

Abstract
Neuropathic pain, epilepsy, insomnia, and tremor disorder may arrive from an increase of intracellular Ca2+ concentration through a dysfunction of T-type Ca2+ channels. Thus, T-type calcium channels could be a target in drug discovery for the treatments of neuropathic pain and epilepsy. From rational drug design approach, a group of 2,5-disubstituted 1,3,4-oxadiazole molecules was synthesized and their selective T-type channel inhibitions were evaluated. The synthetic strategy consists of a short sequence of three reactions: (i) condensation of thiosemicarbazide with acid chlorides; (ii) ring closing by 1,3-dibromo-5,5- dimethylhydantoin; and (iii) coupling with various acid chlorides. 5-Chloro-N-(5- phenyl-1,3,4-oxadiazol-2-yl)thiophene-2-carboxamide (11) was found to selectively inhibit T-type Ca2+ channel over Na+ and K+ channels in mouse dorsal root ganglion neurons and/or human embryonic kidney (HEK)-293 cells and to suppress seizure-induced death in mouse model. Consequently, compound 11 is a useful probe for investigation of physiologic and pathophysiologic roles of the T-channel, and provides a basis to develop a novel therapeutic to treat chronic neuropathic and inflammatory pains.
INTRODUCTION
Voltage-gated calcium channels (VGCCs) are transmembrane, multi-subunit proteins that modulate influx of calcium ions (Ca2+) into the cell in response to membrane depolarization. According to different activation potentials, generally VGCCs can be classified as high voltage-activated (L-, N-, P-/Q- and R-types) and low voltage-activated (T-type) channels.1,2 T-type Ca2+ channels (or T-channels) consist of three distinct channel proteins, Cav3.1, Cav3.2, and Cav3.3 (or α1-G, α1-H, and α1-I) that regulate neural excitability and are involved in pathophysiological disorders, such as neuropathic pain, epilepsy, insomnia, and tremor disorders.3–7 These three sub-types are heterogeneously expressed in the brain and organs such as the heart, vascular smooth muscle, non-vascular smooth muscle, skeletal muscle and others.8–13 The opening of calcium channels leads to an increase of intracellular Ca2+ concentration and subsequent membrane depolarization. This has an effect on several important processes including muscle contraction,13 electrical conduction, neurotransmission, and neuropathic pain.3 The mechanism in neuropathic pains involving over-activation of T-type channel remains unclear and current pharmaco-therapeutics do not adequately control neuropathic pain conditions. In addition, T-type calcium channels also involve in hormone secretion, mechanosensation, and epilepsy.14 Epilepsy leads to disturbances in brain electrical activity and seizure, which is one of the most frequently occurring neurological diseases.15 In rodent models, an increase of T-type current was found in thalamic nurons,16,17 and there was no effect on the amplitude of L-type current, indicating that epilepsy resulted from a selective increase of T-type current. Thus, T-type calcium channel is a good target for the treatments of neuropathic pains and epilepsy.
A number of T-type calcium channel inhibitors have been reported18–25 and representative bioactive molecules are summarized in Figure 1. Mibefradril was a FDA approved anti-hypertension drug and selectively inhibits T-type (IC50=0.1 μM) over L-type (IC50=3 μM) calcium channels.21 However, due to severe drug-drug interactions, it was retracted from market.26 Pyrazine TTA-A8 has been reported to display excellent potency and short-acting selective inhibition of T-type channels and evaluated in human clinical trials in 2010.27 Piperidine Z944 has been reported to selectively inhibit T-type calcium channel (Cav3.2),28 but it showed only weak protective effect on seizures from pentylenetetrazole-induced fatality rat model (33% at 30 mg/Kg).29 Despite the presence of a number of calcium channel inhibitors, satisfactory drugs remain to be developed due to various issues including selectivity, efficacy, and safety profile. Only amlodipine, nicardipine, and nimodipine are FDA-approved drugs for clinical use, and these molecules not only block T-type calcium channels but also L-type and Na- and K-channels.30 Several 1,3,4-oxadiazoles have been reported to act as L- and T-type calcium channel blockers.25,31,32 For instance, through several high throughput screening studies, compound CID3373841 (Figure 1) was found to selectively inhibit T-type calcium channel towards L- and N-type channels.20 Further structural optimization led to a more potent inhibitor CID46943243, which suggested that 1,3,4-oxadiazole scaffold could be a good pharmacophore for the inhibition of T-type calcium channels. To discover potent and selective T-channel inhibitors for the treatment of neuropathic pain, we took a rational drug design approach and report herein the synthesis and selective T-type channel inhibitions of a group of 2,5-disubstituted 1,3,4-oxadiazole molecules.
Figure 1.
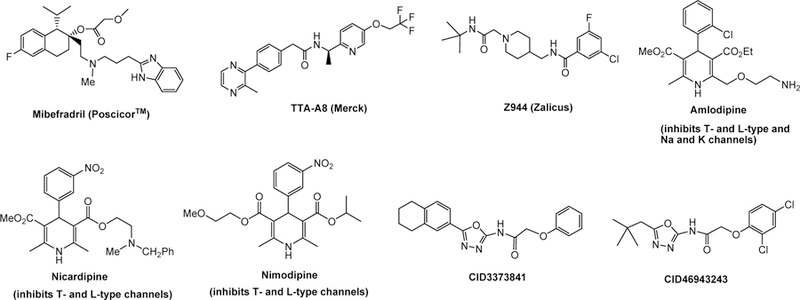
Representative T-type calcium channel blockers
RESULTS AND DISCUSSION
There are some common structural features shared by the reported oxadiazole blockers (Figure 1) and they include (i) all of them have at least one aromatic ring and one or more basic nitrogen atom(s); (ii) some have amide bond(s) and flexible short aliphatic alkane chains; and (iii) a molecular planarity is not required. These are good guidances in the molecular design. Based on these common features, computational modeling of the pharmacophores, and predictive Log P values (partition coefficient), a small library of ten 1,3,4-oxadiazole containing molecules, 6–15, were designed, synthesized and bio-evaluated (Figure 2). A retrosynthesis of 1,3,4-oxadiazole scaffold 1 is depicted in Figure 2 involving the preparations of four representative 2-amino-5-aryl-1,3,4-oxadiazole molecules, 2A–2C and 4, followed by either simple coupling reactions with various acid halides 3A–3D or displacement reactions with 5A or 5B. The amine NH2 function of 2A–2C serves as a nucleophile while the chloroamide function of 4 serves as an electrophile. The 5-substituted 2-amino-1,3,4-oxadiazoles can be made by the amide bond formation of thiosemicarbazide (16) and acid chlorides followed by ring closing through halogenation. Different substituents were installed on C2 and C5 of the 1,3,4-oxadiazole ring to achieve bioactivities. Modification in part A, R1 substituent, focused on different aromatic rings to adjust the molecular lipophilicity. Diverse functional groups were introduced in part B, R2-C=O, to explore the structural features such as rigidity, flexibility, planarity, π−π interaction, and steric hindrance, which may have an effect on potency. 5-Chlorothiophenyl moiety was chosen in R1 or R2 due in part based on the reported bioactivities of the thiophene scaffold including that in the central nervous system.33
Figure 2.
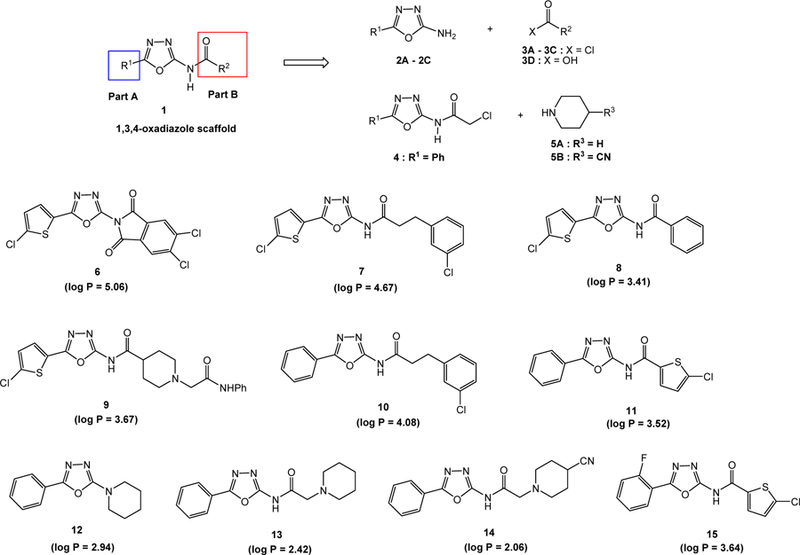
Designed substituted 1,3,4-oxadiazole compounds as T-type calcium channel inhibitors
Predicted log P values of compounds 6–15 were calculated using interactive log P calculator (Molinspiration Cheminformatics 2017; http://www.molinspiration.com/services/logp.html) and the calculated values are described in Figure 2. Except compound 6 with log P value slightly over 5, all other designed 1,3,4-oxadiazole derivatives have log P values between 2 to 5, indicating a suitable hydrophobicity as drug-like molecules.
The syntheses of 2-amino-5-aryl-1,3,4-oxadiazoles34 appear to be general and are depicted in Scheme 1. Hence, coupling of thiosemicarbazide (16) with different acyl chlorides 17A, 3C, and 17B in THF at 25°C gave 1-aryl-3-thiosemicarbazides 18A–18C in 60–100% yields (Scheme 1). The cyclization reactions of 18A–18C were affected by the treatment with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) and potassium iodide under basic conditions to give 1,3,4-oxadiazoles 2A–2C, respectively, in moderate to good yields. A possible mechanism for the cyclization has been reported,35 involving iodination (I2 is generated from DBDMH and KI) of the thioamide moiety followed by elimination of NaSH, H2O and NaI, and annulation from an intramolecular addition reaction of the resulting diimide function by the adjacent amido oxygen. α-Chloroamide 4 was readily prepared in a 79% yield by the coupling of oxadiazole 2B with chloroacetyl chloride.
Scheme 1.
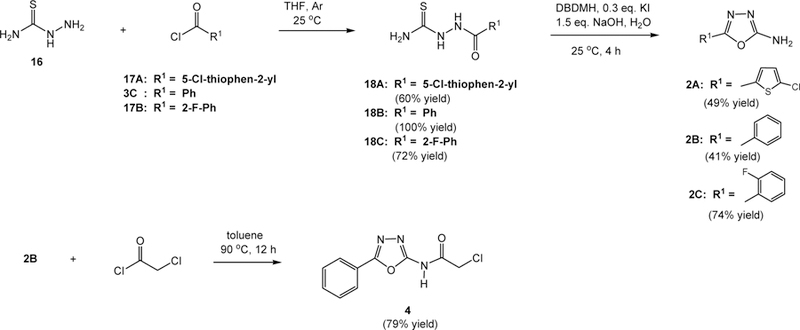
Syntheses of 1,3,4-oxadiazoles 2A–2C and 4
With these four 1,3,4-oxadiazoles, 2A–2C and 4, on hands, the intended 2,5-disubstituted oxadiazoles 6–15 were synthesized and the synthetic reactions are depicted in Schemes 2 and 3. First, amine 2A was condensed with aryl halides 3A–3C, separately, in the presence of pyridine to give oxadiazoles 6–8 in 22, 64, and 30% yield, respectively, along with unidentifiable byproducts and small amounts of starting material, 2-aminooxadiazole 2A. An attempt to improve the yields by changing the reaction solvent to N,N-dimethylformamide failed, and no desired products were obtained. The poor solubility of the products such as 6 and 8 in common organic solvents (diethyl ether, ethyl acetate, and dichloromethane) likely led to the decrease of isolated chemical yields. Oxadiazole 9 was synthesized by the condensation of 2A and carboxylic acid 3D in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-(dimethylamino)pyridine (DMAP). After purification, an 11% yield of 9 and 18% recovery of 2A were isolated. Treatment of 3D with oxalyl chloride failed to produce the corresponding acid chloride. Carboxylic acid 3D was made from the coupling of 4-cyanopiperidine (19) and 2-chloro-N-phenylacetamide (20) in the presence of potassium carbonate in acetonitrile followed by basic hydrolysis of the cyanide group. It was used in the synthesis of 9 as described above without purification.
Scheme 2.
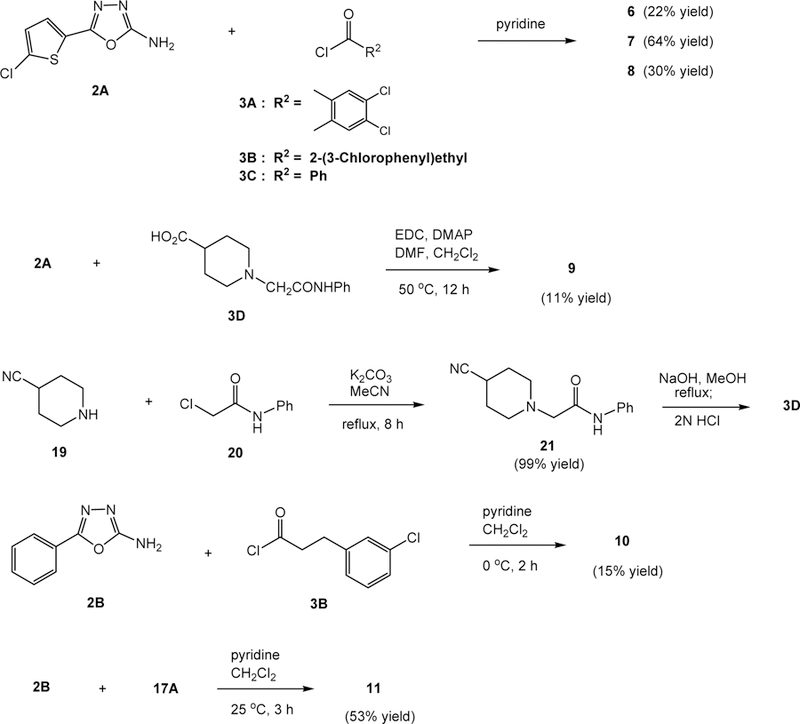
Syntheses of 1,3,4-oxadiazoles 6–11
Scheme 3.
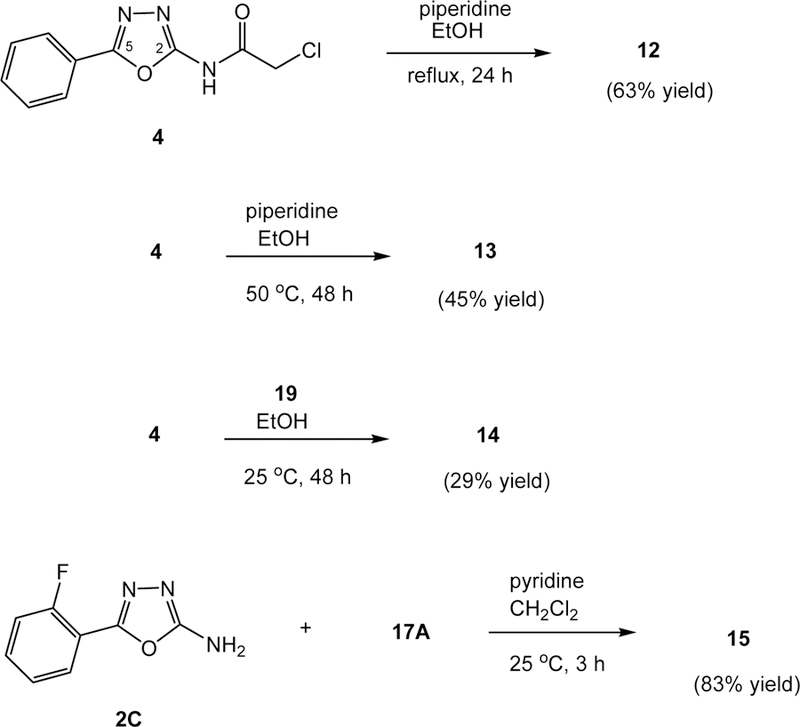
Syntheses of 1,3,4-oxadiazoles 12–15
Oxadizoles 10 and 11, containing phenyl ring at R1, were synthesized in 15% and 53% yield, respectively, from the coupling reactions of 2-amino-5-phenyl-1,3,4-oxadiazole (2B) with 3-(3-chlorophenyl)propanoyl chloride (3B) and 5-chlorothiophene-2-carbonyl chloride (17A) separately in dichloromethane (Scheme 2). Acid chlorides 3B and 17A were prepared from the corresponding carboxylic acids and oxalyl chloride in ethyl acetate in the presence of a catalytic amount of DMF.
Chloroacetamide 4 serves as an electrophile for various substitution reactions through a displacement of the chlorine atom. To our surprise, treatment of 4 with piperidine in refluxing ethanol gave a 63% yield of an unexpected product, 12, in which the chloroacetamide unit was replaced by the piperidine molecule (Scheme 3). The reaction temperature, 82°C, was considered to be the culprit, leading to this unusual displacement reaction. Piperidine likely adds to C2 of the oxadiazole ring providing the conjugate iminoamine anion, which in turn undergoes elimination of the chloroacetamide anion (ClCH2CONH−) producing 2-phenyl-5-piperidyl-1,3,4-oxadiazole (12). To verify such consideration, 4 was treated with piperidine in ethanol at 50°C for 48 h and the expected displacement product 13 was isolated in 45% yield along with unidentifiable byproducts after silica gel column chromatographic separation. There was no reaction at 25°C. To generate a similar analog, compound 4 was allowed to react with 4-cyanopiperidine (19) in ethanol. Interestingly, the reaction took place at 25°C and afforded the expected displacement product, 14, in a 29% yield along with other byproducts. Likely, product 14 undergoes further alkylation reaction with 4 producing the byproducts. The C4-cyano group lowers the pKa value of the NH of 19, which may lead to a faster rate of the SN2 displacement reaction.
Based on the favorable bioactivities found from oxadiazole 11 (vide infra), 2-fluorophenyl analog 15 was synthesized in an 83% yield via a similar coupling reaction of amine 2C and thiophenecarbonyl chloride 17A (Scheme 3). A higher yield in this reaction, comparing with those found in the syntheses of 6–8 and 11, suggests that the fluorine atom in the ortho position of the C5-phenyl ring preventing undesired acylation on the oxadiazole ring.
Oxadiazoles 6 – 15 and a reported T-type calcium channel blocker, Z944 (Figure 1), were first studied their inhibitions of T-Cav3.2 −, Na-, and K-channels, separately, in mouse dorsal root ganglion (DRG) neurons using whole-cell patch clamp recording protocol,29 and results are summarized in Table 1. A few compounds were tested in T-Cav3.2-expressed in human embryonic kidney (HEK)-293 cells from a tetracycline inducible expression system (Invitrogen; see Experimental Section shown below). It has been reported that an over activation of T-channel resulted in the generation of seizure activity,36 hence prolonged latencies to seizures and decreased the fatality rate in seizure mouse model would affirm the inhibition of T-channel activity. The active compounds were consequently evaluated in mouse model for resistance of neuropathic pain by pentylenetetrazole (PTZ)-induced seizures method.36 The inhibition ratio (5th column in Table 1) is the number of mice died among the tested mice, there is no mean or standard error of the mean. For example, if 2 of 6 mice have died, the death ratio would be 33.3% and the inhibition ratio of fatality would be 100−33.3=66.6%. The latency to fatality during the first 20 minutes of testing is also shown in Table 1 (6th column). There are mean and standard error of the mean because each mouse has a value of latency.
Table 1.
In vitro and in vivo bioactivities of 1,3,4-oxadiazoles 6 – 15 and Z944. n = number of experiment; IC50 is half maximal inhibitory concentration; IA stands for rapid inactivating potassium current; IKd stands for delayed rectifier potassium current; NT: not tested.
| 1,3,4-Oxadiazoles | Inhibition (%) of T-Cav3.2- channel in DRG neurons at 1 µM (unless specified) | Inhibition (%) of Na- channel in DRG neurons at 1 µM (unless specified) | Inhibition (%) of K-channel in DRG neurons at 1 µM (unless specified) | Inhibition ratio (%) of fatality in PTZ-model of mice, at 30 mg/Kg | Latency to fatality (minutes) in PTZ model of mice, at 30 mg/Kg |
|---|---|---|---|---|---|
| 6 | 9.8 (3.3 µM) (n=1) | 100 (3.3 µM) (n=1) | −11.6 (IA) −17.8 (IKd) (3.3 µM (n=1) | 0 (n=8) | 1.9±0.3 (n=8) |
| 7 | 20.4±1.9 (n=2) | 82.2 (3.3 μM) (n=1) | −26.0 (IA) −15.6 (IKd) (3.3 µM) (n=1) | 28.6 (n=7) | 13.6±1.8 (n=7) |
| 8 | 24.4±9.6 (n=3) | −71.8 (n=1) | −18.7 (IA) −28.6 (IKd) (n=1) | NT | |
| 11 | 50±9.7 (IC50=0.93 µM) (n=8) | 16.5±8.9 (n=7) | 18.9±11.8 (IA) 24.1±14.6 (IKd) (n=4) | 72.7 (n=11) | 15.3±2.4 (n=11) |
| 12 | 44.2±5.6 64.6±14.1 (in HEK293 cells) (n=2) | 25.7±6.3 (IC50=2.1 µM) (n=5) | 21.9±8.6 (IA) (IC50=19.6 µM) (n = 4) 31.6±5.5 (IKd) (IC50=2.6 µM) (n=4) | NT | |
| 13 | 55.4±28.7 (n=2) | −2.7 (n=1) | 19.9 (IA) 36.5 (IKd) (n=1) | 0 (n=7) | 3.7±1.8 (n=7) |
| 14 | 69.6±6.2 (n=2) | −4.7 (n=1) | 12.9 (IA) 30.0 (IKd) (n=1) | 14 (n=7) | 11.8±2.5 (n=7) |
| Z944 | 74.5±7.8 (0.25 µM, in HEK293 cells) (n=6) | 49.7±6 (IC50=0.51 µM) (n=3) | 34.5±15.0 (IA) 44.9±17.7 (IKd) (n=2) | 33.3 (n=6) | 15.2±3.1 (n=6) |
As described in Table 1 all ten oxadiazoles possess weak to strong inhibitions of T-type calcium channel and particularly molecules 11–14 exhibited strong activities at 1 µM concentration. Molecules 9 and 15 although are active in the inhibition of Ca-channel, 79.5 and 60.9%, respectively, were not investigated further due to its low solubility issue. Compound 10 showed weak inhibition of 5% and was not studied. The three compounds, 9, 10, and 15, are not listed in Table 1. Oxadiazole 6–8 and 11–14 were also studied for their inhibition selectivity towards sodium and potassium channels. Molecules 6 and 7 weakly inhibited Ca-channels with 9.8 and 20.4% inhibition, respectively, and strongly inhibited Na-channel with respective 100 and 82.2%, hence they were not investigated further. Molecule 8 showed weak inhibition, 24.4%, in Ca-channel and negative inhibition in Na- and K-channel. Molecules 12 - 14 although revealed promising Ca-channel inhibition (44.2, 55.4 and 69.6%, respectively), but they also inhibited K-channel and were not pursued further. Compound 11 showed good inhibition (50%) in Ca-channel and lesser potencies in Na- and K-channels (~16–19%). The reported inhibitor, Z944, on the other hand showed a much stronger inhibitory activity against Ca-channel, 74.5% at 0.25 µM. However, it also blocked Na- and K-channels in 49.7 and 34.5%, respectively. Notably, an increase of potassium (K+) current has been shown to decrease the excitability of hippocampal neurons such as CA1 pyramidal neurons, after ischemia.37 Moreover, increases of the amplitude of rapid inactivating potassium currents, IA, and delayed rectifier potassium currents, IKd, in neurons are implicated in the damage of neuronal death after ischemia. Hence, detailed studies of the inhibition of K+ channels by measuring IA and IKd values were carried out and results are listed in Table 1. Interestingly, oxadiazoles 11–14 showed moderate inhibitory activities with IA and IKd values range from 12.9 to 31.6%. The positive control molecule, Z944, also possesses moderate activities with IA and IKd values of 34.5 and 44.9%, respectively. Based on these initial channel inhibitory studies, compounds 6, 7, 11, and Z944 were further bio-evaluated towards their inhibition of seizure-induced mouse model. Compounds 6 and 7 were used as negative controls to verify that the inability in inhibition of Ca-channel resulted in fatality. In this study, fatality rates were calculated as the percentage of mice within each treatment group that died within the 20-min observation period, which revealed 1,3,4-oxadiazole molecules’ ability to inhibit neuropathic pains. As predicted, oxadiazoles 6 and 7 showed none to low suppression (0 and 30%) of seizure-induced death. Compound 11 was the best inhibitor that produced a 72.1% of suppression of seizure-induced death. Under similar conditions, Z944 affected only 33% of inhibition of seizures. Results of the seizure studies indicated that inhibition of T-type calcium channels would lead to inhibition of seizures. Thus, T-type calcium channels could potentially be the biological target for the treatment of neuropathic pain and epilepsy. It appears that aryl and rigid rings in parts A and B (see Figure 1) provide Ca-channel inhibition and reduce neuropathic pain, while aliphatic chain in part B reduces the activities.
CONCLUSION
A series of 1,3,4-oxadiazole derivatives was designed and synthesized as T-type calcium channel blockers. The synthetic sequence is relatively short and should provide a general route for the construction of a library of 1,3,4-oxadiazole molecules for structure-activity relationship study. An unusual displacement reaction of a C2-chloroacetamide unit of oxadiazole 4 by piperidine was found. The synthesized molecules were screened for their ability and selectivity towards inhibition of T-type calcium channel. Two hit compounds, 11 and 15, were found to possess good inhibitory activities on T-type Ca2+ currents and lower activities on voltage-gated Na+ or K+ currents. Enhancement of T-type calcium channel inhibition may be achieved through further structural modification of C5-aryl ring of 1,3,4-oxadiazole scaffold. Studies on seizure-induced mouse model showed that the inhibition of T-type calcium channel could lead to inhibition of seizures or epilepsy. Among various 1,3,4-oxadiazole derivatives, compound 11 was found to be the lead compound for future structural optimization in the treatment of neuropathic pain and epilepsy.
EXPERIMENTAL
Reagent oxalyl chloride was purchased from Fisher Scientific Inc., 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide (EDC) and N,N-dimethylaminopyridine (DMAP) from VWR international LLC, and 5-chlorothiophene-2-carboxylic acid, 1,3-dibromo-5,5-dimethylhydantoin, 3-(3- chlorophenyl)propionic acid, 4-cyanopiperidine, and thiosemicarbazide from Chem-Impex International, Inc. Dry THF and diethyl ether were freshly distilled over sodium-benzophenone under argon. Dichloromethane, toluene, and N,N-dimethyl formamide (DMF) were distilled (under 5 mm Hg of vacuum for DMF) over calcium hydride under argon. 1H NMR (400 MHz) and 13C NMR spectra (100 MHz) spectra were obtained from a 400-MHz Varian NMR spectrometer, and were measured from a solution in CDCl3 unless otherwise mentioned. The chemical shift data for each signal on 1H NMR are given in units of δ relative to TMS (δ=0) or CHCl3 (δ=7.26 ppm). For 13C NMR spectra, the chemical shifts are recorded relative to CDCl3 (δ=77.0 ppm). Mass spectra were taken from an API 2000-triple quadrupole Electrospray ionization-MS/MS mass spectrometer (Applied Biosystems).
N-(Carbamothioylamino)-5-chlorothiophene-2-carboxamide (18A).
To a solution of 1.0 g (6.1 mmol) of 5-chlorothiophene-2-carboxylic acid in 13 mL of distilled EtOAc at 0°C under argon, 0.94 g (7.4 mmol) of oxalyl chloride was added followed by the addition of 2 drops of distilled DMF. The resulting solution stirred at 0°C for 2 h and concentrated to dryness under vacuum to yield 5-chlorothiophene-2-carbonyl chloride (17A), which was used in the subsequent step without purification. To a cold (0°C) suspension of 1.22 g (13 mmol) of thiosemicarbazide (16) in 10 mL of distilled THF under argon, was added slowly a solution of 1.10 g (6.1 mmol) of 17A in 10 mL of THF. The resulting mixture warmed to room temperature and stirred for 12 h. To it, 100 mL of water was added and the precipitated white solids were collected by filtration, washed twice with water, and dried under vacuum to yield 0.87 g (60% yield) of compound 18A as white solids, mp 198.5–199.5°C. - 1H NMR (DMSO-d6): δ 10.50 (s, 1H), 9.37 (s, 1H), 7.22 (d, J = 4.3 Hz, 1H), 7.92 (s, 1H), 7.61–7.76 (m, 2H) ppm. - 13C NMR (DMSO-d6): δ 183.1, 160.8, 137.5, 134.7, 130.5, 129.1 ppm. - MS (positive mode): m/z calcd for C6H6ClN3OS2Na (M+Na)+: 258.0, found 258.3.
1-Benzoyl-3-thiosemicarbazide (18B).
To a cold (0°C) suspension of 4.0 g (43.8 mmol) of thiosemicarbazide (16) in 100 mL of THF under argon was added a solution of 2.8 g (19.9 mmol) of benzoyl chloride (3C) in 20 mL of THF. The resulting reaction mixture was stirred at 25°C for 12 h, concentrated on a rotary evaporator to remove THF, and diluted with 100 mL of water and 10 mL of aqueous NaHCO3 solution. The precipitated white solids collected by filtration, washed with hexane, and dried under vacuum to yield 3.8 g (100% yield) of compound 18B as white solids: mp 115–116°C. - 1H NMR (DMSO-d6): δ 9.36 (s, 1H), 7.79–7.93 (m, 3H), 7.62 (s, 1H), 7.51–7.59 (m, 2H), 7.47 (t, J=7.1 Hz, 2H) ppm. - 13C NMR (DMSO-d6): δ 183.5, 168.6, 134.5, 132.9, 130.4, 129.6 ppm. - MS (positive mode): m/z calcd for C8H9N3OSNa (M+Na)+: 218.05, found 218.1.
1-(2-Fluorobenzoyl)-3-thiosemicarbazide (18C).
To a cold (0°C) suspension of 1.43 g (15.7 mmol) of 16 in 30 mL of THF under argon was added a solution of 1.13 g (7.1 mmol) of 2-fluorobenzoyl chloride (17B) in 15 mL each of THF and CH2Cl2 via cannula. The mixture was stirred at 25°C for 12 h, concentrated on a rotary evaporator to remove most of the solvents, and the precipitated solids were filtered, washed with CH2Cl2 and water, and dried under vacuum to give 1.11 g (72% yield) of 18C as white solids, mp 203–203.5°C. - 1H NMR (DMSO-d6): δ 10.19 (s, 1H), 9.44 (s, 1H), 7.96–7.90 (m, 1H), 7.86–7.80 (m, 1H), 7.56–7.50 (m, 2H), 7.30–7.26 (m, 2H) ppm. - 13C NMR (DMSO-d6): δ 182.8, 164.3, 160.4 (d J = 250 Hz, CF), 134.1, 131.6, 125.2, 122.9, 117.2 ppm. - MS (positive mode): m/z calcd for C8H8FN3OSNa+ (M+Na)+: 236.2, found 236.1.
5-(5-Chlorothiophen-2-yl)-1,3,4-oxadiazol-2-amine (2A).
To a cold (0°C) solution of 0.61 g (3.69 mmol) of KI in 7 mL of water were added 2.9 g (12.3 mmol) of 18A, 20 mL of water, and 4.6 mL of 4 N NaOH solution (18.5 mmol) and the resulting solution was stirred for 5 min. To it, was added 3.87 g (13.5 mmol) of 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) and the solution was stirred at 25°C for 7 h, diluted with 3.6 mL of saturated NaHSO3 aqueous solution, and extracted four times with EtOAc (150 mL each). The combined organic layers were washed with brine, dried (anhydrous Na2SO4), concentrated, and crystallized from EtOAc (80 mL) to yield 1.20 g (49% yield) of compound 2A as white solids, mp 250 – 253°C. - 1H NMR (DMSO-d6): δ 7.33–7.38 (m, 3H), 7.24 (d, J=4.3 Hz, 1H) ppm. - 13C NMR (DMSO-d6): δ 163.5, 152.6, 130.7, 128.3, 126.9, 124.6 ppm. - MS (positive mode): m/z calcd for C6H5ClN3OS (M+H)+ 202.0, found 202.0.
5-Phenyl-1,3,4-oxadiazol-2-amine (2B).
The aforementioned procedure was followed and starting from 3.8 g (19.5 mmol) of compound 18B, 1.3 g (41% yield) of compound 2B was obtained as yellow solids, mp 156–158°C.−1H NMR (DMSO-d6): δ 7.68–7.94 (m, 2H), 7.40–7.62 (m, 3H), 4.90–4.80 (bs, 2H) ppm. - 13C NMR (DMSO-d6): δ 164.9, 156.9, 131.3, 130.2, 126.0, 125.4 ppm. - MS (positive mode): m/z calcd for C8H8N3O (M+H)+ 162.06, found 162.3.
5-(2-Fluorophenyl)-1,3,4-oxadiazol-2-amine (2C).
The aforementioned procedure for the synthesis of 2A was followed. Starting from 1.08 g (5.07 mmol) of 18C, 0.25 g (1.52 mmol), 0.3 g (7.52 mmol) of NaOH, and 1.59 g (5.57 mmol) of DBDMH, after purification using silica gel column chromatography and a mixture of CH2Cl2 and MeOH (10:1) as an eluent, 0.68 g (74% yield) of compound 2C was obtained as light yellow solids, mp 218.5–219.5°C. - 1H NMR (DMSO-d6): δ 7.83 (t, J = 8 Hz, 1H), 7.58–7.53 (m, 1H), 7.42–7.33 (m, 4H) ppm. - 13C NMR (DMSO-d6): δ 165.0, 159.5 (d, J = 251 Hz, CF), 156.9, 133.4, 129.4, 126.0, 118.0, 113.6 ppm. - MS (positive mode): m/z calcd for C8H7FN3O+ (M+H)+ 180.2, found 180.1.
2-Chloro-N-(5-phenyl-1,3,4-oxadiazol-2-yl)acetamide (4).
To a cold (0°C) solution of 0.331 g (2.05 mmol) of compound 2B in 10 mL of toluene under argon, was added 0.2 mL (2.47 mmol) of chloroacetyl chloride and the solution was stirred under reflux for 12 h. The reaction solution was cooled to 25°C, concentrated on a rotary evaporator, and column chromatographed on silica gel using a gradient mixture of CH2Cl2 and MeOH as eluent to give 0.385 g (79% yield of compound 4. - 1H NMR (DMSO-d6): δ 12.35 – 12.18 (bs, 1 H), 8.02–7.82 (m, 2 H), 7.72 – 7.47 (m, 3 H), 4.46 (s, 2 H) ppm. - 13C NMR (DMSO-d6): δ 164.5, 160.7, 157.0, 131.9, 129.6, 126.1, 123.3, 43.2 ppm. - MS (positive mode): m/z calcd for C10H8ClN3O2Na (M+Na)+: 260.6, found 260.4; and C10H9ClN3O2+ (M+H)+: 238.65, found 238.2 (100%).
5,6-Dichloro-2-(5-(5-chlorothiophen-2-yl)-1,3,4-oxadiazol-2-yl)isoindoline-1,3-dione (6).
To a solution of 94 mg (0.35 mmol) of 4,5-dichlorophthalic acid in 4 mL of CH2Cl2 under argon at 0°C, 0.107 g (0.84 mmol) of oxalyl chloride was added dropwise followed by 1 drop of DMF. The resulting solution was stirred under reflux for 3.5 h, cooled to 25°C, and concentrated to dryness to give 95 mg (100% yield) of 4,5-dichlorobenzene-1,2-dioyl dichloride (3A) as white solids. This material was used in the subsequent step without purification. To a suspension of 70 mg (0.35 mmol) of compound 2A in 3 mL of CH2Cl2 under argon at 0°C, were added 82 mg (1.04 mmol) of pyridine and a solution of 95 mg (0.35 mmol) of 3A in 0.5 mL of dichloromethane. The resulting solution was stirred at 25°C for 5 h, and diluted with 200 mL of CH2Cl2 and 30 mL of 10% aqueous NaHCO3 solution. The organic layer was separated and washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluent to give 30 mg (22% yield) of compound 6. - 1H NMR (DMSO-d6): δ 8.48–8.40 (m, 2H), 7.75 (d, J = 4 Hz, 1H), 7.40 (d, J = 4 Hz, 1H) ppm. - 13C NMR (DMSO-d6): δ 161.7, 159.3, 151.2, 138.7, 134.4, 131.1, 131.0, 129.1, 126.6, 122.2 ppm. - MS (positive mode): m/z calcd for C14H4Cl3N3O3SNa (M+Na)+: 421.9, found 422.0.
3-(3-Chlorophenyl)propanoyl chloride (3B).
To a cold (0°C) solution of 46 mg (0.25 mmol) of 3-(3- chlorophenyl)propionic acid in 3 mL of EtOAc under argon were added 38 mg (0.30 mmol) of oxalyl chloride and 1 drop of DMF. The resulting solution was stirred at 0°C for 2 h, concentrated to dryness under vacuum to give 3-(3-chlorophenyl)propanoyl chloride (3B), which was used in the subsequent reaction without purification.
3-(3-Chlorophenyl)-N-(5-(5-chlorothiophen-2-yl)-1,3,4-oxadiazol-2-yl)propanamide (7).
To a suspension of 50 mg (0.25 mmol) of compound 2A in 3 mL of dichloromethane under argon, were added 30 mg (0.38 mmol) of pyridine and a solution of 50 mg (0.25 mmol) of 3B in 0.3 mL of CH2Cl2. The resulting solution was stirred at 25°C for 5 h and diluted with 300 mL of EtOAc and 40 mL of 10% aqueous NaHCO3 solution. The organic layer was separated and washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluents to give 59 mg (64% yield) of compound 7. - 1H NMR (DMSO-d6): δ 7.55 (d, J = 3.9 Hz, 1H), 7.41–7.16 (m, 5H), 2.91 (t, J = 7.4 Hz, 2H), 2.78 (t, J = 7.4 Hz, 2H) ppm. - 13C NMR (DMSO-d6): δ 169.8, 157.0, 155.6, 143.3, 132.9, 132.6, 130.2, 129.1, 128.6, 128.2, 127.1, 126.1, 123.3, 36.8, 29.5 ppm. – MS (positive mode): m/z calcd for C15H11Cl2N3O2SNa (M+Na)+ 390.0, found 389.9.
N-(5-(5-Chlorothiophen-2-yl)-1,3,4-oxadiazol-2-yl)benzamide (8).
To a hot (40°C) solution of 50 mg (0.25 mmol) of compound 2A and 31 μL of pyridine (0.37 mmol) in 5 mL of THF under argon was added 35 mg (0.25 mmol) of benzoyl chloride. The resulting solution was stirred at 40°C for 2 h, diluted with EtOAc, and washed with saturated aqueous NaHCO3 solution and brine, dried (anhydrous Na2SO4), concentrated, and columned chromatographed on silica gel using a gradient mixture of EtOAc and MeOH as eluent to give 23 mg (30% yield) of compound 8. - 1H NMR (DMSO-d6): δ 8.10–7.98 (m, 2H), 7.73–7.59 (m, 3H), 7.59–7.47 (m, 2H), 7.39–7.27 (m, 1H) ppm. - 13C NMR (DMSO-d6): δ 161.3, 133.2, 132.9, 132.7, 132.0, 130.4, 129.5, 128.9 (2C), 128.0, 127.6 (2C), 127.2 ppm. - MS (positive mode): m/z calcd for C13H8CN3O2SNa (M+Na)+ 328.0, found 328.0.
2-(4-Cyanopiperidin-1-yl)-N-phenylacetamide (21).
To a mixture of 3.0 g (17.7 mmol) of 2-chloro-N- phenylacetamide (20) and 4.88 g (35.3 mmol) of anhydrous K2CO3 in 25 mL of distilled MeCN under argon, was added 1.95 g (17.7 mmol) of 4-cyanopiperidine (19). The resulting mixture was stirred under reflux for 8 h, cooled to 25°C, filtered through a layer of Celite, concentrated, and column chromatographed on silica gel using a gradient mixture of CH2Cl2 and MeOH as eluent to give 4.28 g (99% yield) of compound 21. - 1H NMR: δ 8.90–8.80 (bs, 1H), 7.63–7.48 (m, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.09 (t, J=7.8 Hz, 1H), 3.10 (s, 2H), 2.93–2.35 (m, 5H), 2.23–1.81 (m, 4H) ppm. - 13C NMR: δ 168.0, 137.5, 129.2 (2C), 124.4, 121.3, 119.6 (2C), 62.3 (2C), 51.9, 29.1 (2C), 25.6 ppm. - MS (positive mode): m/z calcd for C14H18N3O (M+H)+: 244.0, found 244.5.
1-(2-Oxo-2-(phenylamino)ethyl)piperidine-4-carboxylic acid (3D).
To a solution of 0.138 g (0.57 mmol) of compound 21 in 5 mL of MeOH, was added 2 mL of 3 N NaOH and the solution was stirred under reflux for 12 h. It was cooled to 25°C, concentrated on a rotary evaporator to remove MeOH, neutralized with 2 N HCl solution, and lyophilized to dryness under vacuum to yield compound 3D as a HCl salt together with NaCl as white solids. The crude mixture was used in the subsequent reaction without purification.
1-((Phenylcarbamoyl)methyl)-N-(5-(5-chlorothiophen-2-yl)-1,3,4-oxadiazol-2-yl)piperidine-4- carboxamide (9).
A mixture of 0.15 g (0.57 mmol) of aforementioned compound 3D, 0.114 g (0.57 mmol) of compound 2A, 0.33 g (1.71 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 0.21 g (1.71 mmol) of 4-(N,N-dimethylamino)pyridine (DMAP) in 2 mL of DMF and 2 mL of CH2Cl2 was stirred under argon at 50°C for 12 h. The resulting mixture was cooled to 25°C, diluted with 40 mL of distilled water, adjusted the pH to 3 using 2 N HCl solution, and extracted with CH2Cl2 twice. The combined organic layers were washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of CH2Cl2 and MeOH as eluent to give 28 mg (11% yield) of compound 9 and 20 mg (18% recovery) of starting 2A. Compound 9: - 1H NMR (DMSO-d6): δ 11.90–11.78 (bs, 1H, NH), 9.69 (s, 1H, NH), 7.64 (d, J=8.2 Hz, 2H), 7.57 (d, J=3.9 Hz, 1H), 7.36−7.26 (m, 3H), 7.05 (t, J=7.2 Hz, 1H), 3.15−3.13 (s, 2H), 2.97–2.87 (m, 3H), 2.2 (t, J = 11.1 Hz, 2H), 1.90−1.67 (m, 4H) ppm. - 13C NMR (DMSO-d6): δ 172.8, 168.4, 157.1, 155.7, 144.7, 138.6, 132.6, 129.7, 129.2, 128.7 (2C), 123.7, 119.8 (2C), 62.0, 52.5 (2C), 41.9, 27.9 (2C) ppm. - MS (positive mode), m/z calcd for C20H21ClN5O3S (M+H)+: 446.1, found 446.3.
3-(3-Chlorophenyl)-N-(5-phenyl-1,3,4-oxadiazol-2-yl)propanamide (10).
To a cold (0°C) solution ofc 0.10 g (0.62 mmol) of 2B in 10 mL of CH2Cl2 under argon, were added 75 µL of pyridine and 0.126 g (0.62 mmol) of 3B. The solution was stirred for 2 h, warmed to 25°C, diluted with 20 mL of aqueous NaHCO3, and extracted three times with CH2Cl2. The combined extract was washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluent to give 30 mg (15% yield) of compound 10 as light yellow crystals, mp 201–203°C. - 1H NMR: δ 8.07 (d, J = 8 Hz, 2H), 7.60–7.50 (m, 3H), 7.36 (s, 1H, NH), 7.30–7.20 (m, 4H), 3.15–3.02 (m, 2H), 3.00–2.70 (m, 2H) ppm. - 13C NMR (DMSO-d6): δ 166.0, 164.1, 155.2, 132.8, 133.4, 132.6, 131.1, 130.4, 129.5, 129.2, 128.7, 128.1, 126.9, 35.8, 30.8 ppm. - MS (positive mode): m/z calcd for C17H15ClN3O2 (M+H)+: 329.8, found 330.1 (Cl37 isotope) and 328.0 (Cl35 isotope).
5-Chloro-N-(5-phenyl-1,3,4-oxadiazol-2-yl)thiophene-2-carboxamide (11).
To a cold (0°C) solution of 0.30 g (1.86 mmol) of compound 2B and 0.22 g (2.79 mmol) of pyridine in 10 mL of CH2Cl2 under argon, was added a solution of 0.34 g (1.86 mmol) of 5-chlorothiophene-2-carbonyl chloride (17A) in 5 mL of CH2Cl2. The resulting mixture was stirred at 25°C for 3.5 h, diluted with 40 mL of 10% aqueous NaHCO3 solution, and extracted three times with CH2Cl2. The combined extract was washed with brine, dried (anh. Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluent to give 0.30 g (53% yield) of compound 11. - 1H NMR: δ 8.22 (bs, 1H, NH), 8.12 (d, J=4.3 Hz, 1H), 7.98 (d, J=7.4 Hz, 2H), 7.67–7.49 (m, 3H), 7.06 (d, J=4.3 Hz, 1H) ppm. - 13C NMR: δ 158.3, 154.3, 151.3, 142.1, 137.3 133.3, 129.9, 129.4, 127.2, 127.1, 122.4 ppm. - MS (positive mode): m/z calcd for C13H8ClN3O2SNa (M+Na)+: 328.0, found 327.8.
1-(5-Phenyl-1,3,4-oxadiazol-2-yl)piperidine (12).
To a solution of 0.128 g (0.54 mmol) of 4 in 4 mL of EtOH under argon was added 60 µL (0.65 mmol) of piperidine and the solution was stirred under reflux for 24 h. The solution was cooled to 25°C, added three drops of 2 N NaOH solution, diluted with 50 mL of water, and extracted twice with CH2Cl2. The combined extracts were washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of CH2Cl2 and MeOH as eluent to give 78 mg (63% yield) of compound 12. - 1H NMR: δ 7.98 (d, J=7.8 Hz, 2H), 7.46−7.35 (m, 3H), 3.45 (t, J=5.3 Hz, 4H), 1.69 (d, J=5.1 Hz, 4H), 1.66−1.62 (m, 2H) ppm. - 13C NMR: δ 161.3, 158.9, 130.5, 129.0, 128.4, 126.2, 47.3, 24.9, 23.8 ppm. - MS (positive mode): m/z calcd for C13H16N3O+ (M+H)+: 230.3, found 230.1.
N-(5-Phenyl-1,3,4-oxadiazol-2-yl)-2-(piperidin-1-yl)acetamide (13).
To a solution of 0.13 g (0.55 mmol) of 4 in 3 mL of EtOH under argon was added 60 µL (0.65 mmol) of piperidine and the solution was stirred at 50°C for 48 h. It was cooled to 25°C, added three drops of 2 N NaOH solution, diluted with 50 mL of water, and extracted twice with CH2Cl2. The combined extracts were washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluent to give 70 mg (45% yield) of compound 13. - 1H NMR: δ 8.00 (d, J=6.6 Hz, 2H), 7.53−7.40 (m, 3H), 3.45 (s, 2H), 3.18−3.13 (m, 1H), 2.82 (m, 4H), 1.90 (td, J = 11.2, 5.9 Hz, 2H), 1.84−1.73 (m, 4H) ppm. - 13C NMR: δ 167.6, 161.1, 158.7, 131.3, 128.9, 126.4, 123.8, 61.3, 54.7, 24.8, 22.4 ppm. - MS (positive mode): m/z calcd for C15H19N4O2 (M+H)+: 287.3, found 287.0 (100%).
2-(4-Cyanopiperidin-1-yl)-N-(5-phenyl-1,3,4-oxadiazol-2-yl)acetamide (14).
To a solution of 0.107 g (0.45 mmol) of 4 in 3 mL of EtOH under argon, was added 60 µL (0.54 mmol) of 4-cyanopiperidine (19) and the resulting solution was stirred at 80°C for 24 h. It was cooled to 25°C, added three drops of 2 N NaOH solution, diluted with 50 mL of water, and extracted twice with CH2Cl2. The combined extracts were washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and EtOAc as eluent to give 40 mg (29% yield) of compound 14. - 1H NMR: δ 10.00 (bs, 1H), 7.87 (d, J=7.0 Hz, 2H), 7.43−7.41 (m, 3H), 3.29 (s, 2H), 2.94−2.72 (m, 4H), 2.64 (m, 1H), 2.10−1.95 (m, 4H) ppm. - 13C NMR: δ 167.5, 160.8, 159.5, 130.9, 129.5, 127.0, 124.9, 122.5, 60.5, 56.1, 50.3, 27.4 ppm. - MS (positive mode): m/z calcd for C16H18N5O2+ (M+H)+: 312.3, found 312.2 (100%).
5-Chloro-N-(5-(2-fluorophenyl)-1,3,4-oxadiazol-2-yl)thiophene-2-carboxamide (15).
To a cold (0°C) solution of 60 mg (0.335 mmol) of compound 2C and 40 mg (0.50 mmol) of pyridine in 3 mL of CH2Cl2 under argon, was added a solution of 0.61 mg (0.335 mmol) of 5-chlorothiophene-2-carbonyl chloride (17A) in 1 mL of CH2Cl2. The resulting mixture was stirred at 25°C for 3.5 h, diluted with 15 mL of CH2Cl2 and 8 mL of 10% aqueous NaHCO3 solution, and extracted three times with CH2Cl2. The combined extract was washed with brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of CH2Cl2 and MeOH as eluent to give 90 mg (83% yield) of compound 15 as white solids, mp 150–151°C. - 1H NMR: δ 8.25–8.17 (bs, 1H, NH), 8.18 (d, J=4 Hz, 1H), 7.93 (td, J=8, 2 Hz, 1H), 7.65–7.58 (m, 1H), 7.34 (td, J=8, 2 Hz, 1H), 7.28 (d, J=8 Hz, 1H), 7.07 (d, J=4 Hz, 1H). - 13C NMR: δ 161.0 (d, J=261 Hz, CF), 158.4, 150.8, 142.5, 137.5 135.0, 134.9, 130.4, 129.6, 127.5, 125.1, 117.5, 111.1 ppm. - MS (positive mode), m/z calcd for C13H8ClFN3O2S (M+H)+: 324.7, found 324.0.
Biological studies:
Inhibition of T-channel in DRG neurons.
Mouse dorsal root ganglion (DRG) neurons were prepared from 1–6 month-old C57/BC6 mice. All the procedures related to animal were conducted at AfaSci Research Laboratories and were in strict accordance with NIH guidelines and IACUC approved protocols. After sacrificing the mice by decapitation. the spine was removed and split into two halves from the middle line. Lumbar DRG neurons were collected into a 1.5 mL Eppendorf tube with modified Krebs solution (130 mM NaCl, 10 mM HEPES-Na, 5 mM KCl, 1 mM CaCl2, 10 mM glucose, and 2 mM MgCl2, pH adjusted to 7.35 with 1 N HCl). Subsequently, the DRG neurons were removed for digestion into 0.5 mL of Hank’s balanced salt solution (HBSS) with 1 mg/mL collagenase and 0.5 mg/mL trypsin added. The DRGs were minced with fine scissors and incubated at 35°C for 45–50 min. After removing the HBSS solution, the DRG neurons were dispersed into modified Krebs solution and triturated gentry with 5 fire- polished glass pipettes in gradually shrunk opening until no clumps were visible. The cells were then dispersed onto poly-L-ornithine-coated cover slips and maintained in a modified Krebs solution at 21°C with antibiotics added (0.2 mM streptomycin sulfate, 0.3 mM Penicillin G Sodium, and 0.1 mM Gentamycin.
Preparation of HEK293 cells expressing Cav3.2 channels.
Cav3.2 channels were expressed in human embryonic kidney (HEK)-293 cells using a tetracycline inducible expression system (Invitrogen). Cells were placed in 25-cm2 tissue culture flasks at 37°C, 5% CO2, and 100% relative humidity in D-MEM/F12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12; Gibso) supplemented with fetal bovine serum (FBS, 10% v/v/), sodium pyruvate (0.5 mM, Gibco), penicillin-streptomycin (100 U/mL, 100 µg/mL) and GeneticinR Selective antibiotic (G418; 0.5 mg/mL). Cells were detached from the flask base using a non- enzymatic cell dissociation solution (Cellstripper, Corning), removed and reseeded onto poly-D-lysine coated glass coverslips in 35-mm petri dishes. Cells in dishes were further supplemented with G418 at a final concentration of 1 mg/mL. After 24 hours, expression of Cav3.2 was induced with 1 µg/mL tetracycline. The medium for the HEK-293 cells was composed of D-MEM/F-12 and 15 mM HEPES buffer, with L-glutamine and pyridoxine hydrochloride. It was supplemented with FBS, P/S (penicillin/streptomycin) (Gibco, #15140–122), and 100 mM sodium pyruvate. Similarly, Na- and K- channel expressions in HEK293 cells were prepared.
Patch clamp recordings.
Whole-cell voltage clamp recordings were performed on DRG neurons or cultured HEK293 cells expressing T-type channels (encoded by Cav3.2 channels). All experiments were performed at room temperature. Whole-cell currents were recorded using a MultiClamp 700B amplifier and analyzed using Clampfit of Pclamp software (version 10.4, Molecular Devices, LLC, Sunnyvale, CA, USA). To record calcium currents in HEK293 cells, the external solution was composed (in mM) of 115 choline-Cl, 30 TEA-Cl, 2 CaCl2, 10 glucose and 10 HEPES (pH 7.3–7.4 adjusted with TEA-OH; osmolality about 295 mOsm/kg). The internal solution was composed (in mM) of 125 CsCl, 10 HEPES (acid), 10 EGTA (ethylene glycol tetraacetic acid), 1 CaCl2, 1 MgCl2, 4 MgATP, and 0.3 MgGTP (pH 7.3–7.4 adjusted with CsOH; osmolality about 295 mOsm/kg). Since sodium ions were absent in the external solution, tetrodotoxin was not added. Calcium currents were recorded at a holding potential of −100 mV and then depolarized in 10mV steps of 100 ms duration to activate Cav3.2 expressed in HEK cells. An interpulse interval of 10 seconds allowed the channel recovery from inactivation with stable current recordings. All reagents were purchased from Sigma unless specified otherwise. Test compounds were applied through a rapid solution exchange system with 8 plastic tubings glued into a 27G needle and with opening located closely to the recorded cells. The current responses were normalized to the control. Inhibition percentage was calculated and sigmoidal dose-response curves were generated using KaleidaGraph, XLFit (IDBS, Surrey, UK) or Prism (GraphPad Software, La Jolla, CA, US) for calculation.
Inhibition of seizure in pentylenetetrazol (PTZ)-induced fatality in mice.
It has been reported that over activation of T-channel involves in the generation of seizure activity. Following the reported protocol,36 the pentylenetetrazol (PTZ; 40 mg/kg, by intraperitoneal injection)-induced seizure model in mice (n=6 per group) was used to evaluate the inhibition of seizure. A prolonged latency to seizure and decrease of fatality rate were studied comparing to vehicle [2% DMSO in 0.5% hydroxypropyl cellulose (HPC)]. Fatality latency and rate–the percentage (%) of mice in each treatment group that died within a 20-minutes cutoff of the observational period, were recorded and calculated. Average fatality latency and rate of each treatment group were used to evaluate the molecule’s ability to either prevent or delay the onset of PTZ-induced seizures and death. In all cases, experiments were conducteda in a blind manner with respect to the experimenters.
ACKNOWLEDGEMENTS
Research reported in this publication was, in part, supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number NIH R44 NS086343 (to XSX) and National Science Foundation under award number CHE-1662705 (to DHH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This material was based upon work in part supported by the National Science Foundation under 1826982 (to DHH) for the purchase of a 400-MHz NMR spectrometer.
Footnotes
This manuscript is in celebration of Professor Kaoru Fuji’s 80 birthday and for his dedication to research and education
REFERENCES (AND NOTES)
- 1.Catterall WA, Perez-Reyes E, Snutch TP, and Striessnig J, Pharmacol. Rev, 2005, 57, 411. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA, Cold Spring Harbor Perspectives in Biology, 2011, 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iftinca MC, J. Med. Life, 2011, 4, 126. [PMC free article] [PubMed] [Google Scholar]
- 4.Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, and Perez-Reyes E, Circ. Res, 1998, 83, 103. [DOI] [PubMed] [Google Scholar]
- 5.Monteil A, Chemin J, Bourinet E, Mennessier G, Lory P, and Nargeot J, J. Biol. Chem, 2000, 275, 6090. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, and Lee JH, Nature, 1998, 391, 896. [DOI] [PubMed] [Google Scholar]
- 7.Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, and Perez-Reyes E, J. Neurosci, 1999, 19, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, and Turner RW, Eur. J. Neurosci, 2006, 24, 2581. [DOI] [PubMed] [Google Scholar]
- 9.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA, J. Neurosci, 1999, 19, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vassort G, Talavera K, and Alvarez JL, Cell Calcium, 2006, 40, 205. [DOI] [PubMed] [Google Scholar]
- 11.Cribbs L, Channels (Austin, Tex.), 2010, 4, 447. [DOI] [PubMed] [Google Scholar]
- 12.Fry CH, Sui G, and Wu C, Cell Calcium, 2006, 40, 231. [DOI] [PubMed] [Google Scholar]
- 13.Beam KG and Knudson CM, J. Gen. Physiol, 1988, 91, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilius B, Talavera K, and Verkhratsky A, Cell Calcium, 2006, 40, 81. [DOI] [PubMed] [Google Scholar]
- 15.Ngugi AK, Kariuki SM, Bottomley C, Kleinschmidt I, Sander JW, and Newton CR, Neurology, 2011, 77, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinemann U, Lux HD, and Gutnick MJ, Exp. Brain Res, 1977, 27, 237. [DOI] [PubMed] [Google Scholar]
- 17.Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, and Pape HC, J. Neurosci, 1995, 15, 3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giordanetto F, Knerr L, and Wallberg A Expert Opin. Therapeutic Patents, 2011, 21, 85. [DOI] [PubMed] [Google Scholar]
- 19.Shipe WD, Barrow JC, Yang Z-Q, Lindsley CW, Yang FV, Schlegel K-AS, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, Fox SV, Kraus RL, Marino MJ, Graufelds VK, Vargas HM, Bunting PB, Hasbun-Manning M, Evans RM, Koblan KS, and Renger JJ, J. Med. Chem, 2008, 51, 3692. [DOI] [PubMed] [Google Scholar]
- 20.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Hopkins CR, Xie XS, Conn PJ, Weaver CD, and Lindsley CW, ACS Chem. Neurosci, 2011, 2, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abernethy DR, Am. J. Cardiology, 1997, 80, 4C. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z-Q, Schlege K-Al, Shu Y, Reger TS, Cube R, Mattern C, Coleman PJ, Small J, Hartman GD, Ballard J, Tang C, Kuo Y, Prueksaritanont T, Nuss CE, Doran S, Fox SV, Garson SL, Li Y, Kraus RL, Uebele VN, Taylor AB, Zeng W, Fang W, Chavez-Eng C, Troyer MD, Luk JA, Laethem T, Cook WO, Renger JJ, and Barrow JC, ACS Med. Chem. Lett, 2010, 1, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galemmo R and Hum G, Di-t-butylphenyl piperazines as calcium channel blockers WO2009132454. WO2009CA00580 on 2009–04-28.
- 24.Pajouhesh H, Kaul R, Ding Y, Zhu Y, Zhang L, Chakka N, Grimwood M, Tan J, and Zhou Y, N- Piperidinyl acetamide derivatives as calcium channel blockers Patent number: WO2009146540. US20090420793 on 2009–04-08.
- 25.Bornemeier DA, Cai C, Fors KS, Hagen TJ, Holsworth DD, Jalaie M, Leonard DM, Moody TS, and Take Y, Oxadiazole compounds as calcium channel antagonists Patent number: WO2008050200. WO2007IB03107 on 2007–10-12.
- 26.Krayenbuhl JC, Vozeh S, Kondo-Oestreicher M, and Dayer P, Eur. J. Clinical Pharm, 1999, 55, 559. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z–Q, Schlegel KS, Shu Y, Reger TS, Cube R, Mattern C, Coleman PJ, Small J, Hartman GD, Ballard J, Tang C, Kuo Y, Prueksaritanont T, Nuss CE, Doran S, Fox SV, Garson SL, Li Y, Kraus RL, Uebele VN, Taylor AB, Zeng W, Fang W, Chavez-Eng C, Troyer MD, Luk JA, Laethem T, Cook WO, Renger JJ, and Barrow JC, ACS Med. Chem. Lett 2010, 1, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, Eduljee C, Jiang X, Smith P, Morrison J-L, Jones NC, Braine E, Rind G, Fee-Maki M, Parker D, Pajouhesh H, Parmar M, O’Brien TJ, and Snutch TP Sci. Trans. Med, 2012, 4, 121ra119. [DOI] [PubMed] [Google Scholar]
- 29.Gunaratna MJ, Hua DH, Zou B, Pascual C, Cao W, Zhang M, Weerasekara S, Nguyen TDT; Xiao K, and Xie XS, ARKIVOC, 2019, iii DOI: 10.24820/ark.5550190.p010.752 [DOI] [Google Scholar]
- 30.Kopecky BJ, Liang R, and Bao J, Pflugers Arch 2014, 466, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuiman C, Duran JE, Fors KS, Hagen TJ, Holsworth DD, Jalaie M, Leonard DM, Poel TJ, Quin IJ, and Take Y, Substituted oxadiazole analogs as calcium channel antagonists Patent number: WO2008117148. WO2008IB00645 on 2008–03-10.
- 32.Bankar GR, Nampurath GK, Nayak PG, and Bhattacharya S, Chemico-Biol. Inter, 2010, 183, 327. [DOI] [PubMed] [Google Scholar]
- 33.Deep A, Narasimhan B, Aggarwal S, Kaushik D, and Sharma AK, Cent. Nerv. Syst. Agents Med. Chem, 2016, 16, 58. [DOI] [PubMed] [Google Scholar]
- 34.Matsuno K, Masuda Y, Uehara Y, Sato H, Muroya A, Takahashi O, Yokotagawa T, Furuya T, Okawara T, Otsuka M, Ogo N, Ashizawa T, Oshita C, Tai S, Ishii H, Akiyama Y, and Asai A, ACS Med. Chem. Lett, 2010, 1, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guda DR, Cho HM, and Lee ME, RSC Adv, 2013, 3, 7684. [Google Scholar]
- 36.Xie X, Lancaster B, Peakman T, and Garthwaite J, Pflugers Archiv.: Eur. J. Phys 1995, 430, 437. [DOI] [PubMed] [Google Scholar]
- 37.Zou B, Li Y, Deng P, and Xu DZ, Brain Res 2005, 1033, 78. [DOI] [PubMed] [Google Scholar]