

Abstract
Direct enantioselective α-alkylation of 2-alkylpyridines provides access to chiral pyridines via an operationally simple protocol that obviates the need for prefunctionalization or preactivation of the substrate. The alkylation is accomplished using chiral lithium amides as noncovalent stereodirecting auxiliaries. Crystallographic and solution NMR studies provide insight into the structure of well-defined chiral aggregates in which a lithium amide reagent directs asymmetric alkylation.
Enantioselective alkylation is a fundamentally important transformation in organic synthesis. For enolate-based carbanions, asymmetric alkylations have been historically achieved using covalently attached chiral auxiliaries,1–4 and these reactions have extensive applications in the synthesis of natural products and pharmaceuticals on scales spanning several orders of magnitude.5,6 However, this strategy is not readily applicable to carbanions derived from noncarbonyl precursors. Building on discoveries by Shioiri7 and subsequently Koga,8 we previously demonstrated that chiral lithium amides (CLAs) function as noncovalent stereodirecting reagents enabling highly effective enantioselective transformations of dianionic enediolates derived directly from carboxylic acids.9–11 The asymmetric alkylation is achieved by virtue of mixed aggregation12 between the enediolate and CLA, providing the chiral environment for subsequent functionalization (Scheme 1).13 CLAs derived from amines shown in Scheme 1 produce mixed aggregates with a broad range of organolithium reagents with a remarkably conserved threedimensional architecture.14 Such well-defined aggregation suggests that CLAs can potentially direct the stereochemistry of alkylations for nonenolate monoanionic carbanions for which no covalent chiral auxiliary is feasible.15,16
Scheme 1.
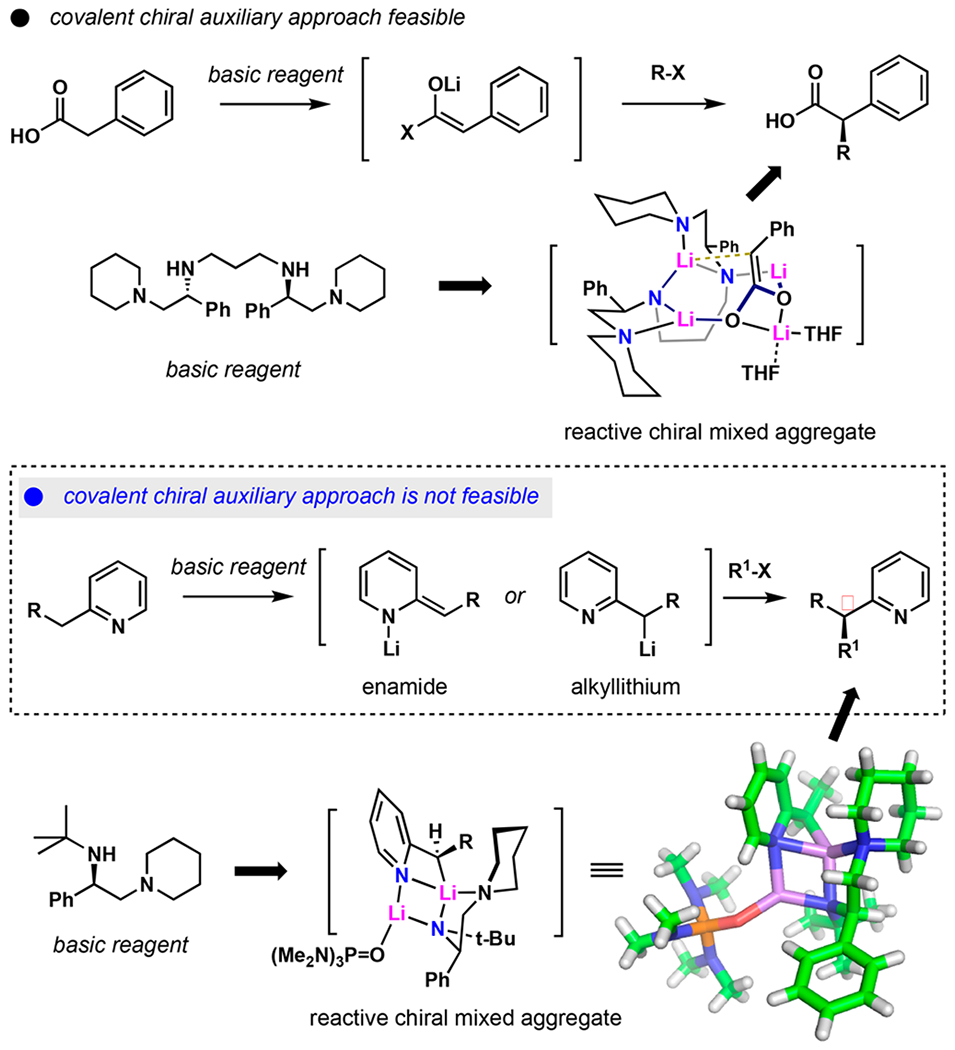
Chiral Lithium Amides in Enolate Alkylation versus Alkylpyridine Alkylation
We became interested in α-alkylation of 2-alkylpyridines for an initial examination of this approach. Pyridines bearing C2-alkyl substituents are privileged ligands in asymmetric catalysis; however, their preparation often involves cumbersome multistep synthesis. Moreover, pyridines are the second most frequently occurring nitrogen-containing heterocycles in pharmaceuticals, with C-2 substitution appearing in more than 60%.17 While several approaches have been developed to access chiral C2-substituted pyridines in some form,18–25 each has its limitations in substrate scope profile, and no direct enantioselective alkylation has been reported.
Herein, we report a general procedure for the direct asymmetric alkylation of C2-alkylpyridines. This approach circumvents the need to incorporate a synthetic handle into the substrate or preactivate the pyridine nucleus prior to alkylation, thus offering an advantage over previously reported methods of accessing chiral pyridines.19–26 We also report isolation and structural characterization of the mixed aggregates between the chiral lithium amide and α-lithio-2-alkylpyridines, supporting a hypothesis for the origin of enantiocontrol.
To identify the optimal reagent for the enantioselective alkylation, several chiral amines were screened first (Table 1). C2-Symmetrical amines (R)-1TA-3TA that gave excellent enantioselectivity in enediolate alkylation provided only moderate level of enantiocontrol (entries 1–3). In contrast, diamines such as R)-1DA-4DA showed an improved enantiomeric ratio (er), with N-tert-butyl-substituted amine (R)-1DA being the best (entries 5–8). Given the importance of additives for aggregation states of organolithium compounds,12 we screened the effect of common lithium ligands. HMPA displayed an enhancement in both conversion (from 22% to 55%) and er (from 87:13 to 97:3, entries 5,10). While LiBr also produced an increased er, the conversion was suppressed due to incomplete lithiation of the substrate. We found that in general, lithiation of 1a was inhibited by lithium compounds, including n-BuLi itself. Toluene proved to be the optimal solvent; ethereal solvents (tetrahydrofuran, Et2O, 1,4-dioxane, 1,2-dimethoxyethane) resulted in no enantioselectivity, while the use of hydrocarbon solvents (hexane, cyclohexane) encountered solubility problems.
Table 1.
Identification of the Optimal Reaction Conditions for Benzylation of 2-Butylpyridinea
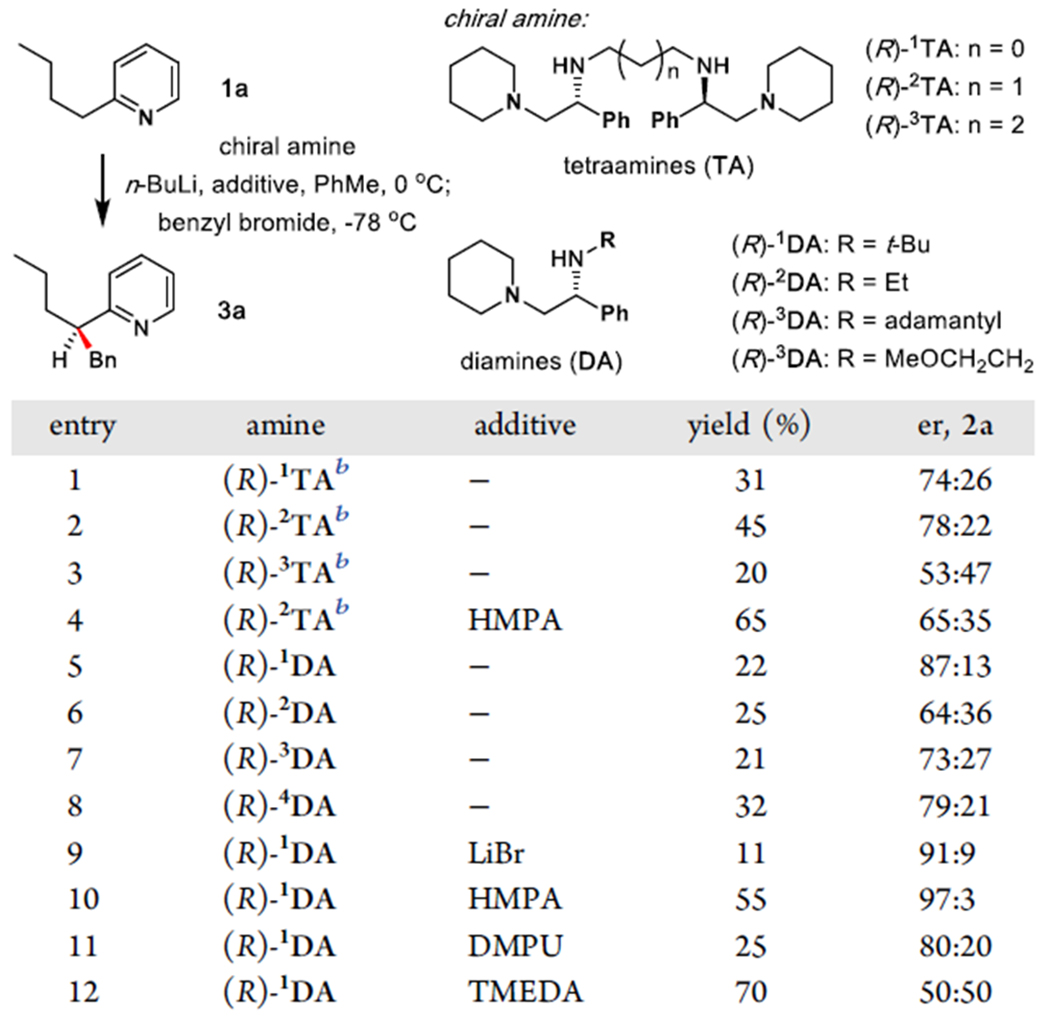 |
Reaction conditions: 0.44 mmol butyl pyridine, 0.90 mmol n-BuLi, 0.45 mmol amine, 0.22 mmol additive, 0.53 mmol BnBr, toluene (5.0 mL), see the Supporting Information for details. Isolated yields are shown. Enantiomeric ratios (er) were determined by high performance liquid chromatography (HPLC) analysis.
3.03 equiv of n-BuLi were used.
High enantioselectivity was obtained upon reaction of 1a with a range of activated electrophiles (Table 2). Methylation proceeded with enantiomer ratio of 93:7 (2a). Allyl bromide and methallyl bromide afforded alkylation products in 94:6 er and 87:13 er, respectively (2b, 2c). High enantioselectivity was observed for a variety of benzylic bromides (2d–2i). Highly reactive heteroaromatic benzylic bromides proved to be effective alkylating agents. 2-Thienyl bromide and 2-(bromomethyl)pyridine reacted rapidly to form 2j in 93:7 er and 2k in 99:1 er. Despite attempted additional refinement of reaction conditions, alkylation with 2-(bromomethyl)-1,3-benzothiazole afforded 2l in 80:20 er.
Table 2.
Scope of Alkyl Halide
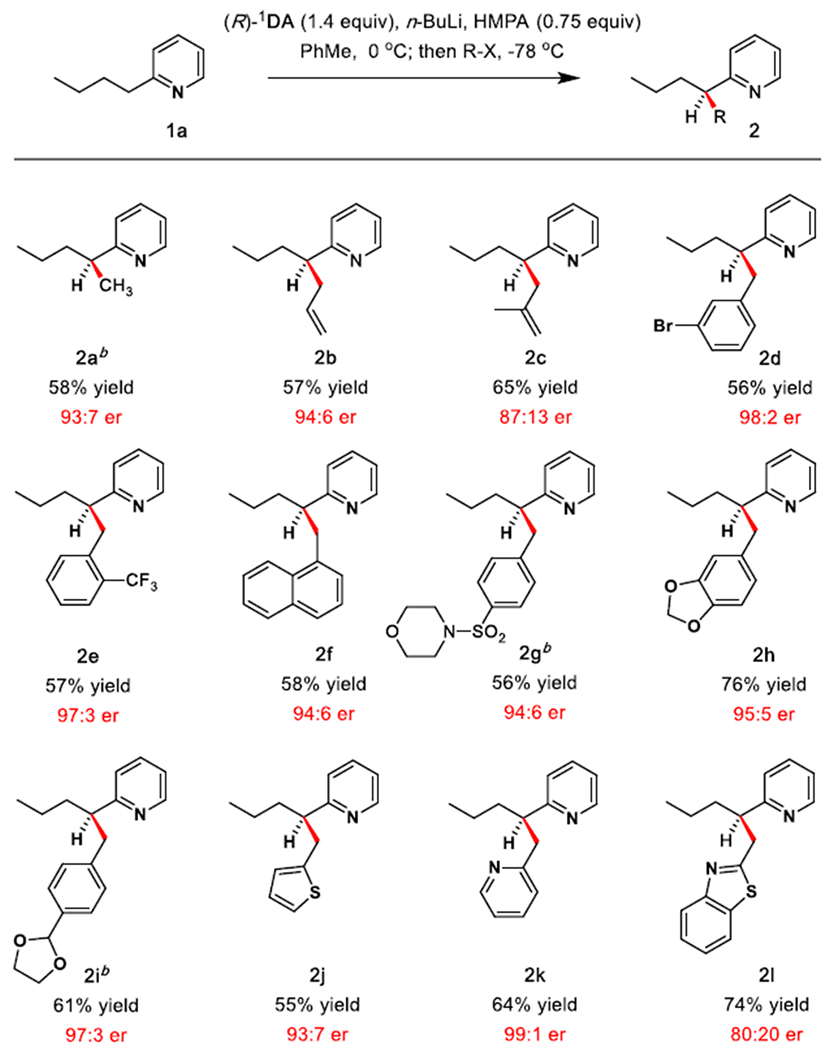 |
Reaction conditions: All reactions were carried out on a 0.44 mmol scale unless otherwise noted. Isolated yields are shown. Results are normalized to bases with the R configuration, enantiomeric ratios (er) measured by HPLC analysis.
0.5 equiv of HMPA and 1.0 equiv of (R)-1DA were used.
The scope of 2-alkylpyridines was investigated by enantioselective benzylation employing the conditions developed for 1a; however, for several substrates improved results were achieved by modifying the stoichiometry of HMPA and the chiral lithium amide (Table 3). Increasing the length of the alkyl chain (3b) produced results similar to those observed during benzylation of 1a. Benzylation of 2-ethylpyridine (1c) occurred in similar yield (76%) but diminished er (85:15) compared to 1a. When the stoichiometry of the chiral amine and HMPA was reduced, the reaction proceeded with greater er (92:8) and somewhat reduced yield (58%).
Table 3.
Scope of 2-Alkyl Pyridines
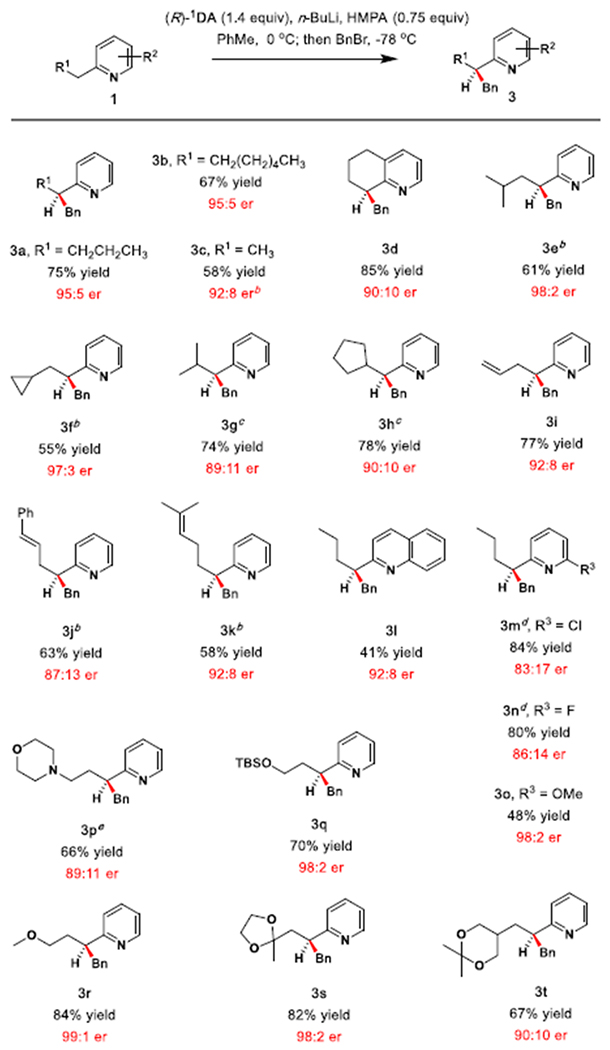 |
Reaction conditions: All reactions were performed on a 0.44 mmol scale unless otherwise noted. Isolated yields are shown. Results are normalized to bases with the R configuration, enantiomeric ratios (er) measured by HPLC analysis.
0.5 equiv of HMPA and 1.0 equiv of (R)-1DA were used.
1.0 equiv of (R)-1DA and 1.25 equiv of HMPA.
1.4 equiv of (R)-1DA and 1.0 equiv of HMPA. LDA used instead of n-BuLi.
1.0 equiv of (R)-1DA and no HMPA.
Alkylation of tetrahydroquinoline 3d proceeded smoothly in very good yield and enantioselectivity. Branching at the γ-position of the C-2 alkyl chain (3e, 3f) had a detrimental impact on enantioselectivity under the standard conditions; however, when the amount of HMPA and chiral lithium amide was reduced, excellent enantioselectivity was observed with only slightly diminished conversion.
Substrates containing β-branching (3g, 3h) were initially alkylated in poor er (<3:1) under the original procedure, but good results were observed by adjusting the quantity of HMPA to 1.25 equiv while lowering the amount of (R)-1DA to 1.0 equiv. Substrates with β,γ-unsaturation were alkylated with poor er (<2:1), but γ,δ and δ,ε-unsaturation was tolerated furnishing 3i and 3j with good enantioselectivity. Pyridine 3k gave results identical to those observed for benzylation of 2-ethylpyridine, 3c. Quinolines are useful compounds in medicinal chemistry.17,26 Quinoline 3l was accessed with very good enantioselectivity by this protocol. Halogen substitution at the 6-position resulted in moderate erosion of enantioselectivity (3m, 3n). In contrast, C6-substitution with a methoxy group enhanced enantioselectivity, furnishing 3o in 98:2 er. The presence of a morpholine group was detrimental, resulting in racemic product under the original conditions. Remarkably, in the absence of HMPA, a strong boost in er to 89:11 was observed (3p), potentially due to internal chelation.
When an ether or ketal are present at the γ-position, an exceptional increase in enantioselectivity is observed (3q, 3r, 3s). Benzylation of 1q also proceeded in very good yield (80%) and excellent er (97:3) in the absence of HMPA when (R)-1DA was replaced with the chiral amide (R)-3TA. Notably, no enhancement was observed upon alkylation of the 1,3-dioxane 3t, in which no internal chelation by the γ-alkoxy group is feasible.
We explored this intriguing enhancement of enantioselectivity by the γ-alkoxy substituents in greater detail, testing additional electrophiles with 1r (Table 4). Methylation occurred in very good yield and excellent enantioselectivity (4a, er 99:1). Alkylation was also successful with less reactive electrophiles. Ethylation of 1a resulted in only 14% conversion, however, under identical conditions 1r reacted with iodo-ethane to furnish 4b in 63% yield and 99:1 er. Reaction with signifi cantly more functionalized N-(4-bromo-methylbenzenesulfonyl)morpholine proceeded smoothly to afford 4c. High er enhancement versus 1a was also observed in the reaction of 1r with 2-(bromomethyl)benzothiazole, which afforded 4d in greater than 99:1 er versus 80:20 for 2l.
Table 4.
Enantioselective Alkylation of 2-(3-Methoxy-1-propyl)pyridine (1r)
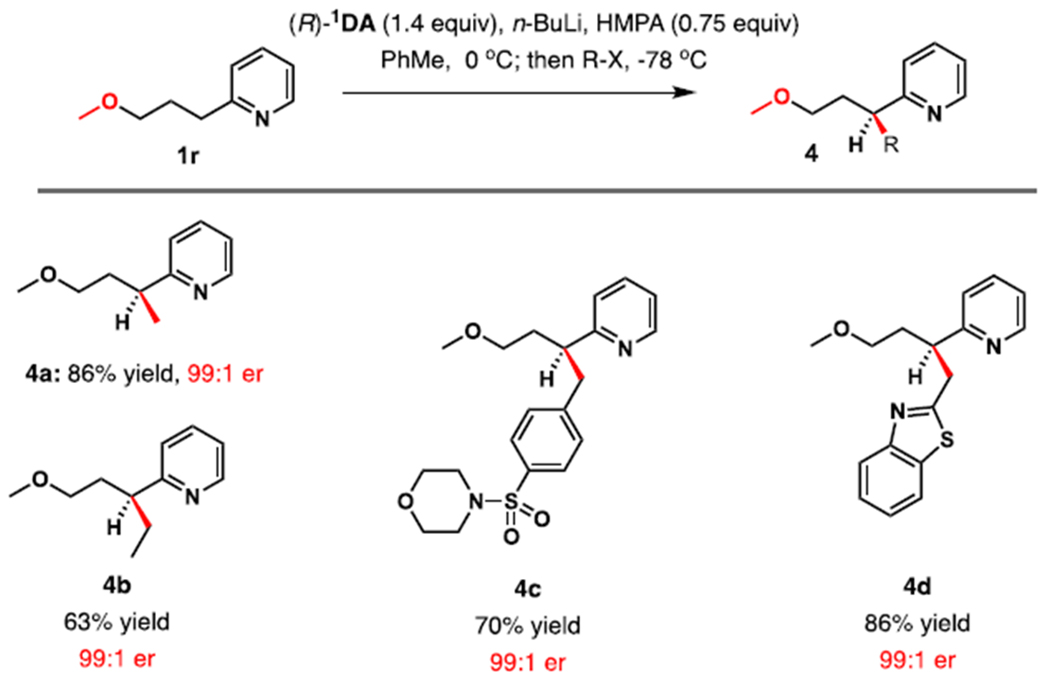 |
Reaction conditions: Reactions were performed on a 0.44 mmol scale unless otherwise noted. Isolated yields are shown. Results are normalized to bases with the R configuration, enantiomeric ratios, determined by HPLC analysis.
Given the growing evidence of the strong correlation between stereocontrol and aggregation states in organolithium chemistry,14,27 a critical part of these investigations was the crystallographic study of the central mixed aggregates involved in the alkylation reaction. We were especially interested in comparing the structures of the more generic 2-alkyl pyridine-derived aggregates to those from 2-(3-methoxy-1-propyl)-pyridine 1r, which afforded a significant boost in enantioselectivity. Initial efforts at crystallization of the reactive aggregate from 2-butylpyridine were unproductive. On the other hand, we succeeded in obtaining high-quality crystals from 2-ethyl pyridine and 1r upon treatment with (R)-1DA, n-BuLi, and HMPA in pentane at −25 °C. Figure 1 shows the rendering of the aggregate structures obtained by X-ray diffraction studies. In both cases, the aggregate has a general structure of (2-Py)(R)CHLi-(R)-Li1DA-HMPA in a 1:1:1 stoichiometry.
Figure 1.
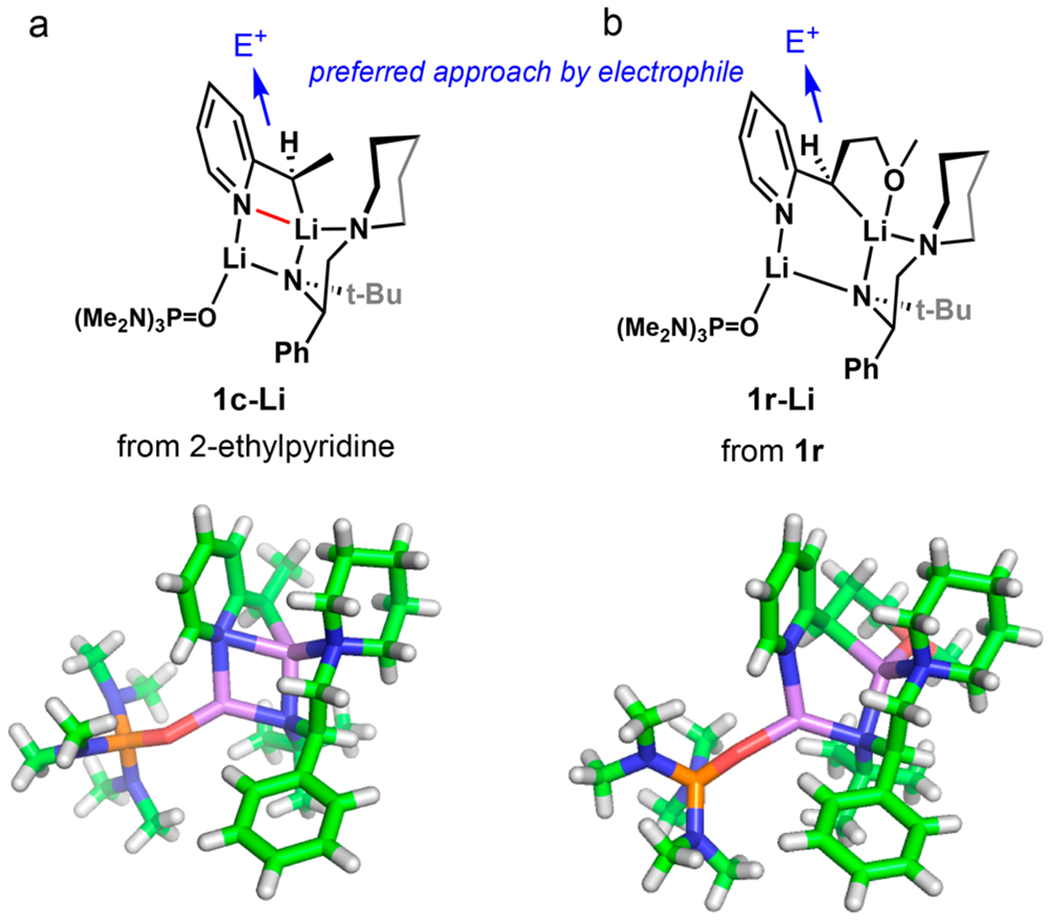
Structures of mixed aggregates determined by X-ray diffraction study of crystals obtained from (a) 2-ethylpyridine (1c-Li); or (b) 1r (1r-Li), (R)-1DA, n-BuLi, and HMPA in pentane.
The structural study revealed significant pyramidalization at the lithiated α-carbon consistent with the sp3 character of an alkyllithium reagent rather than sp2 character of the enolate-like Li enamide tautomer.28–30 Despite the presence of the internally chelating OMe group in 1r, the similarities in arrangement and orientation are striking. The most significant difference between the structures is the disruption of the central four-membered Li–N–Li–N cluster to a six-membered cyclic structure with internal complexation to the OMe group in 1r. We confirmed that the sense of absolute configuration of alkylation products derived from 2-ethyl-pyridine and 1r is identical and consistent with inversion of configuration at C–Li bond in an apparent SE2 mechanism. It is plausible that the 10-fold increase in enantiomer ratio from about 10:1 to 100:1, corresponding to ~1.4 kcal/mol free energy difference in the competing transition structure, is due to the small differences in bond angles; however, it appears to be unlikely. Our current hypothesis for the boost in enantioselectivity observed in alkylation of 1r is that it forms a more stable aggregate minimizing the nonselective background alkylation. This observation is supported by high sensitivity of er to the stoichiometry of HMPA, pointing to a dynamic equilibrium between two or more aggregates of varying reactivity. Racemization by product deprotonation was excluded by deuterium quench experiments, as the product displayed no deuterium incorporation.
Subsequent solution-state studies by 6Li and 31P NMR spectroscopy further corroborate the structures 1c-Li and 1r-Li. For example, the 31P NMR spectrum for 1r-Li has a single resonance at 24.88 ppm (JPLi = 3.50 Hz), while the 6Li spectrum features two resonances at 2.47 (d, JPLi = 3.50 Hz) and 1.64 ppm, in a 1:1 ratio, as expected. These spectra also clearly indicate the presence of a minor isomer (10—15%) at −80 °C. Detailed structural investigations of the dynamic aggregation states along with density functional theory computational examination of the alkylation are the subject of a separate, ongoing study.
The utility of this method for medicinal chemistry applications is illustrated by an expedient enantioselective synthesis of anti-HCMV compound 6 studied by Forge Life Sciences (Scheme 2).31 Direct, asymmetric methylation of 5 afforded the product in 82% yield and 97:3 er. Desilylation, oxidation, and reductive amination with 8-aminoquinoline provided the target structure without erosion of enantiomeric purity.
Scheme 2.
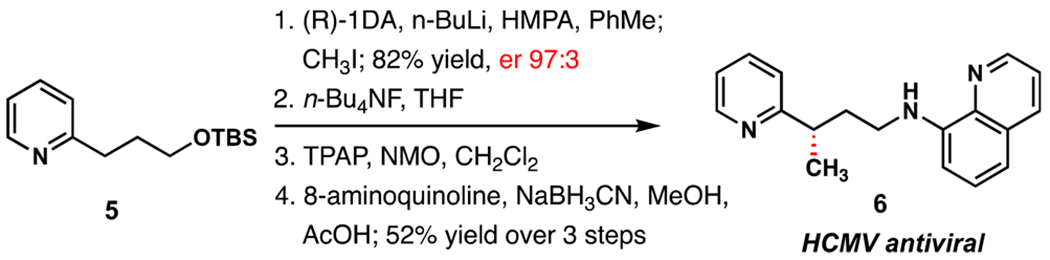
Synthesis on HCMV Antiviral Compound 6 by Direct Asymmetric Alkylation
In summary, an operationally straightforward procedure for asymmetric alkylation of 2-alkylpyridines is described. The chiral amine reagent is readily available in bulk in two simple steps from styrene oxide, and can be readily recovered by aqueous pH-controlled extraction. This reaction validates CLAs as reagents for alkylation of nonenolate-derived organolithium reagents. Valuable insight into the mechanism of enantiocontrol were revealed by X-ray diffraction studies of mixed aggregates obtained by cocrystallization of (R)-1DA with ethylpyridine or 1r upon lithiation with n-BuLi. These structural studies provide a defined view of the intermediate aggregates involved in the alkylation reactions and allow for a structure-based design of new lithium amide reagents for expanded applications in the future.
Supplementary Material
ACKNOWLEDGMENTS
T.W.H. and A.W.C. thank the National Science Foundation (CHE 1361654) for financial support. A.W.C. thanks the Mellichamp Academic Initiative in Sustainability at UCSB for a summer fellowship. This work was supported by the NIH (NIGMS, R01-077379 to A.Z.; R01-131713 to D.B.C.). Dr. Hongjun Zhou is acknowledged for assistance with NMR spectroscopy. Dr. Dmithry Uchenik and the UCSB mass spectroscopy facility are thanked for assistance with mass spectral analysis. Dr. Guang Wu and the UCSB X-ray analytical facility are acknowledged for the assistance with the X-ray diffractometry. S.G. was supported by US-Indo Postdoctoral Fellowships provided by SERB and IUSSTF.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08659.
Full experimental procedures (PDF)
Copies of 1H, 13C NMR spectra (PDF)
Copies HPLC traces (PDF)
Crystallographic data for 1c-Li (CIF)
Crystallographic data for 1r-Li (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Evans DA, Helmchen G, Rüping M In Asymmetric Synthesis—The Essentials; Christmann M, Bräse S, Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp 3–9. [Google Scholar]
- (2).Morales MR; Mellem KT; Myers AG Pseudoephenamine: a practical chiral auxiliary for asymmetric synthesis. Angew. Chem. Int. Ed 2012, 51, 4568–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Roose G Key Chiral Auxiliary Applications; Academic Press: Boston, MA, 2014. [Google Scholar]
- (4).Seyden-Penne J Chiral Auxiliaries and Ligands in Asymmetric Synthesis; John Wiley: New York, 1995. [Google Scholar]
- (5).Dugger RW; Ragan JA; Ripin DHB Survey of GMP bulk reactions run in a research facility between 1985 and 2002. Org. Process Res. Dev 2005, 9, 253–258. [Google Scholar]
- (6).Farina V; Reeves JT; Senanayake CH; Song JJ Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev 2006, 106, 2734–2793. [DOI] [PubMed] [Google Scholar]
- (7).Ando A; Shioiri T Asymmetric synthesis using chiral bases: enantioselective α-alkylation of carboxylic acids. J. Chem. Soc. Chem. Commun 1987, 656–658. [Google Scholar]
- (8).Matsuo J; Koga K Enantioselective alkylation of phenylacetic acid using a chiral bidentate lithium amide as a chiral auxiliary. Chem. Pharm. Bull 1997, 45, 2122–2124. [Google Scholar]
- (9).Stivala CE; Zakarian A Highly enantioselective direct alkylation of arylacetic acids with chiral lithium amides as traceless auxiliaries. J. Am. Chem. Soc 2011, 133, 11936–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lu P; Jackson JJ; Eickhoff JA; Zakarian A Direct enantioselective conjugate addition of carboxylic acids with chiral lithium amides as traceless auxiliaries. J. Am. Chem. Soc 2015, 137, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yu K; Lu P; Jackson JJ; Nguyen T; Alvarado J; Stivala CE; Ma Y; Mack KA; Hayton TW; Collum DB; Zakarian A Lithium enolates in the enantioselective construction of tetrasubstituted carbon centers with chiral lithium amides as noncovalent auxiliaries. J. Am. Chem. Soc 2017, 139, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Reich HJ Role of organolithium aggregates and mixed aggregates in organolithium mechanisms. Chem. Rev 2013, 113, 7130–7178. [DOI] [PubMed] [Google Scholar]
- (13).Ma Y; Stivala CE; Wright AM; Hayton T; Liang J; Keresztes I; Lobkovsky E; Collum DB; Zakarian A Enediolate-Dilithium amide mixed aggregates in the enantioselective alkylation of arylacetic acids: structural studies and a stereochemical model. J. Am. Chem. Soc 2013, 135, 16853–16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma Y; Mack KA; Liang J; Keresztes I; Collum DB; Zakarian A Mixed aggregates of dilithiated Koga tetraamine: NMR spectroscopic and computational studies. Angew. Chem. Int. Ed 2016, 55, 10093–10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Asymmetric alkylation of typical enolates mediated by CLAs:; Imai M; Hagihara A; Kawasaki H; Manabe K; Koga K Catalytic asymmetric benzylation of achiral lithium enolates using a chiral ligand for lithium in the presence of achiral ligand. J. Am. Chem. Soc 1994, 116, 8829–8830. [Google Scholar]
- (16).Frizzle MJ; Nani RR; Martinelli MJ; Moniz GA Asymmetric alkylation of 5-alkyl-2-aminothiazolones using a C2-symmetric chiral tetraamine base. Tetrahedron Lett. 2011, 52, 5613–5616. [Google Scholar]
- (17).Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (18).Trost BM; Thaisrivongs DA Palladium-catalyzed regio-, diastereo-, and enantioselective benzylic allylation of 2-substituted pyridines. J. Am. Chem. Soc 2009, 131, 12056–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yin Y; Dai Y; Jia H; Li J; Bu L; Qiao B; Zhao X; Jiang Z “Conjugate addition-enantioselective protonation of N-aryl glycines to α-branched 2-vinylazaarenes via cooperative photoredox and asymmetric catalysis”. J. Am. Chem. Soc 2018, 140, 6083–6087. [DOI] [PubMed] [Google Scholar]
- (20).Meazza M; Tur F; Hammer N; Jorgensen KA Synergistic diastereo- and enantioselective functionalization of unactivated alkyl quinolines with α,β-unsaturated aldehydes. Angew. Chem. Int. Ed 2017, 56, 1634–1638. [DOI] [PubMed] [Google Scholar]
- (21).Saxena A; Choi B; Lam HW Enantioselective copper-catalyzed reductive coupling of alkenylazaarenes with ketones. J. Am. Chem. Soc 2012, 134, 8428–8431. [DOI] [PubMed] [Google Scholar]
- (22).Best D; Kujawa S; Lam HW Diastereo- and enantioselective Pd(II)-catalyzed additions of 2-alkylazaarenes to N-Boc imines and nitroalkenes. J. Am. Chem. Soc 2012, 134, 18193–18196. [DOI] [PubMed] [Google Scholar]
- (23).Izquierdo J; Landa A; Bastida I; Lopez R; Oiarbide M; Palomo C Base-catalyzed asymmetric α-functionalization of 2-(cyanomethyl)azaarene N-oxides leading to quaternary stereocenters. J. Am. Chem. Soc 2016, 138, 3282–3285. [DOI] [PubMed] [Google Scholar]
- (24).Yu S; Sang HL; Ge S Enantioselective copper-catalyzed alkylation of quinoline N-oxides with vinylarenes. Angew. Chem. Int. Ed 2017, 56, 15896–15900. [DOI] [PubMed] [Google Scholar]
- (25).Yang H; Wang E; Yang P; Lv H; Zhang X Pyridine-directed asymmetric hydrogenation of 1,1-diarylalkenes. Org. Lett 2017, 19, 5062–5065. [DOI] [PubMed] [Google Scholar]
- (26).Kaur K; Jain M; Reddy RP; Jain R Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem 2010, 45, 3245–3264. [DOI] [PubMed] [Google Scholar]
- (27).Harrison-Marchand A; Mongin F Mixed aggregate (MAA): a single concept for all dipolar organometallic aggregates. 1. Structural data.. Chem. Rev 2013, 113, 7470–7562. [DOI] [PubMed] [Google Scholar]
- (28).von Rague Schleyer P; Hacker R; Dietrich H; Mahdi W The eight-membered ring structure of an a-lithio-2,6-dimethylpyridine-tetramethylethylenediamine (TMEDA) dimer. J. Chem. Soc. Chem. Commun 1985, 622–624. [Google Scholar]
- (29).Liddle ST; Clegg W Synthesis and structure of the novel mixed anion-dianion lithium cage compound [(6-LiCH2Py-2-OLi)4(6-CH3Py-2-OLi)2(THF)9]. Chem. Commun 2001, 1584–1585. [DOI] [PubMed] [Google Scholar]
- (30).Wanat RA; Collum DB; Van Duyne G; Clardy J; DePue RT Solid-state and solution studies of lithiated 2-carbomethoxycyclohexanone dimethylhydrazone and lithiated cyclohexanone phenylimine. J. Am. Chem. Soc 1986, 108, 3415–3422. [Google Scholar]
- (31).Remiszevski S; Koyuncu E; Sun Q; Chiang L Anti-hcmv compositions and methods. Patent Application WO 2016077240 A2, 2016
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.