

Abstract
Tissue repair after spinal cord injury (SCI) requires mobilization of immune and glial cells to form a protective barrier that seals the wound and facilitates debris clearing, inflammatory containment, and matrix compaction. This process involves corralling, wherein phagocytic immune cells become confined to the necrotic core surrounded by an astrocytic border. Here, we elucidate a temporally distinct gene signature in injury-activated microglia/macrophages (IAM), which engages axon guidance pathways. Plexin-B2 is upregulated in IAM, which is required for motosensory recovery after SCI. Plexin-B2 deletion in myeloid cells impairs corralling, leading to diffuse tissue damage, inflammatory spillover, and hampered axon regeneration. Corralling begins early and requires Plexin-B2 in both microglia and macrophages. Mechanistically, Plexin-B2 promotes microglia motility, steers IAM away from colliding cells, and facilitates matrix compaction. Our data thus establish Plexin-B2 as an important link that integrates biochemical cues and physical interactions of IAM with the injury microenvironment during wound healing.
Keywords: spinal cord injury, microglia-astrocyte communication, corralling, glial response, neural repair, wound healing, axon regeneration
Introduction
After CNS injury, glial cells are mobilized to form a protective barrier that seals the wound and limits the extent of tissue damage. This barrier also facilitates debris clearing, contains inflammatory cytokines, and promotes wound compaction/matrix reorganization to minimize the scar size and maximize the repair area 1. Building such a barrier involves a process termed corralling wherein phagocytic immune cells become confined to the lesion core surrounded by a narrow rim of reactive astrocytes that separates the necrotic core from the surrounding healthy tissue 2. The corralling is an important step to mitigate secondary tissue injury fueled by inflammatory cytokines, proteases and free radicals released from the lesion core 3. Understanding the signaling pathways that regulate corralling is fundamental to improve neural repair after traumatic brain or spinal cord injury (SCI), as well as other CNS pathologies such as stroke or neurodegenerative disorders.
CNS injury triggers microglial activation within minutes, which is followed by an influx of blood-borne immune cells attracted to chemokines and facilitated by a breach of blood-brain-barrier (BBB) 4. Blood-borne monocytes differentiate into phagocytic macrophages 5, which, together with microglia, constitute the innate immunity responsible for debris clearing and providing a source of trophic and anti-inflammatory factors to promote tissue repair 6,7; but they also release inflammatory cytokines to fuel secondary injury 8. Hence, the innate immune response can both resolve and exacerbate tissue injury. Thus far, the phagocytic and inflammatory functions of microglia and macrophages are better understood; whether they contribute to corralling and wound compaction is not clear, nor are the underlying signaling mechanisms.
Previously, we have demonstrated a role of transmembrane receptor Plexin-B2 during neuroprogenitor migration 9,10. Plexins were originally identified as axon guidance molecules, but subsequently shown to also regulate cell migration and cytoskeletal dynamics through small GTPases during development and in adult physiology 11. Plexin-B2 binds to class IV transmembrane semaphorin ligands (Sema4A-4D, 4F, and 4G) and regulates cellular interactions in a variety of contexts, including vascular development, bone homeostasis, kidney injury, and immune activation 12–21. However, its role in CNS injury is unknown.
Here, we demonstrated that Plexin-B2 is upregulated in injury activated microglia and macrophages (which we termed IAM), and is required for motosensory recovery after SCI. Plexin-B2 regulates corralling and wound compaction by enhancing microglial dispersion, steering them away from colliding cells, and strengthening matrix compaction.
Results
Temporally distinct molecular signatures in activated microglia/macrophages after SCI
Microglia are activated within minutes after CNS injury 4. To accurately capture in vivo gene signatures in IAM, we employed the INTACT method (Isolation of nuclei tagged in specific cell types), based on affinity immunoprecipitation of Sun1-GFP-tagged nuclei (Sun1 being a nuclear envelope protein), which minimizes ex vivo transcriptional artifacts from lengthy tissue dissociation and fluorescence-activated cell sorting (FACS) 22. We bred the INTACT mice with a Cx3cr1CreER allele that expresses tamoxifen (Tam)-inducible Cre recombinase in myeloid lineage 23 (Fig. 1a). We performed dorsal column transection at thoracic level T8, a well-established SCI model, while sham control animals underwent laminectomy only 24,25. To label myeloid cells, Tam was administered at 3 day and 1 day before SCI. Immunohistochemistry (IHC) of the spinal cord tissues at 3 days post-injury (dpi) confirmed the overlap of GFP with myeloid marker IBA1 (Fig. 1b). Spinal cord tissues from 5 mm rostral to 5 mm caudal of the lesion core were collected at 3, 7, and 14 dpi, followed by affinity immunoprecipitation of GFP+ nuclei and RNA extraction. Corresponding tissues from sham control were collected 7 days after surgery. Independent triplicate cDNA libraries were prepared for RNA sequencing (RNA-Seq).
Fig. 1. Gene signatures in activated microglia/macrophages after SCI reveal engagement of axon guidance pathway.
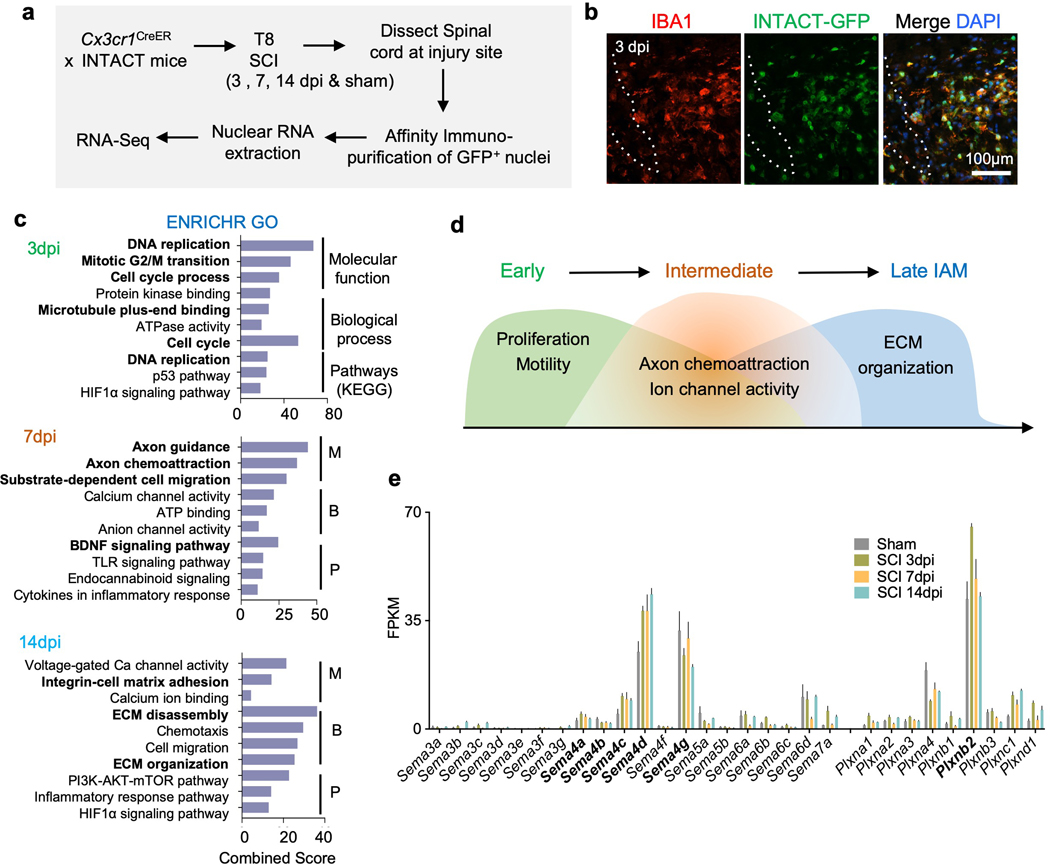
a. Schematic diagram of RNA-Seq using the INTACT method. Animals were injected with tamoxifen at 3 day and 1 day before SCI and affinity immunopurification of GFP-tagged nuclei from spinal cord tissues was performed at three time points after SCI for transcriptome profiling.
b. IHC images show overlap of IBA1 and SUN1-GFP at the lesion site in Cx3cr1CreER INTACT animal 3 days after SCI. Dotted line demarcates the lesion center. Images from 3 independent mice revealed similar results.
c. ENRICHR Gene Ontology (GO) analysis of temporally distinct DEGs in IAM vs. sham controls (n=3 independent mice per timepoint). X-axis represents combined ENRICHR score (log of p-value from Fisher Exact Test multiplied with z-score of deviation from expected rank). M, molecular function; B, biological process; and P, pathways (KEGG).
d. Diagram illustrates temporally distinct cellular processes of IAM in response to SCI as revealed by DEGs.
e. Bar graphs show average expression levels of semaphorin and plexin family members at the indicated timepoints in IAM after SCI as compared to homeostatic microglia/macrophages in sham controls. FPKM, fragments per kilobase of transcript per million mapped reads. Note the increased expression level of Sema4c, 4d, as well as Plxnb2 in IAM.
INTACT RNA-Seq data revealed temporally distinct sets of differentially expressed genes (DEGs) in IAM as compared to homeostatic microglia/macrophages from the sham controls. ENRICHR gene ontology (GO) analysis of the DEGs showed that the early activated genes in 3 dpi IAM were involved in proliferation and motility, while DEGs at 7 dpi mainly concerned migration (e.g. axon guidance, axon chemoattraction, substrate-dependent cell migration, and ion channel activity), and DEGs at 14 dpi were enriched for cell-matrix adhesion and ECM organization GO terms (Fig. 1c). Thus, the molecular signatures of IAM suggested the potential importance of cell motility, axon chemoattraction, and matrix interaction for successful wound repair (Fig. 1d). Further detailed analyses of the IAM transcription programs will be published elsewhere (Wahane et. al., in preparation).
Given the enrichment for axon guidance GO terms in the IAM gene signatures, we examined the expression dynamics of semaphorin and plexin family members, and found high transcriptional levels of Sema4c, 4d, 4g, and Plxnb2 in homeostatic microglia/macrophages, which were further upregulated in IAM, except for Sema4g (Fig. 1e). In subsequent studies, we investigated the role of Plexin-B2 and Sema4c in the innate immunity after SCI.
Upregulation of Plexin-B2 in microglia and macrophages after SCI
To further verify Plexin-B2 induction after SCI, we utilized a targeted gene trap line, termed Plxnb2B, which carries a lacZ reporter inserted after exon 6 of Plxnb2 26 (Fig. 2a). LacZ/X-Gal staining revealed the robust expression of lacZ at the injury site 7 days after T8 dorsal column transection injury (Fig. 2b), but absence of X-gal signals in the littermate controls without the lacZ reporter (Supplementary Fig. 1a). Time course analysis by IHC confirmed a robust upregulation of Plexin-B2 in the lesion area at 3 dpi, which persisted at 7 and 14 dpi, but started to wane by 21 dpi (Fig. 2c).
Fig. 2. Upregulation of Plexin-B2 in myeloid cells after SCI.
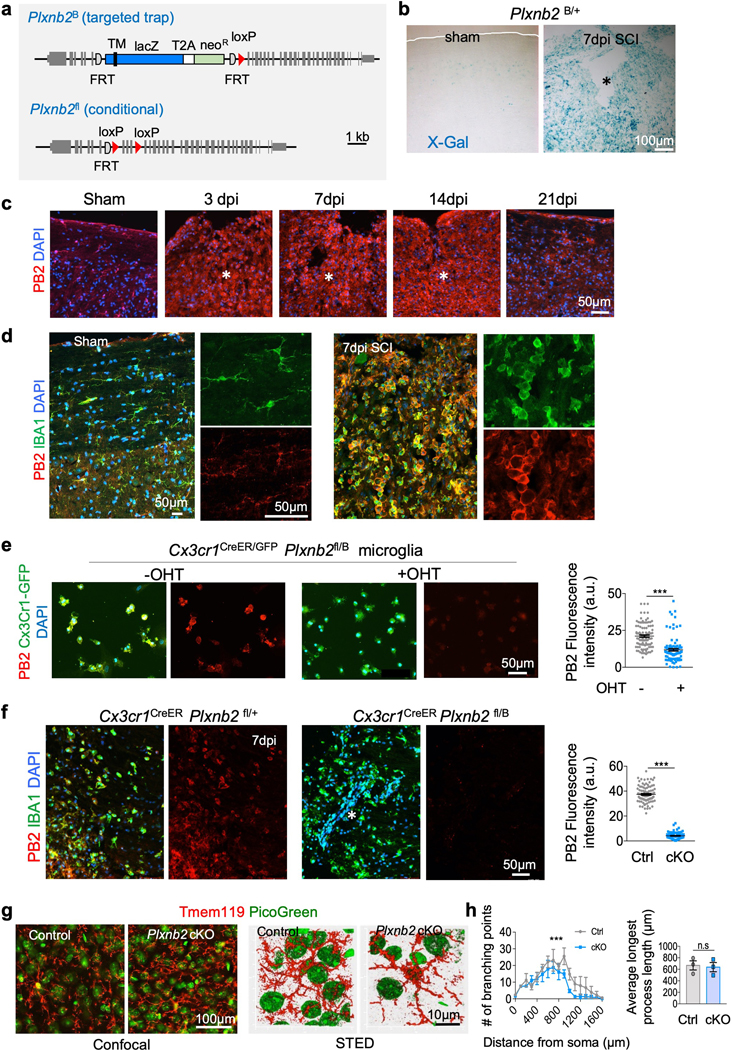
a. Schematics of Plxnb2 mutant alleles. Top, targeted trap null allele (Plxnb2B) that carries an insertion of a lacZ reporter cassette. TM, transmembrane domain. Neo, neomycin resistance gene. Bottom, Plxnb2 conditional allele (Plxnb2fl) that carries loxP sites flanking coding exons 7–9. Grey boxes depict exons. Semi-circles depict FLP recombinase target (FRT) sequences. Red triangles depict loxP sites.
b. X-Gal staining of sagittal sections of thoracic spinal cords from Plxnb2B/+ mice show increased expression of lacZ reporter at 7 days after SCI as compared to sham control. Images from 3 independent mice for each condition showed similar results.
c. IHC shows robust upregulation of Plexin-B2 (PB2) at the lesion site after SCI at 3, 7 and 14 dpi, which started to waned at 21 dpi. DAPI for nuclear counterstaining. n=4 independent mice per timepoint, with similar results obtained.
d. Images of co-immunostaining show co-localization of Plexin-B2 and IBA1 in the spinal cord tissues at baseline and at 7 days after SCI. Note the robust upregulation of Plexin-B2 in IBA1+ cells after SCI. Higher magnification images shown on the right highlight the morphological changes of IBA1+ cells from ramified at baseline to amoeboid after SCI. n=4 independent mice per timepoint with similar results obtained.
e. ICC of primary microglia isolated from Plxnb2fl/B Cx3cr1CreER/GFP mice show Plexin-B2 ablation with hydroxytamoxifen (OHT). Cx3cr1GFP reporter confirmed purity of the primary microglial culture. Right, scatter dot plots of fluorescence intensity for Plexin-B2, n=100 cells for each condition quantified from 10 independent culture wells, unpaired two-tailed Student’s t test, *** p<0.0001. Data represent mean ± SEM.
f. IHC of injured spinal cord tissues at 7 dpi show Plexin-B2 ablation in IBA1+ cells in Plxnb2fl/B Cx3cr1CreER mice as compared to the high expression levels in littermate control (Plxnb2fl/+ Cx3cr1CreER). Animals were treated with tamoxifen from 3 day before until 7 day after SCI. Right, scatter dot plots of fluorescence intensity for Plexin-B2, n=100 cells for each condition quantified from 3 independent mice, unpaired two-tailed Student’s t test, ***p<0.0001. Data represent mean ± SEM.
g. Left, confocal images of cortical tissue of adult mice stained for microglia marker Tmem119. Animals received tamoxifen injection for 14 days (every other day, total 7 injections). PicoGreen for nuclear counterstaining. Right, maximum intensity projection of STED microscopic images show microglia cellular processes. Images obtained from 3 pairs of mice showed similar results.
h. Quantifications show reduced branching points in Plxnb2 cKO microglia as compared to control. Sholl analysis was performed at 100 μm radius intervals (n=17 cells per genotype, from 3 independent pairs of mice). Two-way ANOVA with Bonferroni’s post hoc correction for repeated measures, ***p<0.001). F(DFn, DFd): F(1,640)=83.1 for column factor, F(159,640)=7 for row factor. Microglia from Plxnb2 cKO and control mice displayed comparable average length of the longest process (data were averaged for each animal, n=3 mice for each genotype), Unpaired two-tailed Student’s t test, n.s., p=0.81. Data represent mean ± SEM.
Asterisks in b, c, f denote the lesion core.
We next performed co-immunostaining using cell type-specific markers and found that Plexin-B2 was predominantly induced in activated microglia and influxed blood-borne macrophages, as marked by IBA1 or CD11b (Fig. 2d and Supplementary Fig. 1b-d). Notably, these IAM assumed an amoeboid morphology in contrast to the ramified morphology of homeostatic microglia in the neighboring intact regions. We further utilized Cx3cr1GFP mice, a myeloid reporter line, which verified the upregulation of Plexin-B2 specifically in amoeboid shaped Cx3cr1-GFP+ myeloid cells at 3, 7, and 14 dpi after T8 transection (Supplementary Fig. 1c).
To investigate the functional significance of Plexin-B2 induction in IAM after SCI, we generated mutant mice with Tam-inducible Plxnb2 conditional knockout (cKO) in myeloid cells by crossing a floxed Plxnb2fl allele 10 with a functionally null Plxnb2B allele, as well as the Cx3cr1CreER allele (Fig. 2a). Immunostaining of primary microglia isolated from Plxnb2 cKO mice that also carried the Cx3cr1GFP reporter allele (i.e., Cx3cr1CreER/GFP Plxnb2fl/B) demonstrated Plexin-B2 ablation in Cx3cr1-GFP+ myeloid cells upon treatment with hydroxy-tamoxifen (OHT) (Fig. 2e). The primary cultures of microglia of control and Plxnb2 cKO mice revealed no apparent differences in cell survival, morphology, or growth rate between different genotypes. Stimulation of cultured microglia with lipopolysaccharide (LPS) induced Plexin-B2 upregulation (Supplementary Fig. 1e), indicating that Plexin-B2 may belong to a general microglial gene response to pathological stimuli. In vivo Plexin-B2 ablation was verified in homeostatic microglia (Supplementary Fig. 1f), as well as in IAM 7 days after SCI using Plxnb2 cKO mice that received two Tam injections at 3 day and 1 day before SCI (Fig. 2f).
We first studied the impact of Plexin-B2 deletion on homeostatic microglia in adult mice in a non-injury setting. Immunostaining for Tmem119, a microglia-specific marker, did not reveal overt differences in microglial density in either cortex or spinal cord of adult Plxnb2 cKO mice (Fig. 2g and Supplementary Fig. 1f). As microglia are known to maintain CNS homeostasis by surveying territorial domains through their highly motile processes 27, we compared cellular processes using super-resolution STED microscopy, which revealed less branching points in Plxnb2 cKO microglia by Sholl analysis, although the average length of the longest processes was unchanged from control microglia (Fig. 2g,h). Hence, Plexin-B2 does not appear to be required for microglial survival or proliferation, but is indispensable for maintaining their motile processes in homeostatic state.
Plexin-B2 ablation in myeloid cells impairs motosensory recovery after SCI
To investigate Plexin-B2’s function in IAM, we chose a moderate T8 contusion SCI model that allows spontaneous motosensory recovery over the course of 5 weeks. Specifically, after laminectomy at T8, a calibrated force of 45 kDyn was applied to the spinal cord. We first tested the effect of continuous Plexin-B2 ablation in both resident microglia and infiltrating macrophages by administering Tam every other day starting from 3 day before until 5 week after SCI (Fig. 3a). Plxnb2 cKO mice displayed a significantly worse recovery in walking, hindlimb movement, and hindpaw placement relative to the littermate controls throughout the 5-week recovery course, as quantified by Basso Mouse Scale (BMS) for locomotion 28 (Fig. 3b and Supplementary Videos 1 and 2). Rotarod test for hindlimb and tail balance and ladder walking tests on both regular and irregular interval beams also revealed worse motor functional recovery for cKO mice. Furthermore, von Frey filament tests showed spontaneous recovery of tactile sensory function in control animals, but significant sensory deficits in mutants (Fig. 3c). Of note, both cohorts displayed similar baseline thresholds for tactile sensitivity before SCI (Fig. 3c), and similar body weight during recovery (Supplementary Fig. 2a). This battery of behavioral tests indicated that Plexin-B2 induction in innate immune cells plays a beneficial role for functional recovery after SCI.
Fig. 3. Plexin-B2 induction in IAM is required for functional recovery after SCI.
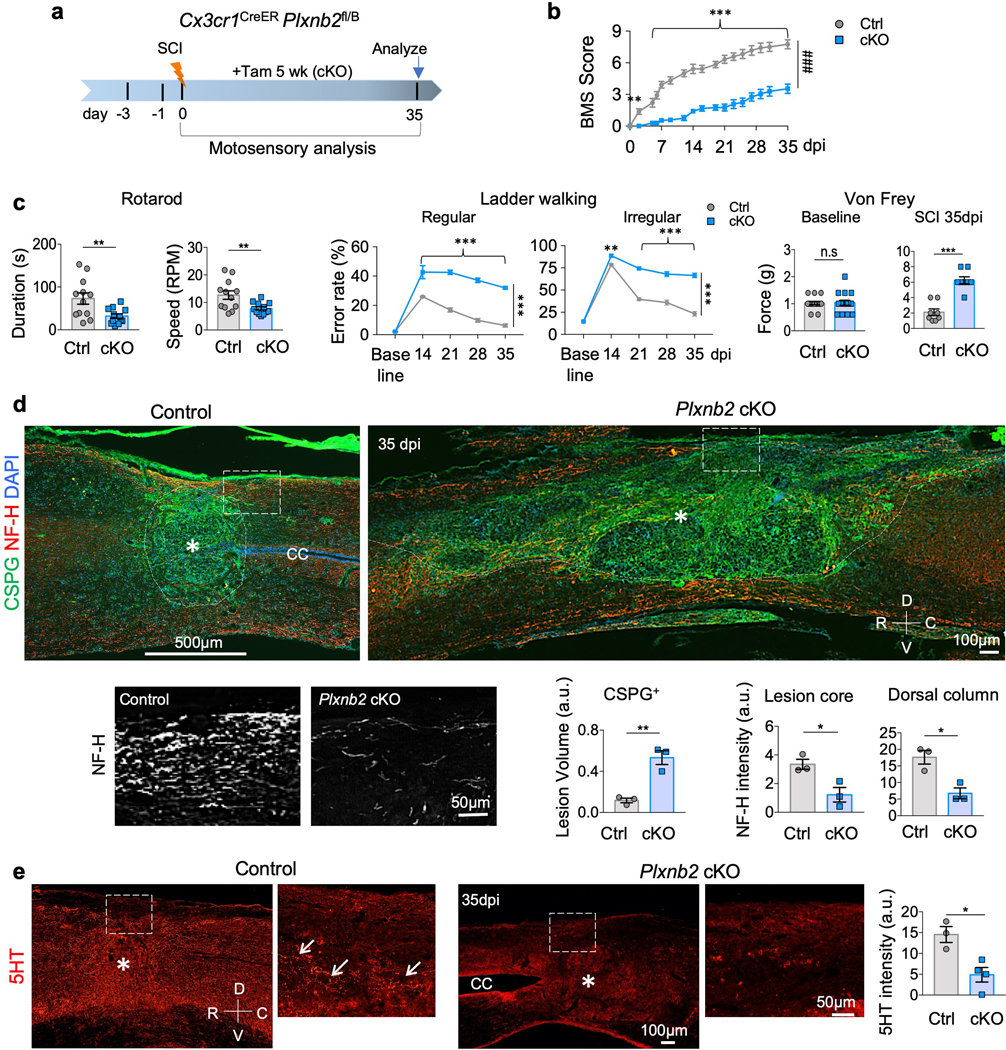
a. Experimental scheme. Tamoxifen injection was started at 3 days before SCI and continued every other day until 5 weeks after SCI.
b. BMS scores during the 5 week recovery after T8 contusion injury demonstrate impaired functional recovery in Plxnb2 cKO mice as compared to littermate controls with the same tamoxifen regimen. n=12 animals per group, two-way ANOVA with Bonferroni post hoc correction for repeated measures, ***p<0.0001, ###p<0.001 for the entire recovery course, F(DFn, DFd): F(1,374)=945.8 for column factor, F(16, 374)=52.11 for row factor. Data represent mean ± SEM.
c. Motosensory behavioral assays by rotarod and ladder walking (regular and irregularly spaced rungs) at 35 dpi demonstrate impaired functional recovery in Plxnb2 cKO mice as compared to littermate controls. Rotarod tests: n=12 animals per group, unpaired two-tailed Student’s t-test, **p=0.011. Ladder walking tests: n=15 animals per group, two-way ANOVA with Bonferroni post hoc correction, ***p<0.0001. For regular, F(DFn, DFd): F(1,140)=312 for column factor, F(4, 140)=106 for row factor. For irregular, F(DFn, DFd): F(1, 140)=369 for column factor, F(4, 140)=315 for row factor. Von Frey filament test was conducted at baseline and at 35 dpi, n=4 animals per group, left and right hindpaws were measured separately, Mann Whitney test, n.s. p=0.99, ***p=0.0005. Data represent mean ± SEM.
d. IHC of sagittal sections of thoracic spinal cords at 35 dpi show larger lesion volume revealed by CSPG and fewer axon fibers at the dorsal column and the lesion center (NH-F) in Plxnb2 cKO mice than littermate controls. n=3 animals per group, unpaired two-tailed Student’s t-test. For lesion volume by CSPG, **p=0.0042. For NF-H intensity, *p=0.027 for lesion core, *p=0.014 for dorsal column. Data represent mean ± SEM.
e. IHC of sagittal sections of thoracic spinal cords at 35 dpi show fewer serotonergic fibers (5HT) at the lesion site in Plxnb2 cKO mice than in littermate controls (white arrows). n=3 for control and n=4 for cKO, unpaired two-tailed Student’s t-test. *p=0.014. Data represent mean ± SEM. R-rostral; C-caudal; D-dorsal; V-ventral; CC-central canal.
Compromised recovery after SCI implies impaired axonal connectivity. Indeed, significantly fewer axon fibers were present in the dorsal column and the lesion core in cKO than in control mice when examined with neurofilament-H (NF-H) immunostaining at 35 dpi (Fig. 3d). There were also fewer descending serotonergic (5HT) axon fibers in the raphespinal tract at the injury site (Fig. 3e). Strikingly, the lesion size appeared much enlarged in mutants, with diffuse deposition of chondroitin sulfate proteoglycans (CSPGs; detected with CS-56 antibody), a family of matrix proteins that can influence axon growth 1,29 (Fig. 3d and Supplementary Fig. 2b,c). We did not find tissue cavitation at the lesion sites in mutant animals, although the central canal in some sections appeared enlarged from tissue distortion due to diffuse injury (Fig. 3e and Supplementary Fig. 2b).
Plexin-B2 induction in IAM is required for corralling and inflammatory confinement
To investigate the underlying mechanisms, we characterized the response of IAM at the injury site. After 5 weeks of recovery, control mice showed signs of successful corralling and injury resolution, with IBA1+ immune cells confined to the lesion core surrounded by a narrow GFAP+ astrocytic border at the injury penumbra (Fig. 4a). There was a clear spatial segregation of these two populations and a well-established physical barrier separating the lesion (occupied by amoeboid-shaped IAM) from the surrounding uninjured neural tissue (containing ramified microglia). In contrast, Plxnb2 cKO mice displayed impaired corralling, with IAM and astrocytes intermingled at the lesion core and the injury penumbra, leading to a defective protective barrier with widespread IBA1+ immune cells well beyond the GFAP+ astrocytic border (Fig. 4a). Furthermore, in mutant mice, wound compaction was compromised, resulting in a larger lesion and unresolved injury (Fig. 4a). Along with disrupted border formation, cells expressing NG2 proteoglycan, i.e. oligodendrocyte precursor cells (OPCs) and pericytes (also PDGFRβ+), had also expanded their territories in mutants (Fig. 4b and Supplementary Fig. 2c). Additionally, whereas control mice showed regressed capillaries (PECAM-1+) at the injury penumbra, mutants showed persistent engorged neo-vasculature in expanded territory at the injury site (Supplementary Fig. 2d).
Fig. 4. Impaired corralling and wound compaction in Plxnb2 cKO after SCI.
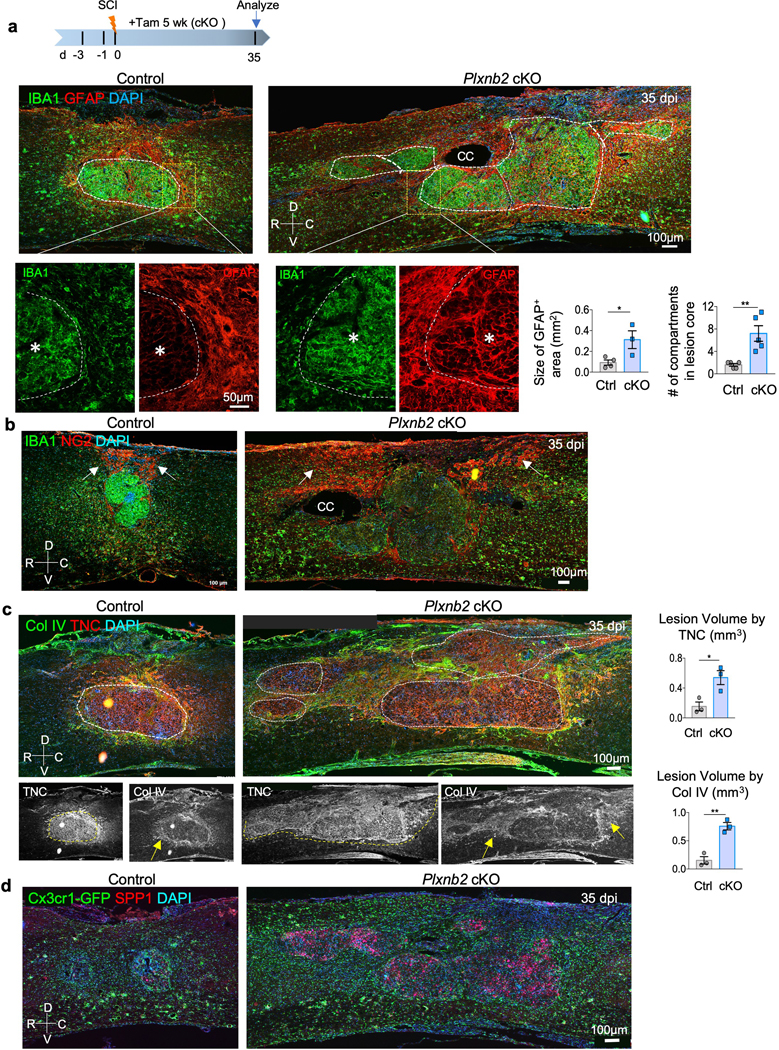
a. Top: experimental scheme. Tamoxifen was injected every other day from 3 days before until 5 weeks after SCI. Bottom: representative IHC images of sagittal sections of thoracic spinal cords show IBA1+ immune cells congregated at the lesion core and surrounded by an astrocytic (GFAP+) border in control animals, but in Plxnb2 cKO animals, they appeared widespread, with multiple gliosis foci in diffuse lesion area as outlined by dashed white lines. Bottom, enlarged images highlight spatial segregation of the two populations at the injury penumbra in control, but intermingling in cKO. R-rostral; C-caudal; D-dorsal; V-ventral; CC-central canal. Asterisks denote the lesion center. Quantifications are shown on right. For the size of GFAP+ area, n=4 mice for control and n=3 for cKO, unpaired two-tailed Student’s t-test, *p=0.035. For the number of gliosis compartment foci, n=5 per genotype, Mann Whitney test, **p=0.0079. Data represent mean ± SEM.
b. IHC of sagittal sections of thoracic spinal cords show expanded territory of NG2+ cells in Plxnb2 cKO animal as compared to the more confined location in the dorsal lesion site in control animal. White arrows point to territory of NG2+ cells.
c. IHC of sagittal sections of thoracic spinal cords show diffuse deposition of matrix proteins at the lesion site in Plxnb2 cKO animals. White dashed lines delineate compacted lesion core in control mice, but multiple gliosis foci in cKO mice. Yellow arrows denote collagen IV deposition in areas surrounding the lesion core, which contains concentrated TNC. Quantification of the lesion volume by TNC and Col IV are shown on the right, n=3 animals per group, unpaired two-tailed Student’s t-test, *p=0.024, **p=0.0029. Data represent mean ± SEM.
d. IHC images of sagittal sections of thoracic spinal cords reveal diffuse pattern of SPP1 at the injury site in Plxnb2 cKO animals. Cx3cr1-GFP reporter also revealed diffuse spread of IAM with amoeboid morphology in cKO mice, whereas in control mice, homeostatic microglia became abundant at the injury penumbra, signifying injury resolution. Images from 3 independent mice per group revealed similar results.
The IAM gene signatures from our INTACT RNA-Seq data indicated the engagement of microglia-matrix interaction and ECM reorganization, particularly at 14 dpi (Fig. 1c,d). Thus, in addition to CSPGs, we also analyzed top upregulated matrix genes identified in IAM. In control mice at 35 dpi, tenascin (TNC) deposition was concentrated at the lesion core, whereas collagen IV (Col IV) was deposited largely at the dorsal wound and the injury penumbra. However, in Plxnb2 cKO mice, both TNC and Col IV were widespread and in disarray, mirroring the highly disorganized spatial pattern of IAM (Fig. 4c). Additionally, the secreted phosphoprotein SPP1 (also known as osteopontin), which is upregulated in microglia during aging and disease 30, was also widespread in cKO mutants (Fig. 4d). Animals with the Cx3cr1-GFP reporter further revealed diffuse distribution of amoeboid-shaped IAM in cKO mutants, reflecting unresolved injury, in contrast to the ramified microglia at the injury penumbra seen in control mice, signifying injury resolution (Fig. 4d). In line with matrix disarray, vinculin, a component of the focal adhesion complex that regulates cell-matrix adhesion and actin polymerization 31, also appeared widespread in mutants, unlike the more confined pattern in control mice reflective of intensified matrix compaction at the lesion border (Supplementary Fig. 3a,b). We further analyzed the expression of 84 ECM genes at the injury site by qRT-PCR, which revealed altered expression profiles of multiple ECM genes in Plxnb2 cKO relative to controls, including Versican (Vcan, a member of the CSPGs), Tnc, Col4a1, Col4a2, Pecam1, and Spp1 (Supplementary Fig. 3c). Altogether, Plexin-B2 induction in IAM facilitates corralling, matrix reorganization, and wound compaction.
Plexin-B2-mediated corralling is important to limit inflammatory spread and regulate cytokine milieu in SCI
Since cellular debris contains a large amount of membrane lipids that are cleared by phagocytotic cells, we performed Oil Red O staining, which revealed that lipid-rich debris was contained in the lesion core at 35 dpi in control animals, but widespread in mutants (Fig. 5a,b and Supplementary Fig. 3d). Concordantly, CD68+ phagocytic cells were congregated at the lesion core in controls, but spilled over beyond the necrotic core in mutants (Fig. 5c,d).
Fig. 5. Impaired containment of phagocytic cells and altered cytokine milieu in Plxnb2 cKO after SCI.
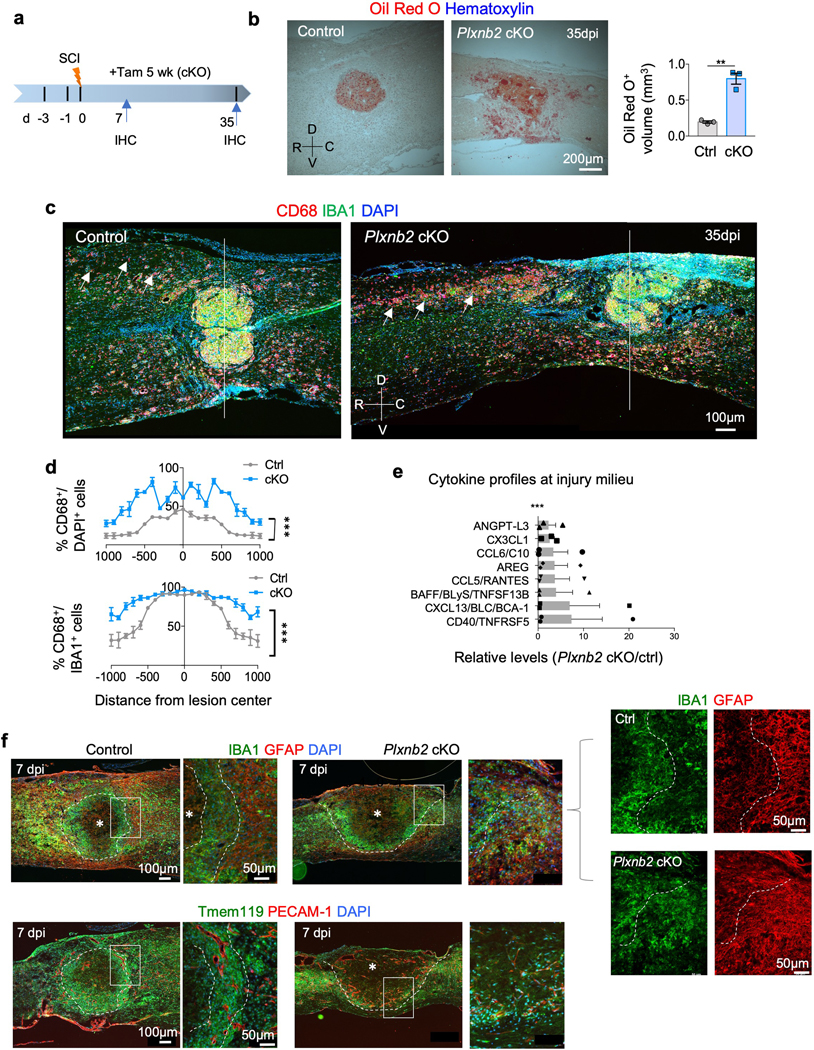
a. Experimental scheme. Tamoxifen was injected every other day from 3 days before until 5 weeks after SCI. IHC was performed at 7 and 35 dpi.
b. Sagittal sections of thoracic spinal cords at 35 dpi reveal widespread Oil Red O lipid staining in Plxnb2 cKO mice as compared to littermate controls, in which lipid debris was contained at the lesion core. Quantifications show calculated Oil Red O+ volume at the injury site, n=3 mice per group, unpaired two-tailed Student’s t-test, **p=0.0015. Data represent mean ± SEM. R-rostral; C-caudal; D-dorsal; V-ventral.
c. Representative IHC images of sagittal sections of thoracic spinal cords show diffuse spread of CD68+ cells and prolonged phagocytosis phenotype of IAM (IBA1+) in Plxnb2 cKO mice even at 5 weeks after SCI (white arrows). Also note impaired corralling of the immune cells and compromised wound compaction in Plxnb2 cKO mice. White line demarcates the lesion center. Images from 3 independent mice for each condition showed similar results.
d. Quantifications show the extent of the spread of phagocytotic cells measured by the prevalence of CD68+ cells among DAPI+ or IBA+ cells at the indicated locations from the lesion center. n=3 mice per group, two-way ANOVA with Bonferroni post hoc correction, ***p<0.0001. For CD68+/DAPI, F(DFn, DFd): F(1, 84)=645.3 for column factor, F(20, 84)=25.83 for row factor. For CD68+/IBA1+, F(DFn, DFd): F(1, 84)=203.4 for column factor, F(20, 84)=32.62 for row factor. Data represent mean ± SEM.
e. Proteome profiler assays show relative levels of upregulated (>2-fold) cytokines or secreted factors at the injury site in Plxnb2 cKO compared to control littermates at 14 dpi after T8 contusion injury. n=3 mice per group, two-way ANOVA with Bonferroni post hoc correction, ***p<0.0001 for the entire profile of 64 proteins on the array. Only proteins with relative levels above the cutoff of 2.0 are plotted. Data represent mean ± SEM.
f. IHC images of consecutive sagittal sections of thoracic spinal cords at 7 dpi (top and bottom panels) stained for the indicated markers reveal early stage corralling phenotype in Plxnb2 cKO animals. Higher magnification images are shown on the right. Top panels show necrotic core (white asterisk) corralled by an inner rim of IBA+ immune cells and an outer rim of GFAP+ astrocytes. White dashed lines delineate the border of spatial segregation between the two populations. Bottom panels show Tmem119+ microglia forming a concentric rim confining the necrotic core in control animals; blood vessels (PECAM-1+) with large lumen size displayed the same concentric orientation. In contrast, in Plxnb2 cKO animals, IBA1+ or Tmem119+ immune cells and GFAP+ astrocytes failed to form concentric rims, resulting in unsealed wound at the dorsal surface and diffuse neo-vasculature with random orientation. Images from 3 independent mice for each group showed similar results.
To compare local cytokines in the injury milieu, we performed ELISA-based proteomic array analyses, which showed upregulated cytokine levels in mutants as compared to controls, including CD40 and CXCL13, signifying an increased inflammatory milieu in mutants (Fig. 5e).
Plexin-B2 ablation did not appear to affect IAM proliferation as shown by EdU pulse studies in Cx3cr1GFP reporter mice (Supplementary Fig. 4a). Phagocytic activity of IAM was also maintained, as demonstrated by time-lapse videos of primary microglia cultures, where control and Plxnb2 cKO microglia phagocytosed fluorescent beads at similar rates and to similar extents (Supplementary Fig. 4b and Supplementary Videos 3 and 4). Plexin-B2 deletion also did not affect upregulation of ApoE and lipoprotein lipase (LPL) in IAM (Supplementary Fig. 4c), two marker genes involved in lipid uptake and metabolism required for microglial scavenging function 32. Finally, in cultures of mixed cortical cells from Plxnb2 cKO mice, treated with or without hydroxy-tamoxifen, the transcription levels of Tnf, Il1b, and C1qa, three cytokines shown to be microglia-derived inducers of inflammatory phenotypes of astrocytes 33, were largely comparable, except for Il1b, which showed a modest decrease in cultures containing Plxnb2 cKO microglia (Supplementary Fig. 4d). Taken together, Plexin-B2 ablation in IAM primarily impairs the spatial segregation of IAM from reactive astrocytes, leading to defective glial barrier and inflammatory spillover.
Functional importance of Plexin-B2 during early innate immune response in SCI
Our next question addressed the timing of Plexin-B2-mediated corralling after SCI. Our initial assumption was that corralling and wound compaction likely took place at a later stage after IAM had completed the main task of debris clearing. Interestingly, we observed a clear spatial segregation of the immune cells and astrocytes already at 7 dpi: IBA1+ cells formed a narrow rim that corralled the edematous, cell-poor necrotic core, and they in turn were surrounded by an outer rim of GFAP+ cells, thus forming a layered ringed structure (Fig. 5f). However, at 3 dpi, IBA1+ and GFAP+ cells were abundantly present at the injury site in close proximity to one another, but not yet spatially segregated (Supplementary Fig. 5a,b).
To distinguish between microglia and macrophages, we used the microglial marker Tmem119, which revealed a delineated concentric pattern of microglia coinciding with that of IBA1+ cells at 7 dpi, indicating a large contribution of microglia in initiating corralling (Fig. 5f and Supplementary Fig. 5c). Remarkably, the concentric pattern of Tmem119+ cells also aligned well with the circumferential pattern of the neo-vasculature (PECAM-1+), with distinctive enlarged lumen size (Fig. 5f and Supplementary Fig. 5c). Concordantly, pericytes (PDGFRβ+) also formed a ringed pattern closely aligned with IBA1+ cells, both surrounded by CSPGs deposited at the injury penumbra (Supplementary Fig. 5d).
By 14 dpi, signs of progressive injury resolution were evident in control mice: IBA1+ cells had shifted towards the necrotic core, while GFAP+ cells remained at the outer rim, and neo-vasculature had regressed with normalized lumen size (Supplementary Fig. 5e; 6a,b). Additionally, by 14 dpi, C1q and phagocytic cells (CD68+) were already contained in the lesion core, matrix proteins such as TNC, Col IV and CSPGs had been reorganized and compacted, and numerous axons (NF-H+) were present in the vicinity of the injury site (Supplementary Fig. 6b-e). Notably, β-catenin displayed a more concentrated pattern at the lesion border, likely reflecting strengthened intercellular adhesion for wound compaction (Supplementary Fig. 6e).
In contrast, the spatial segregation of IBA1+ and GFAP+ cells was severely perturbed in Plxnb2 cKO animals. At 3 dpi, even though IBA1+ cells were recruited to the injury site, their dispersion and physical contact with GFAP+ cells were impaired (Supplementary Fig. 5b). By 7 dpi, IBA1+ or Tmem119+ cells intermingled without clear spatial segregation, and they failed to form a sealed physical barrier, with the dorsal wound completely open (Fig. 5f and Supplementary Fig. 5c,d). Remarkably, the neo-vasculature in the mutants appeared abundant and widespread, and their orientation mirrored the disorganized spatial pattern of IAM (Supplementary Fig. 5c,d). By 14 dpi, wound compaction was compromised, matrix proteins remained widespread and in disarray, and neo-vasculature showed no signs of regression (Supplementary Fig. 5e; 6a-e).
As our longitudinal analysis suggested that Plexin-B2 activity in the early stage of the innate immunity is critical to set the stage for corralling and vascular organization, we studied the mutant phenotypes with different time courses of Tam injection: i) Tam ON for the first 2 or 3 weeks to ablate Plexin-B2 in both microglia and macrophages, followed by 3 or 2 weeks of Tam OFF to allow newly influxed monocytes/macrophages to express Plexin-B2 (Fig. 6a,d), or ii) Tam OFF during the 1st week after injury to allow Plexin-B2 function, followed by Tam ON for the next 4 weeks (Fig. 6e). Of note, monocytes and macrophages exhibit rapid turnover 34 and are constantly replenished from bone marrow stem cells, which are Cx3cr1 negative and thus not affected by Tam treatment 35. Since Tam has a half-life of ~6–12 hour in mice 36, newly influxed macrophages would express Plexin-B2 shortly after Tam withdrawal.
Fig. 6. Plexin-B2 is required during the early stage of innate immune response after SCI.
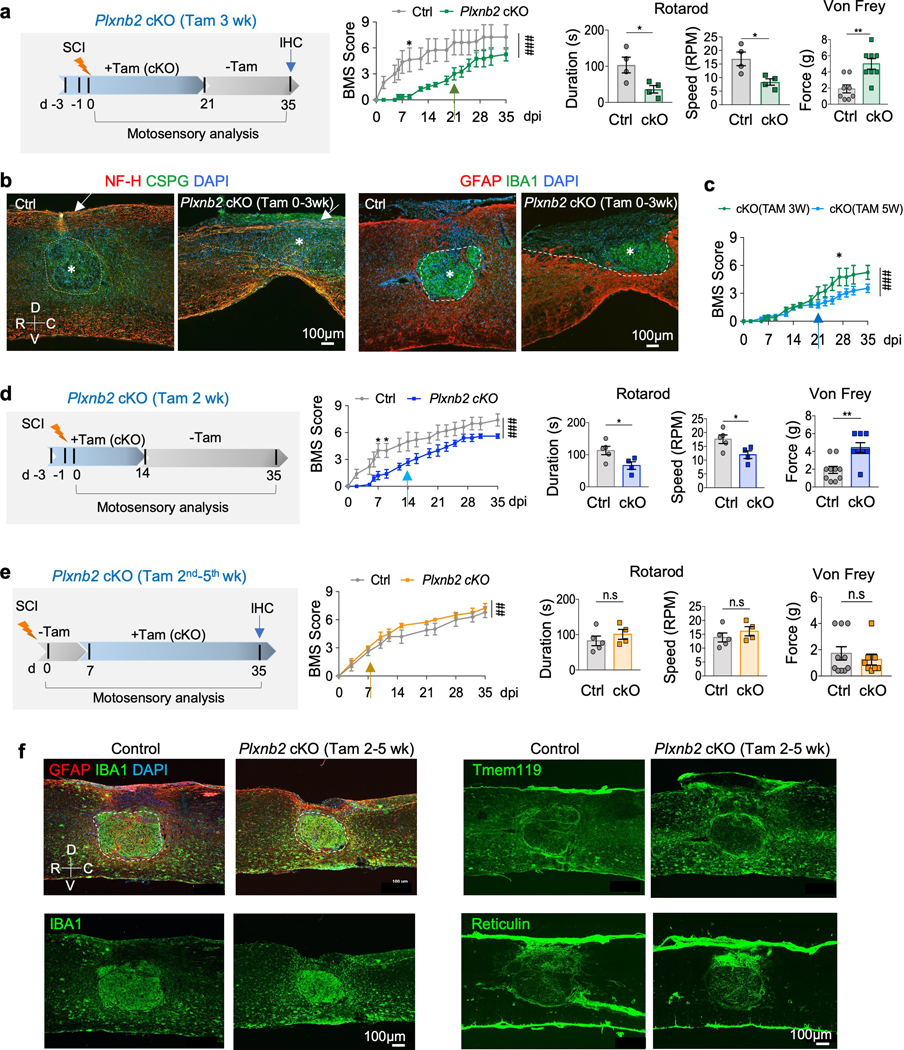
a. Left, experimental scheme (Tam 3 wk). Tamoxifen was injected every other day from 3 days before until 3 weeks after injury, followed by 2 weeks with no tamoxifen injection to allow infiltration of Plexin-B2-expressing macrophages. Right, BMS scores show overall worse recovery of Plxnb2 cKO as compared to the control cohort, n=4 per group, two-way ANOVA with Bonferroni post hoc correction for repeated measures, *p=0.050, ###p<0.001 for the entire recovery course. F(DFn, DFd): F(1, 102)=75.25 for column factor, F(16,102)=7.71 for row factor. Rotarod and von Frey tests at 35 dpi show impaired motosensory recovery for cKO. For rotarod tests: n=4 animals per group, unpaired two-tailed Student’s t-test, *p=0.032 for duration, *p=0.022 for speed. For von Frey tests, n=4 mice per group, left and right hindpaws measured separately, unpaired two-tailed Student’s t-test, **p=0.0059. Data represent mean ± SEM.
b. Left, IHC images of sagittal thoracic spinal cords show larger lesion volume by CSPG staining (outlined by dashed lines) and fewer axon fibers at the dorsal column by NH-F staining (arrows) in Plxnb2 cKO mice (Tam 3 wk) at 35 dpi. Right, IHC images of IBA and GFAP immunostaining show impaired corralling with open wound at the dorsal surface in the mutant animal. Asterisks denote the lesion core. R-rostral; C-caudal; D-dorsal; V-ventral. Images from 3 independent mice per group showed similar results.
c. BMS scores show that the mutant cohort with tamoxifen injection during the first 3 weeks after injury (Tam 3 wk) displayed a partial recovery after 21 dpi when compared to the mutant mice with continuous Tam injections during the entire 5 week duration (Tam 5 wk). n=12 for the Tam 5 wk cohort, n=4 for the Tam 3 wk cohort, two-way ANOVA with Bonferroni post hoc correction, *p=0.019, ###p<0.0001 for the entire recovery course. F(DFn, DFd): F(1, 238)=22.66 for column factor, F(16, 238)=26.56 for row factor. Data represent mean ± SEM.
d. Experimental scheme with tamoxifen injected from 3 days before until 2 weeks after SCI (Tam 2 wk). BMS scores, as well as rotarod and von Frey tests at 35 dpi show impaired motosensory recovery in the mutant relative to the control cohort. n=5 for control, n=4 for Plxnb2 cKO. For BMS, two-way ANOVA with Bonferroni post hoc correction, *p=0.018 and 0.039, respectively, ###p<0.0001 for the entire recovery course. F(DFn, DFd): F(1, 136)=79.72 for column factor, F(16, 136)=24.53 for row factor. For Rotarod tests, unpaired two-tailed Student’s t-test, *p=0.036 for duration, *p=0.035 for speed. For von Frey filament tests, Mann Whitney test, **p=0.0035. Data represent mean ± SEM.
e. Experimental scheme with tamoxifen injected from 2nd to 5th weeks after SCI (Tam 2nd–5th wk). BMS sores, as well as rotarod and von Frey tests at 35 dpi show overall comparable motosensory recovery in both cohorts. n=5 for control, n=4 for Plxnb2 cKO. For BMS, two-way ANOVA with Bonferroni post hoc correction, ##p<0.0099 for the entire recovery course. F(DFn, DFd): F(1, 77)=7.01 for column factor, F(10, 77)=40.51 for row factor. For Rotarod tests, unpaired two-tailed Student’s t-test, n.s., p=0.38 for duration, n.s., p=0.37 for speed. For von Frey filament tests, Mann Whitney test, n.s., p=0.96. Data represent mean ± SEM.
f. IHC images of thoracic spinal cords at 35 dpi for indicated markers show successful corralling in both control and Plxnb2 cKO (Tam 2nd–5th wk) cohorts. White dashed lines outline the lesion core. Note that in both cohorts, Tmem119+ microglia congregated at the injury penumbra, and fibroblasts expressing Reticulin concentrated at the dorsal wound. Images from 3 independent mice per group showed similar results.
We conducted the same battery of motosensory tests, which showed that Plexin-B2 deficiency during the first 3 weeks after injury severely impaired recovery. Likewise, IHC revealed that these mutants had compromised corralling, unsealed wound at the dorsal border, enlarged lesion size (GFAP+ or CSPG+), spillover of inflammatory cells (IBA1+), and fewer NF-H+ axons at the lesion center (Fig. 6a,b and Supplementary Fig. 7a,b). Additionally, lipid debris (Oil Red O+) appeared widespread, so were CD68+ phagocytic cells and matrix proteins, including fibronectin (FN), Col IV and TNC (Supplementary Fig. 7a-c). Consistent with the Tam injection scheme (i.e. Tam 3 wk), at 35 dpi, a portion of the IBA1+ cells expressed high levels of Plexin-B2, representing newly infiltrated macrophages after 21 dpi, while the remaining were negative for Plexin-B2, representing resident microglia and early infiltrated macrophages that had arrived before 21 dpi (Supplementary Fig. 7d). Notably, the BMS scores of this mutant cohort after 21 dpi were improved over the cohort with continuous Plexin-B2 ablation (i.e. Tam 5 wk), although still underperforming relative to the control cohort (Fig. 6c). Hence, Plexin-B2 expression in newly influxed macrophages after 21 dpi partially compensated for the previous Plexin-B2 loss. Similar phenotypes were observed when Tam was delivered only during the first 2 weeks after SCI (Tam 2 wk), further confirming the importance of Plexin-B2 in the early innate immunity (Fig. 6d).
In contrast, mice with normal Plexin-B2 expression in IAM during the 1st week of recovery, followed by cKO in the following 4 weeks (Tam 2nd-5th wk), performed overall at par with control mice in motosensory assays, with even slightly better BMS scores after 14 dpi (Fig. 6e). Consistently, this mutant cohort displayed successful corralling and injury resolution by 35 dpi, and the spatial pattern of fibroblasts expressing reticulin at the injury penumbra was also similar to controls (Fig. 6f). Hence, the initial activity of Plexin-B2 in IAM is sufficient to initiate corralling, thereby setting the stage for subsequent wound compaction; suppressing Plexin-B2 function later had no major detrimental effects on injury resolution.
Plexin-B2 activity in IAM regulates motility, contact inhibition of locomotion and matrix interaction during wound healing
We next investigated the underlying cellular mechanisms. As Plexin-B2 is known to regulate neuroprogenitor migration 9,37–39, we posited that Plexin-B2 might facilitate IAM infiltration and migration at the injury site, both important for initiating the physical contact of IAM with other glia cells, a prerequisite for corralling. To clearly define the injury core for analysis of the extent, timing, and spatial pattern of IAM infiltration, we used here the T8 dorsal column transection SCI model.
Plexin-B2 ablation did not appear to affect recruitment of IAM to the injury site, but significantly reduced IAM dispersion at 3, 7 and 14 dpi, as measured by the spread of IBA1+ cells relative to the lesion border (Fig. 7a,b). The reduced microglial motility in the absence of Plexin-B2 was corroborated by time-lapse videography of co-cultures where primary Cx3cr1-GFP+ microglia were first isolated from Plxnb2 cKO mice and then added onto a lawn of mixed neural cells from the same animal labeled with fluorescent dye CellTracker Red (Fig. 7c). Videos were recorded to track the movement of Cx3cr1-GFP+ microglia with and without hydroxy-tamoxifen over 24 hours, which revealed reduced microglial motility with Plexin-B2 ablation (Supplementary Videos 5 and 6).
Fig. 7. Plexin-B2 promotes microglia dispersion, contact inhibition of locomotion, and focal adhesions.
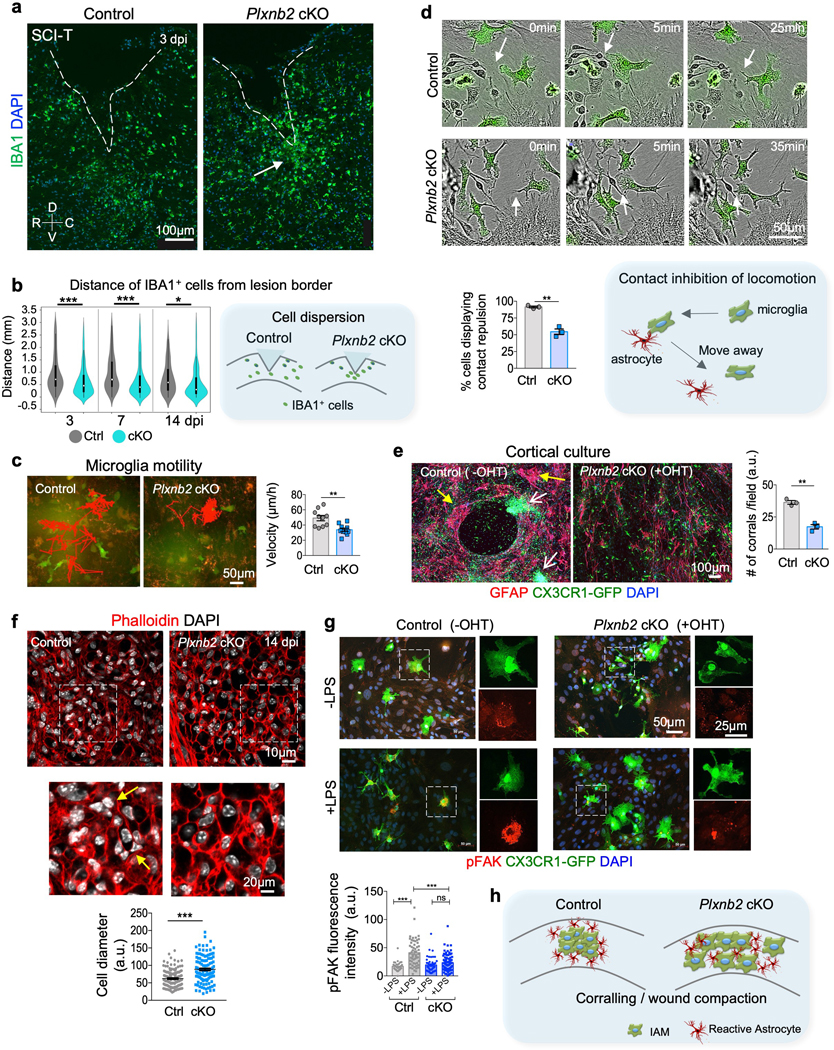
a. IHC images of sagittal sections of thoracic spinal cords at 3 days after T8 dorsal column transection (SCI-T) show even spread of IBA1+ cells at peri-lesion area in control, but more clustered IBA1+ cells in Plxnb2 cKO mice (white arrow). Dashed lines outline lesion border. R-rostral; C-caudal; D-dorsal; V-ventral. Images from 4 independent animals per genotype showed similar results.
b. Violin plots show the distance of IBA1+ cells from the lesion border in control and Plxnb2 cKO mice at 3, 7, and 14 dpi. Quantification scheme is shown on the right. Unpaired two-tailed Student’s t test. For 3 dpi, n=150 cells for control, n=280 cells for cKO, from n=4 animals per group, ***p<0.0001. For 7 dpi, n= 150 cells for control, n=200 cells for cKO, from n=4 animals per group, ***p<0.0001. for 14 dpi: n=150 cells for control, n=200 cells for cKO, from n=3 animals per group, *p=0.020. White dots in violin plots represent median and markers indicate the interquartile range.
c. Frames from time-lapse videos of co-cultures with or without tamoxifen. Primary Cx3cr1-GFP+ microglia were isolated from the cortex of neonatal Plxnb2fl/B Cx3cr1CreER/GFP mice and then added onto a lawn of Cx3cr1-GFP- neural cells from the same mice labeled with CellTracker Red. Red lines trace the movements of a representative microglia over 5 days. For velocity quantifications, n=10 cells from 4 independent animals per group, unpaired two-tailed Student’s t-test, **p=0.0015. Data represent mean ± SEM.
d. Frames from time-lapse videos of mixed cortical cultures from Plxnb2fl/B Cx3cr1CreER/GFP mice, with or without tamoxifen. Microglia were visualized by the Cx3cr1-GFP reporter. Arrows point to microglia that moved away from colliding cells in control conditions, but not in Plxnb2 cKO conditions. Note the time scale between 0 to 25 min for control vs. 0 to 35 min for Plxnb2 cKO microglia. Graphs represent mean ± SEM. To calculate the probability of contact inhibition of locomotion, 60–180 cells were quantified per animal and averaged, n=3 independent animals per group, two-tailed Student’s t test, **p=0.0010. Diagram depicting contact inhibition of locomotion is shown at the bottom right.
e. ICC images of in vitro corralling in cortical cultures from neonatal Plxnb2fl/B Cx3cr1CreER/GFP mice. Microglia were visualized by Cx3cr1-GFP reporter and astrocytes by GFAP. Yellow and white arrows point to spatial segregation of these two populations in control cultures, but intermingling in cultures in which Plxnb2 cKO cKO in microglia was induced by hydroxy-tamoxifen. Quantification is shown on the right. n=3 independent cultures for each condition, unpaired two-tailed Student’s test, **p=0.0011. Data represent mean ± SEM.
f. Images of thoracic spinal cord sections at 14 dpi labeled with phalloidin for cortical F-actin show higher density of glial cells at the lesion core in control as compared to Plxnb2 cKO animals. Magnified images are shown in bottom panels. Quantifications show larger cell diameter at the lesion core in Plxnb2 cKO, n=156 cells for control, n=123 cells for cKO, from 3 independent mice per genotype, unpaired two-tailed Student’s t test, ***p<0.0001. Data represent mean ± SEM.
g. ICC of cortical cultures from neonatal Plxnb2fl/B Cx3cr1CreE/GFP mice, with or without hydroxy-tamoxifen (OHT) treatment. Microglia were visualized with Cx3cr1-GFP. LPS stimulation resulted in upregulation of pFAK in control microglia only. Magnified images of boxed area are shown on the right. Quantifications show fluorescence intensities of pFAK. For control, n=30 cells for -LPS and n=55 cells for + LPS; for cKO, n=45 cells for -LPS and n=93 cells for +LPS. One-way Anova with Tukey’s multiple comparisons post-test. ***p<0.0001. n.s., p=0.12. F(DFn, DFd): F(3, 219)=22.4 for column factor. Data represent mean ± SEM.
h. Diagram depicting the importance of Plexin-B2 in IAM to promote corralling and wound compaction.
In a previous study of bone remodeling, Sema4D was established as a repulsive guidance cue that induces contact inhibition of locomotion in osteoblasts through its receptor Plexin-B1 to steer cells away from osteoclasts 40. Contact inhibition of locomotion is a dynamic process that occurs on a minute-scale whereby a cell that collides with another cell ceases migration before repolarizing and migrating away. To test the hypothesis that Plexin-B2 may mediate contact inhibition of locomotion in IAM, we performed time-lapse videography of cortical cultures to track interactions of Cx3cr1-GFP+ microglia with other neural cells (Supplementary Videos 7 and 8). In control cultures, microglia were highly motile and extended dynamic processes, but upon contact with another cell, they retracted their processes and moved away within minutes (Fig. 7d). The Plxnb2 cKO microglia, on the other hand, displayed diminished propensity to retract their protrusions upon contact with other cells, and maintained contacts for much longer time periods (Fig. 7d).
To further corroborate the corralling defects, we examined the physical interactions between microglia and astrocytes in cortical cultures. After 2 weeks of culture, wildtype microglia and astrocytes exhibited spatial segregation; however, Plxnb2 cKO microglia and wildtype astrocytes remained intermingled (Fig. 7e).
Moreover, in the control animals, wound compaction resulted in cells densely packed at the lesion core, thus minimizing the lesion size while maximizing the repair area (Fig. 7f). In contrast, in Plxnb2 cKO mice, the cell density at the lesion core appeared lower and the average cell size larger, as outlined by phalloidin-stained filamentous actin (F-actin) network. As cell migration and contraction require engagement of focal adhesion, we compared the levels of phospho-focal adhesion kinase (pFAK) in primary microglia with and without LPS stimulation, and found reduced induction of pFAK in Plxnb2 cKO as compared to control cells upon LPS stimulation (Fig. 7g). In summary, Plexin-B2 regulates cytoskeletal dynamics and physical interactions of IAM with other glial cells and with the matrix (Fig. 7h).
Microglia and macrophages both contribute to wound healing after SCI
Finally, we aimed to distinguish the cell-type specific role of Plexin-B2 in microglia vs. macrophages in wound repair. For this purpose, we employed a Tam injection regimen in which animals received three doses of Tam one month before SCI with no subsequent injections (Fig. 8a). As such, Plexin-B2 ablation only occurred in the microglial population but not in peripherally influxed macrophages. We found that this cohort (Tam pre-1m) initially exhibited worse BMS scores relative to control, but no subsequent differences after 14 dpi; and by 35 dpi, rotarod and von Frey filament tests showed no differences in motosensory recovery relative to the control cohort (Fig. 8b). Consistently, IHC revealed that in control animals at 7 dpi, highly proliferative cells (Ki67+) largely overlapped with IBA1+ cells and formed a ring-like structure corralling the necrotic core, which in turn was surrounded by an astrocytic border; but in microglia-only Plxnb2 cKO mice, Ki67+ and IBA1+ cells were congregated in the necrotic core, reflecting reduced dispersion, impaired corralling, and defective glial barrier with unsealed wound at the dorsal surface (Fig. 8c). However, by 35 dpi, wound healing was comparable between the two cohorts (Fig. 8d). Hence, Plexin-B2 signaling in microglia is critical for the early stage of corralling, but Plexin-B2 activity in macrophages was able to largely compensate for the initial delay in wound healing.
Fig. 8. Plexin-B2 activity in both microglia and macrophages contributes to functional recovery after SCI.
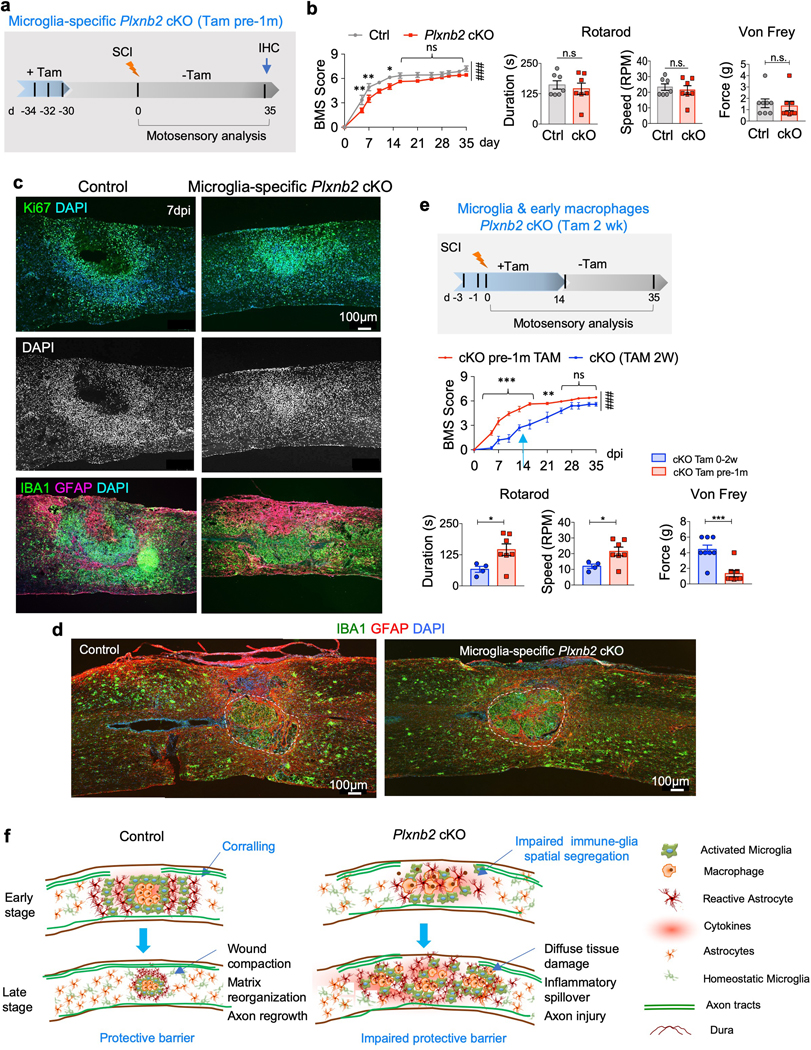
a. Experimental scheme for microglia-specific Plxnb2 cKO with only three tamoxifen injections delivered one month before SCI (Tam pre-1m).
b. BMS scores show worse motor recovery during the first two weeks after injury in Plxnb2 cKO cohort, but a subsequent catch up thereafter, n=8 per group, two-way ANOVA with Bonferroni post hoc correction, **p=0.008, **p=0.0022, and *p=0.031, respectively; ###p<0.0001 for the entire recovery course. F(DFn, DFd): F(1, 156)=47.85 for column factor, F(11, 156)=116.1 for row factor. Rotarod and von Frey tests show no significant differences between the two cohorts at 35 dpi. For Rotarod test, n=7 for each genotype, unpaired two-tailed Student’s t-test, n.s., p=0.59 for duration, n.s., p=0.59 for speed. For von Frey filament tests, n=4 mice per group, left and right hindpaws measured separately, Mann Whitney test, n.s., p=0.081. Data represent mean ± SEM.
c. IHC of sagittal sections of thoracic spinal cords at 7 dpi show sign of early corralling with Ki67+ cells forming a rim corralling cell-poor, necrotic core in control mice, whereas in microglia-specific Plxnb2 cKO mice, Ki67+ cells displayed less dispersion and congregated at the lesion core. DAPI nuclear counterstain highlights spatial pattern of cell density at the injury site. Images from 3 independent mice per group showed similar results.
d. IHC of thoracic spinal cords of both control or microglia-specific Plxnb2 cKO mice display successful corralling and wound compaction at 35 dpi. Images from 3 independent mice per group showed similar results.
e. Comparison of BMS scores during 5 week recovery, and rotarod and von Frey tests at 35 dpi show better motosensory recovery for the cohort with microglia-specific Plxnb2 cKO cohort (Tam pre-1m, n=7, the same cohort as in Fig. 8b) as compared to the Tam 2 wk cohort (n=4, the same cohort as in Fig. 6d). Two-way ANOVA with Bonferroni post hoc correction, ***p=0.0002 for 5 dpi and p<0.0001 for subsequent three timepoints, **p=0.0010, ###p<0.0001 for the entire recovery course. F(DFn, DFd): F(1, 132)=158 for column factor, F(11, 132)=95.4 for row factor. For Rotarod test, n=7 for the Tam pre-1m cohort, n=4 for the Tam 2 wk cohort, unpaired two-tailed Student’s t-test, *p=0.038 for duration, *p=0.038 for speed. For von Frey filament tests, n=4 mice per group, left and right hindpaws measured separately, Mann Whitney test, ***p=0.00050. Data represent mean ± SEM.
f. Diagrams illustrate the importance of Plexin-B2 induction in IAM in mediating early corralling, thereby facilitating debris clearing, inflammation containment, and subsequent matrix reorganization and wound compaction.
To probe whether Plexin-B2’s function in macrophages is critical during the early stage of recovery, we further compared the microglia-only Plxnb2 cKO cohort with the cohort shown earlier in Fig. 6d that received Tam during the first 2 weeks after SCI (Tam 2 wk, i.e. Plexin-B2 was ablated in both microglia and early infiltrating macrophages). The latter cohort overall performed worse by BMS score, particularly during the first 3 weeks, and by 35 dpi, they still lagged in motosensory recovery (Fig. 8e). Hence, Plexin-B2 activity in early influxed macrophages is important in the setting of Plexin-B2 deletion in microglia.
Finally, we investigated recovery and wound healing in mutant animals with deletion of Sema4C, a potential Plexin-B2 ligand. Sema4C KO mutants displayed no overall differences relative to their control littermates in recovery, albeit with slightly worse hindlimb and tail balance by rotarod testing at 35 dpi (Supplementary Fig. 8). These results suggested potentially redundant or compensatory roles of other class IV semaphorins.
Discussion
In CNS injury, microglia and macrophages are known for their phagocytic function and cytokine profiles. Here, we unveiled their additional roles in facilitating corralling and building a protective barrier to seal the wound, orient neo-vasculature, and compact matrix.
Tissue repair relies on coordinated responses from diverse cell types in overlapping phases: an inflammatory phase for debris clearing and cytokine release, a tissue formation phase with precise spatial arrangement of different cell types, and a remodeling phase with capillary regression and matrix compaction 2. The complexity makes it difficult to delineate the sequence of events and distinguish specific roles of each glial population. Previously, reactive astrocytes were presumed to be the main driver for corralling, based on their location in the outer rim. Indeed, an earlier study revealed a requirement of STAT3 in astrocytes for the formation of an astroglial border and corralling 41. The striking phenotypes observed here unveiled an unexpected contribution of IAM for corralling. Clearly, the mutual physical interactions between IAM and astrocytes as mediated by Plexin-B2 are important for their spatial segregation; whether the dysfunction of Plxnb2-deficient IAM would secondarily affect astrocyte reactivity awaits future studies.
Corralling after SCI begins surprisingly early and microglia are largely responsible for the initial formation of a concentric physical barrier, in line with their early activation after SCI, while the peak time of macrophage infiltration occurs at ~7–10 dpi 5. This also agrees with the timeframe of Plexin-B2 induction and its critical function in SCI, as well as previous reports that gliosis is largely completed by two weeks after SCI in adult mice 42. Plexin-B2 does not appear to be required for IAM recruitment, proliferation, phagocytosis, or cytokine release; its main function is to facilitate IAM to form a concentric ring to corral the necrotic core, and to be corralled by an outer rim of reactive astrocytes.
Notably, the concentric ring-like microglial arrangement at the lesion border also influences spatial orientation of neo-vasculature, consistent with microglia being one of the first responders to hypoxia by secreting VEGF 43. The concentric neo-vascular orientation may in turn facilitate corralling by directing blood-borne immune cells towards the lesion core, thus ensuring contained scavenging activity and inflammatory reaction inside the protective barrier. Indeed, the initial cell-poor necrotic core was later replaced by densely packed macrophages filled with lipid debris.
Plexin-B2 regulates several important aspects of cell behavior in IAM, all related to cytoskeletal dynamics: i) cell motility to facilitate IAM dispersion and physical contact with other glial cells to set the stage for corralling, ii) contact inhibition of locomotion, which occurs on a minute-scale to steer IAM away from colliding cells, and iii) strengthening focal adhesions, which have known functions in cell migration, angiogenesis 44, and matrix reorganization/wound compaction. Plexin-B2 is also required for maintaining the motile cellular processes of homeostatic microglia, which is critical for their surveillance function for CNS homeostasis 27. In Drosophila sensory neurons, PlexB forms a complex with integrin β subunit to regulate self-avoidance and tiling of dendritic arbors through surface stabilization of integrin 45; whether Plexin-B2 regulates the tiling behavior of microglia during neurodevelopment awaits future investigation. Here, we only studied Plxnb2 cKO mice up to 4 month of age and did not observe overt behavioral changes, but examining the mutant mice in the context of neurodegeneration would be worthwhile. The cellular source and the specific semaphorins that activate Plexin-B2 in activated IAM remain to be defined. Constitutive Sema4C KO mice displayed comparable wound healing as control mice, indicating either redundant or compensatory mechanisms at play. Other cellular sources of class IV semaphorins should be considered, e.g. Sema4D expressed in oligodendrocytes and Sema4G expressed in neurons 46,47.
Lastly, two recent studies showed that ablation of microglia leads to larger lesions after SCI 48,49, which strengthens the notion that IAM play beneficial roles in wound healing and functional recovery. However, activated macrophages can also trigger axonal dieback or retraction of dystrophic axons through direct physical interaction 50, thus corralling IAM inside the astrocyte border is critical to mitigate their direct contact of with axons.
In summary, our study demonstrates an important role of IAM and Plexin-B2 signaling in promoting corralling and wound compaction. Insights into building a protective physical barrier may aid therapeutic designs for axon regeneration and neural repair after CNS injury.
Methods
Animals.
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Icahn School of Medicine at Mount Sinai. Animals were housed in groups of 5 in a pathogen free barrier facility, in corn bedding lined cages, with pellet chow and water bottles. Tamoxifen was injected in doses of 100 mg/kg, i.p.
Cx3cr1CreER mice were obtained from Jackson Laboratory (B6.129P2(Cg)-Cx3cr1tm2.1(Cre/ERT2)Litt/WganJ; JAX stock #021160). Rosa26-loxP-STOP-loxP-INTACT mice were obtained from Jackson Laboratory (B6;129-Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat/J; JAX stock #021039). Rosa26INTACT/+ Cx3cr1CreER/+ mice were healthy and fertile. The Cx3cr1GFP/+ reporter line was obtained from Jackson Laboratory (B6.129P-Cx3cr1tm1Litt/J; JAX stock # 005582). Mice with a Plxnb2 targeted trapping mutation with lacZ reporter (Plxnb2B), and mice with the Plxnb2fl conditional allele have been derived from the EUCOMM mouse line Plxnb2tm1a(EUCOMM)Wtsi in our laboratory 10,26. Mice were bred on a C57BL/6J genetic background.
Spinal cord injury model.
Six to eight week old mice of mixed gender of the indicated genotypes were used for our SCI studies. Mice were anesthetized by isofluorane inhalation (Baxter Healthcare, NDC 10019–360-40). For dorsal column transection, the procedure was performed as previously described 24. Briefly, the lamina of T8 spinal segment was exposed and removed, and the dorsal column was transected bilaterally using iris microscissors (Fine Science Tools, 15000–00), with maximum depth reaching ~0.8 mm. For sham control, only T8 laminectomy was performed.
For T8 contusion injury, after T8 laminectomy, an infinite horizon impactor (IH-0400, Precision Systems and Instrumentation) was used to deliver an impact force of 45 kDyn to the spinal cord. All animals received subcutaneous injection of 1 ml of saline, 10 mg/kg of Baytril and 0.05 mg/kg of buprenorphine every day for the first week following surgery. Bladder expression was performed for animals undergoing T8 contusion injury twice a day for the duration of the experiments.
Motosensory behavioral analysis.
Mice were housed on a 12 hr light/dark cycle with ad libitum access to food and water. All animals were acclimated to the testing room or apparatus for 1 hr prior to behavioral testing. Blind scoring was performed to ensure that observers were unaware of genotypes or treatments.
For BMS scoring, hindlimb locomotor performance was evaluated according to the open-field Basso Mouse Scale (BMS) 51, with scores ranging from 0 (complete paralysis) to 9 (normal mobility); each score represents a distinct motor functional state. Mice were randomly separated into groups without any preferences, while verifying that the average starting score was similar in all groups. Locomotor activity in an open field was monitored every other day by placing the mouse for 5 min at the center of an enclosure made of molded plastic with a smooth, non-slippery floor. Before each evaluation, mice were carefully examined for signs of peritoneal infection, wound in hindlimbs, tail or foot autophagia. Animals that showed a difference of more than 2 score points between left and right hindlimbs were excluded from the experimental analysis.
For rotarod test, an accelerating rotarod was used to measure gross motor capability and coordination by accelerating the rod from 0 to 40 rpm. Each animal was subjected to one practice trial, followed by two test trials, with an interval of 20 min between trials. The latency to fall was averaged from the two test trials per animal.
For regular/irregular horizontal ladder test, paw placement was assessed during walking while mice were placed on a horizontal ladder with regular/irregularly placed 3 mm diameter rungs. The mouse home cage was located at the end of the ladder. Mice were video-recorded for at least 6 consecutive runs for each trial (3 min). Side and bottom views of the animal (mirror and direct view) were used to detect foot-faults (drag, slip, foot-falls) and to analyze for hindlimbs coordination.
Von Frey filament test was used to determine tactile sensory recovery after SCI 52,53. Mice were placed on a metal mesh and the plantar surface (glabrous) of the hindpaw was stimulated with a set of calibrated von Frey filaments (0.004 – 8 g). Each filament was applied five consecutive times against the lateral area of the paw. Hindpaw withdrawal or licking was marked as a positive response. A positive response in three out of five repetitive stimuli was defined as the von Frey threshold. Mice were tested at baseline before surgery and at the indicated time points after SCI. For each animal, left and right hindpaws were tested separately.
Affinity immunopurification of GFP-tagged nuclei (INTACT).
Affinity immunopurification of SUN1-GFP-tagged nuclei was performed as previously described 22. Spinal cords (from 5 mm rostral to 5 mm caudal to the injury center) were rapidly dissected on ice, and pooled from three animals for each sample. Nuclei were separated from cellular debris using an iodixanol gradient, wherein nuclei were separated out at the interphase between the 30/40% phases. We obtained between 1.5 – 2 million nuclei per purification, which were then incubated with 10 μl of 0.2 mg/ml rabbit monoclonal anti-GFP antibody (Life Technologies G10362). ProteinG Dynabeads (Invitrogen 10004D) were added to this mixture, incubated for 20 min for magnetic separation of GFP-positive nuclei, and the solution was then passed through a 20 μm filter (CellTrics, Sysmex Partec GmbH 04–0042-2315). All steps were performed at 4°C.
Total nuclei yields and the percentage of GFP+ nuclei were calculated as previously described 22. Specifically, from the initial ~2 million nuclei obtained per preparation, an aliquot was saved and stained with DAPI. About 9% nuclei from this fraction were GFP+, as counted by either fluorescence microscopy or using hemocytometer chambers. In the final bead-bound fraction, we obtained 95–98% purity of GFP+ cells. These nuclei were stored at −80°C for subsequent nuclear RNA extraction, qRT-PCR, and RNA-Seq.
RNA extraction, library preparation, and RNA-sequencing.
GFP+ bead-bound nuclei were resuspended in buffer RLT for RNA extraction using the RNeasy Micro Kit, along with the recommended on-column DNase digestion (Qiagen 74004). RNA quality and concentrations were measured using an Agilent Bioanalyzer (Agilent RNA 6000 Pico, 5067–1513) and samples having RIN Scores higher than 7 were used. Total RNA was converted to cDNA and amplified using the Ovation RNA-Seq System V2 (Nugen 7102). All samples underwent SPIA Amplification (single primer isothermal amplification) (Ovation RNA-Seq System V2, NuGen, 7102–08) prior to library preparation (NEBNext Ultra DNA Library Prep Kit for Illumina, NEB, 7370S). Three independent samples for each condition were barcoded using the NEBNext Index primers (NEB E7335S), pooled together as one sample, and run on the Illumina platform HiSeq2500 (Rapid Mode-2×50bp) (Macrogen). Next-generation RNA-sequencing generated 40–80 million paired 50 base reads per library.
Quantitative RT-PCR.
For qRT-PCR, cDNA synthesis was performed using the SuperScript III First Strand Synthesis System (Invitrogen 18080–051). Quantitative PCR was performed using PerfeCTa SYBR Green FastMix Rox (QuantaBiosciences) in an ABI 7900HT qPCR system (Applied Biosystems). Gapdh was used as housekeeping gene. Primers used were as follows: Tnf-forward: CCCTCACACTCAGATCATCTTCT, Tnf-reverse: GCTACGACGTGGGCTACAG; Il1b-forward: GAAATGCCACCTTTTGACAGTG, Il1b-reverse: TGGATGCTCTCATCAGGACAG; C1qa-forward: AAAGGCAATCCAGGCAATATCA, C1qa-reverse: TGGTTCTGGTATGGACTCTCC.
Bioinformatics analysis for differential gene expression.
Quality of sequencing reads was assessed using fastQC 54. Reads were mapped against the mouse genome (GRCm38) and rRNA sequences using ContextMap version 2.7.9 55, using BWA 56 as short read aligner and default parameters. Number of read fragments per gene were determined from the mapped RNA-Seq reads using featureCounts (strand-specific for stranded libraries, non-strand-specific otherwise) 57. Gene expression was quantified in terms of fragments per kilobase of exons per million mapped reads (FPKM).
Differential gene expression analysis was performed using edgeR 58. For each pairwise differential gene expression analysis, only the genes with at least an average of 10 reads per sample were evaluated. p-values were adjusted for multiple testing using the method by Benjamini and Hochberg 59 and genes with an adjusted p-value <0.01 were considered significantly differentially expressed.
ENRICHR GO analysis.
For gene ontology (GO) analyses, differentially expressed gene (DEG) lists were entered into the web-version of Enrichr software (http://amp.pharm.mssm.edu/Enrichr/) 60. All scales from the ENRICHR database are depicted as ‘Combined Score’ 61, computed by taking log of the p-value from the Fisher Exact Test and multiplying it with the z-score of the deviation from the expected rank.
Immunohistochemistry.
Animals were sacrificed and perfused using ice-cold 4% PFA (Thermo Scientific AC416785000). Spinal cords at the injury site were dissected, post-fixed overnight at 4°C, and embedded in OCT (Tissue-Tek) (Thermo Fisher 4585). 12 μm thick sagittal sections were collected on SuperFrost+ slides (VWR 48311–703) and stored at -20°C. The sections were washed with 1x PBS, incubated with blocking buffer with 5% normal donkey serum (Jackson Immunoresearch 017–000-121) and 0.3% Triton X-100 (Acros Organics 9002–93-1) in PBS for one hour at room temperature prior to adding primary antibodies (details below) diluted in antibody-dilution buffer with 1% BSA (Fisher BioReagents BP9703100) and 0.3% Triton X-100 in PBS, and incubated overnight at 4°C. Subsequently, AlexaFluor 488-, AlexaFluor 594-, AlexaFluor 647-conjugated secondary antibodies (1:300, Jackson ImmunoResearch) were added and DAPI (1:1000, Invitrogen D1306) was used for counterstaining.
Primary antibodies and their dilutions: anti-5-HT (Rabbit, 1:200, ImmunoStar 20080), aCaspase 3 (Rabbit, 1:200, RnD Systems AF835), ApoE (Goat, 1:1000, Millipore 178479), CD11b (Goat, 1:300, MyBioSource MBS420973), CD68/Macrosialin (Rat, 1:200, BioRad MCA1957GA), Collagen IV-a1 (Mouse, 1:800, Sigma C1926), CSPG (CS-56) (Mouse, 1:100, Sigma C8035), DAPI (1:1000, Invitrogen D1306), Fibronectin (Rabbit, 1:400, EMD Millipore AB2033), GFAP (Mouse, 1:1000, EMD Millipore MAB360), GFP (Chicken, 1:1000, Aves Lab GFP-1020), IBA1 (Rabbit, 1:500, Wako 019–19741), IBA1 (Goat, 1:500, Novus NB100–1028), IL-1a (Rabbit, 1:400, Abcam ab7632), Ki67 (Rabbit, 1:200, Abcam ab15580), Ki67 (Mouse, 1:200, BD Biosciences 556003), LPL (Rabbit, 1:500, Bioss bs-1973R), N-Cadherin (Mouse, 1:500, BD Biosciences 610920), NeuN (Mouse, 1:400, EMD Millipore MAB377), NeuN (Chicken, 1:400, Aves Lab NUN), NF-H (Chicken, 1:1000, EMD Millipore AB5539), NG2 (Rabbit, 1:200, EMD Millipore AB5320), Olig2 (Rabbit, 1:500, EMD Millipore AB9610), P2ry12 (Rat, 1:100, Biolegend 848002), PDGFRβ (Rabbit, 1:100, Abcam ab32570), PECAM-1/CD31 (Rabbit, 1:300, Abcam ab28364), pFAK (Tyr397) (Rabbit, 1:400, Thermo Fisher 44–624G), Phalloidin (1:200, Thermo Fisher A12380), Plexin-B2 (Armenian Hamster, 1:800, eBioscience eBio3E7), Reticulin (Rat, 1:500, Abcam ab51824), SPP1/Osteopontin (Rabbit, 1:200, ProteinTech 25715–1-AP), β-Catenin (Mouse, 1:300, BD Biosciences 610153), Tenascin (Rabbit, 1:100, Merck Millipore AB19011), Tmem119 (Rabbit, 1:500, Abcam ab209064), Vimentin (Chicken, 1:500, Novus Biologicals NB300–223), Vinculin (Mouse, 1:400, Thermo Fisher NB6001293).
Images were captured with Zeiss microscopes (AxioCamMRc). Tiled images were merged using Photoshop CS6. Immunofluorescence intensity analyses were performed as described 62. In brief, the mean intensity in microglia was calculated after subtracting background intensity.
For consistency, midline sagittal sections of the spinal cords were used for most analyses. To measure the distribution of microglia, the horizontal distance of each individual IBA1+ cells from the lesion border was measured in ImageJ software and plotted using violin plot 63 (Python). To measure the scatter of CD68+ cells, a vertical line through lesion center was drawn, and the numbers of cells was counted in consecutive 100 μm areas from the midline.
LacZ staining.
β-galactosidase reporter expression was identified histochemically in spinal cord sections by X-Gal assay. X-gal staining solution was prepared by adding X-Gal (dissolved in dimethylformamide) at 1 mg/ml to staining buffer (0.02% Igepal, 0.01% sodium deoxycholate, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 2 mM MgCl2 diluted in 0.1 M PBS (pH 7.3)). Spinal cord sections were prepared as described above and incubated overnight in X-gal staining solution. Bright field images were captured with Zeiss microscopes (AxioCamMRc).
EdU pulse study.
EdU solution (10 mM, Click-iT EdU Alex Fluor 488 Imaging Kit, Invitrogen #C10337, 100 μl /10 g body weight) was injected intraperitoneally 3 hours before tissue collection. Before EdU detection, slides were air-dried for 1 hour. Click-iT reaction cocktail (1x Click-iT reaction buffer, CuSO4, Alexa Fluor azide, and 1x reaction buffer additive) was prepared and applied to slides for 30 min, washed with PBS for 2 min, followed by IHC procedure for the indicated markers.
Lesion volume.
Sagittal 12 μm thick sections of injured spinal cords were stained for the indicated markers to estimate scar size. For each mouse, all sagittal spinal cord sections were collected, stained, imaged and measured. The positive area of every section for Oil Red O, CSPG, TNC, or Col IV were measured by ImageJ, respectively. To calculate lesion volume, the sum of positive areas was calculated and then multiplied by 12 μm: v = ∑ area * 12.
STimulated Emission Depletion Microscopy (STED).
Animals were perfused and tissues harvested, post-fixed and embedded in OCT as described above for immunofluorescence. Samples were cut at 25 μm thickness and collected on SuperFrost+ Slides. Samples were stained for immunofluorescence staining using anti-TMEM119 (microglia-specific) and a secondary antibody recommended for STED imaging (anti-rabbit IgG Atto 594, Sigma, 77671–1ml-F). Counterstaining was performed with Pico-Green (Quant-iT PicoGreen dsDNA Reagent, Invitrogen, P7581). Samples were mounted using the ProLong Diamond Antifade Mountant (Invitrogen P36965) and Micro Cover Glasses, Rectangular #1.5 (VWR, 48393–251). Sections were imaged using the Leica TCS SP8 STED 3x microscope (Mount Sinai Microscopy core) and images were analyzed with ImageJ software.
Sholl analysis.
Sholl analysis was carried out as described 64,65. In brief, z-stack confocal images were condensed into a maximum intensity projection image (Zen software) over which concentric circles were drawn, centered on the soma with 100 μm interval for every circle (Sholl analysis, Image J). For each microglia, the number of intersections of microglia branches with each circle were plotted to create the Sholl plot. The length of the longest branch from each cell were also plotted.
RT2 Profiler PCR Arrays.
Fourteen days after surgery (laminectomy or SCI contusion), injured spinal cord regions were collected, and total RNA was extracted with the RNeasy Micro kit (Qiagen 74004). Total RNA was then converted to cDNA using the RT2 First Strand Kit, and cDNA derived from 10 μg of RNA was used for the RT2 Profiler PCR Arrays (QIAGEN, PAMM-013ZA-2). Heatmaps were plotted using GraphPad Prism 7.0.
Proteome profiler assay (Cytokine Array).
Fourteen days after surgeries (laminectomy or T8 contusion), injured regions of spinal cords were collected, and protein was extracted with Illustra triplePrep Kit (GE healthcare). 100 μg of protein lysates were used for the cytokine array (Proteome Profiler antibody assays-Mouse Cytokine Array, R&D Systems ARY028). Images were captured with a Gel Doc XR system (Bio-Rad), and analyzed with ImageJ.
Microglia culture.
Forebrains from Plxnb2 cKO P2 pups were harvested in DPBS on ice and tissue was minced using a scalpel blade. The tissue pieces were transferred into 15 ml tubes with 1 ml DPBS using sterile plastic transfer pipettes. Samples were spun down at 1,200 rpm for 5 minutes, the DPBS discarded and 1 ml of 0.25% Trypsin EDTA (Invitrogen 25200072) was added to the tissue pellet and gently mixed. The tube was incubated in a 37°C water bath for 8 minutes, and tissue triturated gently using a 200 μl micropipette tip to obtain single cell solution. 10 ml of warm DMEM/F12 complete (DMEM/F12 supplemented with 10% FBS and 1x Pen-Strep (Thermo Fisher 15140122) was added to the pellet to dilute and inhibit the trypsin, cells were spun at 1,200 rpm for 5 minutes and resuspended again in DMEM. Cells were then seeded onto high attachment 6 well Primaria plates (Corning 08–772-4J) at a density of 1.5 million cells per well. Medium was changed once every week and cells were kept in culture for up to 4 weeks. Typically, 2-week old cultures were used for our experiments.
To separate microglia from neurons/astrocytes, 1 ml of 0.25% Trypsin-EDTA was added to the 6-well plate and incubated at 37°C for 5 minutes. Neurons and astrocytes would delaminate first, leaving the highly adherent microglia behind. Supernatant was removed and either used for subsequent experiments or discarded. Another 1 ml 0.25% Trypsin-EDTA was added per well to detach microglia, by incubating cells at 37°C for 10 minutes. This method yielded ~90% pure microglia with minimal neural cell contamination.
Cortical cultures and in vitro corralling assay.
For mixed-cell cortical cultures, cortical tissues from neonatal Plxnb2fl/B Cx3cr1CreER/GFP pups were harvested and dissociated as single cells as described above. Mixed cell cultures were plated onto Primaria plates with DMEM/F12 supplemented with 10% FBS and 1% Pen-Strep.
For microglia motility assays, primary microglia (Cx3cr1-GFP+) were first cultured alone, and then detached and replated onto a lawn of Cx3cr1-GFP- cells (neurons, astrocytes) that were detached early during the preparation as described above and labeled with CellTracker Red. IncuCyte Zoom (Sartorius) was used for real time cell imaging, and cell motility was tracked using ImageJ.
For in vitro corralling assays, cortical cells were isolated from Plxnb2fl/B Cx3cr1CreER/GFP P2 pups and cultured as mixed cell cultures in laminin-coated 4-well chamber slides, in DMEM/F12 supplemented with 10% FBS and 1% Pen-Strep, with 50% media change once a week. At the end of 2 weeks, cultures were fixed with 4% formaldehyde, washed thrice with PBS, and subjected to immunocytochemistry.
Phagocytosis assay.
pHrodo Red Zymosan Bioparticles (Invitrogen P35364) were added at a concentration of 10 μg/well to a lawn of primary microglia obtained as described above, and allowed to settle for 30 minutes. Wells were imaged at 10x magnification for red fluorescence and phase contrast, every hour for 24 hours using IncuCyte Zoom.
MtrackJ for microglia motility.
To measure microglia motility, the Cx3cr1-GFP+ primary microglia were cultured in IncuCyte Zoom and time-lapse images were captured every 30min for 5 days. Individual microglia were tracked for their migration with the MTrackJ plugin of ImageJ 66. The velocity of individual microglial cell soma (μm/h) was calculated as the traveled distance (μm) divided by the observation period of 120 hours.
Contact inhibition of locomotion (CIL).
To determine probability of CIL of microglia, Cx3cr1-GFP-tagged primary microglia were tracked in cortical cultures in IncuCyte incubator and time-lapse images were captured every 5 min for 5 hours. Visual inspection of microglial protrusion retraction upon colliding with other cell types were determined within ~30 min after contact 67,68. The CIL percentage was calculated by dividing the number of microglia showing retraction within 30 min after making contact by the total number of microglia that had made contact with other cells.
Statistical analysis.
All samples were randomly allocated into experimental groups at the start of each individual study. Mice of the same genotype were randomly assigned to each experiment at the beginning of the study. Blind scoring was performed for in vivo studies. Data collection and analysis for in vitro experiments were not performed blind to the conditions of the experiments. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in previous publications 24,25,69. For each data set, Shapiro-Wilk test (Prism 7, GraphPad Software) was performed to determine parametric (p> 0.05) vs. non-parametric samples (p<0.05 in the Shapiro-Wilk test). For parametric data, F-test was conducted to compare variances (Prism 7). For samples with similar variance (p>0.05 in the F-test), unpaired t-test (two-sided) was used. For samples with significantly different variances (p < 0.05 in the F-test), unpaired two-tailed t-test with Welch’s correction was performed. For statistical comparison between non-parametric vs. parametric data sets, or between 2 non-parametric data sets, Mann-Whitney test was performed, which does not require the assumption of normal distribution (Prism 7). For comparison of multiple groups, one-way ANOVA with Tukey’s multiple comparisons post-test were performed. For studies with repeated measures, two-way ANOVA repeated measures (RM) followed by post hoc Bonferroni test were performed. Outliers were excluded based on Grubbs’ outlier test (GraphPad, p<0.05). The mean values are presented along with standard error of the mean (SEM) as error bars. *, **, and *** denote p<0.05, p<0.01, and p<0.001, respectively.
Supplementary Material
Acknowledgements
We thank all members of the Zou and Friedel laboratories for constructive comments. The work was supported by grants from NIH/NINDS (R01/R56 NS073596), Craig H Neilsen Foundation (#476516), and New York State Spinal Cord Injury Research Board (DOH01-C32242GG, -C33268GG) to H.Z., NIH/NINDS (R01 NS092735) to R.H.F, NIH/NINDS (NS086444, NS093537), NIH/NIA (U01 AG046170) to B.Z., and National Institute on Drug Abuse (PPGPOIDAO8227) to V.Z. Additional fellowship support was provided by the Chinese Scholarship Council to X. Z. and New York State Spinal Cord Injury Research Board (DOH01-C32634GG) to S.W. M-S.F., M.K. and C.C.F. were supported by grants FR2938/7–1 and CRC 1123 (Z2) from the Deutsche Forschungsgemeinschaft (DFG) to C.C.F. We would also like to thank Drs. Zhenyu Yue and Yuanxi Zhang for assisting in rotarod tests, and Mount Sinai Microscopy CoRE for assisting STED imaging.
Footnotes
Accession codes
RNA-Seq data files were deposited at the National Center for Biotechnology Information (NCBI) GEO repository under accession number GSE113566.
Declaration of Interests: The authors declare no competing interests.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. RNA-Seq data files were deposited at the National Center for Biotechnology Information (NCBI) GEO repository under accession number GSE113566.
References
- 1.Silver J. & Miller JH Regeneration beyond the glial scar. Nat Rev Neurosci 5, 146–156 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Burda JE & Sofroniew MV Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Courtine G. & Sofroniew MV Spinal cord repair: advances in biology and technology. Nat Med 25, 898–908 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Beck KD, et al. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133, 433–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popovich PG & Jones TB Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends Pharmacol Sci 24, 13–17 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Hines DJ, Hines RM, Mulligan SJ & Macvicar BA Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia 57, 1610–1618 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Carlson SL, Parrish ME, Springer JE, Doty K. & Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol 151, 77–88 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Friedel RH, et al. Plexin-B2 controls the development of cerebellar granule cells. J Neurosci 27, 3921–3932 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daviaud N, Chen K, Huang Y, Friedel RH & Zou H. Impaired cortical neurogenesis in plexin-B1 and -B2 double deletion mutant. Dev Neurobiol 76, 882–899 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Nakada M, et al. Molecular targets of glioma invasion. Cell Mol Life Sci 64, 458–478 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurrapu S. & Tamagnone L. Transmembrane semaphorins: Multimodal signaling cues in development and cancer. Cell Adh Migr 10, 675–691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koropouli E. & Kolodkin AL Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr Opin Neurobiol 27, 1–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumanogoh A. Semaphorins - A Diversity of Emerging Physiological and Pathological Activities, (Springer; Japan, Tokyo, 2015). [Google Scholar]
- 15.Kumanogoh A. & Kikutani H. Immunological functions of the neuropilins and plexins as receptors for semaphorins. Nat Rev Immunol 13, 802–814 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Negishi-Koga T. & Takayanagi H. Bone cell communication factors and Semaphorins. Bonekey Rep 1, 183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasterkamp RJ Getting neural circuits into shape with semaphorins. Nat Rev Neurosci 13, 605–618 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Sakurai A, Doçi CL, Doci C. & Gutkind JS Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer. Cell Res 22, 23–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran TS, Kolodkin AL & Bharadwaj R. Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol 23, 263–292 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Worzfeld T. & Offermanns S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discov 13, 603–621 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Xia J, et al. Semaphorin-Plexin Signaling Controls Mitotic Spindle Orientation during Epithelial Morphogenesis and Repair. Dev Cell 33, 299–313 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Mo A, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkhurst CN, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuboyama T, et al. HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci Rep 7, 8641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parikh P, et al. Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc Natl Acad Sci U S A 108, E99–107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier V, et al. Semaphorin 4C and 4G are ligands of Plexin-B2 required in cerebellar development. Mol Cell Neurosci 46, 419–431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kettenmann H, Hanisch UK, Noda M. & Verkhratsky A. Physiology of microglia. Physiol Rev 91, 461–553 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Basso DM, et al. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23, 635–659 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Bradbury EJ, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416, 636–640 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bays JL & DeMali KA Vinculin in cell-cell and cell-matrix adhesions. Cell Mol Life Sci 74, 2999–3009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keren-Shaul H, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Furth R. Origin and turnover of monocytes and macrophages. Curr Top Pathol 79, 125–150 (1989). [PubMed] [Google Scholar]
- 35.Fogg DK, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Robinson SP, Langan-Fahey SM, Johnson DA & Jordan VC Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab Dispos 19, 36–43 (1991). [PubMed] [Google Scholar]
- 37.Deng S, et al. Plexin-B2, but not Plexin-B1, critically modulates neuronal migration and patterning of the developing nervous system in vivo. J Neurosci 27, 6333–6347 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirschberg A, et al. Gene deletion mutants reveal a role for semaphorin receptors of the plexin-B family in mechanisms underlying corticogenesis. Mol Cell Biol 30, 764–780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha B, Ypsilanti AR, Boutin C, Cremer H. & Chedotal A. Plexin-B2 regulates the proliferation and migration of neuroblasts in the postnatal and adult subventricular zone. J Neurosci 32, 16892–16905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deb Roy A, et al. Optogenetic activation of Plexin-B1 reveals contact repulsion between osteoclasts and osteoblasts. Nat Commun 8, 15831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wanner IB, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33, 12870–12886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaur C, Rathnasamy G. & Ling EA Biology of Microglia in the Developing Brain. J Neuropathol Exp Neurol 76, 736–753 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Zhao X. & Guan JL Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev 63, 610–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meltzer S, et al. Epidermis-Derived Semaphorin Promotes Dendrite Self-Avoidance by Regulating Dendrite-Substrate Adhesion in Drosophila Sensory Neurons. Neuron 89, 741–755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellver-Landete V, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun 10, 518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brennan FH, Hall JCE, Guan Z. & Popovich PG Microglia limit lesion expansion and promote functional recovery after spinal cord injury in mice. bioRxiv, 410258 (2018). [Google Scholar]
- 50.Busch SA, Horn KP, Silver DJ & Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci 29, 9967–9976 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basso DM, et al. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. Journal of neurotrauma 23, 635–659 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Descalzi G, et al. Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci Signal 10(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitsi V, et al. RGS9–2--controlled adaptations in the striatum determine the onset of action and efficacy of antidepressants in neuropathic pain states. Proc Natl Acad Sci U S A 112, E5088–5097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andrews S. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010). [Google Scholar]
- 55.Bonfert T, Kirner E, Csaba G, Zimmer R. & Friedel CC ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics 16, 122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H. & Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK & Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benjamini Y. & Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B 57, 289–300 (1995). [Google Scholar]
- 60.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jensen EC Quantitative analysis of histological staining and fluorescence using ImageJ. The Anatomical Record 296, 378–381 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Hintze JL & Nelson RD Violin plots: a box plot-density trace synergism. The American Statistician 52, 181–184 (1998). [Google Scholar]
- 64.Longair MH, Baker DA & Armstrong JD Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27, 2453–2454 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Morrison HW & Filosa JA A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. Journal of neuroinflammation 10, 782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meijering E, Dzyubachyk O. & Smal I. Methods for cell and particle tracking in Methods in enzymology, Vol. 504 183–200 (Elsevier, 2012). [DOI] [PubMed] [Google Scholar]
- 67.Roy AD, et al. Optogenetic activation of Plexin-B1 reveals contact repulsion between osteoclasts and osteoblasts. Nature communications 8, 15831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scarpa E, et al. A novel method to study contact inhibition of locomotion using micropatterned substrates. Biology open 2, 901–906 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finelli MJ, Wong JK & Zou H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 33, 19664–19676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.