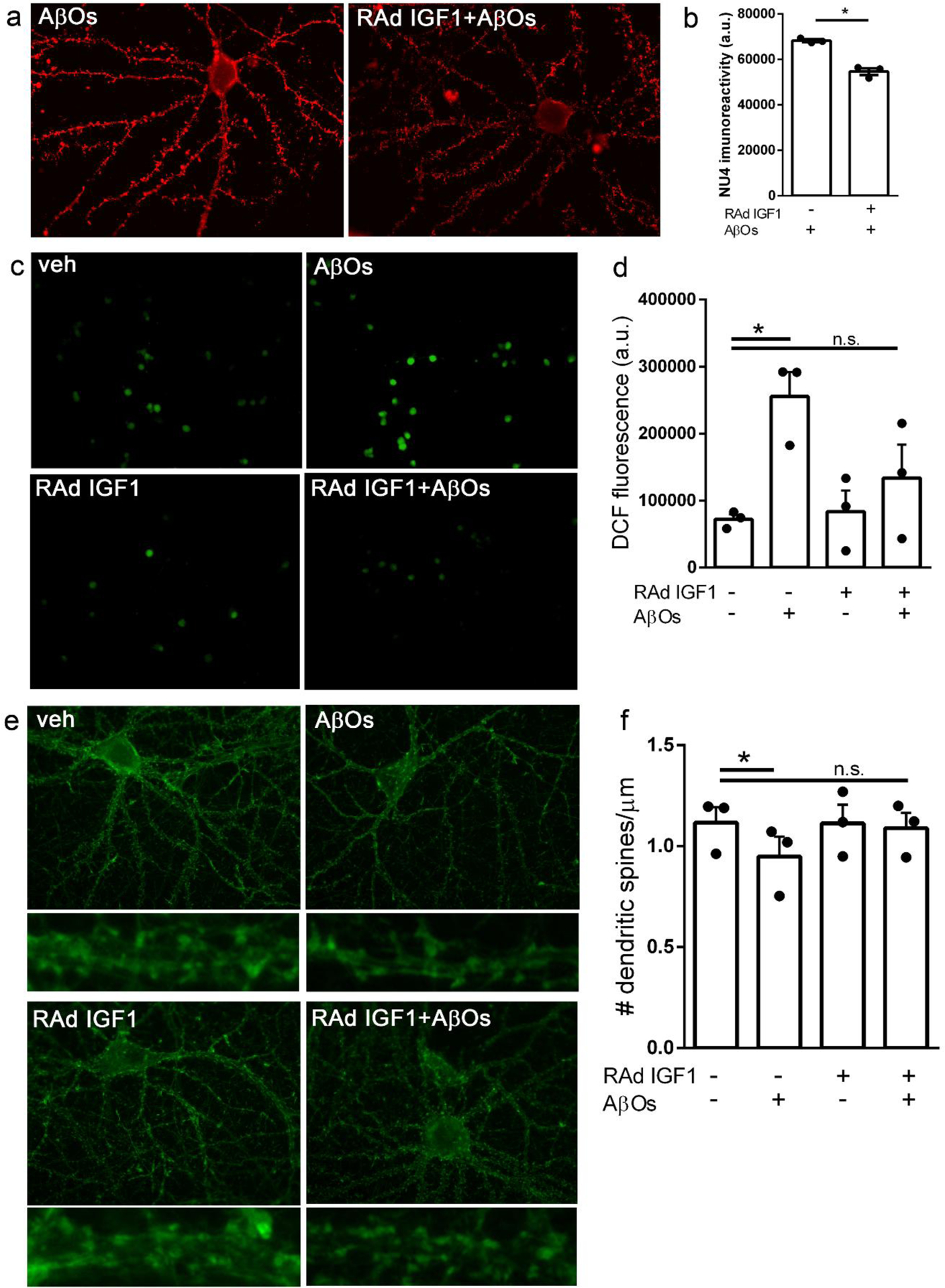
RAd-IGF1 protects hippocampal neurons in culture from AβO-induced toxicity. Hippocampal neuronal cultures were transduced with RAd-IGF1 3 days prior to exposure to 500 nM AβOs (or vehicle). (a) Representative images for AβO binding to hippocampal neurons and (b) quantification of immunofluorescence levels (NU4 immunoreactivity). *p < 0.05, Student’s paired t test. (c) Representative DCF fluorescence images and (d) quantification of ROS levels by DCF integrated fluorescence intensity. *p < 0.05, two-way ANOVA followed by Dunnettś post-test. (E) Representative images of phalloidin-labeled dendritic spines in hippocampal neurons (insets below each panel represent zoom images of dendrite segments) and (F) quantification of the number of dendritic spines per μm of dendrite segment. *p < 0.05, Two-way ANOVA followed by Dunnettś post-test.
Next, we evaluated the potential of RAd-IGF1 to prevent AβO-induced neuronal oxidative stress [34, 40, 42]. Whereas exposure to AβOs increased neuronal ROS levels by about three-fold, this effect was blocked in cultures transduced by RAd-IGF1 (Fig. 2c,d).
We then investigated the effect of RAd-IGF1 transduction on dendritic spine loss induced by AβOs. Hippocampal neurons exposed to AβOs exhibited reduced dendritic spine density compared to vehicle-treated neurons, in line with our previous reports [34, 37, 41]. In contrast, no reduction in spine density was verified in cultures transduced by RAd-IGF1 (Fig. 2e,f). These results show that RAd-IGF1 transduction is protective against AβO-induced neurotoxicity in hippocampal neurons.
RAd-IGF1 prevents AβO-induced memory impairment in mice
To determine whether transduction by RAd-IGF1 could prevent AβO-induced memory impairment, we delivered RAd-IGF1 via i.c.v. 5 days prior to i.c.v. infusion of AβOs in mice. Mice that received an i.c.v infusion of AβOs (10 pmol) failed the Novel Object Recognition (NOR) memory test both 24 hours and 7 days after infusion, consistent with our previous reports [31–35]. Interestingly, mice transduced by RAd-IGF1 were protected against memory impairment induced by AβOs in the NOR task (Fig. 3).
Fig. 3.
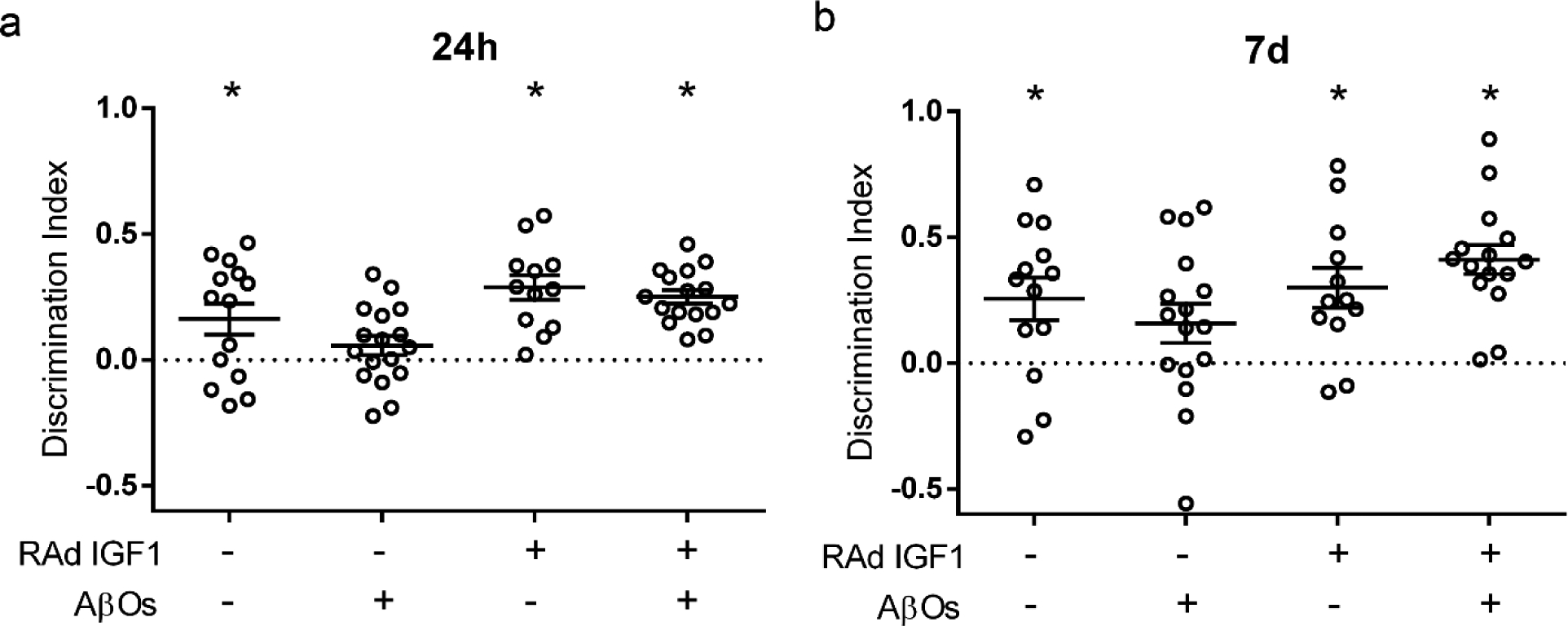
RAd-IGF1 prevents memory loss in AβO-infused mice. Discrimination index in the Novel Object Recognition (NOR) memory task. Swiss mice were i.c.v.-infused with vehicle or AβOs (10 pmol) and tested in the NOR task 24 hours (a) or 7 days (b) after infusion. *p< 0.05, Column statistics (one-sample t-test) comparing the discrimination index to chance level (0).
Discussion
We first characterized IGF1 and IGF1R levels in hippocampal postmortem tissue from AD patients and healthy controls. For this, we separated the <10 kDa free, biologically active IGF1 from >10 kDa protein-bound inactive IGF1 [45]. This revealed a 60% lower ratio of active/inactive IGF1 in AD compared to control hippocampi. We further found an increase in IGF1Rα in AD hippocampi. Steen et al. (2005) reported decreased IGF1 and IGF1R detected by immunohistochemistry in AD hippocampi, in line with our finding of reduced mature IGF1 in AD brains [27]. However, in contrast with lower IGF1R immunoreactivity in AD reported by Steen and co-workers (2005), we observed increased IGF1R levels in AD hippocampi by Western immunoblotting. Our findings are in line with the report by Moloney et al. (2010) of increased IGF1R in the temporal cortex of AD patients [48].
Similar to findings in human brain tissue, hippocampi from APP/PS1 transgenic mice displayed a trend of decreased active/inactive IGF1 ratio. Moreover, APP/PS1 mice showed a 30% increase, albeit not statistically significant, in IGF1R levels compared to WT. Whether the increase in IGF1R is part of the pathophysiology of AD or a compensatory response triggered by reduced IGF1 remains unclear. Of note, Talbot et al. (2012) reported that AD patients present brain IGF1 resistance [49]. Moreover, Trueba-Sáiz et al. (2013) showed that IGF1R phosphorylation in response to environmental enrichment is diminished in the hippocampi of APP/PS1 mice [50]. These observations could explain the increase in IGF1R we have found.
Attempts to determine whether IGF1 signaling is protective or detrimental in AD have yielded controversial results, with a number of studies suggesting that inhibition of IGF1 signaling is protective against AD progression in mouse models [50–58]. Since less is known regarding the role of IGF1 signaling in disease onset [18], we aimed to investigate the possible role of IGF1 before the full-blown establishment of the disease in mice. To this end, we infused mice i.c.v. with AβOs, a model that allows investigation of the acute consequences of AβO toxicity. Similar to our findings in human and APP/PS1 hippocampi, a decreased ratio between active/inactive IGF1 was verified in the hippocampi of AβO-infused mice. Interestingly, hippocampal IGF1R levels were decreased in AβO-infused mice, suggesting that IGF1R levels could vary with disease progression. One hypothesis is that AβOs induce an acute reduction in both active IGF1 and IGF1R, leading to reduced IGF1 signaling in the hippocampus, which then leads to a more chronic increase in IGF1R as a compensatory response mechanism.
Local brain production of IGF1 declines with age, and AD patients present lower levels of brain IGF1 [27, 50, current findings]. We thus evaluated the therapeutic potential of increasing local IGF1 production in the brain by delivery of an adenoviral vector (RAd-IGF1) to drive the expression of IGF1. To investigate whether increasing IGF1 levels could be protective against AβO toxicity, we used in vitro and in vivo models transduced with RAd-IGF1. In vitro, we observed that transduction by RAd-IGF1 reduced AβO binding to neurons in hippocampal cultures, and prevented both neuronal oxidative stress and loss of dendritic spines induced by AβOs. This is in line with an early report by Dore et al. (1997) describing IGF1 as a neuroprotective factor against Aβ toxicity [51], and with results of Pitt et al. (2017) showing that astrocyte-derived IGF1 induces endocytosis and extracellular release of AβOs bound to neurons, and is protective against AβO-induced neurotoxicity [59].
Next, we tested the RAd-IGF1 vector in AβO-infused mice to determine the effect of brain expression of IGF1 on memory. Previous studies have indicated a tropism of RAds for ependymocytes and that the ependymal route is an effective approach to increase IGF1 levels in the cerebrospinal fluid using RAd-IGF1 [60, 61]. Our finding that 3 month-old mice transduced with RAd-IGF1 via the ependymal route were protected against memory impairment induced by i.c.v. infusion of AβOs indicates a protective role of IGF1 on AβO-induced memory loss. Interestingly, IGF1 deficiency has been linked to increased brain accumulation of Aβ in APP/PS1 mice [62], as wells as to cognitive impairment and mood disorders [63]. Moreover, Carro et al. (2006) have shown that IGF1 enhances cognition, decreases Aβ levels and astroglial activation in the brains in APP/PS2 mice [53]. Although the role of IGF1 in AD is still incompletely understood, our results indicate that IGF1 protects mice against acute AβO toxicity, suggesting that it could be neuroprotective in the early stages of the disease.
In conclusion, our results show that the IGF1/IGF1R pathway is affected in AD hippocampi as well as in the hippocampi of mouse models of AD. Results further establish that adenoviral-mediated expression of IGF1 protects hippocampal neurons from AβO toxicity and prevents memory impairment induced by AβOs in mice. Our findings point to RAd-mediated gene therapy as an approach to boost brain levels of IGF1 and prevent neuronal damage and brain dysfunction in AD.
Acknowledgments
This work was supported by grants from the National Institute for Translational Neuroscience (INNT/Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) to STF and a travel award from International Brain Research Organization (IBRO/PROLAB) to MPCR and STF; and a grant #PICT15-1998 from the Argentine Agency for Science and Technology (ANPCyT) to PCR. MCS and JTSF are pre-doctoral fellows supported by CNPq and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), respectively. MFZV is a post-doctoral fellow supported by the Argentine Research Council (CONICET). YPRF is an undergraduate student and ASS is a post-doctoral fellow supported by CNPq and CAPES/FAPERJ, respectively. We thank donors and their families/caregivers for their contribution to this study, Dr. Mauricio M. Oliveira for insightful discussions and Dr. MMO and Ms. Juliette López Hanotte for help with experiments.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- [1].Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2 (7):a006338. doi: 10.1101/cshperspect.a006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG (2015) Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci 9:191. doi: 10.3389/fncel.2015.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8 (6):595–608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de la Monte SM (2012) Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72 (1):49–66. doi: 10.2165/11597760-000000000-00000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].De Felice FG (2013) Connecting type 2 diabetes to Alzheimer’s disease. Expert Rev Neurother 13 (12):1297–1299. doi: 10.1586/14737175.2013.864824 [DOI] [PubMed] [Google Scholar]
- [6].De Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10 (1 Suppl):S26–32. doi: 10.1016/j.jalz.2013.12.004 [DOI] [PubMed] [Google Scholar]
- [7].De Felice FG, Ferreira ST (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63 (7):2262–2272. doi: 10.2337/db13-1954 [DOI] [PubMed] [Google Scholar]
- [8].Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM (1996) Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 39 (11):1392–1397 [DOI] [PubMed] [Google Scholar]
- [9].Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53 (9):1937–1942. doi: 10.1212/wnl.53.9.1937 [DOI] [PubMed] [Google Scholar]
- [10].Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, Kanba S, Kiyohara Y (2011) Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology 77 (12):1126–1134. doi: 10.1212/WNL.0b013e31822f0435 [DOI] [PubMed] [Google Scholar]
- [11].Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, Chiang CH, Huang PH, Chen TJ, Lin SJ, Chen JW, Chan WL (2014) Diabetes mellitus and the risk of Alzheimer’s disease: a nationwide population-based study. PLoS One 9 (1):e87095. doi: 10.1371/journal.pone.0087095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR, Schellenberg GD, Cherrier MM, Craft S (2006) Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging 27 (3):451–458. doi: 10.1016/j.neurobiolaging.2005.03.016 [DOI] [PubMed] [Google Scholar]
- [13].Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B (2012) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 69 (1):29–38. doi: 10.1001/archneurol.2011.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, Cholerton B, Plymate SR, Arbuckle M, Craft S (2013) Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J Alzheimers Dis 35 (4):789–797. doi: 10.3233/JAD-122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, Callaghan M, Arbuckle M, Behl C, Craft S (2015) Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis 44 (3):897–906. doi: 10.3233/JAD-141791 [DOI] [PubMed] [Google Scholar]
- [16].Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, Dahl D, Caulder E, Neth B, Montine TJ, Jung Y, Maldjian J, Whitlow C, Friedman S (2017) Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J Alzheimers Dis 57 (4):1325–1334. doi: 10.3233/JAD-161256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wrigley S, Arafa D, Tropea D (2017) Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front Cell Neurosci 11:14. doi: 10.3389/fncel.2017.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gubbi S, Quipildor GF, Barzilai N, Huffman DM, Milman S (2018) 40 YEARS of IGF1: IGF1: the Jekyll and Hyde of the aging brain. J Mol Endocrinol 61 (1):T171–T185. doi: 10.1530/JME-18-0093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ashpole NM, Sanders JE, Hodges EL, Yan H, Sonntag WE (2015) Growth hormone, insulin-like growth factor-1 and the aging brain. Exp Gerontol 68:76–81. doi: 10.1016/j.exger.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zemva J, Schubert M (2014) The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: possible therapeutic implications. CNS Neurol Disord Drug Targets 13 (2):322–337 [DOI] [PubMed] [Google Scholar]
- [21].Tham A, Nordberg A, Grissom FE, Carlsson-Skwirut C, Viitanen M, Sara VR (1993) Insulin-like growth factors and insulin-like growth factor binding proteins in cerebrospinal fluid and serum of patients with dementia of the Alzheimer type. J Neural Transm Park Dis Dement Sect 5 (3):165–176 [DOI] [PubMed] [Google Scholar]
- [22].Mustafa A, Lannfelt L, Lilius L, Islam A, Winblad B, Adem A (1999) Decreased plasma insulin-like growth factor-I level in familial Alzheimer’s disease patients carrying the Swedish APP 670/671 mutation. Dement Geriatr Cogn Disord 10 (6):446–451. doi: 10.1159/000017188 [DOI] [PubMed] [Google Scholar]
- [23].Ostrowski PP, Barszczyk A, Forstenpointner J, Zheng W, Feng ZP (2016) Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS One 11 (5):e0155733. doi: 10.1371/journal.pone.0155733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van Exel E, Eikelenboom P, Comijs H, Deeg DJ, Stek ML, Westendorp RG (2014) Insulin-like growth factor-1 and risk of late-onset Alzheimer’s disease: findings from a family study. Neurobiol Aging 35 (3):725 e727–710. doi: 10.1016/j.neurobiolaging.2013.08.014 [DOI] [PubMed] [Google Scholar]
- [25].Westwood AJ, Beiser A, Decarli C, Harris TB, Chen TC, He XM, Roubenoff R, Pikula A, Au R, Braverman LE, Wolf PA, Vasan RS, Seshadri S (2014) Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 82 (18):1613–1619. doi: 10.1212/WNL.0000000000000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vidal JS, Hanon O, Funalot B, Brunel N, Viollet C, Rigaud AS, Seux ML, le-Bouc Y, Epelbaum J, Duron E (2016) Low Serum Insulin-Like Growth Factor-I Predicts Cognitive Decline in Alzheimer’s Disease. J Alzheimers Dis 52 (2):641–649. doi: 10.3233/JAD-151162 [DOI] [PubMed] [Google Scholar]
- [27].Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis 7 (1):63–80 [DOI] [PubMed] [Google Scholar]
- [28].Morris JC (1993) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43 (11):2412–2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- [29].Ferretti REL, Damin AE, Brucki SMD, Morillo LS, Perroco TR, Campora F, Moreira EG, Balbino ES, Lima M, Battela C, Ruiz L, Grinberg LT, Farfel JM, Leite REP, Suemoto CK, Pasqualucci CA, Rosemberg S, Saldiva PHN, Jacob-Filho W, Nitrini R (2010) Post-Mortem diagnosis of dementia by informant interview. Dement Neuropsychol 4 (2):138–144. doi: 10.1590/S1980-57642010DN40200011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13 (2):159–170. doi: 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- [31].Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST (2013) Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci 33 (23):9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espirito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG (2013) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab 18 (6):831–843. doi: 10.1016/j.cmet.2013.11.002 [DOI] [PubMed] [Google Scholar]
- [33].Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST (2013) Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry 18 (10):1053–1054. doi: 10.1038/mp.2012.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brito-Moreira J, Lourenco MV, Oliveira MM, Ribeiro FC, Ledo JH, Diniz LP, Vital JFS, Magdesian MH, Melo HM, Barros-Aragao F, de Souza JM, Alves-Leon SV, Gomes FCA, Clarke JR, Figueiredo CP, De Felice FG, Ferreira ST (2017) Interaction of amyloid-beta (Abeta) oligomers with neurexin 2alpha and neuroligin 1 mediates synapse damage and memory loss in mice. J Biol Chem 292 (18):7327–7337. doi: 10.1074/jbc.M116.761189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lourenco MV, Frozza RL, de Freitas GB, Zhang H, Kincheski GC, Ribeiro FC, Goncalves RA, Clarke JR, Beckman D, Staniszewski A, Berman H, Guerra LA, Forny-Germano L, Meier S, Wilcock DM, de Souza JM, Alves-Leon S, Prado VF, Prado MAM, Abisambra JF, Tovar-Moll F, Mattos P, Arancio O, Ferreira ST, De Felice FG (2019) Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med 25 (1):165–175. doi: 10.1038/s41591-018-0275-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL (2008) Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 29 (9):1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL (2009) Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A 106 (6):1971–1976. doi: 10.1073/pnas.0809158106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL (2007) Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem 100 (1):23–35. doi: 10.1111/j.1471-4159.2006.04157.x [DOI] [PubMed] [Google Scholar]
- [39].Herenu CB, Cristina C, Rimoldi OJ, Becu-Villalobos D, Cambiaggi V, Portiansky EL, Goya RG (2007) Restorative effect of insulin-like growth factor-I gene therapy in the hypothalamus of senile rats with dopaminergic dysfunction. Gene Ther 14 (3):237–245. doi: 10.1038/sj.gt.3302870 [DOI] [PubMed] [Google Scholar]
- [40].De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL (2007) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282 (15):11590–11601. doi: 10.1074/jbc.M607483200 [DOI] [PubMed] [Google Scholar]
- [41].Batista AF, Forny-Germano L, Clarke JR, Lyra ESNM, Brito-Moreira J, Boehnke SE, Winterborn A, Coe BC, Lablans A, Vital JF, Marques SA, Martinez AM, Gralle M, Holscher C, Klein WL, Houzel JC, Ferreira ST, Munoz DP, De Felice FG (2018) The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J Pathol 245 (1):85–100. doi: 10.1002/path.5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saraiva LM, Seixas da Silva GS, Galina A, da-Silva WS, Klein WL, Ferreira ST, De Felice FG (2010) Amyloid-beta triggers the release of neuronal hexokinase 1 from mitochondria. PLoS One 5 (12):e15230. doi: 10.1371/journal.pone.0015230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Abramoff MD, Magalhaes PJ and Ram SJ (2004) Image Processing with ImageJ. Biophotonics International, 11, 36–42. [Google Scholar]
- [44].de Godoy MA, Saraiva LM, de Carvalho LRP, Vasconcelos-Dos-Santos A, Beiral HJV, Ramos AB, Silva LRP, Leal RB, Monteiro VHS, Braga CV, de Araujo-Silva CA, Sinis LC, Bodart-Santos V, Kasai-Brunswick TH, Alcantara CL, Lima A, da Cunha ESNL, Galina A, Vieyra A, De Felice FG, Mendez-Otero R, Ferreira ST (2018) Mesenchymal stem cells and cell-derived extracellular vesicles protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-beta oligomers. J Biol Chem 293 (6):1957–1975. doi: 10.1074/jbc.M117.807180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lewitt MS, Boyd GW (2019) The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor-Binding Proteins in the Nervous System. Biochem Insights 12:1178626419842176. doi: 10.1177/1178626419842176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ferreira ST, Klein WL (2011) The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem 96 (4):529–543. doi: 10.1016/j.nlm.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Viola KL, Klein WL (2015) Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129 (2):183–206. doi: 10.1007/s00401-015-1386-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C (2010) Defects in IGF-1 receptor, insulin receptor and IRS-½ in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31 (2):224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- [49].Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122 (4):1316–1338. doi: 10.1172/JCI59903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Trueba-Saiz A, Cavada C, Fernandez AM, Leon T, Gonzalez DA, Fortea Ormaechea J, Lleo A, Del Ser T, Nunez A, Torres-Aleman I (2013) Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl Psychiatry 3:e330. doi: 10.1038/tp.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dore S, Kar S, Quirion R (1997) Rediscovering an old friend, IGF-I: potential use in the treatment of neurodegenerative diseases. Trends Neurosci 20 (8):326–331 [DOI] [PubMed] [Google Scholar]
- [52].Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I (2002) Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med 8 (12):1390–1397. doi: 10.1038/nm793 [DOI] [PubMed] [Google Scholar]
- [53].Carro E, Trejo JL, Gerber A, Loetscher H, Torrado J, Metzger F, Torres-Aleman I (2006) Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol Aging 27 (9):1250–1257. doi: 10.1016/j.neurobiolaging.2005.06.015 [DOI] [PubMed] [Google Scholar]
- [54].Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, Kelly JW, Dillin A (2010) Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell 9 (2):126–134. doi: 10.1111/j.1474-9726.2009.00541.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Freude S, Schilbach K, Schubert M (2009) The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease. Curr Alzheimer Res 6 (3):213–223 [DOI] [PubMed] [Google Scholar]
- [56].Gontier G, George C, Chaker Z, Holzenberger M, Aid S (2015) Blocking IGF Signaling in Adult Neurons Alleviates Alzheimer’s Disease Pathology through Amyloid-beta Clearance. J Neurosci 35 (33):11500–11513. doi: 10.1523/JNEUROSCI.0343-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Puig KL, Kulas JA, Franklin W, Rakoczy SG, Taglialatela G, Brown-Borg HM, Combs CK (2016) The Ames dwarf mutation attenuates Alzheimer’s disease phenotype of APP/PS1 mice. Neurobiol Aging 40:22–40. doi: 10.1016/j.neurobiolaging.2015.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].George C, Gontier G, Lacube P, Francois JC, Holzenberger M, Aid S (2017) The Alzheimer’s disease transcriptome mimics the neuroprotective signature of IGF-1 receptor-deficient neurons. Brain 140 (7):2012–2027. doi: 10.1093/brain/awx132 [DOI] [PubMed] [Google Scholar]
- [59].Pitt J, Wilcox KC, Tortelli V, Diniz LP, Oliveira MS, Dobbins C, Yu XW, Nandamuri S, Gomes FCA, DiNunno N, Viola KL, De Felice FG, Ferreira ST, Klein WL (2017) Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Abeta oligomers. Mol Biol Cell 28 (20):2623–2636. doi: 10.1091/mbc.E17-06-0416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Herenu CB, Sonntag WE, Morel GR, Portiansky EL, Goya RG (2009) The ependymal route for insulin-like growth factor-1 gene therapy in the brain. Neuroscience 163 (1):442–447. doi: 10.1016/j.neuroscience.2009.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pardo J, Uriarte M, Console GM, Reggiani PC, Outeiro TF, Morel GR, Goya RG (2016) Insulin-like growth factor-I gene therapy increases hippocampal neurogenesis, astrocyte branching and improves spatial memory in female aging rats. Eur J Neurosci 44 (4):2120–2128. doi: 10.1111/ejn.13278 [DOI] [PubMed] [Google Scholar]
- [62].Poirier R, Fernandez AM, Torres-Aleman I, Metzger F (2012) Early brain amyloidosis in APP/PS1 mice with serum insulin-like growth factor-I deficiency. Neurosci Lett 509 (2):101–104. doi: 10.1016/j.neulet.2011.12.048 [DOI] [PubMed] [Google Scholar]
- [63].Zegarra-Valdivia JA, Santi A, Fernandez de Sevilla ME, Nunez A, Torres Aleman I (2019) Serum Insulin-Like Growth Factor I Deficiency Associates to Alzheimer’s Disease Co-Morbidities. J Alzheimers Dis 69 (4):979–987. doi: 10.3233/JAD-190241 [DOI] [PubMed] [Google Scholar]