

SUMMARY
Intracellular protein filaments are ubiquitous for cellular functions, but forming bona fide biomimetic intracellular filaments of small molecules in living cells remains elusive. Here, we report the in situ formation of self-limiting intracellular filaments of a small peptide via enzymatic morphological transition of a phosphorylated and trimethylated heterochiral tetrapeptide. Enzymatic dephosphorylation reduces repulsive intermolecular electrostatic interactions and converts the peptidic nanoparticles into filaments, which exhibit distinct types of cross-β structures with either C7 or C2 symmetries, with the hydrophilic C-terminal residues at the periphery of the helix. Macromolecular crowding promotes the peptide filaments to form bundles, which extend from the plasma membrane to nuclear membrane and hardly interact with endogenous components, including cytoskeletons. Stereochemistry and post-translational modification (PTM) of peptides are critical for generating the intracellular bundles. This work may offer a way to gain lost functions or to provide molecular insights for understanding normal and aberrant intracellular filaments.
Enzymatic morphological transition leads to the in situ formation of self-limiting intracellular peptide filaments in live cells. Illustrating that enzymatic reaction and post-translational modification (PTM) control the intermolecular interactions of molecular assemblies to generate artificial monodispersed filaments of small molecules in a highly dynamic and crowded intracellular environment, this work byFeng et al. highlights the critical role of multiple PTMs in the peptides and provides molecular insights for understanding normal and aberrant intracellular filaments
INTRODUCTION
Noncovalent filaments of proteins are ubiquitous and play many critical roles in cells, such as myosin filaments for contraction,1 actin filaments for maintaining cell shapes,2 microtubules for cell division,3 and intermediate filaments for supporting the nuclear envelope.4 Moreover, filaments of aberrant proteins are known to associate with human diseases,5 such as tau filaments of Alzheimer diseases.6 Forming in the heterogenous, dynamic cellular environment, these protein filaments have stimulated the efforts to generate noncovalent filaments of synthetic molecules for understanding and mimicking the assembling behaviors and functions of the endogenous protein filaments. While using the self-assembly7 of synthetic molecules (i.e., lipids,8 saccharides,9 peptides10–12) to form noncovalent filaments has been successful in vitro13–16 or in well-confined conditions, the generation of intracellular filaments of synthetic molecules from self-assembly in living cells remains a challenge. Because cells are inherently out of equilibrium,17 it is challenging to carry out self-assembly, a thermodynamic equilibrium process, of synthetic molecules inside cells. Moreover, the cytosol is dynamic, heterogenous, and highly crowded with proteins,18 which would affect the self-assembly in a rather unpredicted manner.
One particularly promising class of self-assembling materials for mimicking protein structures and functions are peptides64,65,66. Because of the versatile chemistry, relevance to biological signals, and programmable conformations,73 self-assemblies of peptides have found applications in tissue engineering,19 therapeutics,20,21 and bioimaging.22 Despite the successful formation of filament of peptide in vitro and various applications in vivo, direct formation and real-time imaging of self-assembled filament of peptide in live cells is still extremely challenging. Moreover, the molecular understanding of the structures and dynamics of short peptides at very low concentration (0.2 mg/mL) in aqueous solution has yet to be achieved. Considering that the intracellular protein filaments are regulated by post-translational modification (PTM)23 and endogenous enzymatic reactions,24 we have integrated self-assembly with enzymatic reaction as a biomimetic approach25,26 for controlling intermolecular interactions in complex environment, to create intracellular noncovalent filaments.
Here, we report the enzymatic formation of self-limiting intracellular filaments of a synthetic peptide (Scheme 1). Using cryoelectron microscopy (cryo-EM), fluorescent microscopy, electron tomography (ET), molecular dynamics, and molecular engineering, we elucidated the formation of intracellular filaments of a trimethylated tetrapeptide in live cells. The cryo-EM structural determination reveals that peptide self-assembles into two distinct types of cross-β structures that possess either C7 or C2 symmetries, as revealed by helical reconstructions using IHRSR.27 Atomistic molecular dynamics simulations suggest that water and ions are present in the central pore of the filament and provide stabilization to the filament structure; MD simulations also find that phosphorylation of the peptide leads to reduced stability of the filament, agreeing with that filament formation requires dephosphorylation. Forming inside cells, the peptide filaments, exhibiting monodispersed diameters, pack as twist bundles and extend from the plasma membrane to nuclear membrane. Being orthogonal to endogenous cytoskeletons and hardly interacting with other endogenous components, the filaments are cytoskeleton-like and able to impede cell migration. Cell-free experiment and coarse-grained (CG) molecular simulations support the macromolecular crowding enables the bundling of the filaments. Moreover, molecular engineering confirmed the importance of the stereochemistry and PTM of peptides for forming intracellular bundles, which also underscores the inherent difference between in vitro and in vivo conditions. Using enzymatic morphological transition to generate artificial intracellular filaments of small molecules in a highly dynamic and crowded intracellular environment, this work may offer a way to gain lost functions of intermediate filaments28 or to provide valuable insights for understanding pathogenic filaments of protein or peptides.
Scheme 1. Schematic Illustration of Intracellular Conversion of Nanoparticles.

Schematic illustration shows the intracellular conversion of the nanoparticles of 1P-KMe3 to bundles of intracellular filaments of 1-KMe3 via enzymatic morphological transition, the molecular structures of 1P-KMe3 and 1-KMe3, and the cryo-EM construction, and molecular dynamics simulations of peptide filament of 1-KMe3.
RESULTS
Molecular Design and Enzymatic Morphological Transition
The peptide, 1P-KMe3 (Scheme 1), consists of a polarity sensitive fluorescent dye (nitrobenzoxadiazole [NBD]), a self-assembling D-peptide backbone (D-Phe-D-Phe),29 a phosphatase cleavage site (D-phosphotyrosine), and a C-terminal trimethyl-L-lysine. Such a design allows alkaline phosphatase (ALP)30 to convert 1P-KMe3 to 1-KMe3 (Figure 1A) and to initiate the self-assembly and morphological transition. We prepared the unnatural amino acids Fmoc-D-Tyr(PO3H2)-OH and Fmoc-Lys(Me)3-OH, and synthesized 1P-KMe3 with Fmoc solid-phase peptide synthesis (SSPS).31 After using liquid chromatography to purify the crude product, we obtained 1P-KMe3. After being treated with ALP for 24 h in PBS buffer (pH 7.4), 1P-KMe3 becomes dephosphorylated and turns into 1-KMe3. According to negatively stained TEM (Figure S1), while 1P-KMe3 self-assembles to form nanoparticles around 15 ± 3 nm, enzymatic dephosphorylation results in filaments with monodispersed diameters around 6 ± 1 nm. The partially aligned filament in the TEM images indicates that the interfilamental interactions favor the formation of bundles.
Figure 1. Structure and Intermolecular Interactions of the Peptide Filaments of 1-KMe3.

(A) A cryo-EM image of type 1 (yellow arrow) and type 2 (red arrow) 1-KMe3 filaments.
(B) Average power spectra of type 1 filaments of 1-KMe3.
(C) 3D reconstruction of type 1 filaments of 1-KMe3 from cryo-EM images.
(D) Atomic model of the type 1 fibril with cross-β structure.
(E) Top views of the cross-section of the EM density of type 1 filament and the stick representation of the peptides.
(F and G) CPK model and chemical structures of one layer of the filament at the cross-section (E).
Structures of the Filaments
To elucidate the structure of the filaments made of 1-KMe3, we used cryo-EM to determine the structure of these filaments (Figures 1A and S2). There are two types of filaments (types 1 and 2). The helical symmetries of both types of filaments were found by trial-and-error until recognizable peptide-like features were seen. Possible helical symmetries were calculated from the averaged power spectrum of boxed filaments (Figure 1B). Finally, the type 1 filaments were determined to have C7 symmetry (rise 4.9Å, twist 2.3 degrees) while the type 2 filaments to have C2 symmetry (rise 4.9Å, twist 2.7 degrees).
For the type 1 filament with C7 symmetry, we were able to reach ~4.3Å resolution by helical reconstruction using IHRSR,27 as judged by a model:map Fourier shell correlation (FSC) (Figure S3). The reconstructed fibril density shows 1-KMe3 molecule stacked in a parallel cross-β structure (Figures 1C–1E; Figure S4; Video S1). As shown in the CPK model (Figure 1F), extensive aromatic-aromatic interactions contribute to the self-assembly of 1-KMe3. With the N-terminal attached NBD motifs pointing to the center and the hydrophilic C-terminal residues (D-tyrosine and L-trimethyllysine) at the periphery of the helix, the hydrophobic residues (D-Phe-D-Phe) constitute the middle ″rings″ of the helix (Figure 1G). It is possible that water molecules are trapped in the center, but after imposing 7-fold symmetry this density is uninterpretable. An atomic model was built with cross-β restraints (Table S1). The model is most favorable as judged by the model:map real-space correlation coefficient (Figure S5), although at this resolution there is still some ambiguity of the EM map hand as well as the hydrogen bonds between cross-β layers.
For the type 2 filaments with C2 symmetry, we were able to reach ~5–6Å resolution by helical reconstruction. The EM map clearly shows that each asymmetric unit has three 1-KMe3 molecules (Figure S6) so it forms a pseudo C6 structure within one cross-β layer. At such resolution, the ambiguity of EM map hand and the difficulties of many possible peptide orientations within the asymmetric unit prevent us from building a reliable atomic model.
Formation of Intracellular Filaments
NBD allows confocal laser scanning microscopy (CLSM) to directly visualize the formation of the filaments in vitro and in vivo.8 In vitro CLSM images show (Figure 2A) that the solution of 1P-KMe3 (200 μM) contains a few fluorescent puncta in a largely dark background, suggesting that 1P-KMe3 forms the loose nanoparticles, as revealed by TEM (Figure S1), which equilibrate with monomeric 1P-KMe3 at this concentration. Upon the treatment of ALP (2 U/mL) over 24 h, the solution with scatter fluorescent spots transforms to a bright fibrous network of 1-KMe3 (Figures S7 and S8). This result confirms that 1P-KMe3 and 1-KMe3 are suitable for fluorescent imaging of the enzymatic formation of the peptide filaments. We next used CLSM to visualize the growth of filaments in live Saos-2 cells, an osteosarcoma cell line32 that overexpresses tissue nonspecific ALP. Being incubated with 1P-KMe3 for 1 h, Saos-2 cells exhibit bright fluorescent droplets, which are likely made of nanoparticles of 1P-KMe3 in the cytosol but mostly near the cell membrane (Figure 2B). Although the detailed mechanism of the endocytosis of the nanoparticles of 1P-KMe3 remains to be elucidated, we speculate that nanoparticles of 1P-KMe3 are able to interact the ALP enriched in lipid rafts of the Saos2 cells, thus inducing cav-eolin-mediated endocytosis.33 Moreover, the D-peptide backbone of 1P-KMe3 prevents its proteolysis in lysosomal environment for its eventually lysosome escape. From 1 h to 2 h, short fluorescent fibers start to grow from the edge of cells, indicating the transformation of the nanoparticles to the filaments in live cells. At 12 h incubation, a network of interconnecting nanofibers has developed in the cytoplasm of the cells (Video S2). From 12 to 24 h, the density of the nanofibers increases considerably (Figures 2B and 2C; Video S3), confirming the formation of intracellular filaments. Inhibiting the activity of the ALP of Saos-2 cells by a known inhibitor (DQB)34 prevents the formation of intracellular filaments and results in fluorescent droplets, which further diminish at a higher concentration of the inhibitor (Figures 2D and S9). To validate the enzymatic transformation in live cells, we quantified the conversion of 1P-KMe3 to 1-KMe3 after incubating the Saos-2 cell with 1P-KMe3. We found that 45%, 60%, 72%, 83%, and 91% of 1P-KMe3 molecules were dephosphorylated after 0.5, 1, 2, 12, and 24 h incubation, respectively (Figure 2E). This result agrees with the gradual growth of the intracellular filaments (Figure 2B). After incubating the Saos-2 cells with fresh culture medium, the intracellular fibrous network dissociates (Figure 2F) to become a few fluorescent puncta, confirming that the intracellular filaments are noncovalent and formed by self-assembly. The above results confirm that enzymatic dephosphorylation converts the droplets in the cytosols to intracellular filaments overtime in live Saos-2 cells. These results, to the best of our knowledge, fill the gap of generating and direct visualizing intracellular artificial filaments in live cells.
Figure 2. Formation of the Intracellular Filaments.
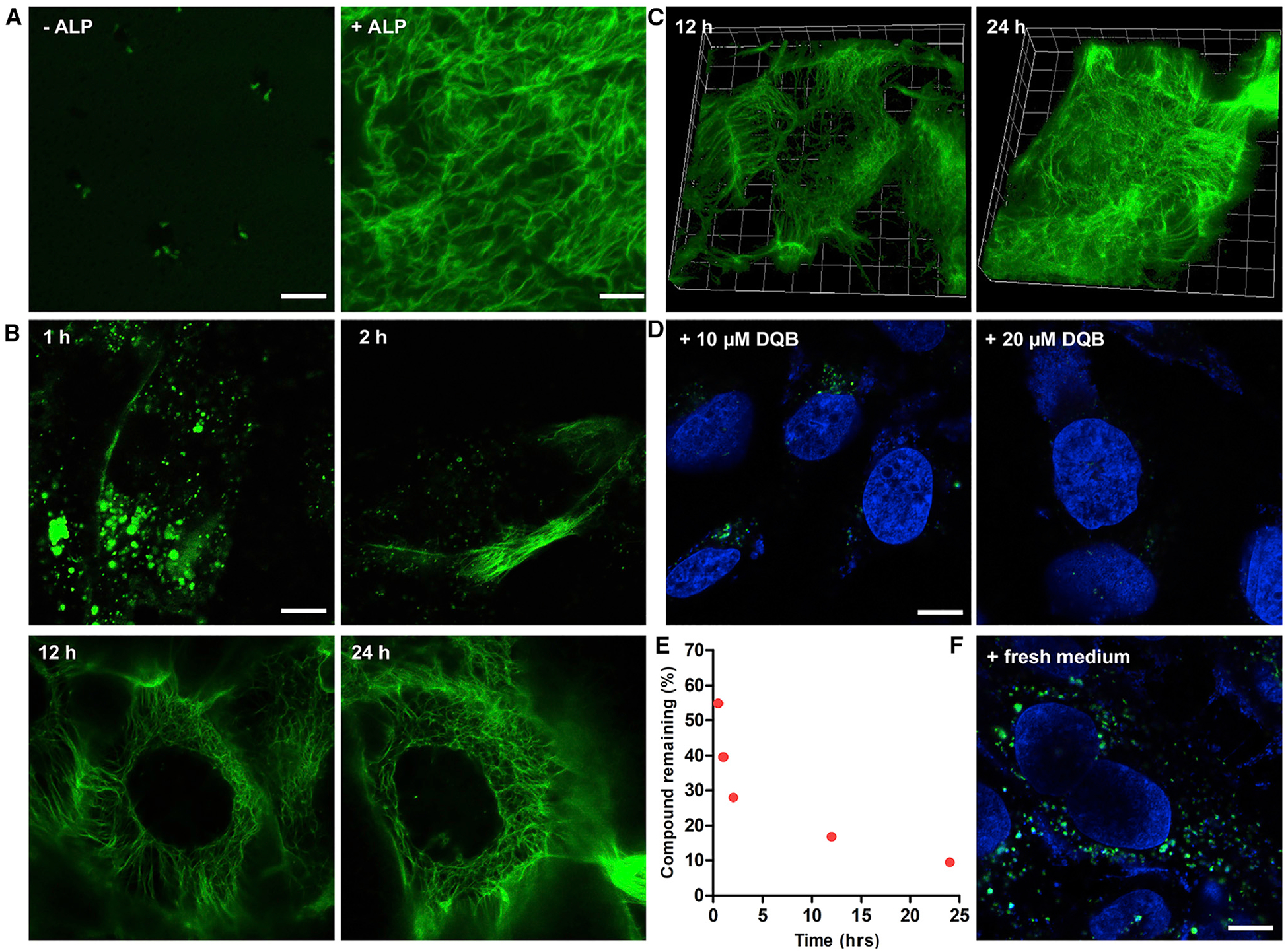
(A) CLSM images of nanostructures formed before and after adding ALP (2 U/mL) to the solution of 1P-KMe3 (200 μM). Scale bars, 5 μm.
(B) CLSM images of Saos-2 cells treated with 1P-KMe3 (200 μM) at different time. Scale bars, 10 μm.
(C) Three-dimensional CLSM image of Saos-2 cells treated with 1P-KMe3 (200 μM) for 12 or 24 h.
(D) CLSM images of Saos-2 cells treated with 1P-KMe3 (200 μM) and co-incubated with ALP inhibitor (DQB) for 24 h. Scale bars, 10 μm.
(E) Percentage of 1P-KMe3 remaining after treating with Saos-2 cells over the time course of 24 h.
(F) CLSM images of Saos-2 cells treated with 1P-KMe3 (200 μM) for 12 h and then incubated with fresh culture medium for 24 h. Scale bars, 10 μm.
The intracellular networks of the 1-KMe3 filaments resemble the appearance of cytoskeletons. To examine their relationship with the endogenous cytoskeletal filaments, we performed immunofluorescent staining of major cytoskeletal protein filaments, such as actin filaments, microtubules, and intermediate filaments (i.e., vimentin and lamin) after incubating the Saos-2 cells with 1P-KMe3 for 24 h (Figures 3 and S10). Despite forming microtubule-like radiating fibers from the perinuclear region toward the cell periphery, the 1-KMe3 filaments hardly overlap with microtubules, indicating little interaction between the 1-KMe3 filaments and the microtubules in the cells. Although exhibiting different spatial distribution patterns from that of the F-actin inside the cells, the presence of the 1-KMe3 filaments in cytoplasm causes the aggregation of F-actin, suggesting that the 1-KMe3 filaments likely increasing intracellular viscosity and indirectly disrupt the F-actin dynamics, which play a role in cell motility.35 Additionally, the 1-KMe3 networks hardly colocalize with cytoplasmic intermediate filaments vimentin or nuclear intermediate filament lamin. These results suggest that the intracellular filament networks formed by 1-KMe3 are orthogonal to the endogenous cytoskeleton filaments of the Saos-2 cells. Although it is hard to rule out completely the interaction between the 1-KMe3 filaments and other proteins, the intracellular formation of the 1-KMe3 filaments is driven by enzyme-instructed self-assembly than by association with other proteins.
Figure 3. The Networks of the Synthetic Peptide Filament Being Orthogonal to Cytoskeletons.
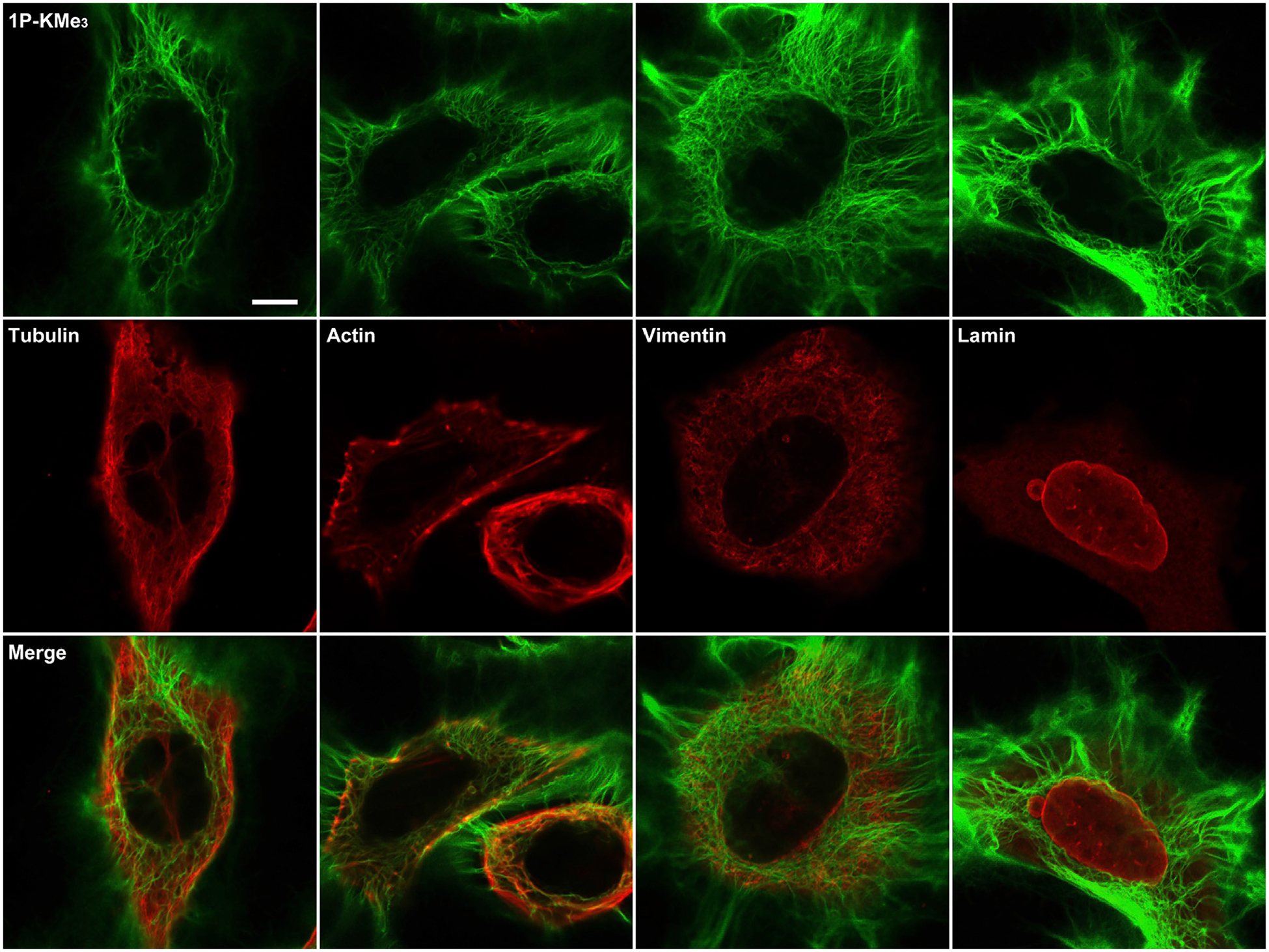
Immunofluorescent images (antibody staining for cytoskeleton proteins [red]) of the Saos-2 cells incubated with 1P-KMe3 (200 μM) for 24 h. Scale bars, 10 μm.
ET of the Intracellular Filament Bundles
Providing 3D organization of cellular fractionations, ET allows for the investigation of cellular architecture of complexes and supramolecular assemblies with structural dynamics in their natural, cellular environment.36,37 To directly investigate the artificial filaments in the cellular environment and their cellular location, we acquired montages of overlapping, high-magnification TEM images of a whole Saos-2 cell after treating it with 1P-KMe3 for 24 h and imaged with CLSM (Figure S11). In contrast to the case of the untreated Saos-2 cells (Figures 4A and S12), bundles of filaments appear abundantly inside the treated cells (Figures 4B and S13–S15). Spreading from the plasma membrane to the nuclear membrane (Figure 4C), these bundles have a various diameter (35–130 nm) and length. The 3D electron tomographic reconstruction (Figure 4D) confirms that these bundles are made up of clusters of intertwining filaments, agreeing with the in vitro TEM results. The morphology and diameters of these bundles differ from those of cytoskeletons (25 nm microtubule fibers and closely packed parallel actin filaments). Extending through the entire cytoplasm, the 1-KMe3 bundles of filaments are in proximity to but not in contact with different organelles (e.g., nucleus, endoplasmic reticulum, and mitochondria) (Figures S13 and S14). Moreover, compared with the small and few vacuoles in untreated Saos-2 cells (Figure S12), large endocytic vacuoles (Figures 4B, 4C, and S15) are scattered throughout the cytoplasm of the 1P-KMe3-treated cells, indicating the formation of intracellular filaments involving endocytosis.38
Figure 4. Electron Microscopy Images of the Filament Bundles of the Peptide (1-KMe3) inside the Cells.
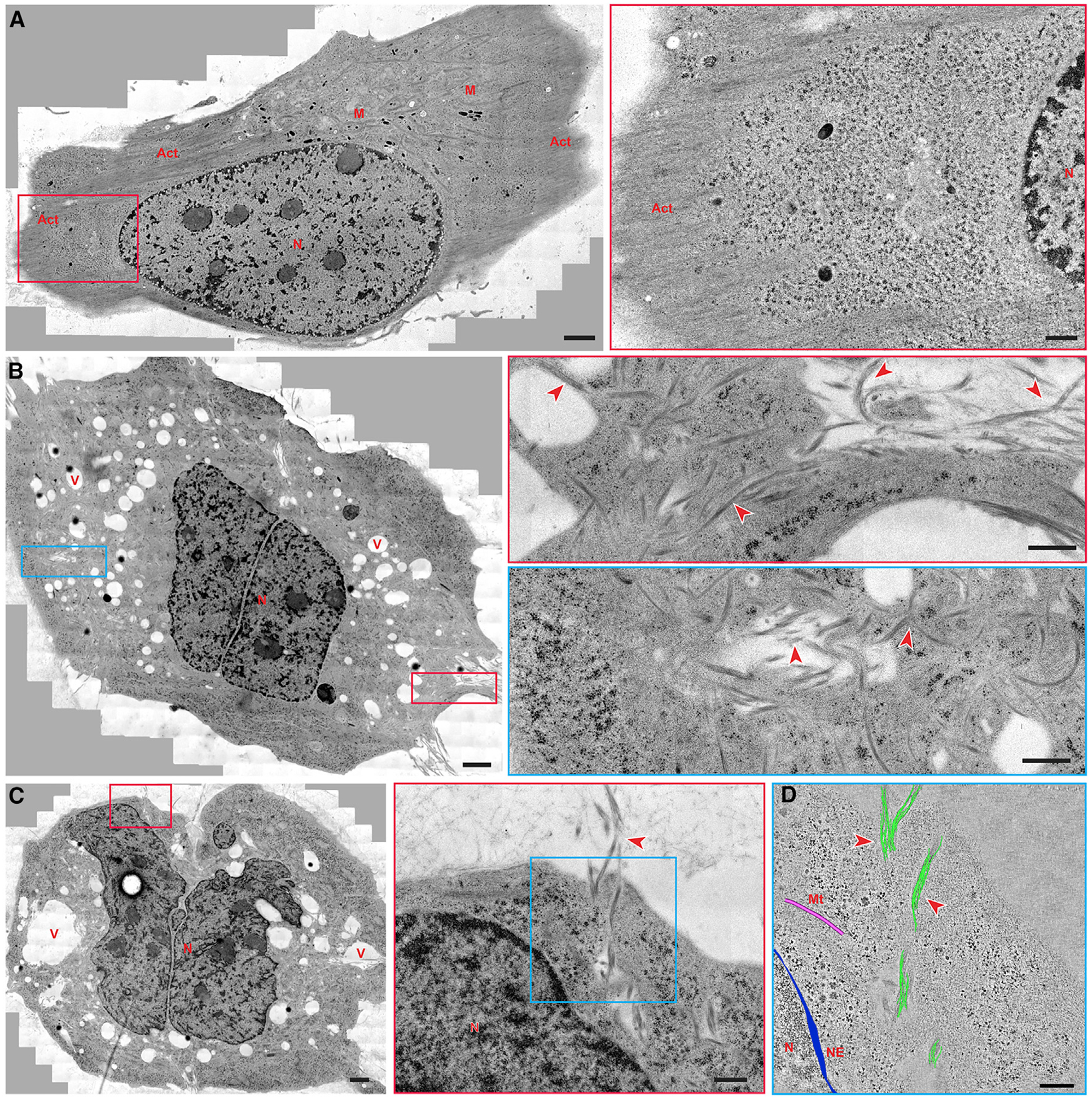
(A) TEM image of a whole Saos-2 cell (wild type, untreated) and higher-magnification electron micrograph of the red boxed area.
(B) TEM image of a treated Saos-2 cell (1P-KMe3 200 μM, 24 h) and higher-magnification electron micrographs of the red boxed area and the blue boxed area.
(C) TEM image of another treated Saos-2 cell (1P-KMe3 200 μM, 24 h) and higher-magnification electron micrograph of the red boxed area.
(D) 3D reconstruction models of the filament bundles (green), microtubules (pink), and nuclear envelope (blue) on an electron tomographic image of the blue boxed area in (C).
Solid red arrowheads indicate bundles of artificial filament; N, nucleus; M, mitochondria; Act, actin; V, vacuoles; Mt, microtubules; NE, nuclear envelope. Scale bars in (A)–(C), 2000 nm, in the boxed area in (A)–(C), 500 nm, and in (D), 250 nm.
Since the 1-KMe3 filaments affect the actin dynamics, we employed a wound closure assay to determine their effect on altering cell motility (Figures S16 and S17). While the control cells migrate significantly to cross the scratch, the cells treated with 1P-KMe3 (200 μM) hardly migrate, agreeing with the modulation of dynamics of F-actin. The addition of 1P-KMe3 at a lower concentration (100 μM), which is unable to form the intracellular filaments (Figure S18), scarcely affects the migration of the cells, further confirming that the 1-KMe3 filaments impede the cell migration.
Forming Bundles of the Filaments Due to Crowding
Macromolecular crowding in cellular environments is known to promote protein aggregation and phase separation.39,40 To mimic the crowding in cytoplasm, we added crowding agent polyethylene glycol (PEG, 20 kDa), which induces attractive interactions through depletion effects,41 into the solution of 1P-KMe3. In the presence of PEG, bundles of filaments were formed via enzyme-instructed assembly. The formation of bundles depends on the degree of crowding (or excluded volume); while the addition of 20% and 10% PEG (w/v) induces the thick bundles of filaments with an average diameter of 56 ± 12 and 45 ± 10 nm, respectively, the presence of 5% PEG (w/v) results in thinner bundles (28 ± 6 nm) with randomly oriented single filaments. The effect of crowding on the formation of filament bundles is also studied by using CG molecular dynamics simulations. The results confirm that rigid filaments form thick bundles only in a highly crowded environment, as observed in experiments (Figure 5B and Figures S37–S39), which correspond to the formation of filament bundles within crowded cellular environments (Figure 4). Details and discussions of the CG simulations are included in Notes S3 and S4. While there could be a kinetic effect that prevent these fibers from coming together, the helicity of the filament also contributes the self-limiting of the diameters of the bundles.42 In addition, we note that the individual fiber features a slight twist as illustrated in Figure 1E. Therefore, it is conceivable that to maximize the interaction between two fibers, they are not aligned perfectly, which likely leads to packing defects that limit the growth of the bundle beyond specific size. Alternatively, fibers might undergo additional twist to maximize the interactions between fibers; the accumulation of such twisting strain will also limit the growth of the bundle.
Figure 5. Formation of Bundles of Filaments in Crowded Environments.
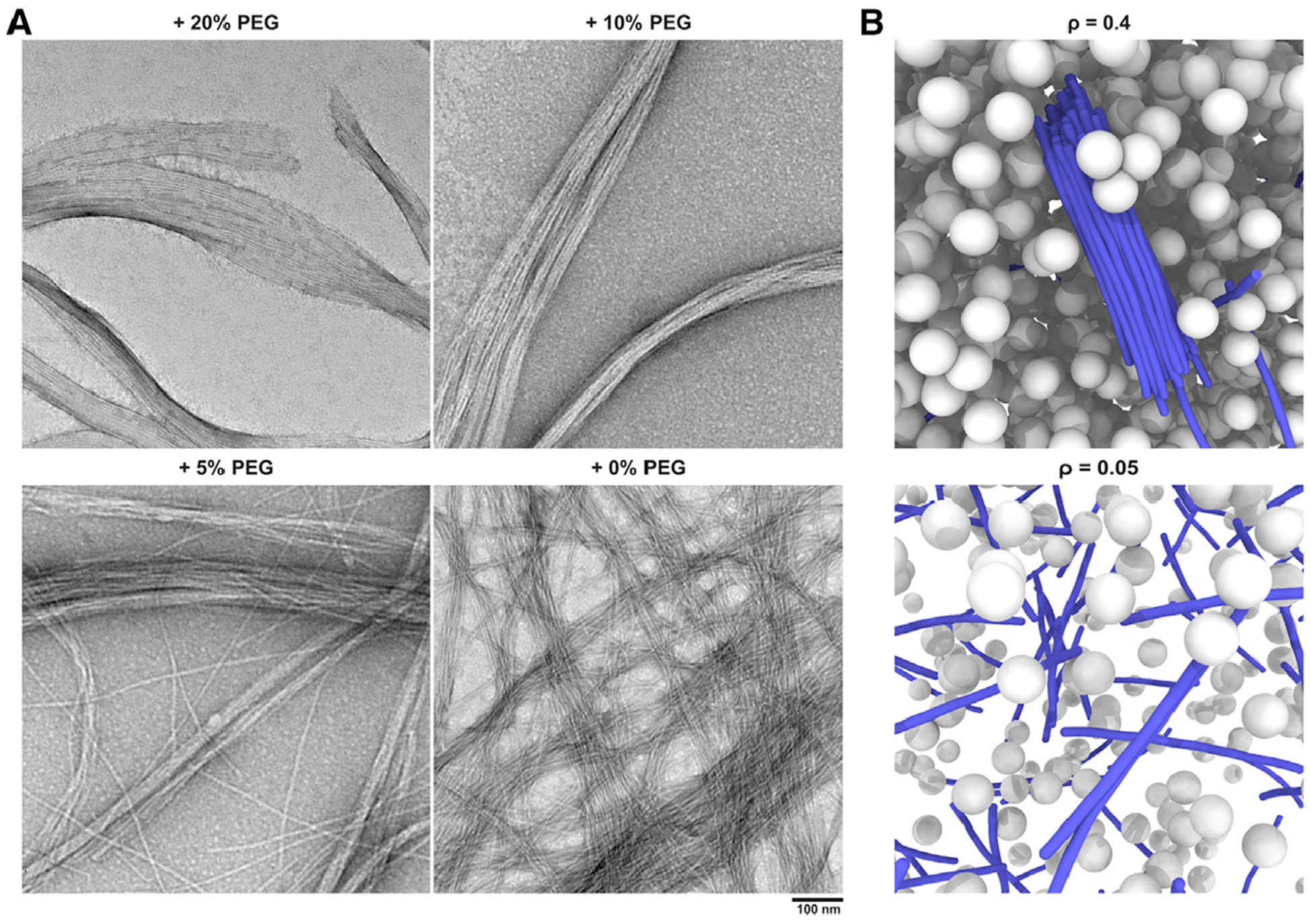
(A) TEM image of nanostructures formed after adding ALP (2 U/mL) to the solution of 1P-KMe3 (200 μM) for 24 h, in the presence of 0%, 5%, 10%, and 20% (w/v) PEG 20000. Scale bar, 100 nm.
(B) MD simulation snapshots of coarse-grained (CG) model with different volume fractions of crowders. Thick filament bundles are only observed in highly crowded environment.
Atomistic Molecular Dynamics Simulations
Using the cryo-EM structural model of the 1-KMe3 filament, we conducted atomistic molecular dynamics simulations to further probe the intermolecular interactions and the effect of phosphorylation on the structural stability of the filament. An infinite filament is modeled with periodic boundary condition and solvated with explicit water molecules and ions using the CGenFF, Charmm36 protein force fields, and CharmmGUI,43–45 and molecular dynamics simulations are then conducted at the timescale of ~100 ns to probe the effect of tyrosine phosphorylation on the structural stability of the filament as well as the potential presence of water and ions in the core of the filament; the latter issue is particularly interesting because the electron density of the pore region is difficult to interpret at the current EM resolution.
During the MD simulation, the length of the filament increases due to thermal fluctuation, and the structure becomes less ordered compared to the initial EM model (Figure 6A). The magnitude of length increases, and the rate of extension depends on the conformation of the peptide bond between NBD and the rest of the peptide as well as the phosphorylation state of the tyrosine residue. As discussed in more detail in the Supplemental Information (Note S1; Figure S36), the filament that has the least extension features a trans peptide bond for the linkage between NBD and the rest of the unphosphorylated peptide. Analysis of neighboring peptide-peptide interactions (Figures S33 and S34) suggests that both intra-layer and inter-layer peptide interactions are dominated by favorable van der Waals interactions, and the magnitude of interaction is stronger for the inter-layer peptides due to favorable side-chain stacking interactions, whereas repulsive electrostatic interactions are observed between neighboring NBD moieties and adjacent methylated lysine residues. Therefore, the phosphorylation of tyrosine, which further increases the apparent charge of the peptide leads to destabilized filament, which is manifested as substantially larger filament extension observed in the 1P-KMe3 simulations (Figure 6B).
Figure 6. All-Atom Molecular Dynamics Simulations.

(A) Initial structure and a snapshot after 50 ns of MD simulation of the 1-KMe3 filament with fion = 0.5; fion specifies the number of ions per layer of peptide orthogonal to the filament axis. Water and ions are omitted for clarity, and black bars represent periodic boundary of the simulation box.
(B) Elongated length of the filament after 120 ns of MD simulation for different systems (1-KMe3 and 1P-KMe3 with different number of placed ions in the filament core). Error bars represent standard deviation.
(C) Distribution of water (red) and cations (yellow) within the filament (gray) in the initial structure and a snapshot after 50 ns of MD simulation of the 1-KMe3 filament with fion = 0.5.
(D) The number of water molecules that are maintained within the filament at the end of 120 ns of MD simulation for different systems. Error bars represent standard deviation.
The error bars in (B) and (D) are obtained using the last 10 nanoseconds of three independent 120 ns MD simulations.
An interesting observation from the MD simulation is that a significant number of water molecules are absorbed in the filament (Figure 6D); as shown in Supplemental Information (Figures S35 and S36), these include water molecules initially placed in the central pore of the filament as well as water molecules that penetrate into the filament during the MD simulation (Figure 6C). This is not unexpected because the filament core is lined by a group of NBD moieties, which feature substantial dipoles that repel each other (Figure S33) and need to be stabilized by water molecules; in addition, the tyrosine side chain and the C-terminal carboxylate can also be stabilized by water, providing additional driving force for water penetration into the filament (Note S2).
To test whether ions (e.g., Na+) can also occupy the filament core to provide further stabilization of the NBD moieties, we initiated independent simulations with different numbers of ions in the filament core (0.5 to 1 ion per peptide layer). During the simulation, some ions diffuse out of the filament (Figure S36), and only a modest number of ions remain in the interior of the filament (Figure 6C). Nevertheless, the number of water molecules adsorbed in the filament and thus the filament length extension are affected by the amount of ions (Figures 6B and 6D).
DISCUSSION
This work demonstrates a strategy to generate artificial intracellular filaments via enzyme-controlled intracellular morphological transition. Many works have reported the self-assembly or enzymatic self-assembly in well-controlled conditions. To the best of our knowledge, this is the first example to direct formation of self-limiting filaments of peptides inside live cells and observe their formation directly by CLSM and EM. Because of heterogeneity, dynamics, and protein crowding inside cells, using short peptide (<6) to mimic protein filaments, as a form of biomaterials, in live cells has been unsuccessful. It is rather unexpected that such uniform filaments form in cytosol, a highly sophisticated and dynamic medium. Besides the enzymatic reactions, the formation of the intracellular bundles of filaments is controlled by multiple factors, such as the behaviors of the peptide assemblies, the stability of the peptides, PTM, and the activities of enzymes (or cell types) (Scheme S1; Figure S19). For example, replacing trimethyl-L-lysine in 1P-KMe3 to trimethyl-D-lysine generates 2P-KMe3, which only forms pericellular filament on Saos-2 cells (Figure S20). Incubating the 3P-KMe3, in which all the amino acid residues are L-enantiomers, with cells results in weak fluorescence on live cells, likely due to the proteolysis of the peptide (Figure S21). Although 2P-KMe3 and 3P-KMe3, in the presence of PEG, both form bundles of filaments via enzyme-instructed assembly (Figure S22), none of them form intracellular filaments. This result highlights the inherent difference between the intracellular environment and cell-free condition. Moreover, removing the methyl-groups from trimethyl-lysine (1P-K) disturbs the filaments structures and forms aggregates of nanoparticles (Figure S23). The requirement of methylation at lysine, a PTM, of the peptide for forming intracellular filaments is unexpected, but it indicates PTM as an important feature for designing peptide assemblies. In addition, we also treated 1P-KMe3 with stromal cells (HS-5) and glioblastoma cells (T98G) and only observed fluorescent dots in the cytosol of these cells (Figures S24 and S25), suggesting that the high expression of ALP inside Saos2 is essential for the formation of intracellular filaments. Notably, all the filaments formed by enzymatic dephosphorylation exhibit monodispersed diameters, suggesting enzymatic self-assembly likely is a mechanism used by nature to generate self-limiting nanostructures.46
A considerable number of small peptides are known to form filaments (or nanofibers) in vitro, but the atomistic information of the molecular arrangement in the filament is limited. This work reveals the molecular arrangement of short peptides in the filament at 4.3Å resolution, representing a significant advance for understanding the molecular interactions that stabilize the filaments of ultrashort peptides. Atomistic molecular dynamics simulations further highlighted the importance of water and ions in the center of the filament to the structural stability. These results are valuable for designing peptide filaments for functions.
In the past, it has not been possible to generate intracellular filament bundles of small molecules using enzymatic reactions. This inability likely originates from two aspects: (1) before the enzymatic reaction, the small precursor molecules diffuse easily even in the crowded cytosol, and (2) after the enzymatic reaction, the small molecules become less soluble and is difficult to diffuse in the crowded cytosol to form large assemblies. The formation of nanoparticles by 1P-KMe3 likely limits the diffusion of the precursors in the highly crowded cytosol prior to the dephosphorylation, thus resulting in high local concentration of 1-KMe3 after the enzymatic reaction for the robust formation of the filaments. The transition from nanoparticles to filaments likely is resulted from successive enzymatic dephosphorylation. Such morphological transition allows artificial intracellular filaments to modulate intracellular viscosity.47 This work also confirms that the essence of intracellular phase transition is enzymatic reaction(s), which agree with the use of enzymatic switch to control phase transition.48 Although the distribution of molecular filaments in live cells is orthogonal to other cellular cytoskeletons, such filaments prevent the migration of cancer cells. Since cell migration is a key feature of metastasis that contributes to the failure of cancer therapy, this result underscores the potential applications of the intracellular filaments in controlling cell mechanics49,50 or selectively targeting tumors.20
EXPERIMENTAL PROCEDURES
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bing Xu (bxu@brandeis.edu).
Materials Availability
All unique or stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and Code Availability
Data supporting the findings of this study are available within the paper and the Supplemental Information or are available from the lead contact upon reasonable request. The accession numbers for the filaments of 1-KMe3 reported in this paper are EMDB: EMD-22051 and PDB: 6X5I.
Materials and Synthesis
Descriptions of the materials, peptide synthesis and characterization are available in the Supplemental Experimental Procedures, Figures S26–S29, and Tables S2 and S3.
Cryo-EM and Image Processing
After 6-time dilution, 4.5 μL of 1-KMe3 sample was applied to discharged lacey carbon grids and plunge frozen using a Vitrobot Mark IV (FEI). Grids were imaged in a Titan Krios at 300 keV at NCI cryo-EM facility and recorded with a K2 direct electron detector at 1.06Å per pixel, with 25 ″fractions″ per image. Each fraction represented a dose of ~2 electrons/Å2. All the images were first motion corrected and dose weighted by MotionCorr v2.151, and the first fraction was removed. Then, a total of 1,107 images were selected that were free from drift or astigmatism, contained visible virus filaments, and had a defocus range from 0.5 to 3.0 μm determined by CTFFIND3.52 The SPIDER software package53 was used for subsequent steps. The contrast transfer function (CTF) was corrected by multiplying each image by the theoretical CTF, both reversing phases where they need to be reversed and improving the signal-to-noise ratio. This multiplication of the images by the CTF is actually a Wiener filter in the limit of a very poor SNR. The program e2helixboxer within EMAN254 was used for boxing long filaments from the micrographs. Overlapping 384-pixel long segments, with a 7 px shift between adjacent boxes (~1.5 times the axial rise per subunit), were extracted from these long filaments and then padded to 384 × 384 pixel (108,886 segments for type 1 filaments; 65,221 for type 2 filaments). The CTF determination and particle picking came from the integrated images (all 24 fractions after motion correction), while the segments used for the initial alignments and reconstruction came from the first 10 fractions accounting for a total dose of ~20 electrons/Å2. The determination of the helical symmetry was by trial and error, searching for a symmetry that yielded recognizable peptide-like feature. The IHRSR algorithm55 was used for the helical reconstructions. The map was further sharpened using a negative B factor of 100. The final resolution of the map was estimated by d9956 and model:map FSC if a model is available.
Model Building
The atomic model of type 1 filament was built de novo. First, the map corresponding to a single cross-β layer was segmented from the filament density map in Coot.57 Then, seven copies of the peptide molecules were manually docked into the density map and refined against the EM map using PHENIX.58 Two possible peptide orientation and two EM volume hands were considered, and the solution with the best real-space correlation coefficient (RSCC) was picked. Then, a filament model was generated from this and then refined against the whole EM map. Four different restraints of the cross-β hydrogen bonds were screened, and the solution with the best RSCC was picked. The statistics are listed in Table S1.
Confocal Microscopy
Saos-2 cells were seeded at 1.5 × 105 cells in a 3.5 cm confocal dish for 24 h to allow attachment. After incubating with 100 μM or 200 μM 1P-KMe3, Saos-2 cells were washed with live-cell image solution (Life Technologies A14291DJ) for 3 times and stained with 1.0 μg/ml Hoechst 33342 for 10 min at 37°C in dark. Finally, the cells were washed three times and kept in the live-cell imaging solution for imaging using Zeiss LSM 880 confocal microscopy at the lens of 63× with oil.
Immunofluorescence of Cytoskeleton Proteins
Saos-2 cells were seeded at 1.5 × 105 cells in a 3.5 cm confocal dish for 24 h to allow attachment. After incubating with 200 μM 1P-KMe3 for 24 h at 37°C in a humidified atmosphere of 5% CO2, the cells were washed by PBS for three times and then fixed by 4% formaldehyde for 15 min at 37°C. Following a three-time wash with PBS, the cells were incubated in 1% BSA/10% normal goat serum/0.3M glycine in 0.1% PBS-Tween for 1 h to permeate the cells and block non-specific protein-protein interactions. After another three time wash by PBS buffer, the cells are treated with primary antibodies overnight at 4°C. The secondary antibody is (Alexa Fluor 647 goat anti-rabbit (or mouse) immunoglobulin G (IgG) (H+L) used at the concentration of 2 μg/ml for 1 h. Finally, Hochest 33342 was used to stain the cell nucleus.
Cell-Sample Preparation for Transmission Electron Microscopy and ET
The methods for TEM of cells and ET used were performed as previously described.59 Briefly, the cells were seeded on Aclar film discs of 1.5 mm diameter and 51 μm thick, mounted on a Lab-Tek II chambered cover glass using Matrigel. Before seeding the cells, the discs were UV sterilized and incubated in supplement-free McCoy’s 5A medium for 2 h. Then, the Saos-2 cells were seeded at 2.0 × 105 cells, chosen to obtain 70%–90% confluent cultures one day after seeding, in an incubator at 37°C and 5% CO2. After 24 h, the cells were treated with 200 μM 1P-KMe3 for 24 h, while the control cells were treated with complete culture medium. Then, the cells were washed with supplement-free culture medium and imaged using Zeiss LSM 880 confocal microscopy. Then, the Aclar discs with cells were quickly transferred into the aluminum platelets (type A) for high-pressure freezing, filled with 150 mM sucrose (as a cryoprotectant) in growing medium with no supplement and covered with the flat side of another platelet (type B), creating a cavity of 0.1 mm depth. Samples were rapidly frozen using a Leica HPM-100 high-pressure freezer (Leica Microsystems, Vienna, Austria). High pressure freezing was followed by freeze substitution using the Leica AFS-2 automatic freeze-substitution device. Dehydration and fixation occurred at low temperatures (starting at −90°C) over 5 days in a solution containing 1% osmium tetroxide, 0.5% anhydrous glutaraldehyde, and 2% water in anhydrous acetone. Afterward, the temperature was raised to 4°C, and cells on disks were infiltrated and embedded in EMbed 812/Araldite 502-Resin at RT. Polymerization of was done at 60°C for a few days.
Transmission Electron Microscopy of Cells
Multiple ultrathin sections (of 70–80 nm) were cut on a Reichart Ultracut E, collected on Formvar coated copper slot grids, post-stained with uranyl acetate (saturated solution) and Reynold’s lead citrate, and initially imaged at a JEOL JEM-1200EX TEM with a 1k CCD camera (GATAN) in order to get overview maps of the sections and localize the cells of interest. Then, for high-resolution images of these cells, we imaged them at a 200 kV Tecnai F20 intermediate voltage TEM (FEI, Inc., Hillsboro, OR, USA) with a 4k CCD camera (GATAN), at 19,000× magnification (1.12 nm pixel size). For large overviews of the cells at medium magnification, we acquired montages of overlapping images in an automated fashion using the microscope control software SerialEM.60
ET
After collecting and analyzing TEM images, the grids with the sections and cells of interest were prepared for ET by coating both sides with 10-nm colloidal gold fiducials that was previously incubated for 30 min in 5% BSA.61 We acquired dual-axis tilt series of the areas of interest by tilting the sample from −60° to +60°, with 1° increments using the microscope control software SerialEM60 on a Tecnai F20 (200 keV) and the 2K × 2K CCD camera. Using the gold fiducial markers of the tilt series images in etomo from the IMOD software package,62,63 we generated 3D tomographic reconstructions, on which, later on, and we did 3D modeling of our structures of interest.
All Atom Molecular Dynamics Simulation
The cryo-EM structural model is used as the initial structure for the 1-KMe3 fiber simulation. The fiber has approximately C7 symmetry, and a small helical twist of ~2.34° per peptide; thus, 22 layers of peptides are included such that periodic condition can be maintained along the filament axis across the boundary of unit cells (i.e., 22 × 2.34°~360°/7). The peptide is described with the CHARMM36m force field, and the NBD moiety is described with CGenFF.43,44 The filament is solvated with TIP3P water and ions using CHARMM-GUI45; the ion concentration is close to be 0.15 M, which is the physiological condition. The initial simulation box is of dimension 14 × 14 × 10.8 nm3 and contain ~208K atoms. Gromacs simulation package is used for the atomistic simulations.67
To explore the effect of phosphorylation on the structural stability of the fiber, we phosphorylate the Tyr residue in each peptide, leading to the 1P-KMe3 simulation models. Since 1P-KMe3 was observed not to form filaments in the experiment, we hypothesize that the 1P-KMe3 fiber is structurally less stable in the simulations than the 1-KMe3 fiber models.
Electrostatic interactions are calculated using particle-mesh-Ewald68 with a grid size ~1 Å. Van der Waals interactions are calculated with force-switch with a cutoff distance of 1.2 nm and a switching distance of 1.0 nm. The Nose-Hoover thermostat69,70 and Parrinello-Rahman barostat71 are used to control the temperature and pressure at 300 K and 1 bar, respectively. Semi-isotropic pressure coupling is used since the filament is continuous across the periodic boundary along the filament axis. Bonds with hydrogen are constrained using the LINCS algorithm72 to allow an integration time step of 2 fs. Three independent simulations are performed for all of the model filaments and the simulations are propagated for at least 120 ns (see Table S4).
The cryo-EM structural model has the cis configuration for the peptide bond between NBD and the phenylalanine residue (Figure S30). The cis configuration for a peptide bond is rather unusual, although it has been observed near the end of a peptide. Nevertheless, since it is difficult to distinguish the cis from the common trans conformer at the EM resolution, we have also constructed a trans fiber model in which the cis peptide bond is rotated into the trans configuration. This is done by allowing other dihedral angles relevant to the NBD residue to adjust to avoid overlaps with other atoms.
As illustrated in Figure S31, adjusting the conformation of the first peptide bond leads to structural changes in the NBD moieties, which lean further downward compared to the EM structural model. As a result, the volume of the center cavity is slightly larger in the trans filament compared to the cis model. For example, when water molecules are introduced into the system, more water molecules (Nwat = 156) are included for the trans fiber compared to the cis model (Nwat =112) with fion = 0.
Another technical detail concerns the placement of water and ions in the filament core. At the current EM resolution, the density of the core is difficult to interpret. Since the core is lined with NBD, which is highly polar, it is likely water and ions are present. Water molecules are introduced into the pore by superimposing water boxes at the bulk water density with the filament structure and removing water molecules within 2.8 Å from any protein atom. As to ions, we place a varying number of Na+ ions in the core; this is characterized by fion, the number of ion(s) per peptide layer, and we tested the values of 0, 0.5, 0.72, and 1.0. When ions are placed in the filament core, the same number of water is removed.
To investigate the favorable structure of the peptide in solution, we simulate a single trans peptide in solution up to 250 ns with four independent simulations and calculate Ramachandran plot for all four peptide bonds (Figure S32). The initial dihedral angles obtained from the cryo-EM structure are marked as yellow star. As shown in the Figure S32, the equilibrium structure of peptide in solution is rather different from the cryo-EM structure. Therefore, for the peptides to self-assemble into the highly ordered filaments, significant structural transition of individual peptides as well as the subtle balance between favorable inter-peptide interactions and unfavorable entropic loss are required.
CG Simulations
To investigate crowding effect on the bundling of filaments, we perform CG Langevin dynamics (LD) simulations by using a generic polymer model. The LAMMPS simulation package is used for the CG simulations.73
Each polymer chain is modeled as a bead-spring chain that is composed of 32 monomers of diameter σ and mass m; crowders are modeled as spherical particles of diameter 4 σ and mass 64m. Neighboring monomers are connected by the harmonic bonding potential Ub(r) = k(r−r0)2, in which the reference distance r0 is set to 1.2 σ. The force constant is taken to be k = 1000 kB/σ2, where kB and T denote the Boltzmann constant and simulation temperature, respectively. The harmonic angular potential Ub(θ) = kangle(θ− θ0)2 is also applied with a kangle of kBT/ θ02, in which θ0 = π
The non-bonded interactions between monomers of polymer are described by a truncated and shifted Lennard-Jones potential; the cutoff distance is 2.5 σ. Other non-bonded interactions between polymer-crowder and crowder-crowder are taken to be purely repulsive and described by the Weeks-Chandler-Andersen (WCA) potential74 with the same well-depth parameter ε as that for the polymer monomer-monomer interactions.
The Langevin dynamics (LD) simulations are conducted with the Velocity-Verlet integrator and an integration time step of 0.002 τ, where τ is reduced time unit (τ = (mσ/ kBT)0.5). All of our simulation box is cubic with a dimension L = 128σ. Periodic boundary condition is applied to all directions. The number of polymer molecules is set to be 128. The volume fraction of the crowder varies from 0.01 to 0.40, where ρ = (4/3) πσc3N/L3and N is the number of crowder particles.
Supplementary Material
HIGHLIGHTS.
Enzymatic morphological transition constructs intracellular peptide filaments
Peptide assemblies form monodispersed filaments in the dynamic cellular environment
Cryo-EM construction reveals the distinct types of cross-β structures of filaments
Phosphorylation of peptides controls the intermolecular interactions of filaments
ACKNOWLEDGMENTS
This research was, in part, supported by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E and by NIH CA142746 (to B.X.), GM122510 (to E.H.E.), F99CA234746 (to Z.F.), and NSF DMR-2011846. The computational work was supported by the NSF grant CHE-1829555 to Q.C. and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1A6A1A03024940). Computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number ACI-1548562, are greatly appreciated; part of the computational work was performed on the Shared Computing Cluster which is administered by Boston University’s Research Computing Services (www.bu.edu/tech/support/research/).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrp.2020.100085.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Needham J, Kleinzeller A, Miall M, Dainty M, Needham DM, and Lawrence ASC (1942). Is muscle contraction essentially an enzyme-substrate combination? Nature 150, 46–49. [Google Scholar]
- 2.van den Ent F, Amos LA, and Löwe J (2001). Prokaryotic origin of the actin cytoskeleton. Nature 413, 39–44. [DOI] [PubMed] [Google Scholar]
- 3.Mitchison T, and Kirschner M (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. [DOI] [PubMed] [Google Scholar]
- 4.Gerace L, and Blobel G (1980). The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19, 277–287. [DOI] [PubMed] [Google Scholar]
- 5.Chuang E, Hori AM, Hesketh CD, and Shorter J (2018). Amyloid assembly and disassembly. J. Cell Sci 131, jcs189928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitesides GM, and Grzybowski B (2002). Self-assembly at all scales. Science 295, 2418–2421. [DOI] [PubMed] [Google Scholar]
- 8.Onogi S, Shigemitsu H, Yoshii T, Tanida T, Ikeda M, Kubota R, and Hamachi I (2016). In situ real-time imaging of self-sorted supramolecular nanofibres. Nat. Chem 8, 743–752. [DOI] [PubMed] [Google Scholar]
- 9.Lee SS, Fyrner T, Chen F,Álvarez Z, Sleep E, Chun DS, Weiner JA, Cook RW, Freshman RD, Schallmo MS, et al. (2017). Sulfated glycopeptide nanostructures for multipotent protein activation. Nat. Nanotechnol 12, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar M, Ing NL, Narang V, Wijerathne NK, Hochbaum AI, and Ulijn RV (2018). Amino-acid-encoded biocatalytic self-assembly enables the formation of transient conducting nanostructures. Nat. Chem 10, 696–703. [DOI] [PubMed] [Google Scholar]
- 11.Draper ER, Eden EGB, McDonald TO, and Adams DJ (2015). Spatially resolved multicomponent gels. Nat. Chem 7, 848–852. [DOI] [PubMed] [Google Scholar]
- 12.Wu D, Sinha N, Lee J, Sutherland BP, Halaszynski NI, Tian Y, Caplan J, Zhang HV, Saven JG, Kloxin CJ, and Pochan DJ (2019). Polymers with controlled assembly and rigidity made with click-functional peptide bundles. Nature 574, 658–662. [DOI] [PubMed] [Google Scholar]
- 13.Bera S, Mondal S, Xue B, Shimon LJW, Cao Y, and Gazit E (2019). Rigid helical-like assemblies from a self-aggregating tripeptide. Nat. Mater 18, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui H, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ, and Stupp SI (2010). Spontaneous and x-ray-triggered crystallization at long range in self-assembling filament networks. Science 327, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frederix PWJM, Scott GG, Abul-Haija YM, Kalafatovic D, Pappas CG, Javid N, Hunt NT, Ulijn RV, and Tuttle T (2015). Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem 7, 30–37. [DOI] [PubMed] [Google Scholar]
- 16.Omosun TO, Hsieh MC, Childers WS, Das D, Mehta AK, Anthony NR, Pan T, Grover MA, Berland KM, and Lynn DG (2017). Catalytic diversity in self-propagating peptide assemblies. Nat. Chem 9, 805–809. [DOI] [PubMed] [Google Scholar]
- 17.Brangwynne CP, Tompa P, and Pappu RV (2015). Polymer physics of intracellular phase transitions. Nat. Phys 11, 899–904. [Google Scholar]
- 18.Dobson CM (2004). Chemical space and biology. Nature 432, 824–828. [DOI] [PubMed] [Google Scholar]
- 19.Smith DJ, Brat GA, Medina SH, Tong D, Huang Y, Grahammer J, Furtmüller GJ, Oh BC, Nagy-Smith KJ, Walczak P, et al. (2016). A multiphase transitioning peptide hydrogel for suturing ultrasmall vessels. Nat. Nanotechnol 11, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng Z, Han X, Wang H, Tang T, and Xu B (2019). Enzyme-Instructed Peptide Assemblies Selectively Inhibit Bone Tumors. Chem 5, 2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudalla GA, Sun T, Gasiorowski JZ, Han H, Tian YF, Chong AS, and Collier JH (2014). Gradated assembly of multiple proteins into supramolecular nanomaterials. Nat. Mater 13, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang G, Ren H, and Rao J (2010). A biocompatible condensation reaction for controlled assembly of nanostructures in living cells. Nat. Chem 2, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snider NT, and Omary MB (2014). Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat. Rev. Mol. Cell Biol 15, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desai A, and Mitchison TJ (1997). Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol 13, 83–117. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z, Gu H, Fu D, Gao P, Lam JK, and Xu B (2004). Enzymatic formation of supramolecular hydrogels. Adv. Mater 16, 1440–1444. [Google Scholar]
- 26.Hu BH, and Messersmith PB (2003). Rational design of transglutaminase substrate peptides for rapid enzymatic formation of hydrogels. J. Am. Chem. Soc 125, 14298–14299. [DOI] [PubMed] [Google Scholar]
- 27.Egelman EH (2014). Ambiguities in helical reconstruction. eLife 3, 04969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omary MB, Coulombe PA, and McLean WHI (2004). Intermediate filament proteins and their associated diseases. N. Engl. J. Med 351, 2087–2100. [DOI] [PubMed] [Google Scholar]
- 29.Reches M, and Gazit E (2003). Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 300, 625–627. [DOI] [PubMed] [Google Scholar]
- 30.Millán JL (2006). Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology (Wiley; ). [Google Scholar]
- 31.Merrifield RB (1963). Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc 85, 2149–2154. [Google Scholar]
- 32.Rodan SB, Imai Y, Thiede MA, Wesolowski G, Thompson D, Bar-Shavit Z, Shull S, Mann K, and Rodan GA (1987). Characterization of a human osteosarcoma cell line (Saos-2) with osteoblastic properties. Cancer Res. 47, 4961–4966. [PubMed] [Google Scholar]
- 33.Parton RG, Joggerst B, and Simons K (1994). Regulated internalization of caveolae J. Cell Biol. 127, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, et al. (2009). Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J. Med. Chem 52, 6919–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollard TD (2003). The cytoskeleton, cellular motility and the reductionist agenda. Nature 422, 741–745. [DOI] [PubMed] [Google Scholar]
- 36.Lucić V, Förster F, and Baumeister W (2005). Structural studies by electron tomography: from cells to molecules. Annu. Rev. Biochem 74, 833–865. [DOI] [PubMed] [Google Scholar]
- 37.Lee MJ, Mantell J, Hodgson L, Alibhai D, Fletcher JM, Brown IR, Frank S, Xue WF, Verkade P, Woolfson DN, and Warren MJ (2018). Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat. Chem. Biol 14, 142–147. [DOI] [PubMed] [Google Scholar]
- 38.Huotari J, and Helenius A (2011). Endosome maturation. EMBO J. 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellis RJ (2001). Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci 26, 597–604. [DOI] [PubMed] [Google Scholar]
- 40.Ribeiro SS, Samanta N, Ebbinghaus S, and Marcos JC (2019). The synergic effect of water and biomolecules in intracellular phase separation. Nat. Rev. Chem 3, 552–561. [Google Scholar]
- 41.Zhou HX, Rivas G, and Minton AP (2008). Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys 37, 375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall DM, Bruss IR, Barone JR, and Grason GM (2016). Morphology selection via geometric frustration in chiral filament bundles. Nat. Mater 15, 727–732. [DOI] [PubMed] [Google Scholar]
- 43.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, and Mackerell AD Jr. (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD Jr. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo S, Kim T, Iyer VG, and Im W (2008). Software news and updates - CHARNIM-GUI: A web-based grraphical user interface for CHARMM. J. Comput. Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y, Nguyen TD, Yang M, Lee B, Santos A, Podsiadlo P, Tang Z, Glotzer SC, and Kotov NA (2011). Self-assembly of self-limiting monodisperse supraparticles from polydisperse nanoparticles. Nat. Nanotechnol 6, 580–587. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura H, Lee AA, Afshar AS, Watanabe S, Rho E, Razavi S, Suarez A, Lin YC, Tanigawa M, Huang B, et al. (2018). Intracellular production of hydrogels and synthetic RNA granules by multivalent molecular interactions. Nat. Mater 17, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Liang G, Wang L, and Xu B (2006). Using a kinase/phosphatase switch to regulate a supramolecular hydrogel and forming the supramolecular hydrogel in vivo. J. Am. Chem. Soc 128, 3038–3043. [DOI] [PubMed] [Google Scholar]
- 49.Fletcher DA, and Mullins RD (2010). Cell mechanics and the cytoskeleton. Nature 463, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geng J, Li W, Zhang Y, Thottappillil N, Clavadetscher J, Lilienkampf A, and Bradley M (2019). Radical polymerization inside living cells. Nat. Chem 11, 578–586. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, and Cheng Y (2013). Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- 53.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, and Leith A (1996). SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol 116, 190–199. [DOI] [PubMed] [Google Scholar]
- 54.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- 55.Egelman EH (2000). A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy 85, 225–234. [DOI] [PubMed] [Google Scholar]
- 56.Afonine PV, Klaholz BP, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, and Urzhumtsev A (2018). New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Cryst. Sec. D Struct. Biol 74, 814–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Cryst. Sect. D Biol. Cryst 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 58.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. Sect. D Biol. Cryst 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao Y, Berciu C, Kuang Y, Shi J, Nicastro D, and Xu B (2013). Probing nanoscale self-assembly of nonfluorescent small molecules inside live mammalian cells. ACS Nano 7, 9055–9063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- 61.Iancu CV, Tivol WF, Schooler JB, Dias DP, Henderson GP, Murphy GE, Wright ER, Li Z, Yu Z, Briegel A, et al. (2006). Electron cryotomography sample preparation using the Vitrobot. Nat. Protoc 1, 2813–2819. [DOI] [PubMed] [Google Scholar]
- 62.Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- 63.Mastronarde DN (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol 120, 343–352. [DOI] [PubMed] [Google Scholar]
- 64.(2020)URSGDTZKNLF/D SEPIRG
- 65.JRFZRGBIOHGOIL(2125)
- 66.KJZFBKLFJEAOL(2000)
- 67.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindah E (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. [Google Scholar]
- 68.Darden T, York D, and Pedersen L (1993). Particle mesh Ewald: An N$log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- 69.Nosé S (1984). A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys 81, 511–519. [Google Scholar]
- 70.Hoover WG (1985). Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Physiol 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- 71.Parrinello M, and Rahman A (1980). Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett 45, 1196–1199. [Google Scholar]
- 72.Hess B, Bekker H, Berendsen HJC, and Fraaije JGEM (1997). LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472. [Google Scholar]
- 73.Plimpton S (1995). Fast parallel algorithms for short-range molecular dynamics. J. Comp. Physiol 117, 1–19. [Google Scholar]
- 74.Weeks JD, Chandler D, and Andersen HC (1971). Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys 54, 5237–5247. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the paper and the Supplemental Information or are available from the lead contact upon reasonable request. The accession numbers for the filaments of 1-KMe3 reported in this paper are EMDB: EMD-22051 and PDB: 6X5I.