

Abstract
Cleft palate (CP) is the second most common congenital birth defect. The etiology of CP is complicated, with involvement of various genetic and environmental factors. To investigate the gene regulatory mechanisms, we designed a powerful regulatory analytical approach to identify the conserved regulatory networks in humans and mice, from which we identified critical microRNAs (miRNAs), target genes and regulatory motifs (miRNA–TF–gene) related to CP. Using our manually curated genes and miRNAs with evidence in CP in humans and mice, we constructed miRNA and transcription factor (TF) co-regulation networks for both humans and mice. A consensus regulatory loop (miR17/miR20a–FOXE1–PDGFRA) and eight miRNAs (miR-140, miR-17, miR-18a, miR-19a, miR-19b, miR-20a, miR-451a and miR-92a) were discovered in both humans and mice. The role of miR-140, which had the strongest association with CP, was investigated in both human and mouse palate cells. The overexpression of miR-140-5p, but not miR-140-3p, significantly inhibited cell proliferation. We further examined whether miR-140 overexpression could suppress the expression of its predicted target genes (BMP2, FGF9, PAX9 and PDGFRA). Our results indicated that miR-140-5p overexpression suppressed the expression of BMP2 and FGF9 in cultured human palate cells and Fgf9 and Pdgfra in cultured mouse palate cells. In summary, our conserved miRNA–TF–gene regulatory network approach is effective in detecting consensus miRNAs, motifs, and regulatory mechanisms in human and mouse CP.
Keywords: cleft palate, feed-forward loop, miR-140, regulatory motif, regulatory network, transcription factor
Introduction
Cleft palate (CP) is the second most common congenital birth defect with prevalence of approximately 1 in 700 live births worldwide [1]. CP occurs when there is any failure in palate development. The etiology of CP is complicated, with multiple genetic and environmental factors and their interactions. CP significantly affects the quality of life through speech impediments, feeding disorders, frequent ear infections and hearing problems [2]. While various genetic studies have been conducted to identify CP-associated genes [3–5], epigenetic factors including noncoding RNAs [e.g. microRNAs (miRNAs)] and their gene regulatory networks still remain largely unknown.
Transcription of a protein-coding gene is regulated at the transcriptional level [e.g. via transcription factors (TFs)] and posttranscriptional level (e.g. via miRNAs). miRNAs are small noncoding RNA with a length of 21–22 nucleotides. More than 2000 miRNAs have been discovered in humans, many of which have been associated with different physiological and pathological conditions. miRNAs act as repressors of target genes at the posttranscriptional level. Therefore, they can modulate the expression of their target genes in the regulatory networks [6–8]. Several miRNAs have been associated with birth defects. For example, miR-140 plays a critical role in palate morphogenesis through the suppression of Pdgfra in zebrafish [9]. miR-200b regulates the expression of genes encoding Smad2, E-cadherin and Snail during palatogenesis in mice and ectopic miR-200b expression results in increased apoptosis and decreased cell proliferation during palatal fusion [10]. miR-17-92 miRNA cluster regulates expression of T-box transcriptional regulators in craniofacial development [11]. The expression of the miR-17-92 cluster is regulated by BMP signaling and TF AP-2α. miR-17-92 mutant embryos display severe midfacial defects, including mandibular hypoplasia, cleft lip and CP. While several miRNAs have been identified in palate development and CP, miRNA–gene regulatory interactions, along with their corresponding functions, are rarely studied.
A large number of miRNAs are expressed during palate development [12]; however, it is still unclear which miRNAs play a crucial role in CP and their mechanism. A typical gene’s transcription is regulated by TFs. TFs control the transcriptional rate from DNA to mRNA by binding to the TF-binding sites in the promoter region of the target gene [13]. The TFs and miRNAs can be co-regulated under at least three scenarios: (1) expression of miRNAs is regulated by TFs at their promoter regions [14], (2) TFs and miRNAs may mutually regulate each other by forming feedback loops (FBLs), and (3) both miRNAs and TFs may regulate their common target genes together and create feed-forward loops (FFLs).
Network analysis using motif (e.g. miRNA–TF–gene) finding (e.g. FFLs and FBLs) has demonstrated effectiveness in construction of regulatory networks. From these networks, critical regulators (miRNAs and TFs) and their interactions may provide insights into the underlying molecular mechanisms of the disease or cellular condition [15]. For example, miRNA–TF co-regulation, an important FFL unit, has helped researchers identify critical miRNAs, TFs and their regulatory motifs in various diseases, such as schizophrenia [6], glioblastoma multiforme [16], non-small cell lung cancer [17], ovarian cancer [18], colorectal cancer [15] and osteosarcoma [19].
A large number of genetic mutations have been reported in human and mouse CP [20]. In this study, using candidate CP-related miRNAs, TFs and genes, we constructed miRNA–TF co-regulatory networks and performed network analysis to obtain consensus hub nodes, modules and motifs involved in CP. Genes hereafter refer to non-TF non-miRNA genes. Motifs are composed of two or more nodes (TF, miRNA, gene) connected by interactions. ‘Hub’ denotes nodes that are highly connected in the network, reflecting a more critical role in regulation. Through comparison of CP-related miRNA–TF networks between humans and mice, we found that miR-140 might be a conserved regulator in both human and mouse CP. We further experimentally investigated the role of miR-140-5p and miR-140-3p in cell proliferation and downstream gene regulation in cultured human and mouse palate cells. This study provides guidance on construction of a conserved miRNA–TF–gene network from which critical regulators can be identified and their function can be further investigated across organisms.
Results
CP-associated genes, TFs, miRNAs, regulatory pairs and FFLs
Figure 1 summarizes the overall analytical framework. We carefully collected and curated genes associated with CP in humans and mice through our previous study (Supplementary Tables S1 and S2) [21]. There were 131 human genes and 252 mouse genes previously reported in CP. Among them, human CP-associated genes were subcategorized into 25 TF genes and 106 non-TF genes. With extensive literature search, 46 miRNAs, which possibly regulate these CP-related genes, were collected (Supplementary Note S1). Next, we estimated their possible interactions: 184 miRNA–gene interactions, 76 miRNA–TF interactions, 474 TF–gene interactions, and 68 TF–miRNA interactions (Materials and methods; Table 1). Furthermore, these interactions formed three types of motifs (Figure 1C): 126 type I motifs, 80 type II motifs and 17 type III motifs (Table 2; Supplementary Tables S3–S5). In mice, we collected 75 TF genes, 177 non-TF genes and 30 miRNAs that were associated with CP in various mouse studies (Supplementary Note S2) [21]. We estimated interactions among these molecules: 326 miRNA–gene interactions, 176 miRNA–TF interactions, 1773 TF–gene interactions and 150 TF–miRNA interactions (Table 3). The number of the three types of motifs were 807 (type I), 269 (type II) and 108 (type III) (Table 4; Supplementary Tables S6–S8).
Figure 1.
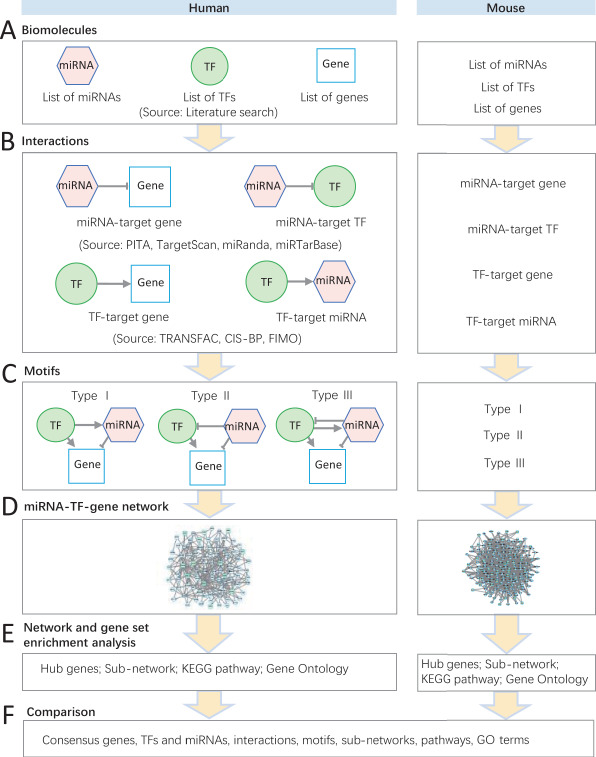
Flowchart of miRNA–TF regulatory network analysis for CP in humans and mice. (A) Three lists of miRNAs, TFs and genes manually curated from literature in humans and mice. (B) Different types of interactions between TFs, miRNAs and genes. (C) Three types of regulatory motifs (type I, type II and type III). (D) The integrated miRNA–TF–gene regulatory network. (E) The hub gene selection, subnetwork identification and gene set enrichment analysis of network genes. (F) The consensus miRNAs/TFs/genes, subnetworks, motifs, pathways and GO terms shared between humans and mice.
Table 1.
Summary of the integrated regulatory relationships among human CP-related miRNAs, TFs and genes
| Relationship | No. of pairs | No. of miRNAs | No. of genes | No. of TFs | Methoda |
|---|---|---|---|---|---|
| miRNA–gene | 184 | 29 | 61 | 0 | A |
| miRNA–TF | 76 | 25 | 0 | 20 | A |
| TF–gene | 474 | 0 | 129 | 18 | B |
| TF–miRNA | 68 | 24 | 0 | 12 | B |
| Total | 802 | 29 | 129 | 24 |
aIn the Method column, A indicates regulation relationships were supported by at least two of the following four databases (miRanda, miRTarBase, PITA and TargetScan), and B indicates FIMO was used for identification of TF–target relationship.
Table 2.
Summary of human three-node FFLs and FBLs
| Type | No. of nodes | No. of pairs | ||||||
|---|---|---|---|---|---|---|---|---|
| Motifs | TFs | miRNAs | Genes | miRNA–gene | miRNA–TF | TF–gene | TF–miRNA | |
| I | 126 | 9 | 17 | 44 | 89 | 0 | 73 | 35 |
| II | 80 | 11 | 22 | 35 | 67 | 32 | 64 | 0 |
| III | 17 | 3 | 4 | 16 | 17 | 134 | 17 | 4 |
| Total | 223 | 24 | 24 | 50 | 117 | 32 | 112 | 35 |
Table 3.
Summary of the integrated regulatory relationships among mouse CP-related miRNAs, TFs and genes
| Relationship | No. of pairs | No. of miRNAs | No. of genes | No. of TFs | Methoda |
|---|---|---|---|---|---|
| miRNA–gene | 326 | 3127 | 113 | 0 | A |
| miRNA–TF | 176 | 26 | 0 | 54 | A |
| TF–gene | 1773 | 0 | 250 | 48 | B |
| TF–miRNA | 150 | 26 | 0 | 28 | B |
| Total | 2425 | 30 | 250 | 66 |
aIn the Method column, A indicates regulation relationships were supported by at least two of the following four databases (miRanda, miRTarBase, PITA and TargetScan), and B indicates FIMO was used for identification of TF–target relationship.
Table 4.
Summary of mouse three-node FFLs and FBLs
| Type | No. of nodes | No. of pairs | ||||||
|---|---|---|---|---|---|---|---|---|
| Motifs | TFs | miRNAs | Genes | miRNA–gene | miRNA–TF | TF–gene | TF–miRNA | |
| I | 807 | 20 | 23 | 109 | 266 | 0 | 428 | 113 |
| II | 269 | 27 | 19 | 78 | 178 | 75 | 173 | 0 |
| III | 108 | 10 | 10 | 59 | 99 | 16 | 83 | 16 |
| Total | 1184 | 33 | 27 | 110 | 301 | 75 | 507 | 113 |
To assess the consensus (i.e. conserved) CP-related molecules and regulation in humans and mice, we only used the shared genes and miRNAs with homologues in both organisms. For miRNAs, we used miRbase [22] to obtain homologues. For genes (both TFs and non-TFs), we used the NCBI HomoloGene database (https://www.ncbi.nlm.nih.gov/homologene, Release 68) [23]. There were eight miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, miR-92a, miR-140 and miR-451a), 14 TF genes (BMP2, FOXE1, FOXF2, GRHL3, IRF6, MSX1, PAX3, PAX9, SOX9, SP8, TBX1, TBX22, TFAP2A and VAX1) and 23 non-TF genes (BMP4, BMP7, COL2A1, FGF10, FGF18, FGF8, FGF9, FGFR1, FGFR2, GABRB3, GAD1, JAG2, MN1, NOG, PDGFRA, PTCH1, ROR2, RYK, SUMO1, TCOF1, TGFB1, TGFB3 and WNT5A) that were identified as CP-related molecules with homologues in humans and mice (Figures 1 and 2A). This data process indicated 28% (37/131) of human CP genes and 14% (37/252) of mouse CP genes could be matched (e.g. evidence in both organisms). We hypothesized that these genes would be more likely to play similar roles in CP across humans and mice, that is, conserved function. The results also indicated that most of the CP-related genes (at least 72%) are not shared between humans and mice. This might be caused by factors such as different types of genetic studies, organism-specific regulation and different human and mouse genomes and gene annotations. Note that the total number of CP-related genes (n = 131) and miRNAs (n = 46) in humans is smaller than that of the genes (n = 252) and miRNAs (n = 30) in mice.
Figure 2.
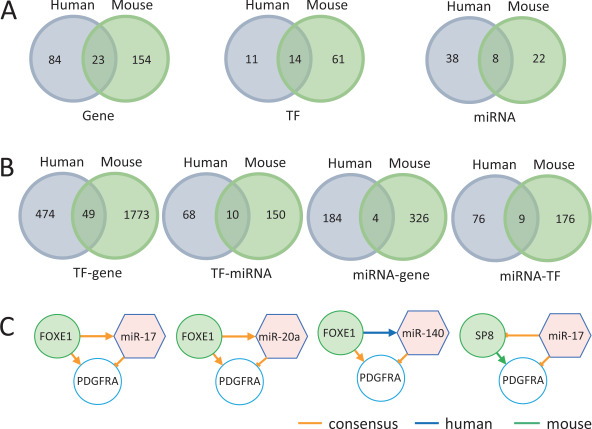
The distribution and intersections of genes, TFs and miRNAs and their pairwise interactions between humans and mice. (A) The distribution and intersections of genes, TFs and miRNAs between humans and mice. (B) The distribution and intersection of TF–gene, TF–miRNA, miRNA–gene and miRNA–TF between humans and mice. (C) The motifs with interactions unique or consensus in humans and mice.
We performed pathway enrichment analysis of the consensus TF and non-TF genes (n = 37) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) [24] annotations. The significantly enriched pathways included mitogen-activated protein kinase (MAPK) (hsa04010, FDR = 1.37 × 10−5; mmu04010, FDR = 6.57 × 10−6), transforming growth factor-β (TGF-β) (hsa04350, FDR = 2.84 × 10−5; mmu04350, FDR = 1.53 × 10−5), Rap1 (hsa04015, FDR = 3.19 × 10−4; mmu04015, FDR = 1.64 × 10−4), Ras (hsa04014, FDR = 4.11 × 10−4; mmu04014, FDR = 2.05 × 10−4), Hippo (hsa04390, FDR = 4.41 × 10−4; mmu04390, FDR = 2.29 × 10−4) and phosphatidylinositol 3′-kinase-Akt (PI3K-Akt) (hsa04151, FDR = 4.72 × 10−4; mmu04151, FDR = 2.29 × 10−4) signaling pathways (Supplementary Tables S9 and S10). Here, FDR refers to the false discovery rate calculated by the Benjamini–Hochberg multiple-test correction procedure [25]. Interestingly, among these enriched signaling pathways, all except one (Rap1 pathway) have been reported in CP previously [1, 10, 26–28]. For Rap1 pathway, a previous study suggested that both RAP1B and RAP1A might interfere with the mitogen-activated protein kinase/extracellular-signal-regulated kinase (MEK/ERK) pathway that causes the Kabuki syndrome. This syndrome may present with CP [29].
We next examined the consensus pairwise interactions within the trimolecule motifs (e.g. TF–gene, TF–miRNA, miRNA–gene and miRNA–TF) between humans and mice. There were 49 consensus TF–gene interactions, 10 consensus TF–miRNA interactions, 4 consensus miRNA–gene interactions and 9 consensus miRNA–TF interactions (Figure 2B; Supplementary Table S11).
We next compared the trimolecule motifs shared between humans and mice. There were two consensus motifs (miRNA–TF–gene): miR17–FOXE1–PDGFRA and miR20a–FOXE1–PDGFRA (Figure 2C). These two motifs had the same TF and gene with same regulatory interactions, but the miRNAs were different. Interestingly, miR-17 and miR-20a share the same seed sequence (AAAGUG) [30] so that both can suppress PDGFRA expression by binding to the target sites in the 3′ untranslated regions. Therefore, these two motifs actually belong to a single motif cluster (miR17/miR20a–FOXE1–PDGFRA). Interestingly, our analytical procedure detected them without having this prior information (i.e. seed sequences). With this scenario, the two miRNAs, miR-17 and miR-20a, could act concurrently to regulate other genes and affect CP development.
Human-specific miRNA–TF–gene regulatory network and its features
We constructed the human-specific miRNA–TF–gene regulatory network by integrating all three types of human motifs (Supplementary Figure S1). This network consisted of 87 nodes (24 miRNAs, 13 TFs and 50 genes) and 296 edges. Using the MCC (maximal clique centrality) [31] score obtained through the cytoHubba plugin (version 0.1), we identified 10 hub nodes from this network, including four miRNAs (miR-27b, MCC = 44; miR-497, MCC = 40; miR-424, MCC = 38; miR-20a, MCC = 28), and six TFs (FOXE1, MCC = 122; SP8, MCC = 74; TFAP2A, MCC = 56; RUNX2, MCC = 32; RARA, MCC = 32; SOX9, MCC = 28). For each hub TF or miRNA, target genes were obtained (Supplementary Table S12), and then, KEGG pathway enrichment analysis of its target genes was done using WebGestalt online tool [32]. The target genes of three hub TFs (FOXE1, SP8 and TFAP2A) and three hub miRNAs (miR-20a, miR-424 and miR-497) were significantly enriched in several pathways that are closely related to the CP-specific biological processes (Figure 3A). The results from functional enrichment analysis of the target genes implied that these hub regulators might play essential roles in CP. For example, the target genes (n = 18) of TF SP8 were found to be significantly enriched in the Rap1 (FDR = 1.87 × 10−4), Ras signaling (FDR = 2.08 × 10−4) and MAPK pathways (FDR = 3.03 × 10−4). SP8 is known to be associated with CP [33]. For miRNAs, target genes of two miRNAs (miR-424 and miR-497) were significantly enriched in the PI3K-Akt signaling pathway (FDR = 8.77 × 10−5 and FDR = 2.32 × 10−6, respectively), Ras signaling pathway (FDR = 1.11 × 10−5 and FDR = 2.20 × 10−7, respectively) and MARK (FDR = 1.80 × 10−5 and FDR = 4.17 × 10−7, respectively) signaling pathway, all important in palate development. The miR-20a targets were enriched in the Rap1 (FDR = 4.90 × 10−2) and Ras (FDR = 4.90 × 10−2) signaling pathways. Three pathways (PI3K-Akt, Ras and MAPK) have been previously reported to be associated with CP [34, 35]. This suggests that these three miRNAs (miR-497, miR-424 and miR-20a) might be new candidates of CP-associated miRNAs.
Figure 3.
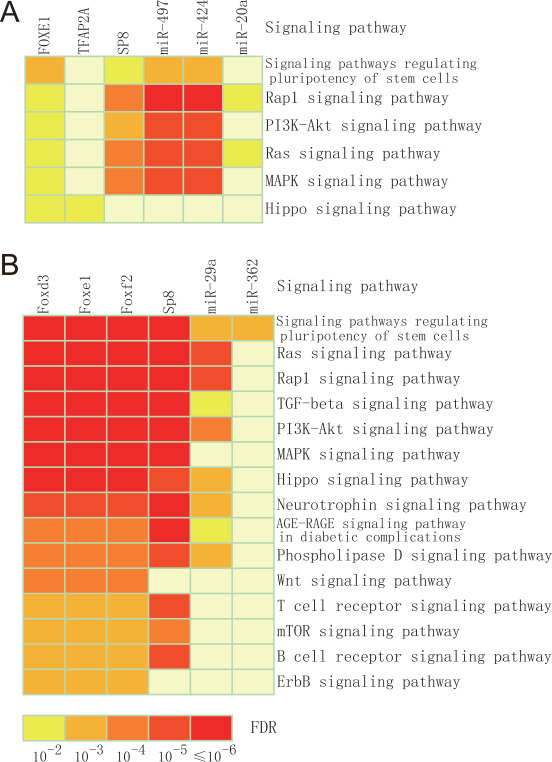
KEGG pathway enrichment analysis. (A) KEGG pathway enrichment analysis of targets of TF and miRNAs that are related to human CP. (B) KEGG pathway enrichment analysis of targets of TF and miRNAs related to mouse CP.
Identification of potential human CP-causing TFs and miRNAs in the Wnt pathway
Previous studies indicate that Wnt pathway plays important roles in CP [36]. For example, mutations in WNT3 are associated with the tetra-amelia syndrome, including CP [1]. A single-nucleotide polymorphism (dbSNP rs7205289) located in pre-miR-140 sequence was attributed to the susceptibility to CP by impacting miR-140 processing [37]. The previous studies have suggested that TFs and miRNAs in the Wnt pathway might be causative regulators of CP [1].
Based on the KEGG pathway database [24], our miRNA–TF–gene regulatory network could recruit five genes in the Wnt pathway: AXIN2, WNT10A, WNT11, WNT3A and WNT5A. We constructed a subnetwork by integrating all motifs that involved the five genes. The subnetwork included 17 nodes (seven miRNAs, five TFs and five genes) and 31 interactions (Figure 4A). FOXE1, RUNX2, TFAP2A, miR-155 and miR-410 were direct upstream regulatory molecules of WNT3A and WNT5A, which have been reported to be involved in midfacial development [38]. The network analysis suggested some of these molecules and interactions might be related to such functions.
Figure 4.
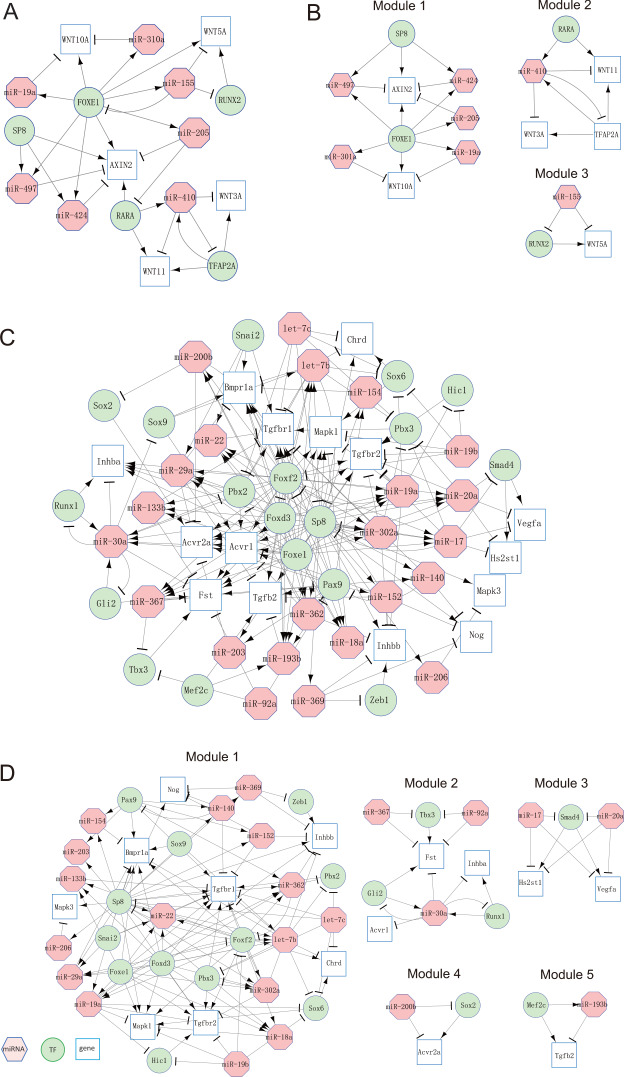
miRNA–TF–gene regulatory network in humans and mice. (A) Network consisting of miRNA–TF–gene motifs related to human CP. (B) Extracted network modules from the miRNA–TF–gene network in humans. (C) Network consisting of miRNA–TF–gene motifs related to mouse CP. (D) Extracted network modules from the miRNA–TF–gene network in mice.
Using the Markov cluster algorithm (MCL) [39] with default setting (inflation = 2.5), we obtained three network modules from the corresponding miRNA–TF–gene regulatory network (Figure 4B). Each module was composed of a set of miRNAs, TFs, and target genes, which were adjacent in terms of network topology. Among these modules, two TFs (SP8 and RARA) and one miRNA (miR-155) were considered the most promising CP nodes. The direction of the network interactions stated that these three regulatory molecules (SP8, RARA, and miR-155) can modulate the others at the transcriptional or posttranscriptional level (Figure 4A). In fibroblasts from individuals with nonsyndromic cleft lip with/without CP, miR-155 is upregulated by more than 2-fold change [40], whereas its downregulation represses RUNX2 expression [41].
Mouse-specific miRNA–TF–gene regulatory network and its features
In parallel with the regulatory networks built in humans, we constructed the mouse-specific miRNA–TF–gene regulatory network by integrating all three types of mouse motifs (types I, II and III) (Supplementary Figure S2). This network consisted of 170 nodes (27 miRNAs, 33 TFs and 110 genes) and 996 interactions (edges). Using MCC scores, 10 hub nodes were extracted, which included six miRNAs (miR-200b, miR-203, miR-29a, miR-30a, miR-362 and miR-367) and four TFs (Foxe1, Foxf2, Foxd3 and Sp8). For each hub TF and miRNA, molecular targets were identified (Supplementary Table S13). Next, we performed KEGG pathway enrichment analysis in these target genes using WebGestalt tool [32]. A set of four hub TFs (Foxd3, Foxe1, Foxf2 and Sp8) and two hub miRNAs (miR-29a and miR-362) were found, of which the corresponding target genes were significantly enriched in various pathways that are closely associated with CP-specific biological processes (Benjamini–Hochberg’s FDR < 0.05) (Figure 3B). The results from functional enrichment of these target genes implied that these hub regulators might play essential roles in mouse CP. The target genes of SP8 were found to be significantly enriched in the PI3K-Akt, Ras, TGF-β, Rap1, MAPK, Hippo and Wnt pathways (FDRs = 3.90 × 10−11, 1.62 × 10−10, 8.62 × 10−10, 3.61 × 10−9, 3.79 × 10−9, 2.35 × 10−5 and 2.67 × 10−3, respectively), whereas SP8 was already identified to be associated with CP in mice [33]. Interestingly, similar features of SP8 were also found in humans.
Targets of miR-29a were enriched in the Ras, TGF-β, PI3K-Akt and Hippo signaling pathways (FDRs =3.21 × 10−5, 1.52 × 10−2, 3.75 × 10−4 and 7.35 × 10−4, respectively). These pathways are important in palate development. The targets of miR-362 were enriched in the signaling pathways regulating pluripotency of stem cells (FDR = 3.52 × 10−3). All of these pathways have been previously reported to be associated with CP [34]. Taken together, our results strongly suggest that miR-29a and miR-362 might play their roles in CP through their regulatory mechanisms.
Identification of potential mouse CP-causing TFs and miRNAs in the TGF-β signaling pathway
The TGF-β signaling pathway plays a critical role in regulating palatogenesis [26]. We investigated miRNAs, TFs and genes in the TGF-β signaling pathway in mouse CP (Figure 4C). We found that five miRNAs (miR-17, miR-200b, miR-20a, miR-367 and miR-92a) directly regulated CP-related genes; therefore, their functions related to CP need to be further investigated. For example, in module 3 (Figure 4D), both miR-17 and miR-20a repressed Smad4 (a consensus CP-related TF) and Hs2st1 and Vegfa (two consensus CP-related genes). Similarly, in module 4, miR-200b repressed Sox2 and Acvr2a (Figure 4D). miR-200b is a key regulator of Snail and Smad2, both of which are important for palatogenesis [10]. miR-17-92-mediated TGFB1 induction suppresses proliferation through the TGF-β signaling pathway [27]. Furthermore, miR-367 inhibits expression of various p63 isoforms (miR-302/367 and miR-203) that are critical for both lip and palate development [42].
Conserved miRNA–TF–gene regulatory network in human and mouse CP
In our comparison of human miRNA–TF–gene regulatory network with that of mice, we obtained a consensus (overlapped) subnetwork (Figure 5). This overlapped network consisted of 8 miRNAs, 7 TFs and 27 genes. These miRNAs, TFs and genes would be high priority for further study.
Figure 5.
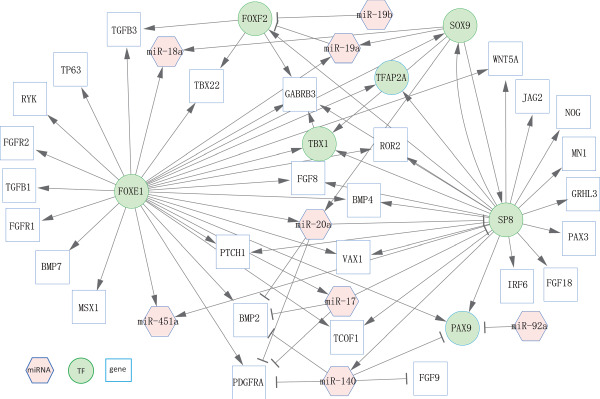
The consensus subnetwork between humans and mice. This subnetwork was extracted from the whole human miRNA–TF–gene regulatory network and mouse miRNA–TF–gene regulatory network.
Among the eight miRNAs in the overlapped subnetwork, miR-140 contained the highest outdegree (4) and one indegree (outdegree and indegree refer to the edges with direction in the network). miR-140 regulated three genes (BMP2, FGF9 and PDGFRA) and one TF (PAX9). miR-20a had the second highest outdegree (3). It regulated two target genes (BMP2 and PDGFRA) and one TF (SP8). miR-17 had an outdegree of 3—it regulated two genes (BMP2 and PDGFRA) and one TF (SP8). Interestingly, two TFs (FOXE1 and SP8) had the strongest interactions with other targets in the consensus subnetwork (degree = 28 and 21, respectively). Importantly, FOXE1 regulated several miRNAs (miR-17, miR-18a, miR-19a, miR-20a and miR-451a), while SP8 regulated miR-140 in the network (Figure 5).
We conducted Gene Ontology (GO) term enrichment analysis of TFs and genes in the consensus subnetwork. Our analysis revealed that six of top 10 GO biological process terms were related to palate development or organ morphogenesis. These six terms are face morphogenesis (GO:0060325, FDR = 7.57 × 10−10), palate development (GO:00600215, FDR = 2.57 × 10−9), organ morphogenesis (GO:00098875, FDR = 1.97 × 10−8), embryonic limb morphogenesis (GO:00303265, FDR = 2.18 × 10−8), inner ear morphogenesis (GO:00424725, FDR = 5.75 × 10−8) and multicellular organism development (GO:00072755, FDR = 6.85 × 10−8) (Supplementary Table S14).
The regulatory mechanism of miR-140 in human and mouse palate cells
We predicted the regulatory mechanism of miR-140 in CP (Figure 6A), according to our networks (Figure 5) and published literature (for details, see Discussion). miR-140 might be involved in palate development through the regulation of various protein-coding genes: BMP2, FGF9, PAX9 and PDGFRA.
Figure 6.
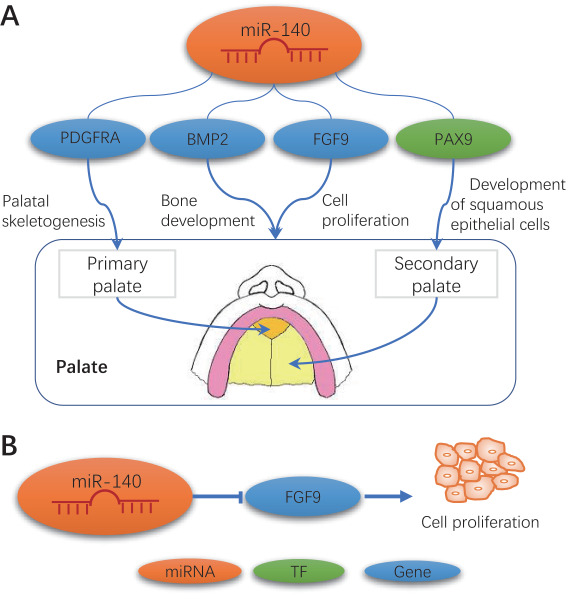
A proposed regulation mechanism of miR-140 involving in humans and mice. (A) miR-140 regulates genes related to CP or palate development. (B) miR-140 mediates cell proliferation by regulating FGF9.
To validate the role of miR-140-3p and miR-140-5p in cell proliferation, cell proliferation assays with either miR-140-3p or miR-140-5p mimic were conducted in cultured human and mouse palate cells. miR-140-5p, but not miR-140-3p, suppressed cell proliferation in both human and mouse palate cells (Figure 7). To investigate the anticorrelational gene regulation by miR-140-3p or miR-140-5p, quantitative real-time PCR (RT-PCR) analyses were conducted for the predicted target genes (BMP2, FGF9, PAX9 and PDGFRA) after treatment with either miR140-3p or miR-140-5p mimic in human and mouse palate cells (Figure 8). As expected, treatment with miR-140-3p mimic failed to suppress the expression of target genes except PAX9 in human palate cells. By contrast, miR-140-5p mimic suppressed the expression of BMP2 and FGF9 in human palate cells and Fgf9 and Pdgfra in mouse palate cells.
Figure 7.
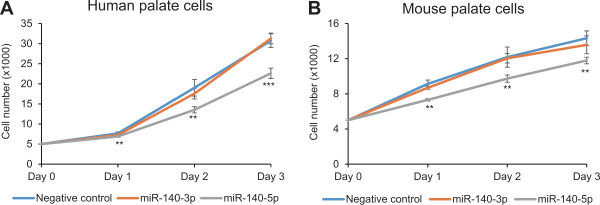
Effect of miR-140-3p and miR-140-5p on cell proliferation in human and mouse palate cells. (A) Cell proliferation assay of human palate cells with miR-140-3p, miR-140-5p and control mimics. **p < 0.01, ***p < 0.001. (B) Cell proliferation assay of mouse palate cells with miR-140-3p, miR-140-5p and control mimic. **p < 0.01.
Figure 8.
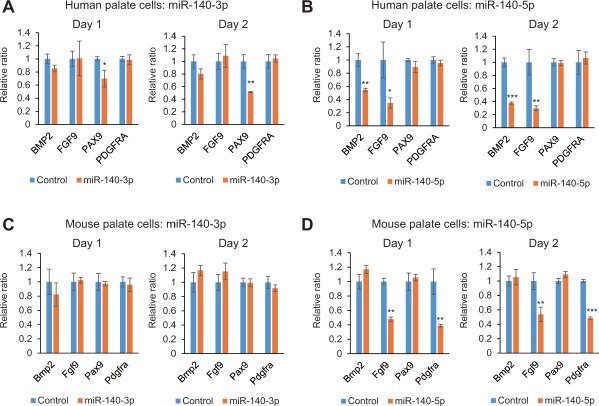
Downstream target gene regulation by miR-140-3p and miR-140-5p in cultured human and mouse palate cells. (A–D) Quantitative RT-PCR analyses of the indicated genes after treatment with miR-140-3p (A and C) or miR-140-5p (B and D) in human and mouse palate cells. *p < 0.05, **p < 0.01, ***p < 0.001.
Our previous study indicated that the FGF9–PITX2 signaling cascade regulates cranial neural crest cell proliferation during palate formation [28]. In this study, we found that miR-140-5p inhibited cell proliferation through the suppression of FGF9 in cultured palate cells (Figure 6B).
Discussion
In this study, we constructed miRNA–TF–gene networks using potential CP-related TFs, genes and miRNAs, which were manually curated. Conserved networks or subnetworks were extracted from human and mouse CP-related networks. Based on analysis of such conserved networks, eight miRNAs and two consensus regulatory loops were identified that might play critical roles in CP etiology. The potential miR-140 regulatory mechanism in CP was further investigated. Our follow-up bioinformatics analyses and wet lab experiments supported that such a conserved miRNA–TF–gene network approach is powerful in detecting critical miRNAs, motifs and regulatory mechanisms that are shared between human and mouse CP. Therefore, this work provides a general procedure for the construction of a conserved miRNA–TF–gene regulatory network, which can be applied to other disease or phenotype.
Both miRNAs and TFs are important regulators, and each of them may regulate many target genes in cellular systems. To better reflect the specific regulation in the complex regulatory network, we defined three types of regulatory motifs. After extracting the interactions among miRNAs, TFs and genes, we obtained motifs in humans and mice (Tables 2 and 4). Interestingly, three consensus type I motifs (miR17–FOXE1–PDGFRA, miR20a–FOXE1–PDGFRA and miR140–FOXE1–PDGFRA) and one consensus type II motif (miR17–SP8–PDGFRA) were commonly highlighted through the comparison between human and mouse regulatory networks (Figure 2C). These consensus motifs in different species are more likely to be functional in CP. Although the number of motifs in humans and mice are significantly different, our analysis revealed several consensus signaling pathways (Supplementary Figure S3).
Conservation can be measured in several ways, leading to different results. To control the false positive rate, we used stringent criteria to extract conserved genes, TFs, miRNAs and motifs from the whole regulatory networks. First, the homologous protein-coding genes and TFs between humans and mice were extracted from the most recent version of NCBI HomoloGene database (https://www.ncbi.nlm.nih.gov/homologene, Release 68). Guided by a tree built from sequence similarity, HomoloGene uses a Blastp program to compare sequences and the matched proteins from input organisms and to restrict distance metrics to keep unlikely orthologs from being organized together. Second, for miRNAs, we used miRbase to obtain homologues [22]. miRbase assigned the same number to homologous miRNA loci in different species (i.e. has-miR-140 and mmu-miR-140 are homologous) [22]. Finally, we extracted the motifs that have the same homologous genes, TFs and miRNAs from both humans and mice and considered them as conserved motifs.
Based on both human and mouse analyses, miR-140, miR-17 and miR-20a appeared to be quite relevant, and all had reasonably high centrality in the conserved subnetwork (Figure 5). The roles of miR-17 and miR-20a in CP or palate development have already been investigated in a previous study [11]. In that study, the authors discovered that a deletion of the miR-17-92 cluster (encoding miR-17, miR-18a, miR-19a/b, miR-20a and miR-92a) in mice resulted in craniofacial malformations including CP. Moreover, their work demonstrated that the miR-17-92 cluster directly regulated T-box factors, which have critical roles during craniofacial development. Therefore, we hypothesized that the third miRNA, miR-140, might play critical roles in CP. We performed experiments to test this hypothesis, and our results supported this hypothesis.
We provided a comparative study of the miRNA–TF–gene regulatory network between humans and mice and identified the overlapped subnetwork consisting of 8 miRNAs, 7 TFs and 27 genes. These miRNAs, TFs and genes might act as the bridge components for linking human and mouse CP. Among the eight miRNAs belonging to the overlapped subnetwork, miR-140 is the miRNA that had the largest outdegree. miR-140 regulated four nodes in the subnetwork, including three target genes (BMP2, FGF9 and PDGFRA) and one TF (PAX9).
In zebrafish, the function of miR-140 in CP has been verified. miR-140 overexpression in zebrafish causes a cleft between lateral elements of the ethmoid plate that is a structural analogue of the amniote palate found in the higher vertebrates, while downregulation produces an abnormal shape of the palate [9]. Such function can only be inferred in humans and mice, but it has not been confirmed by experiments yet. Recent genetic studies have shown that miR-140 might also be associated with the etiology of CP in humans [37]. First, a genetic association study illustrated that a single-nucleotide polymorphism (rs7205289:C > A) in a precursor of miR-140 (pre-miR-140) contributed to the susceptibility of nonsyndromic CP by affecting the processing of miR-140. An allele of rs7205289, having higher frequency in the patients, was associated with a decrease of miR-140-5p expression as well as an increase of miR-140-3p expression. In mice, miR-140 regulates cartilage development and homeostasis [43]. In this respect, it is very interesting to know that miR-140 null mice exhibit shorter palatal bones (submucous CP) [43].
Based on our consensus networks and published studies [9, 44–49], we deduced the regulatory mechanism of miR-140 in CP (Figure 6A). miR-140 might be involved in palate development through the regulation of various protein-coding genes: BMP2, FGF9, PAX9 and PDGFRA. The interaction between miR-140-5p and PDGFRA is conserved across species [9], it has been observed that miR-140 targets PDGFRA in palatal skeletogenesis in zebrafish [9]. BMP2 expression is identified in both the epithelium and mesenchyme of the anterior region of developing palatal shelves [49]. miR-140-5p regulates bone development by inhibiting BMP2 expression [48]. FGF9 has been identified as a direct target for miR-140-5p [44]. miR-140-5p suppresses cancer cell proliferation, migration as well as invasion through IGF1R in non-small cell lung cancer, along with targeting two genes, TGFBR1 and FGF9 [45].
In mice, nonkeratinized squamous epithelium shields the soft palate, ventral surface of the tongue, inner cheeks, the floor of mouth and inner lips. Keratinized squamous epithelium is found in the gingiva and hard palate as well as areas of the dorsal surface of the tongue. Pax9 promotes development of squamous epithelial cells [46]. An indispensable role of Pax9 in the organs originated from neural crest cells was demonstrated by the absence of teeth as well as cleft secondary palate in Pax9-deficient mice [50]. Bmp2 has been validated as a direct miR-140 target through luciferase assays in 3T3 cells [47, 48]. Fgf9 has been identified as a direct target of miR-140-5p [44].
While miR-140-3p failed to inhibit cell proliferation in both human and mouse palate cells, PAX9 expression was significantly reduced in human palate cells by 30% on day 1 and 50% on day 2 (Figure 8). It suggests that 50% reduction of a single gene may not be sufficient to inhibit cell proliferation. Indeed, mice with heterozygous deletion of Fgf9 show normal development. By contrast, treatment with miR-140-5p mimic suppressed expression of two genes (BMP2 and FGF9 in humans and Fgf9 and Pdgfra in mice) by more than 50% each. Although these double knockout mice have not been studied yet, these multiple mutations may cause CP.
This study has several limitations. (1) Incomplete and highly heterogenous data. We used the most available CP-related set of molecules to build networks, but this set was not complete. To explore the interaction relationships among these molecules, we used highly heterogenous data. (2) Prediction of miRNA and TF targets may include false positive or negative results. Although we used stringent criterion to come up with the results, some of interactions might still be false. (3) Validation. It was hard to verify all regulatory networks. Due to the scope of this work, we only validated the regulation of miR-140. However, our analysis is the first to systematically examine the miRNA–TF–gene regulation in CP by a comparative regulatory network approach and using the available data in both humans and mice.
Materials and methods
Manual curation of human and mouse CP genes
The list of CP genes was acquired through systematic literature review using three databases, Medline (Ovid), PubMed (NLM) and Embase (Ovid), with the inclusion criteria being a description of genes significantly associated with CP reported as an original research article in the English language. Articles that did not describe genetic mutations or were not an original research article were excluded. Scopus (Elsevier) and Mouse Genome Informatics databases were used to retrieve any genes missed in the systematic review. The details of methods and results from systematic review for mouse CP genes were reported by Suzuki et al. [21], and similar strategies were applied to human gene curation [70]. After careful manual curation, a list of 131 human genes (25 TFs and 106 non-TF genes) and another list of 252 mouse genes (75 TFs and 177 non-TF genes) were obtained. These genes had either mutations or association reported in individuals or mouse strains with CP and were considered as candidate genes for human or mouse CP.
Manual curation of human and mouse miRNAs
Forty-six human miRNAs were collected from eight articles [11, 12, 37, 40, 51–54]. The detailed description of the sources for individual miRNA is provided as Supplementary Note S1. Similarly, 30 mouse miRNAs were collected from eight articles [10, 11, 42, 43, 55–57] (Supplementary Note S2; Supplementary Figures S4 and S5). One of these three following conditions was used to consider miRNAs associated with CP: (1) the miRNA targets CP-associated genes (obtained from genome-wide association studies), (2) differentially expressed miRNAs between CP and control samples and (3) differentially expressed miRNAs in different developmental stages in the palates.
Identification of miRNA–TF and miRNA–gene interactions
The possible target genes of miRNAs were downloaded from four target prediction databases: PITA (Version 6), miRanda (Release August 2010), miRTarBase (Release 7.0) and TargetScan (Release 7.1) [58–61]. miRNA–target pairs that existed in at least two canonical miRNA–target databases above were retained for further analysis.
Identification of TF–target interactions
The promoter sequences (−1000 to +200 bp of the transcription start site) of human and mouse GENCODE protein-coding genes as well as precursor miRNAs (human, wgEncodeGencodeBasic V27; mouse, wgEncodeGencodeBasic VM16, respectively) were collected from the UCSC Table Browser [62] as described by Sun et al. [16]. Meanwhile, TF motifs were identified from TRANSFAC® Professional version (Release 2016.1) [63]. For each TF, we chose the TF motifs having the highest informative content as well as the TF motifs containing directed/inferred evidence obtained from the Catalog of Inferred Sequence Binding Preferences [64]. The position weight matrix of these TF motifs was mapped to the promoter sequences using Find Individual Motif Occurrences program (FIMO) [65]. The significant hits having a P-value of <1 × 10−5 that occurred within the promoter of GENCODE genes were retained.
Identification of motifs (FFLs and FBLs)
After obtaining the overall interactions, three types of motifs were extracted by using our computer program written in C (MotifPredictor, publicly available at https://www.uth.edu/bioinfo/software.htm and https://github.com/emanlee/MotifPredictor). CoMoFinder [66] was also used, and it generated essentially the same results as the MotifPredictor (Supplementary Figure S6). Among those three motifs, two were FFLs (types I and II), and the remaining one was a FBL (type III) (Figure 1C). In a type I motif, the TF regulates its targeted miRNA as well as targeted protein-coding gene (non-TF gene) at the transcriptional level, whereas the employed miRNA regulates the targeted protein-coding gene at the posttranscriptional level. In a type II motif, the TF regulates its targeted protein-coding gene at the transcriptional level, while the miRNA regulates its targeted protein-coding gene and the TF at the posttranscriptional level. Finally, in a type III motif, both the TF and miRNA regulate their consensus targeted protein-coding gene, whereas the miRNA and TF mutually regulate each other.
Regulatory network and identifying overlapped FFLs
A regulatory network was built by integrating all the motifs obtained from the previous analysis for humans and for mice, respectively. Next, the two networks were integrated, and then, a consensus subnetwork was selected. Network visualization and analysis was by Cytoscape software version 3.6 (http://cytoscape.org/) [67].
Network analysis
After constructing the network, centrality measure and MCC were applied through the cytoHubba plugin (version 0.1) implemented in Cytoscape (version 3.6) [31]. A robust and highly efficient network clustering algorithm, MCL [39], was applied for extracting modules from the miRNA–TF–gene regulatory networks.
KEGG pathway and GO term enrichment analysis
To find the pathways enriched in the target genes and TFs, WebGestalt webtool [32] was used for gene set enrichment analysis with KEGG pathway annotations. We also used the DAVID online tool for GO term annotations [68].
Cell proliferation assay
Human embryonic palatal mesenchymal (HEPM) cells were obtained from the American Type Culture Collection (CRL-1486), and cultured in minimum essential medium Eagle-α modification supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and l-glutamine. Primary mouse embryonic palatal mesenchymal (MEPM) cells were obtained at embryonic day (E) E13.5 and cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% FBS, penicillin/streptomycin, l-glutamine, nonessential amino acid and β-mercaptoethanol. Cells were plated in 96-well cell culture plates at a density of 5000 per well and treated with a mimic for negative control, miR-140-3p and miR140-5p (mirVana miRNA mimic, ThermoFischer Scientific) using the TransIT-X2 system (Mirus Bio LLC, Madison, WI) for HEPM cells and lipofectamine RNAiMAX transfection reagent (ThermoFischer Scientific) for MEPM cells, according to the manufacturer’s protocol (3 pmol of mimic and 0.3 μl of transfection reagent in 100 μl each medium per well), respectively. Cell proliferation was determined using the cell counting kit 8 (Dojindo Molecular Technology, Gaitherburg, MD) (n = 6 per group), as previously described [69].
Quantitative RT-PCR
Total RNAs were isolated from cultured HEPM and MEPM cells treated with a mimic for target miRNAs for 1 and 2 days (n = 3 per group) using the QIAshredder and RNeasy mini extraction kit (QIAGEN), as previously described [69]. The PCR primers used are listed in Supplementary Table S15.
Statistical analysis
Statistical significance of the differences between two groups was estimated using a two-tailed Student’s t-test. Multiple comparisons were made via one-way analysis of variance with Tukey–Kramer post hoc test. A P-value of <0.05 was considered statistically significant. For all groups, data are represented as mean ± SD.
Key Points
We designed a conserved miRNA–TF–gene network approach to detecting consensus miRNAs, TFs and regulatory motifs in complex disease or phenotype across organisms.
We pinpointed eight miRNAs (miR-140, miR-17, miR-18a, miR-19a, miR-19b, miR-20a, miR-451a and miR-92a) having strong association with CP in both humans and mice.
We identified one consensus regulatory motif (miR17/miR20a–FOXE1–PDGFRA) in human and mouse CP.
miR-140-5p overexpression inhibited cell proliferation through the suppression of predicted target genes in cultured human and mouse palate cells.
Supplementary Material
Acknowledgments
We thank Musi Zhang for the technical assistance and the members in Bioinformatics and Systems Medicine Laboratory for the useful discussion.
Aimin Li is an assistant professor in the Shaanxi Key Laboratory for Network Computing and Security Technology, School of Computer Science and Engineering, Xi’an University of Technology, Xi’an, Shaanxi, 710048, China. He is also a visiting scientist in Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston.
Peilin Jia is an assistant professor in the Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston. She co-directs the Bioinformatics and Systems Medicine Laboratory.
Saurav Mallik is a bioinformatics post-doctoral fellow in the Bioinformatics and Systems Medicine Laboratory, Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston.
Rong Fei is an associate professor in the Shaanxi Key Laboratory for Network Computing and Security Technology, School of Computer Science and Engineering, Xi’an University of Technology, China.
Hiroki Yoshioka is a post-doctoral fellow in the Department of Diagnostic & Biomedical Sciences, School of Dentistry, The University of Texas Health Science Center at Houston.
Akiko Suzuki is a senior research associate in the Department of Diagnostic & Biomedical Sciences, School of Dentistry, The University of Texas Health Science Center at Houston.
Junichi Iwata is an associate professor in the Department of Diagnostic & Biomedical Sciences, School of Dentistry, The University of Texas Health Science Center at Houston.
Zhongming Zhao is a professor in the Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston. He directs the Bioinformatics and Systems Medicine Laboratory.
Funding
This work was partially supported by National Institutes of Health grants [R01LM012806 (to Z.Z.), R01DE026767 (to J.I.), R03DE026208 (to J.I.), R03DE026509 (to J.I.), R03DE028340 (to J.I.), R03 DE027393 (to Z.Z.), R03DE028103 (to Z.Z.) and R03DE027711 (to P.J.)]. Publication charges for this article have been funded by authors’ academic fund. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Dixon MJ, Marazita ML, Beaty TH, et al. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 2011;12(3):167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mossey PA, Little J, Munger RG, et al. Cleft lip and palate. The Lancet 2009;374(9703):1773–85. [DOI] [PubMed] [Google Scholar]
- 3. Yu Y, Zuo X, He M, et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat Commun 2017;8:14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cai Y, Patterson KE, Reinier F, et al. Copy number changes identified using whole exome sequencing in nonsyndromic cleft lip and palate in a Honduran population. Birth Defects Res 2017;109(16):1257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heron EA, Connolly S. Review of statistical methodologies for the detection of parent-of-origin effects in family trio genome-wide association data with binary disease traits. Brief Bioinform 2014;16(3):429–48. [DOI] [PubMed] [Google Scholar]
- 6. Guo A-Y, Sun J, Jia P, et al. A novel microRNA and transcription factor mediated regulatory network in schizophrenia. BMC Syst Biol 2010;4(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang W, Mitra R, Lin C-C, et al. Systematic dissection of dysregulated transcription factor–miRNA feed-forward loops across tumor types. Brief Bioinform 2015;17(6):996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li A, Qin G, Suzuki A, et al. Network-based identification of critical regulators as putative drivers of human cleft lip. BMC Med Genomics 2019;12(s1):119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eberhart JK, He X, Swartz ME, et al. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet 2008;40(3):290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shin J-O, Lee J-M, Cho K-W, et al. MiR-200b is involved in Tgf-β signaling to regulate mammalian palate development. Histochem Cell Biol 2012;137(1):67–78. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Bai Y, Li H, et al. MicroRNA-17-92, a direct Ap-2α transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet 2013;9(9):e1003785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S, Sun C, Meng Y, et al. A pilot study: screening target miRNAs in tissue of nonsyndromic cleft lip with or without cleft palate. Exp Ther Med 2017;13(5):2570–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 2012;13(9):613–26. [DOI] [PubMed] [Google Scholar]
- 14. Somel M, Guo S, Fu N, et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res 2010;20(9):1207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang H, Luo J, Liu C, et al. Investigating microRNA and transcription factor co-regulatory networks in colorectal cancer. BMC Bioinformatics 2017;18(1):388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun J, Gong X, Purow B, et al. Uncovering microRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput Biol 2012;8(7):e1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitra R, Edmonds MD, Sun J, et al. Reproducible combinatorial regulatory networks elucidate novel oncogenic microRNAs in non-small cell lung cancer. RNA 2014;20(9):1356–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao M, Sun J, Zhao Z. Synergetic regulatory networks mediated by oncogene-driven microRNAs and transcription factors in serous ovarian cancer. Mol Biosyst 2013;9(12):3187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Poos K, Smida J, Nathrath M, et al. How microRNA and transcription factor co-regulatory networks affect osteosarcoma cell proliferation. PLoS Comput Biol 2013;9(8):e1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki A, Sangani DR, Ansari A, et al. Molecular mechanisms of midfacial developmental defects. Dev Dyn 2016;245(3):276–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki A, Abdallah N, Gajera M, et al. Genes and microRNAs associated with mouse cleft palate: A systematic review and bioinformatics analysis. Mech Dev 2018;150:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2013;42(D1):D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sayers EW, Agarwala R, Bolton EE, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res 2019;47(D1):D23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2011;40(D1):D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995;57:289–300. [Google Scholar]
- 26. Xu X, Han J, Ito Y, et al. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-β/BMP signaling during tooth and palate development. Dev Cell 2008;15(2):322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L, Shi JY, Zhu GQ, et al. MiR-17-92 cluster regulates cell proliferation and collagen synthesis by targeting TGFB pathway in mouse palatal mesenchymal cells. J Cell Biochem 2012;113(4):1235–44. [DOI] [PubMed] [Google Scholar]
- 28. Iwata J, Tung L, Urata M, et al. Fibroblast growth factor 9 (FGF9)-pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor β (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. J Biol Chem 2012;287(4):2353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bögershausen N, Tsai I-C, Pohl E, et al. RAP1-mediated MEK/ERK pathway defects in kabuki syndrome. J Clin Invest 2015;125(9):3585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008;133(2):217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chin C-H, Chen S-H, Wu H-H, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014;8(4):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Vasaikar S, Shi Z, et al. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017;45(W1):W130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kasberg AD, Brunskill EW, Potter SS. SP8 regulates signaling centers during craniofacial development. Dev Biol 2013;381(2):312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu M, Xie A. Selection of key genes and regulators associated with Treacher Collins syndrome based on expression profiling analysis. Int J Clin Exp Med 2017;10(5):8481–91. [Google Scholar]
- 35. Suzuki S, Marazita ML, Cooper ME, et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. The American Journal of Human Genetics 2009;84(3):406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuan G, Singh G, Chen S, et al. Cleft palate and Aglossia result from perturbations in Wnt and hedgehog signaling. Cleft Palate Craniofac J 2017;54(3):269–80. [DOI] [PubMed] [Google Scholar]
- 37. Li L, Meng T, Jia Z, et al. Single nucleotide polymorphism associated with nonsyndromic cleft palate influences the processing of miR-140. Am J Med Genet A 2010;152(4):856–62. [DOI] [PubMed] [Google Scholar]
- 38. Chiquet BT, Blanton SH, Burt A, et al. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum Mol Genet 2008;17(14):2212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 2002;30(7):1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schoen C, Glennon JC, Abghari S, et al. Differential microRNA expression in cultured palatal fibroblasts from infants with cleft palate and controls. Eur J Orthod 2017;40(1):90–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Komori T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 2010;339(1):189–95. [DOI] [PubMed] [Google Scholar]
- 42. Warner DR, Mukhopadhyay P, Brock G, et al. Micro RNA expression profiling of the developing murine upper lip. Dev Growth Differ 2014;56(6):434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyaki S, Sato T, Inoue A, et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev 2010;24(11):1173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang H, Fang F, Chang R, et al. MicroRNA-140-5p suppresses tumor growth and metastasis by targeting transforming growth factor β receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology 2013;58(1):205–17. [DOI] [PubMed] [Google Scholar]
- 45. Kai Y, Peng W, Ling W, et al. Reciprocal effects between microRNA-140-5p and ADAM10 suppress migration and invasion of human tongue cancer cells. Biochem Biophys Res Commun 2014;448(3):308–14. [DOI] [PubMed] [Google Scholar]
- 46. Bobryshev YV, Orekhov AN, Chistiakov DA. MicroRNAs in esophageal adenocarcinoma: functional significance and potential for the development of new molecular disease markers. Curr Pharm Des 2015;21(23):3402–16. [DOI] [PubMed] [Google Scholar]
- 47. Nicolas FE, Pais H, Schwach F, et al. mRNA expression profiling reveals conserved and non-conserved miR-140 targets. RNA Biol 2011;8(4):607–15. [DOI] [PubMed] [Google Scholar]
- 48. Hwang S, Park S-K, Lee HY, et al. miR-140-5p suppresses BMP2-mediated osteogenesis in undifferentiated human mesenchymal stem cells. FEBS Lett 2014;588(17):2957–63. [DOI] [PubMed] [Google Scholar]
- 49. Zhang Z, Song Y, Zhao X, et al. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002;129(17):4135–46. [DOI] [PubMed] [Google Scholar]
- 50. Peters H, Neubüser A, Kratochwil K, et al. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 1998;12(17):2735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rattanasopha S, Tongkobpetch S, Srichomthong C, et al. PDGFRa mutations in humans with isolated cleft palate. Eur J Hum Genet 2012;20(10):1058–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li D, Zhang H, Ma L, et al. Associations between microRNA binding site SNPs in FGFs and FGFRs and the risk of non-syndromic orofacial cleft. Sci Rep 2016;6: 31054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J, Zou J, Li Q, et al. Assessment of differentially expressed plasma microRNAs in nonsyndromic cleft palate and nonsyndromic cleft lip with cleft palate. Oncotarget 2016;7(52): 86266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma L, Xu M, Li D, et al. A miRNA-binding-site SNP of MSX1 is associated with NSOC susceptibility. J Dent Res 2014;93(6):559–64. [DOI] [PubMed] [Google Scholar]
- 55. Mukhopadhyay P, Brock G, Pihur V, et al. Developmental microRNA expression profiling of murine embryonic orofacial tissue. Birth Defects Res A Clin Mol Teratol 2010;88(7):511–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cao H, Yu W, Li X, et al. A new plasmid-based microRNA inhibitor system that inhibits microRNA families in transgenic mice and cells: a potential new therapeutic reagent. Gene Ther 2016;23(6):527–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ries R, Yu W, Holton N, et al. Inhibition of the miR-17-92 cluster separates stages of palatogenesis. J Dent Res 2017;96(11):1257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chou C-H, Chang N-W, Shrestha S, et al. miRTarBase 2016: updates to the experimentally validated miRNA–target interactions database. Nucleic Acids Res 2015;44(D1):D239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. John B, Enright AJ, Aravin A, et al. Human microRNA targets. PLoS Biol 2004;2(11):e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kertesz M, Iovino N, Unnerstall U, et al. The role of site accessibility in microRNA target recognition. Nat Genet 2007;39(10):1278–84. [DOI] [PubMed] [Google Scholar]
- 61. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120(1):15–20. [DOI] [PubMed] [Google Scholar]
- 62. Casper J, Zweig AS, Villarreal C, et al. The UCSC genome browser database: 2018 update. Nucleic Acids Res 2017;46(D1):D762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matys V, Kel-Margoulis OV, Fricke E, et al. TRANSFAC® and its module TRANSCompel®: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 2006;34(suppl_1):D108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Weirauch MT, Yang A, Albu M, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014;158(6):1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 2011;27(7):1017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liang C, Li Y, Luo J, et al. A novel motif-discovery algorithm to identify co-regulatory motifs in large transcription factor and microRNA co-regulatory networks in human. Bioinformatics 2015;31(14):2348–55. [DOI] [PubMed] [Google Scholar]
- 67. Saito R, Smoot ME, Ono K, et al. A travel guide to Cytoscape plugins. Nat Methods 2012;9(11):1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008;4(1):44. [DOI] [PubMed] [Google Scholar]
- 69. Suzuki A, Pelikan RC, Iwata J. WNT/β-catenin signaling regulates multiple steps of myogenesis by regulating step-specific targets. Mol Cell Biol 2015;35(10):1763–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Suzuki A, Li A, Gajera M, Abdallah N, Zhang Z, Zhao Z, Iwata J. MicroRNA-374a, -4680, and -133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Medical Genomics 2019;12(1):93–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.