

Abstract
A series of novel imidazobenzodiazepine analogs of the lead chiral ligand SH-053–2’F-S-CH3 (2), an α2/α3/α5 (Bz)GABA (A)ergic receptor subtype selective ligand, which reversed PCP-induced prepulse inhibition (PPI) of acoustic startle, were synthesized. These chiral (S)-CH3 ligands are targeted for the treatment of schizophrenia and depression. These new ligands were designed by modifying the liable ester functionality in 2 to improve the metabolic stability, cytotoxicity, and activity as compared to 2. Based on the data to date, the most promising ligands are the N-cyclopropyl amide GL-I-55 (8c) and the methyl bioisostere GL-I-65 (9a). The in vitro metabolic stability, cytotoxicity and in vivo locomotor effects are described in this report. Based on these results, 8c and 9a are the most promising for further in vivo pharmacology.
Keywords: schizophrenia, depression, GABAA receptor, SH-053–2’F-S-CH3, bioisosteres, metabolism
Graphical Abstract
Introduction
Schizophrenia is one of the most complex central nervous system (CNS) disorders known. Approximately 1% of the population worldwide is affected by this disease. The symptoms fall into three broad categories: positive symptoms including hallucinations and delusions; negative symptoms such as the blunted affect, social dysfunction and lack of motivation or pleasure in daily life, and cognitive abnormalities. For instance, in the disruption of cognition, symptoms such as impairments in working memory, verbal memory, attention and executive deficits, which can lead to severe emotional distress, are prominent.1 The treatments commonly used to manage positive symptoms are typical and atypical antipsychotic drugs, which target and block the over-activation of dopamine receptors2 in patients with schizophrenia. In fact, the drugs prescribed, to date, only reduce the positive symptoms in the majority of schizophrenia patients. They are often not effective especially in patients with negative symptoms and cognitive abnormalities.3 Moreover, the antipsychotics in many patients induce adverse effects such as tolerance, addiction, sedation, drug-resistance, weight gain and liver toxicity.4 The lack of proper medications and limited efficient psychosocial therapy together increase the psychological and financial burden rapidly not only for the patients and their family but also for society as a whole. Consequently, there is an urgent clinical need for novel drug candidates that can address all aspects of schizophrenia including the alleviation of the negative and cognitive symptoms with decreased side effects.
Accumulating evidence has long suggested that the disruption of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA) activity may play a critical role in the pathophysiology of schizophrenia, as well as depression.5,6 The GABAA receptor (GABAAR) is a pentameric ligand-gated chloride ion channel,7 which is located in the membrane of neuronal cells. It consists of a total 19 different subunits in the human brain (α1–6, β1–3, γ1–3, δ, ε, π, θ, ρ1–3),8 the configuration of which determine ligand affinity, ion flux, membrane resistance, depolarization and other properties of the membrane.9 The minimum requirement to provide a benzodiazepine receptor-mediated GABAAR ion channel is the combination of five subunits, which contain two α-, two β- and one γ- subunit.10 GABAARs contain the binding sites of a series of important clinical drugs, such as benzodiazepines, neurosteroids, and barbiturates.11 The benzodiazepines, which are classical psychoactive drugs such as diazepam, bind at the extracellular interface of the α+γ2-subunits which results in increasing efficacy of GABA regarding chloride ion flux.12 Subtype-specific ligands exhibit different CNS effects depending on the specific α1–6β2/3γ2 subtype involved.13 Principally, the α1-containing GABAARs, are associated with the sedative, ataxia, anticonvulsant, anterograde amnesia, and addictive effects of benzodiazepine allosteric modulators of GABAARs.14 The α2/3-subunit-containing GABAARs mediate the anxiolytic, anticonvulsant, antinociceptive effects and perhaps muscle relaxant effect at higher doses.15 Whereas the relatively minor population of α5-containing GABAARs, which have been described in the previous investigation, are implicated in cognition, learning and memory processes without motor effects or anxiolysis.16–19 The ligands have also been implicated in reversing the adverse effects in animal models of schizophrenia and depression. Therefore, novel α5 or α2/3/5 subtype selective benzodiazepine ligands that are nearly silent or exhibit no efficacy at α1 GABAARs may have the potential to reduce the negative symptoms and improve cognitive brain function with little or no sedative, amnesic or ataxia effects for patients with schizophrenia, as well as effects to alleviate the symptoms of depression.
It is well known that asymmetry plays an essential role in physiological processes; therefore, a large number of drugs are chiral molecules, in which one enantiomer of a drug may have the desired beneficial effect, while the other enantiomer may cause serious adverse effects.20,21 This was observed with thalidomide.22 Based on our earlier established BZD/GABAA pharmacophore model23 that was developed by ligand binding data (see Supplementary material), a series of chiral molecules have been designed, synthesized and studied. Ligand SH-053–2’F-R-CH3 (1) was the lead stereospecific GABAAR α5-subtype selective positive allosteric modulator (PAM) because of the chiral (R)-methyl group at the C-4 position of imidazobenzodiazepine (IBZD), as shown in Figure 1. This resulted in increased potency at α5 GABAA receptor subtypes accompanied by a concomitant decrease at the α1, α2, and α3 subtypes. This α5-selective ligand has been reported to successfully alleviate the hyperactivity of the dopamine system induced by amphetamine in the methylazoxymethanol acetate model (MAM) of schizophrenia.24,25 Moreover, this ligand 1 in an animal behavioral model of depression exhibited anxiolytic and antidepressant effects in female mice that were exposed to unpredictable chronic mild stress (UCMS) in a sex-dependent manner. The effects on male mice in the same paradigm were very weak or none at all.26 Furthermore, analogs of 4(R)-CH3 enantiomer 1, such as methyl amide MP-III-022,27 the carboxylic acid SH-053–2’F-R-CH3-acid28, etc., which are more potent at the α5-subtype than 1, have also been investigated and undergone a number of behavioral studies.29,30
Figure 1.

Structures of enantiotopic chiral lead imidazobenzodiazepine (IBZD) ligands 1 and 2.
In order to study how the chirality at the C-4 position of IBZD could affect the pharmacological response, the enantiomer of 1, SH-053–2’F-S-CH3 (2) was synthesized.18 As expected, these two enantiomers exhibited vastly different GABAergic efficacy and binding affinity at Bz/GABA(A) receptor sites. Ligand 2 was found to be an α2-, α3-, α5-β3γ2 subtype selective, while 1 was an α5-subtype selective ligand. Since 1 and 2 both target the α5-subunit, they have been assayed in several behavioral models. Examination of the results revealed 2 exerted anxiolytic effects in both rhesus monkeys31 and rats32, while 1 had only a minimal anxiolytic effect when much higher doses were applied. Interestingly 2 also successfully mitigated the amphetamine-induced hyperactivity in a poly (I:C) animal model of schizophrenia.33
The results of these studies indicated that both enantiomers may be useful in the treatment of the symptoms of schizophrenia, while the α2/3/5 agonist 2 exhibited the potential for treatment of conditions comorbid with schizophrenia, such as anxiety, which commonly coexists in patients with schizophrenia. Unfortunately, during other studies, ligand 2 was shown to be metabolized at a very high rate in vitro in the presence of human liver microsomes (HLM);34 3% of 2 remaining after incubation at 37 °C for 30 minutes, while ligand 1 was stable in HLM with 100% remaining under the same conditions, as presented in Table 1. The principal aim of this research was to improve the metabolic stability of the ester functional group while retaining the properties potentially useful for the treatment of schizophrenia and depression. Schizophrenia and some forms of depression (bipolar I disorder and perhaps major depressive disorder) are linked both genetically and etiologically. Herein we present the synthesis and characterization of novel analogs of SH-053–2’F-S-CH3 (2) including esters, thioesters, amides, carboxylic acids and the ester bioisostere oxadiazoles at the C(3) position together with modifications at the C(8) position. The newly designed ligands were prepared and investigated in in vitro metabolism studies, cytotoxicity assays and in vivo in mice with a rotarod study to identify ligands with a lower systematic clearance rate, low toxicity, little or no sedative/ataxia activity, which might be useful in the management of the symptoms schizophrenia23 and depression.25 This go/no-go point is important for the development of a safe treatment for schizophrenia and depression that can occur at any age.
Table 1.
In vitro liver microsomal stability of 1 and 2
| Ligand | Liver microsomal stability (%-remaining) Conditions: 4 μM compound, 37 °C, 30 minutes |
|||
|---|---|---|---|---|
| HLMa | DLMb | MLMc | RLMd | |
| SH-053-2'F-R-CH3 (1) | 100.0 | 90.3 | 59.1 | 5.4 |
| SH-053-2'F-S-CH3 (2) | 2.3 | 90.7 | 66.5 | 6.0 |
Human liver microsomes (HLM)
dog liver microsomes (DLM)
mouse liver microsomes (MLM)
rat liver microsomes (RLM).
Results and Discussion
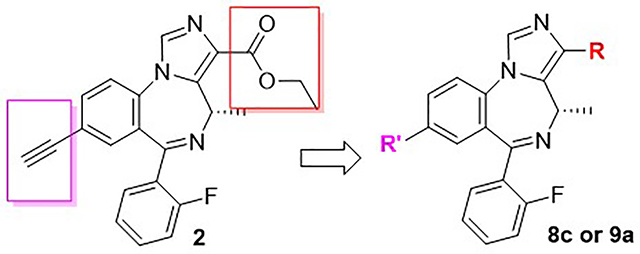
Previously, evidence from the ligand-based GABAAR pharmacophore/receptor model and SAR23,35 have indicated that subtle changes in the IBZD substituents will affect the BzR/GABA(A)R subtype selectivity dramatically. For instance, the loss of the chiral methyl group at the C(4) position will decrease the α5-subtype efficacy while the efficacy at α2/α3-β3γ2 GABA(A)ergic subtypes will increase dramatically. Efficacy at the α1 subtype will increase with a 2’-F substituent on the C(6) pendent phenyl ring, but the 2’-N and 2’-H analogs remain α2/α3 subtype selective.30 It has also been shown that the α5 selectivity can be modified by the presence of different lipophilic groups at the C(8) position.19 Analogs, which have been optimized at the C(3) position, demonstrated higher affinity and potency at α5-36 or α2/3-37–39 subtypes leading to better and longer physiological effects. For these reasons, the C(3) and C(8) positions are considered to be the optimal regions for ligand improvement. To evaluate the modification of the lead compound 2 at these two positions, a series of novel analogs were designed, synthesized and characterized, as summarized in Figure 2.
Figure 2.

Chiral analogs and bioisosteres of the lead compound 2.
As mentioned earlier, the C(8)-acetylene and C(4) methyl function were found to enhance the α5-subtype selectivity, therefore a cyclopropyl group, which is commonly incorporated into biologically active materials, was introduced into the parent compound 2 as a substituent to extend the SAR. As the result of its flexible rotation at the C(8) position, it should fill a similar volume of electron density as the ethynyl moiety. Consequently, it might exhibit parallel physiological effects and enhanced subtype selectivity due to similar interactions in the lipophilic binding pocket (L2, see Figure S1 in Supplementary material for the model).22 Moreover, the metabolism by liver microsomes would be expected to be different for a cyclopropyl substituent as compared to an ethynyl functional group. Hence, the C(8)-substituted cyclopropyl analog GL-I-78 (3) was prepared based on this hypothesis. The cyclopropyl ligand 3 was synthesized from cyclopropyl boronic acid via a Suzuki-coupling reaction40 with the aryl bromide SH-I-48B (4b), whose synthesis had been previously described. 17 The coupling reaction was carried out in the presence of potassium phosphate and 10 mol% Pd-catalyst, bis(triphenylphosphine)palladium(II) diacetate, in toluene at 100 °C for 12 hours in good yield (82%), as shown in Scheme 1.
Scheme 1.

Synthesis of 8-cyclopropyl-4-(S-CH3) imidazobenzodiazepine 3 from bromide 4b.17
Based on the results described previously, the ethyl ester 2 was rapidly degraded in the HLM, which suggests modification of the ester might play an essential role in metabolism, as well as efficacy, as compared to the lead compound 2. Therefore, a series of esters, which have different alkyl groups were synthesized. The synthesis of the C(8)-bromo methyl ester MP-III-059 (4a) and the C(8)-ethynyl methyl ester MP-III-021 (5a) were achieved in the presence of sodium methoxide in methanol in good yields, as depicted in Scheme 2. The majority of the reactions were carried out via a transesterification reaction41 with pieces of freshly cut lithium rod in the presence of alcohols in THF at 40 °C for 0.5 hours to generate the corresponding alkoxides. To this solution, ester 2 was added, which had been dissolved in dry THF, and full conversion was observed after 15 to 30 minutes. New ester analogs generated via this general route were: propyl ester GL-I-32 (5b), isopropyl ester GL-I-31 (5c), t-butyl ester GL-I-30 (5d) and 2-methyl butyl ester GL-I-33(5e), respectively. The yields were good to excellent (77–97%), as illustrated in Scheme 3. However, the trifluoroethyl ester GL-I-36 (5f), and the cyclopropyl ester GL-I-38 (5g) were not obtained under these conditions. As a result, both analogs were prepared by an alternate procedure. The ester 2 was saponified to form the carboxylic acid, SH-053–2’F-S-CH3-acid (6), with sodium hydroxide in ethanol at reflux for 1 hour in nearly quantitative yield (99%) after acidic workup, as illustrated in Scheme 4. The subsequent reactions for the synthesis of ester 5f and 5g were conducted with thionyl chloride in dichloromethane at reflux to give the acyl chloride, which was converted into ester 5f and 5g with 2,2,2-trifluoroethanol and cyclopropanol, respectively. Additionally, it must be pointed out that the acid 6 was synthesized not only because it was an important intermediate for the majority of the analogs, but also because the enantiomer (R)-CH3 carboxylic acid in the C(4)R-isomer series significantly increased the α5-subtype selectivity and potency.27 It was hoped that the acid 6 in the C(4)S-isomer series might affect a similar increase on the selectivity and potency at the α2/α3/α5-subtype.
Scheme 2.

Synthesis of methyl esters 4a and 5a from ethyl ester 4b or 2, respectively.17
Scheme 3.

Synthesis of esters 5b-5e.40
Scheme 4.

Synthesis of analogs 5f, 5g, 7, 8a-8f.
In a previous study, it was shown that a heteroatom attached directly to the carbonyl at C(3) would affect the GABAAR subtype selectivity. For example, a thioester at C(3) in the 2’N series exerted no efficacy at the α1-subtype even at a higher concentration in comparison with the corresponding oxygen substituted ester, but with a slightly higher preference for the α3-subtype.42 Therefore, it was decided to replace the oxygen atom in lead compound 2 at the C(3) position with a sulfur atom. The thioethyl ester GL-I-77 (7) was synthesized from the carboxylic acid 6 following the general procedure as mentioned below in Scheme 4 with ethanethiol as the nucleophile.
Besides the slight changes in steric effects as compared to lead ester 2, the amide analogs are well-known stable replacements for the labile ester moieties. The amides were considered to be important replacements for ester functions to improve the in vivo stability, as well as the bioavailability when compared to the stability of esters. To evaluate the potential activity of the amides in regard to future SAR, a series of amide analogs 8a-8f were synthesized to enable a comparison between esters and amides in regard to in vitro metabolic stability, cytotoxicity and in vivo locomotor activity. The methyl amide MP-III-023 (8a) was prepared by the direct amidation of ethyl ester 2 in the presence of methylamine in dichloromethane in a sealed tube. As indicated in Scheme 4, the rest of the amide analogs were synthesized from carboxylic acid 6 via an acyl chloride intermediate, using the corresponding primary or secondary amine. This process gave the ethyl amide GL-I-43 (8b), cyclopropyl amide GL-I-55 (8c), isopropyl amide GL-I-57 (8d), t-butyl amide GL-I-41 (8e) and dimethyl amide GL-I-54 (8f), respectively, in good to excellent yields (71–88%).
Ester bioisosteres have been utilized commonly in drug design in order to render them metabolically stable with less adverse effects and provide better clinical candidates.43 The previous results indicated that replacement of ester functions by substituted 1,2,4-oxadiazoles36,44 significantly increased the in vitro and in vivo stability, as well as slightly improved the hydrophilicity of the ligands.19 Moreover, the efficacy at BZD receptors was enhanced as compared to the corresponding ester ligands as well.19 Encouraged by this previous evidence, the substituted 1,2,4-oxadiazoles 9a-9c were designed and synthesized from lead 2. The condensation of the ethyl ester 2 and corresponding amidoxime in the presence of sodium hydride in THF was employed to prepare the C(3)-substituted oxadiazole ligands. This route was successfully applied previously with non-chiral ligands.43 However, the desired oxadiazoles with the C(4) chiral methyl group were only obtained in moderate yield (30–40%). The reason was the formation of a byproduct (50–60% yield), in which the C(5)-C(6) imine bond isomerized to the C(4)-C(5) position. As a result, the chirality at the C(4) position was lost. To overcome this problem, the amount of sodium hydride, which would readily lead to isomerization of the imine bond, was reduced from 4 equivalents to 1.1 equivalents. In addition, the amidoxime was stirred longer with NaH before the ester was added to the mixture. This process gave methyl oxadiazole GL-I-65 (9a), ethyl oxadiazole GL-I-66 (9b) and isopropyl oxadiazole GL-I-81 (9c) in improved yields of 79–85%, respectively, as depicted in Scheme 5.
Scheme 5.

Synthesis of oxadiazoles 9a-9c.
To explore the extent of flexibility of the hydrogen-bond acceptor in the binding pocket designed H1 in the pharmacophore/receptor model (see Figure S1 in Supplementary material),22,36 it was decided to synthesize the linear nitrile at the C(3) position, which was sometimes employed to increase water solubility of a ligand and has been used to reduce the possible oxidative metabolism by the liver in clinical studies.45 The nitrile MP-III-018.A (10) was produced from the ester 2 via reaction with dimethyl aluminum amine46 in xylene at 80 °C in 75% yield, as shown in Scheme 6. The dimethylaluminum amine was prepared freshly by adding trimethyl- aluminum into a solution of ammonia-saturated dichloromethane.45
Scheme 6.

Synthesis of the C(3)-nitrile 10.
The characterization of the lead compound 2 was being carried out simultaneously with the synthesis and assessment of the novel analogs. Due to the ability of 2 to potentiate the α5 containing GABAAR as well as the α2 and α3 subtypes, while inactive at the α1 containing GABAAR, which mediates sedation and other adverse effects, ligand 2 was evaluated in behavioral models in direct comparison with its enantiomer 1. For cataleptic behavior in the schizophrenia studies,47 the antipsychotic, haloperidol elicited a significant cataleptic response. In contrast, both C(4)-methyl substituted ligands 1 and 2 did not show any signs of catalepsy in comparison to the vehicle, as illustrated in Figure 3. Moreover, in the prepulse inhibition (PPI) assay, which was used as a classical animal model of schizophrenia, the S-isomer 2 significantly reversed the PPI deficit induced by phencyclidine hydrochloride (PCP), an N-methyl-D-aspartate (NMDA) antagonist.48 Ligand 1 did antagonize the deficit compared to vehicle but failed to reach a significant effect at the same dosage, as depicted in Figure 4. However, PPI results with the R-isomer 1 were trending toward significance but would have required a larger number of animals. Additionally, in the 6 Hz electroshock (3 seconds duration) assay, both ligands were evaluated for their ability to protect against epileptic seizures. As shown in Figure 5, ligand 2 was able to protect mice from the electroshock-induced seizures for all time points (1/4, 1/2, 1, 2, and 4 hours), which was predicted due to the activation of α2 and α3 subtypes (Figure 5A) as well as α5 subtypes. However, the R-enantiomer (α5) 1 was not effective at any time at a relatively higher dose in the 6 Hz paradigm. For the rotarod (n=8) assay, neither 1 nor 2 produced significant ataxia at 100 mg/kg i.p. in mice (Figure 5B). This is in complete agreement with the poor efficacy at α1β3γ2 Bz/GABAergic subtypes previously reported.30
Figure 3.

Impact of 1 or 2 treatment (30 mg/kg, ip) on cataleptic behavioral. Haloperidol, but not 1 nor 2 produced a significant cataleptic response at 30, 60, and 120 minutes post-injection. The data are expressed as the mean (+SEM) time (sec) subjects spent with both front paws on an elevated bar. *p<0.001, the difference from controls receiving vehicle alone (see Supplementary material for details).
Figure 4.

Impact of 1 or 2 treatment (30 mg/kg, IP) on prepulse inhibition. Ligand 2, but not 1 reversed the PPI deficits induced by PCP when given one hour before testing. The data are expressed as the mean (+SEM) percent prepulse inhibition of the startle response to an auditory stimulus when preceded by a nonstartle-eliciting auditory cue at 5, 10, and 15 decibels. Disruption of prepulse inhibition was obtained in rats by an acute injection of phencyclidine (PCP·HCl, 1.5 mg/kg, SC) ten minutes prior to testing. *p<0.05, difference from controls receiving phencyclidine with 0 mg/kg drug pretreatment. (see Supplementary material for details).
Figure 5.

Impact of 1 or 2 treatment on anticonvulsant effects. A) Mice were dosed i.p. with either 1 (150 mg/kg) or 2 (100 mg/kg) and a 6 Hz shock by a corneal electrode was administered at various time points, and the animal observed for convulsions. Ligand 2 was able to protect some subjects while 1 was not able to protect mice from electroshock-induced seizures. B) In the rotarod assay, mice were dosed i.p. with 1 or 2 (100 mg/kg) 30 minutes prior to testing on a rotating rod (6 rpm). Mice that fell 3 times during a 1-minute trial were deemed toxic (ataxic or sedated). (see Supplementary material for details)
The parent compound 2 and its analogs were assayed for metabolic stability in vitro with HLM and mouse liver microsomes (MLM) to identify the most stable analogs and the possible trend of steric effects in respect to stability. Moreover, it was important to evaluate stability in both species with respect to applied animal models and future human treatment with such a diverse array of functional groups at C(3). The results of this study are shown in Table 2.
Table 2.
In vitro In vitro microsomal stability of SH-053-2'F-S-CH3 (2) analogs.
| Entry | Structure at C(3) with C(8)-ethynyl | Microsomal stability (HLM) after 2 hr | Microsomal stability (MLM) after 2 hr | |||
|---|---|---|---|---|---|---|
| Half-life (min) | % remaining | Half-life (min) | % remaining | |||
| 2 | ester | CO2CH2CH3 | < 5b | < 5.0b | 155 ± 9d | 78.0 ± 0.2d |
| 3 | CO2CH2CH3e | < 5b | < 5.0b | 46 ± 3 | 16.0 ± 0.4 | |
| 4a | CO2CH3f | 10 ± 1d | 2.0 ± 0.3d | 116 ± 13 | 41.0 ± 0.6 | |
| 4b | CO2CH2CH3f | < 5b | < 5.0b | < 5b | < 5.0b | |
| 5a | CO2CH3 | 119 ± 21d | 43.0 ± 0.4d | 216 ± 28 | 62.0 ± 0.5 | |
| 5b | CO2CH2CH2CH3 | < 5c | < 5.0c | 24 ± 0d | 12.0 ± 0.2d | |
| 5c | CO2CH(CH3)2 | 57 ± 2d | 47.0 ± 0.2d | 227 ± 16d | 83.0 ± 0.1d | |
| 5d | CO2C(CH3)3 | 514 ± 115d | 89.0 ± 0.2d | 196 ± 20d | 78.0 ± 0.2d | |
| 5f | CO2CH2CF3 | < 5a | < 5.0a | < 5b | < 5.0b | |
| 5g | CO2-cyclopropyl | < 5a | < 5.0a | 19 ± 2d | 11.0 ± 0.2d | |
| 6 | acid | COOH | 358 ± 56 | 74.0 ± 0.4 | 139 ± 11d | 54.0 ± 0.3d |
| 7 | thioester | COSCH2CH3 | 101 ± 19 | 28.0 ± 0.3 | 46 ± 6 | 9.0 ± 0.5 |
| 8a | amide | CONHCH3 | 1780 ± 39 | 92.0 ± 0.3 | 845 ± 5 | 86.0 ± 0.2 |
| 8b | CONHCH2CH3 | 1978 ± 50 | 93.0 ± 0.3 | 1961 ± 46 | 92.0 ± 0.3 | |
| 8c | CONH-cyclopropyl | 1625 ± 803d | 89.0 ± 0.4d | 320 ± 34d | 72.0 ± 0.3d | |
| 8d | CONHCH(CH3)2 | 107 ± 10 | 38.0 ± 0.5 | 28 ± 0 | 75.0 ± 0.3 | |
| 8e | CONHC(CH3)3 | 205 ± 14 | 63.0 ± 0.3 | 142 ± 9 | 52.0 ± 0.3 | |
| 8f | CON(CH3)2 | 59 ± 5d | 41.0 ± 0.9d | 24 ± 2d | 19.0 ± 0.9d | |
| 9a | 1,2,4-oxadiazole | 3-CH3 | 866 ± 213 | 86.0 ± 0.3 | 443 ± 66 | 81.0 ± 0.4 |
| 9b | 3-CH2CH3 | 563 ± 123 | 80.0 ± 0.4 | 213 ± 17 | 63.0 ± 0.3 | |
| 9c | 3-CH(CH3)2 | 2584 ± 1920 | 91.0 ± 0.3 | 500 ± 109 | 79.0 ± 0.5 | |
| 10 | nitrile | CN | 41 ± 5 | 9.0 ± 0.3 | 58 ± 6 | 17.0 ± 0.6 |
Compound was not detected after 10 minutes.
Compound was not detected after 20 minutes.
Compound was not detected after 30 minutes.
Compound was assessed at 1-hour incubation time.
C(8)-cyclopropyl.
C(8)-bromo. (see Supplementary material for details)
The lead compound 2, as mentioned before, was found to be rapidly metabolized on HLM with less than 5% remaining after 20 minutes, while 2 exhibited good stability for MLM with 78% remaining after 1 hour. Among the ester analogs, 3, 4a, 4b, 5a-d, 5f and 5g an increasing trend in metabolic stability in HLM was observed as the size of the alkyl chain increased. However, esters bearing a linear 2–3 carbon chain (4a, 5a, 5b, 5f, and 5g), regardless of the functional group at the C(8) position (-bromo, -cyclopropyl or -ethynyl), were metabolized very rapidly with less than 5% remaining after 10, 20, and 30 minutes in HLM. A similar trend was observed for MLM, however, the C(8)-bromo and -cyclopropyl replacement of the ethynyl group decreased the metabolic stability (see 2 vs 3, 4b; 4a vs 5a). It is important to point out that the introduction of an ester function with a longer carbon side chain or a terminal CF3 group reduced the metabolic stability significantly, as shown in Table 1. Both ligands 4b and 5f were not detected anymore after 20 minutes in HLM or MLM, respectively. Most of the esters are only slightly more stable in MLM, as compared to HLM, which suggests that longer lipophilic linear alkyl chains in the ester functional group should be avoided in the ligand design. The carboxylic acid 6 was slightly more stable compared to the esters. The stability of thioethyl ester 7 for HLM was improved slightly compared to ethyl ester 2, which suggests a sulfur atom in place of an oxygen atom would provide greater metabolic stability. Electronegativity might play a role here. The nitrile 10 was metabolized at a rapid rate with only 9% and 16% of 10 remaining after two hours in HLM and MLM, respectively. As expected, the amide analogs remarkably enhanced the metabolic stability in both HLM and MLM. Unlike the esters, the trend regarding steric effects on the rate of metabolism of the amides was opposite to the esters; bulkier substituents were less metabolically stable. Presumably, bulkier esters are more lipophilic and react faster with P450 enzymes of the liver microsomal extract. The ethyl amide 8b exhibited the best stability with more than 92% of 8b remaining after 2 hours, as well as the longest half-life for both types of liver microsomes among all the amides. The isopropyl amide 8d was the only ligand, whose metabolism was a little inconsistent, as a result, the isopropyl ester 5c and amide 8d shared a similar metabolic pattern. As expected, the replacement of the ester moiety with the oxadiazole bioisosteres 9a-9c significantly improved the metabolic stability over a two-hour period of incubation in both HLM and MLM. It is notable that the isopropyl oxadiazole 9c exhibited the longest half-life (2584 minutes) as compared to all the analogs investigated in the SH-053–2’F-S-CH3 (2) series. In brief, it is clear that the amides and substituted oxadiazole functionality at the C(3) position provide the desired ligands with longer half-life values, as compared to other functional groups. Further evaluation of these analogs involved a study of potential cytotoxicity in both HEPG2 (liver) and HEK293 (kidney) cell lines, as summarized in Table 3.
Table 3.
In vitro cytotoxicity of SH-053-2'F-S-CH3 (2) analogs.
| Entry | Structure at C(3) with C(8)-ethynyl | HEK293 LD50 (μM)a | HEPG2 LD50 (μM)a | |
|---|---|---|---|---|
| 2 | ester | CO2CH2CH3 | 28.0 ± 4.1 | 73.5 ± 14.4 |
| 3 | CO2CH2CH3b | 58.0 ± 6.0 | 87.0 ± 6.6 | |
| 4a | CO2CH3c | 98.5 ± 7.9 | 136.8 ± 6.3 | |
| 4b | CO2CH2CH3c | 187.0 ± 49.7 | >400 | |
| 5a | CO2CH3 | 68.2 ± 4.3 | 136.6 ± 6.3 | |
| 5b | CO2CH2CH2CH3 | 53.2 ± 4.4 | 56.3 ± 4.0 | |
| 5c | CO2CH(CH3)2 | 48.9 ± 4.4 | 39.9 ± 2.8 | |
| 5d | CO2C(CH3)3 | 12.3 ± 1.6 | 21.4 ± 1.9 | |
| 5e | CO2C(CH3)2CH2CH3 | 17.2 ± 1.6 | 17.1 ± 1.4 | |
| 5f | CO2CH2CF3 | 138.1 ± 31.8 | >400 | |
| 5g | CO2-cyclopropyl | 35.8 ± 3.3 | 85.1 ± 3.8 | |
| 6 | acid | COOH | >400 | >400 |
| 7 | thioester | COSCH2CH3 | 20.5± 4.7 | 30.2 ± 9.7 |
| 8a | amide | CONHCH3 | >200 | >200 |
| 8b | CONHCH2CH3 | 95.5 ± 12.6 | 65.4 ± 7.3 | |
| 8c | CONH-cyclopropyl | 63.8 ± 3.0 | 93.5 ± 5.7 | |
| 8d | CONHCH(CH3)2 | 80.2 ± 11.1 | 56.9 ± 4.8 | |
| 8e | CONHC(CH3)3 | 33.6 ± 3.0 | 43.4 ± 4.0 | |
| 8f | CON(CH3)2 | >400 | >400 | |
| 9a | 1,2,4-oxadiazole | 3-CH3 | 69.3± 2.3 | >180 |
| 9b | 3-CH2CH3 | 46.9 ± 6.6 | 35.1 ± 3.7 | |
| 9c | 3-CH(CH3)2 | 32.5 ± 7.6 | 21.7 ± 1.8 | |
| 10 | nitrile | CN | 81.6 ± 6.7 | 57.1 ± 5.6 |
Compounds were incubated at different concentrations with the specific cells for 48 h, followed by detection of cell viability using a Cell-Titer Glo (Promega). The results were normalized using DMSO (negative control) and 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (150 mM in DMSO final concentration, positive control). Data were acquired by three independent experiments carried out in quadruplet.
C(8)-cyclopropyl.
C(8)-bromo. (see Supplementary material for details)
Examination of the cell viability assay indicated that ligands with a C-3 carboxylic acid substituent of S-isomer analog 6 and dimethyl amide 8f were non-toxic even at 400 μM (LD50 > 400 μM) in the presence of HEK293 and HEPG2 cells. Compounds C(8)-bromo ethyl ester 4b and trifluoroethyl ester 5f were non-toxic at doses higher than 400 μM (LD50 > 400 μM) for the HEPG2 cells, and exhibited an LD50 of 187 μM and 138 μM in HEK293 cells, respectively. Methyl amide 8a showed no toxicity at concentrations below 200 μM for both cell lines. Esters 3, 4a, 5a, 5b, amides 8b-8d, methyl oxadiazole 9a and nitrile 10, were safe at lower concentrations with LD50 values of >50 μM for both cell lines. Usually, these ligands potentiate the GABAARs at nanomolar concentrations, consequently far below the measured LD50 values of 50 μM. Compounds with moderate cytotoxicity (30 μM <LD50 <50 μM) were isopropyl ester 5c, cyclopropyl ester 5g, t-butyl amide 8e and ethyl oxadiazole 9b. Ligands that exhibited pronounced cytotoxicity (LD50 <30 μM) for HEK293 kidney cells were ethyl ester 2, t-butyl ester 5d, 2-methyl butyl ester 5e, thioethyl ester 7, and isopropyl oxadiazole 9c. These data revealed that increasing the size of the substituent of the oxadiazole moiety may increase cytotoxicity; most of the esters are considered to be cytotoxic if given at very high doses (much higher than any therapeutic doses), unless a different group at C(8) was introduced (see 3, 4a, and 4b). Trifluoroethyl ester was fairly cytotoxic perhaps because the trifluoroethanol group is a much better leaving group. In vitro toxicity can be used as a measure to estimate in vivo toxicity, however, variations can occur. For instance, lead compound 2 exhibited a fairly low LD50 value, however it was not toxic in a number of pharmacological assays in the mice, rats and primates.23–25, 30–32 The cytotoxicity assays indicated which analogs were least cytotoxic but were not potent enough (nM) to rule out further pharmacological studies in models of schizophrenia or depression.
To assess the possibility of undesired CNS side effects (α1-mediated), a locomotor coordination study of each analog was conducted by placing mice on a rotating rod for a maximum of 3 minutes after oral administration by gavage at a dose of 40 mg/kg (Figure 6). The mice were also observed for loss of righting reflex, an indication of undesired CNS effects. Most of the mice treated with compounds exhibited no sedation or ataxia, nor loss of righting reflection vs the control diazepam. Most of the compounds exhibited sensorimotor steadiness at all three time-points, which indicated no sedative/ataxic effects. However, some amides did exbihit some sensorimotor deficits. Methyl amide 8a and ethyl amide 8b effected severe motor impairment, while the cyclopropyl amide 8c and dimethyl amide 8f exhibited only minor impairment. The bulkier isopropyl amide 8d and t-butyl amide 8e did not show any motor impairment. These results suggested that these particular amides, which produced sedation/ataxia, are sedating presumably, because of some efficacy at the α1-GABAAR subtype. In regard to the pharmacological effects of the amide side chain, it is obvious that a smaller alkyl chain promoted a more sedating effect, which should not result from metabolism nor cytotoxicity since these two assays indicated that the amides with smaller substituents were highly stable and non-toxic.
Figure 6.

Effect of compounds on sensorimotor coordination. (see Supplementary materials for details)
After examination of all results for the series of C(4)-S-CH3 substituted enantiomers described above, the best two compounds, which exhibited excellent stability, minimal or no toxicity and did not exhibit motor impairments, were cyclopropyl amide GL-I-55 (8c) and methyl oxadiazole GL-I-65 (9a). Moreover, it was suggested that the bioisosteres GL-I-66 (9b) and GL-I-81 (9c), which were very metabolically stable and exhibited no sensorimotor impairment, will need to be administrated at low concentrations in behavioral studies to avoid adverse chronic side effects. Amides 8a and 8b exerted excellent stability and no cytotoxicity, however, these two ligands were sedating at 40 mg/kg p.o. Esters (2–5g) including the lead compound 2 exhibited either cytotoxicity or were metabolically less stable in the presence of HLM and MLM. Thioester 7, isopropyl amide 8e and nitrile 10 were not metabolically stable enough to encourage further studies. Carboxylic acid 6 exhibited excellent properties in all three assays, however, the acid functionality will prevent the penetration of the blood brain barrier which was demonstrated for the R enantiomer.27 This property is not suitable for the treatment of schizophrenia and depression. Cognitive deficits developed in these two disease states are still poorly managed with current drugs. Although the data is only preliminary, S-CH3 ligand 9a demonstrated procognitive effects (C+) in the Y-maze paradigm. The S-CH3 ligand 8f was active in the antidepressant assay (forced swim test) and procognitive (A++ and C++) assay.50 However, 8f was metabolized fairly rapidly in HLM as shown in Table 2. To date, these two ligands along with 8c in this S-enantiomeric series seemed to be the best candidates for additional preclinical behavioral testing, while in the R-CH3 series (α5 subtype selective series) a number of ligands have promising pharmacological effects for the treatment of schizophrenia and depression.23–25,31,35,50
Conclusions
It is clear that ligand design to target specific GABAAR subtypes without adverse effects is very difficult, in part, due to the absence of a crystal structure of the α1–6β3γ2 ion channel, as well as the diversity of the receptor subunit expression in different regions of the brain. Although a crystal structure has been reported for a pentameric GABA ion channel (5 beta subunits form the ion pore), which is an important result going forward, however, the lack of a benzodiazepine binding site in this structure renders it ineffective for ligand design.49 In the present research, a number of ligands have been successfully designed and synthesized with the C(4) S-CH3 stereochemistry. This series of esters, amides, and bioisosters of the lead compound 2 are devoid of the liable ester functionality at C(3) to enhance metabolic stability. The synthesis of these analogs, importantly, were inexpensive and straightforward with improved yields over the previous routes.43 More importantly, these reactions can be easily scaled up to gram quantities. The novel ligands 8c and 9a are metabolically stable in the presence of human and mouse liver microsomes and exhibit excellent cytotoxicity (LD50) values. Furthermore, they do not induce sedative effects, which is a major problem of classical benzodiazepine drugs. Taken together, this research of the S-CH3 enantiomers have provided several novel ligands that possess good safety and metabolic profiles for further in vivo behavioral testing in animal models in both schizophrenia and depression.23–25, 31–32 The in vivo activity of 8c and 9a in models of schizophrenia and depression is ongoing and preliminary studies look very promising.50
Experimental Section
General
Unless specified, all reactions were performed in oven-dried round-bottom flasks or a screw-cap test tube or heavy walled pressure vessel under an argon atmosphere, if required. All organic solvents were anhydrous or purified when necessary by standard methods and purchased from commercial suppliers, as well as all the chemicals. Reactions were monitored by TLC on plates from Dynamic Adsorbents, Inc. under a UV light. The purification of some analogs was completed by flash chromatography on silica gel (230–400 mesh, Dynamic Adsorbents). The melting points were determined on a Stuart melting point SMP3 apparatus manufactured by Barloworld Scientific US Ltd. The optical rotation values were recorded on a Jasco DIP-370 Digital Polarimeter. The 1H and 13C NMR spectra were obtained on Bruker Spectrospin 300 MHz and GE 500 MHz instruments, in CDCl3, or DMSO. Chemical shifts were reported in δ (ppm) relative to TMS as an internal standard. Multiplicities are presented as follows: singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), multiplet (m). The purity of all analogs was determined to be > 95%, by HPLC-MS performed on a Shimadzu LCMS-2020 and HRMS was performed on a Shimadzu LCMS-IT-TOF at the Milwaukee Institute for Drug Discovery in the Shimadzu Laboratory for Advanced and Applied Analytical Chemistry.
(S)-Ethyl-8-cyclopropyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-78, 3)
To a solution of the bromo ethyl ester 4b (5.0 g, 11.3 mmol) in toluene (100 mL) and water (14.5 mL), cyclopropyl boronic acid (4.9 g, 57.0 mmol), potassium phosphate (10.3 g, 48.6 mmol) and bis(triphenylphosphine)palladium(II) diacetate (0.85 g, 1.13 mmol) were added under argon. A reflux condenser was attached and the mixture was degassed under vacuum with argon; this process was repeated four times. The mixture was stirred and heated to 100°C. After 12 h the reaction was completed on analysis by TLC (silica gel) and it was then cooled to rt. Water (10 mL) was added and the mixture was extracted with EtOAc (3 × 15 mL), after which the filtrate was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The black residue which resulted was purified by a wash column (silica gel, EtOAc/hexane 7:3) to afford the desired 8-cyclopropyl-imidazobenzodiazepine 3 as a white solid (3.74 g, 82%): mp 106–108 °C; [α]D25 = +65.34 (c 0.81, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.88 (s, 1H), 7.54 (t, J = 7.0 Hz, 1H), 7.48 – 7.32 (m, 2H), 7.19 (t, J = 7.6 Hz, 2H), 7.08 – 6.85 (m, 2H), 6.63 (q, J = 6.9 Hz, 1H), 4.34 (dd, J = 14.4, 7.1 Hz, 2H), 2.11 (d, J = 7.0 Hz, 1H), 1.91 – 1.72 (m, J = 4.0 Hz, 1H), 1.37 (t, J = 7.1 Hz, 3H), 1.23 (d, J = 7.0 Hz, 2H), 0.95 (d, J = 8.1 Hz, 2H), 0.59 (dd, J = 10.1, 4.6 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 164.15 (s), 163.09 (s), 160.10 (d, JC-F = 250.1 Hz), 143.86 (s), 141.56 (s), 134.84 (s), 132.06 (s), 131.65 (s), 131.54 (s), 131.20 (d, JC-F = 1.3 Hz), 129.16 (d, JC-F = 12.8 Hz), 129.08 (d, JC-F = 9.4 Hz), 128.44 (s), 127.85 (s), 124.30 (d, JC-F = 3.3 Hz), 121.93 (s), 115.97 (d, JC-F = 21.6 Hz), 60.58 (s), 50.03 (s), 15.02 (s), 14.62 (s), 14.42 (s), 9.88 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H23FN3O2 404.1769; found 404.1766.
(S)-Methyl-8-bromo-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (MP-III-059, 4a)
The ethyl ester SH-053–2’F-S-CH3 2 (2.0 g, 4.52 mmol) was dissolved in methanol (120 mL). Sodium methoxide (0.98 g, 18.1 mmol) was added in one portion and the solution was heated to reflux. The reaction mixture was monitored on analysis by TLC (silica gel, EtOAc/hexane 4:1) until the starting material had been consumed. This took about 30 min. The reaction mixture was cooled to rt and then quenched with a saturated aq solution of NaHCO3 (20 mL). Water (50 mL) was then added to the solution and the methanol was removed under reduced pressure. The product was extracted with EtOAc (3 × 100 mL) and the organic layers were combined, washed with brine (50 mL) and dried (Na2SO4). The solution was concentrated under reduced pressure. The solid, which resulted, was purified by flash column chromatography (silica gel, EtOAc/hexane 4:1) which provided pure methyl ester 4a as an off-white solid (1.68 g, 87% yield): mp 194–195 °C; [α]D25 = −4.55 (c 0.22, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.92 (s, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.57 (t, J = 7.0 Hz, 1H), 7.51 – 7.36 (m, 3H), 7.23 (t, J = 7.5 Hz, 1H), 7.02 (t, J = 9.3 Hz, 1H), 6.67 (q, J = 7.2 Hz, 1H), 3.89 (s, 3H), 2.13 (d, J = 7.3 Hz, 1H), 1.26 (d, J = 7.3 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 163.29 (s), 162.70 (s), 160.06 (d, JC-F = 251.3 Hz), 141.56 (s), 134.89 (s), 133.57 (s), 133.01 (s), 132.18 (s), 132.07 (s) 131.18 (d, JC-F = 2.0 Hz), 131.10 (s), 129.22 (s), 128.35 (d, JC-F = 12.5 Hz), 124.56 (d, JC-F = 3.4 Hz), 123.69 (s), 120.98 (s), 116.21 (d, JC-F = 21.4 Hz), 51.86 (s), 50.04 (s), 14.86 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C20H16BrFN3O2 428.0404; found 428.0402.
(S)-Methyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (MP-III-021, 5a)
The ethyl ester SH-053–2’F-S-CH3 2 (150 mg, 0.387 mmol) was dissolved in MeOH (20 mL). Sodium methoxide (84 mg, 1.55 mmol) was added in one portion and the solution was heated to reflux. The reaction mixture was monitored by analysis by TLC (silica gel, EtOAc/hexane 4:1) until the starting material had been consumed. This took about 30 min. The reaction mixture was cooled to rt and then quenched with a saturated aq solution of NaHCO3 (4 mL). Water (10 mL) was then added to the solution and the methanol was removed under reduced pressure. The methyl ester 5a was then extracted with EtOAc (3 × 40 mL). The organic layers were combined, washed with brine (10 mL) and dried (Na2SO4). The solution was concentrated under reduced pressure. The solid, which resulted, was purified by flash column chromatography (silica gel, EtOAc/hexane 4:1) which provided pure methyl ester 5a as an off-white solid (117 mg, 81% yield): mp 119–120 °C; [α]D25 = +6.00 (c 2.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.96 (s, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.59 (t, J = 9.5 Hz, 2H), 7.49–7.40 (m, 2H), 7.29–7.21 (m, 1H), 7.04 (t, J = 9.3 Hz, 1H), 6.69 (q, J = 7.2 Hz, 1H), 3.92 (s, 3H), 3.16 (s, 1H), 2.16 (d, J = 7.3 Hz, 1H), 1.29 (d, J = 7.1 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 163.71 (s), 161.17 (s), 161.16 (d, JC-F = 251.1 Hz), 141.47 (s), 135.49 (s), 135.01 (s), 134.46 (s), 134.13 (s), 132.30 (d, JC-F = 7.1 Hz), 131.30 (s), 129.30(s), 129.21 (s), 128.58 (d, JC-F = 9.4 Hz), 124.58 (d, JC-F = 3.2 Hz), 122.33 (s), 121.83 (s), 116.40 (d, JC-F = 21.2 Hz), 81.31 (s), 79.90 (s), 51.93 (s), 49.88 (s), 14.92 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C22H17FN3O2 374.1299; found 374.1307.
General procedure for the synthesis of esters (5b-5e)
The ethyl ester SH-053–2’F-S-CH3 2 (200 mg, 0.52 mmol) was stirred in dry THF (20 mL) in an oven-dried flask under an argon atmosphere at 40 °C. Simultaneously, the corresponding anhydrous alcohol R2-OH (5 mL) was stirred in a separate oven-dried flask under argon at 40 °C. Small pieces of freshly cut Li rod (excess) were quickly added to the dry alcohol solution and the suspension was stirred for 30 min under argon. The ethyl ester solution was then added to the alcohol/lithium mixture and the reaction was monitored on analysis by TLC (silica gel) until the starting material had been consumed. This took 30–45 min. The reaction mixture was then quenched with a saturated aq solution of NaHCO3 aq (15 mL) and the product was extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine (20 mL), dried (Na2SO4) and the solvent was removed under reduced pressure. The solid, which resulted, was purified by flash column chromatography to afford the pure corresponding esters 5b-5e.
(S)-Propyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-32, 5b)
The ester 5b was prepared from 2 following the general procedure for esters with dry 1-propanol. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 7:3) to yield the pure propyl ester 5b as a white powder (0.17 g, 82%): mp 204–205 °C; [α]D25 = +5.26 (c 0.19, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.95 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.59 (dd, J = 15.9, 7.7 Hz, 2H), 7.46 (dd, J = 14.0, 6.5 Hz, 2H), 7.31 – 7.22 (m, 1H), 7.05 (t, J = 9.2, 1H), 6.71 (q, J = 6.6 Hz, 1H), 4.42 – 4.22 (m, 2H), 3.16 (s, 1H), 1.94 – 1.76 (m, 2H), 1.29 (d, J = 6.8 Hz, 3H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.23 (s), 163.02 (s), 160.11 (d, JC-F = 250.9 Hz), 141.64 (s), 135.18 (s), 134.88 (s), 134.54 (s), 133.93 (s), 131.95 (d, JC-F = 8.3 Hz), 131.19 (s), 129.63 (s), 129.59 (s), 128.66 (d, JC-F = 11.6 Hz), 124.49 (d, JC-F = 3.4 Hz), 122.21 (s), 121.62 (s), 116.21 (d, JC-F = 21.4 Hz), 81.43 (s), 79.70 (s), 66.38 (s), 50.14 (s), 22.09 (s), 14.88 (s), 10.49 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H21FN3O2 402.1612; found 402.1615.
(S)-Isopropyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-31, 5c)
The isopropyl ester 5c was prepared from 2 following the general procedure for esters with dry isopropanol. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 3:7) to yield the pure isopropyl ester 5c as a white powder (0.20 g, 97%): mp 231–232 °C; [α]D25 = +14.89 (c 0.94, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.93 (s, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.58 (dd, J = 16.6, 7.7 Hz, 2H), 7.44 (dd, J = 14.2, 6.6 Hz, 2H), 7.25 (t, J = 7.5 Hz, 1H), 7.04 (t, J = 9.2 Hz 1H), 6.70 (q, J = 7.0 Hz, 1H), 5.36 – 5.20 (m, 1H), 3.15 (s, 1H), 1.40 (dd, J = 9.4, 6.4 Hz, 6H), 1.28 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.19 (s), 162.52 (s), 160.12 (d, JC-F = 251.9 Hz), 141.63 (s), 135.16 (s), 134.76 (s), 134.57 (s), 133.92 (s), 131.92 (d, JC-F = 8.7 Hz), 131.18 (s), 129.87 (s), 129.62 (s), 128.73 (d, JC-F = 6.0 Hz), 124.48 (d, JC-F = 3.4 Hz), 122.20 (s), 121.60 (s), 116.19 (d, JC-F = 21.5 Hz), 81.44 (s), 79.67 (s), 68.26 (s), 50.14 (s), 21.95 (s), 14.85 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H21FN3O2 402.1612; found 402.1610.
(S)-tert-Butyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-30, 5d)
The t-butyl ester 5d was prepared from 2 following the general procedure for esters with dry t-butanol. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 3:7) to yield the pure t-butyl ester 5d as a white powder (0.21 g, 96%): mp 214–215 °C; [α]D25 = + 15.29 (c 0.85, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.92 (s, 1H), 7.69 (d, J = 7.3 Hz, 1H), 7.65 – 7.51 (m, 2H), 7.45 (dd, J = 15.0, 6.9 Hz, 2H), 7.28 – 7.21 (m, 1H), 7.05 (t, J = 9.3 Hz, 1H), 6.67 (q, J = 6.9 Hz, 1H), 3.15 (s, 1H), 1.63 (s, 9H), 1.28 (d, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.17 (s), 162.24 (s), 160.12 (d, JC-F = 250.7 Hz), 141.19 (s), 135.14 (s), 134.69 (s), 134.58 (s), 133.91 (s), 131.91 (d, JC-F = 8.2 Hz), 131.16 (s), 130.85 (s), 129.59 (s), 128.73 (d, JC-F = 13.1 Hz), 124.49 (d, JC-F = 3.5 Hz), 122.22 (s), 121.50 (s), 116.21 (d, JC-F = 21.4 Hz), 81.51 (s), 79.60 (s), 50.23 (s), 28.33 (s), 21.97 (s), 14.85 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C25H23FN3O2 416.1769; found 416.1768.
(S)-tert-Pentyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-33, 5e)
The 2-methylbutyl ester 5e was prepared from 2 following the general procedure for esters with dry 2-methylbutan-2-ol. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 1:1) to yield the pure 2-methylbutyl ester 5e as a white powder (0.17 g, 77%): mp 94–96 °C; [α]D25 = +34.15 (c 0.41, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.92 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.66 – 7.50 (m, 2H), 7.45 (dd, J = 16.3, 8.6 Hz, 2H), 7.29 – 7.19 (m, 1H), 7.05 (t, J = 9.2 Hz, 1H), 6.66 (q, J = 7.2 Hz, 1H), 3.15 (s, 1H), 1.98 (q, J = 7.4 Hz, 2H), 1.59 (s, 6H), 1.28 (d, J = 3.6 Hz, 3H), 0.99 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.17 (s), 162.15 (s), 160.11 (d, JC-F = 250.2 Hz), 141.12 (s), 135.15 (s), 134.68 (s), 133.89 (s), 131.96 (s), 131.85 (s), 131.16 (d, JC-F = 1.6 Hz), 130.80 (s), 129.58 (s), 128.71 (d, JC-F = 13.2 Hz), 124.49 (d, JC-F = 3.4 Hz), 122.24 (s), 121.49 (s), 116.20 (d, JC-F = 21.5 Hz), 84.17 (s), 81.48 (s), 79.61 (s), 50.24 (s), 33.47 (s), 25.91 (s), 25.73 (s), 14.91 (s), 8.47 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C26H25FN3O2 430.1925; found 430.1928.
(S)-8-Ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylic acid (SH-05 3–2’F-S-CH3-acid, 6)
The ethyl ester SH-053–2’F-S-CH3 2 (5.0 g, 12.91 mmol) was dissolved in EtOH (200 mL), after which sodium hydroxide pellets (4.1 g, 103.25 mmol) were added to the solution. This reaction mixture was heated to 55 ˚C for 0.5 h and the EtOH was removed under reduced pressure. The remaining aq solution was stirred at 0 ˚C for 10 min and then aq HCl (1 M) was added dropwise to the solution until the pH was 5 (pH paper). A pale white precipitate which formed, was left in the solution for 10 min and was then collected by filtration, washed with cold water and the aq layer also allowed to stand at rt for 10 h to yield additional acid 6. The combined solids were dried under vacuum for 7 h to provide pure acid 6 as a white powder (4.6 g, 99%): mp 217–218 °C; [α]D25 = −14.58 (c 0.48, CHCl3); 1H NMR (300 MHz, DMSO-d6): δ 8.36 (s, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.55 (dt, J = 7.8, 6.5 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.21 (t, J = 9.3 Hz, 2H), 6.61 (d, J = 7.6 Hz, 1H), 4.36 (s, 1H), 1.13 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, DMSO-d6): δ 164.63 (s), 162.92 (s), 159.85 (d, JC-F = 242.7 Hz), 141.08 (s), 136.73 (s), 135.57 (s), 134.68 (d, JC-F = 2.7 Hz), 133.19 (s), 133.14 (s), 132.64 (d, JC-F = 6.1 Hz), 131.89 (s), 129.36 (d, JC-F = 2.3 Hz), 128.85 (d, JC-F = 8.2 Hz), 125.14 (d, J = 3.0 Hz), 123.98 (s), 121.15 (s), 116.39 (d, JC-F = 20.3 Hz), 83.42 (s), 81.97 (s), 49.82 (s), 15.02 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C21H15FN3O2 360.1143; found 360.1142.
General procedure for the synthesis of esters/thioester/amides (5f, 5g, 7, 8b-8f)
A mixture of the acid SH-053–2’F-S-CH3-acid 6 (200 mg, 0.56 mmol), thionyl chloride (0.407 mL, 5.6 mmol) and dry DCM (20 mL) was placed in an oven dried round bottom flask under argon. This suspension, which formed, was allowed to reflux at 60 ˚C for 1 h under argon. The absence of the starting material was confirmed on analysis by TLC (silica gel). The organic solvent and excess thionyl chloride were removed under reduced pressure on a rotovapor. This procedure was repeated five times with dry DCM (15 mL). The yellow residue, which remained was dissolved in dry DCM (20 mL) and cooled to 0 ˚C for 10 min under argon. Then the appropriate nucleophilic alcohol/thiol/amine (5.6 mmol), followed by triethylamine (0.78 mL, 5.6 mmol) was added to the reaction mixture at 0˚C and the mixture individually was then allowed to warm to rt and stirred for 1–2 h. After the completion of the reaction by TLC (silica gel), the solvent was removed under reduced pressure. The residue was treated with ice cold water (15 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was washed with brine (20 mL) and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by column chromatography to yield the corresponding pure esters, thioesters or amides described below.
(S)-2,2,2-Trifluoroethyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (GL-I-36, 5f)
The trifluoroethyl ester 5f was prepared from carboxylic acid 6 following the general procedure for esters/thioester/amides with dry 2,2,2-trifluoroethanol as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 2:3) to yield the pure trifluoroethyl ester 5f as a white powder (0.20 g, 82%): mp 226–227 °C; [α]D25 = +9.43 (c 0.53, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.98 (s, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 7.9 Hz, 2H), 7.50 – 7.36 (m, 2H), 7.23 (t, J = 7.3 Hz, 1H), 7.01 (t, J = 8.9 Hz, 1H), 6.60 (q, J = 7.0 Hz, 1H), 4.88 – 4.54 (m, 2H), 3.16 (s, 1H), 1.26 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.41 (s), 161.09 (s), 160.07 (d, JC-F = 250.9 Hz), 143.06 (s), 135.54 (s), 135.29 (s), 134.19 (s), 133.89 (s), 132.08 (d, JC-F = 8.1 Hz), 131.16 (s), 129.61 (s), 128.45 (d, JC-F = 12.7 Hz), 127.72 (s), 124.53 (d, JC-F = 3.2 Hz), 123.04 (q, JC-F = 276.5 Hz), 122.31 (s), 121.95 (s), 116.20 (d, JC-F = 21.4 Hz), 81.27 (s), 79.99 (s), 60.15 (q, JC-F = 36.9 Hz), 50.10 (s), 14.70 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C23H16F4N3O2 442.1173; found 442.1176.
(S)-Cyclopropyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxyl- ate (GL-I-38, 5g)
The cyclopropyl ester 5g was prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry cyclopropanol as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 3:2) to yield the pure cyclopropyl ester 5g as a white powder (0.16 g, 71%): mp 230–231 °C; [α]D25 = +11.38 (c 1.23, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.92 (s, 1H), 7.67 (d, J = 7.4 Hz, 1H), 7.55 (d, J = 7.9 Hz, 2H), 7.49 – 7.32 (m, 2H), 7.22 (t, J = 7.0 Hz, 1H), 7.00 (t, J = 8.6 Hz, 1H), 6.64 (q, J = 6.6 Hz, 1H), 4.38 – 4.21 (m, 1H), 3.15 (s, 1H), 1.24 (d, J = 5.6 Hz, 3H), 0.87 (d, J = 7.1 Hz, 2H), 0.77 (d, J = 5.5 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 163.77 (s), 163.25 (s), 160.06 (d, JC-F = 250.6 Hz), 141.87 (s), 135.20 (s), 134.93 (s), 134.39 (s), 133.88 (s), 131.97 (d, JC-F = 9.0 Hz), 131.15 (s), 129.58 (s), 129.04 (s), 128.60 (d, JC-F = 11.4 Hz), 124.49 (d, JC-F = 3.1 Hz), 122.25 (s), 121.69 (s), 116.17 (d, JC-F = 21.5 Hz), 81.37 (s), 79.85 (s), 50.02 (s), 49.32 (s), 14.82 (s), 5.22 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H19FN3O2 400.1456; found 400.1453.
(S)-S-Ethyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carbothioate (GL-I-77, 7)
The thioester 7 was prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry ethanethiol as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 2:3) to yield the pure thioethyl ester 7 as a white powder (0.18 g, 79%): mp 210–211 °C; [α]D25 = +21.43 (c 0.98, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.91 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.64 – 7.50 (m, 2H), 7.48 – 7.38 (m, 2H), 7.24 (t, J = 7.3 Hz, 1H), 7.02 (t, J = 9.1 Hz, 1H), 6.65 (q, J = 14.0, 6.8 Hz, 1H), 3.16 (s, 1H), 3.09 – 2.89 (m, 2H), 1.33 (t, J = 7.4 Hz, 3H), 1.26 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 187.81 (s), 163.16 (s), 160.11 (d, JC-F = 250.3 Hz), 138.17 (s), 135.44 (s), 135.17 (s), 134.48 (s), 134.34 (s), 133.95 (s), 131.97 (d, JC-F = 8.9 Hz), 131.23 (s), 129.66 (s), 128.61 (d, JC-F = 11.7 Hz), 124.49 (d, JC-F = 3.4 Hz), 122.19 (s), 121.76 (s), 116.17 (d, JC-F = 21.5 Hz), 81.41 (s), 79.78 (s), 49.85 (s), 22.56 (s), 14.79 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C23H19FN3OS 404.1227; found 404.1229.
(S)-8-Ethynyl-6-(2-fluorophenyl)-N,4-dimethyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxamide (MP-III-023, 8a)
The ethyl ester SH-053–2’F-S-CH3 2 (200 mg, 0.52 mmol) was added to a sealed vessel fitted with a septum at −30 °C and treated with methyl amine (10 mL, 33% wt solution in EtOH). The vessel was sealed with a screw-cap and stirred at 50 °C for 18 h. The solution was then cooled to rt and the methyl amine and ethanol were removed under reduced pressure. The residue, which resulted, was purified by flash column chromatography (silica gel, EtOAc/hexane 3:2) to afford the pure methyl amide 8a as a pale white powder (0.15g, 79%): mp 136–137 °C; [α]D25 = −11.54 (c 2.34, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.84 (s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.64 (t, J = 7.0 Hz, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.44 (dd, J = 14.1, 7.1 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 7.17 (s, 1H), 7.03 (t, J = 9.3 Hz, 1H), 6.91 (q, J = 6.5 Hz, 1H), 3.15 (s, 1H), 2.97 (d, J = 5.0 Hz, 3H), 1.27 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 163.41 (s), 162.86 (s), 161.81(d, JC-F = 250.8 Hz), 138.68 (s), 135.31 (s), 134.58 (s), 134.14 (s), 133.46 (s), 132.28 (s), 132.26 (d, JC-F = 6.4 Hz), 131.47 (s), 129.47 (s), 128.36 (d, JC-F = 10.0 Hz), 124.54 (d, JC-F = 3.5 Hz), 122.24 (s), 121.69 (s), 116.28 (d, JC-F = 21.5 Hz), 81.42 (s), 79.76 (s), 49.66 (s), 25.67 (s), 15.06 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C22H18FN4O 373.1459, found: 373.1462.
(S)-N-Ethyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxamide (GL-I-43, 8b)
The ethyl amide 8b was prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry ethylamine as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 1:1) to yield the pure ethyl amide 8b as a white powder (0.18 g, 88%): mp 208–209 °C; [α]D25 = −13.33 (c 0.6, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.97 (s, 1H), 7.69 (dd, J = 16.7, 7.9 Hz, 2H), 7.58 (d, J = 8.2 Hz, 1H), 7.46 (dd, J = 13.4, 7.2 Hz, 2H), 7.27 (t, J = 7.4 Hz, 2H), 7.04 (t, J = 9.3 Hz, 1H), 6.93 (q, J = 7.2 Hz, 1H), 3.55 – 3.37 (m, 2H), 3.16 (s, 1H), 1.29 (d, J = 7.2 Hz, 3H), 1.23 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 162.82 (s), 162.51 (s), 160.10 (d, JC-F = 250.5 Hz), 138.79 (s), 134.97 (s), 134.70 (s), 133.88 (s), 133.39 (s), 131.82 (d, JC-F = 4.8 Hz), 131.75 (s), 131.35 (d, JC-F = 1.5 Hz), 129.76 (s), 128.84 (d, JC-F = 11.9 Hz), 124.45 (d, JC-F = 3.4 Hz), 122.06 (s), 121.37 (s), 116.06 (d, JC-F = 21.5 Hz), 81.54 (s), 79.55 (s), 49.88 (s), 33.71 (s), 15.02 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C23H20FN4O 387.1614; found 387.1614.
(S)-N-Cyclopropyl-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carbox- amide (GL-I-55, 8c)
The cyclopropyl amide 8c as prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry cyclopropylamine as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 1:1) to yield the pure cyclopropyl amide 8c as a white powder (0.19 g, 86%): mp 225–226 °C; [α]D25 = −11.77 (c 0.17, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.00 (s, 1H), 7.69 (dd, J = 16.5, 8.1 Hz, 2H), 7.58 (d, J = 8.3 Hz, 1H), 7.46 (dd, J = 14.0, 6.8 Hz, 3H), 7.27 (t, J = 7.3 Hz, 1H), 7.04 (t, J = 9.3 Hz, 1H), 6.93 (q, J = 7.2 Hz, 1H), 3.17 (s, 1H), 2.85 (dq, J = 10.6, 3.6 Hz, 1H), 1.30 (d, J = 7.3 Hz, 3H), 0.89 – 0.73 (m, 2H), 0.69 – 0.50 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 164.02 (s), 162.83 (s), 160.12 (d, JC-F = 249.8 Hz), 138.93 (s), 134.96 (s), 134.66 (s), 133.91 (s), 133.36 (s), 131.82 (d, JC-F = 8.3 Hz), 131.59 (s), 131.37 (s), 129.79 (s), 128.84 (d, JC-F = 11.4 Hz), 124.46 (d, JC-F = 3.3 Hz), 122.04 (s), 121.42 (s), 116.07 (d, JC-F = 21.5 Hz), 81.53 (s), 79.53 (s), 49.89 (s), 22.09 (s), 15.00 (s), 6.55 (s), 6.51 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H20FN4O 399.1616; found 399.1618
(S)-8-Ethynyl-6-(2-fluorophenyl)-N-isopropyl-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxa- mide (GL-I-57, 8d)
The isopropyl amide 8d was prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry isopropylamine as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 2:3) to yield the pure isopropyl amide 8d as a white powder (0.16 g, 74%): mp 238–239 °C; [α]D25 = −6.19 (c 2.1, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.81 (s, 1H), 7.62 (dd, J = 17.0, 7.9 Hz, 2H), 7.51 (d, J = 8.3 Hz, 1H), 7.46 – 7.33 (m, 2H), 7.21 (t, J = 7.5 Hz, 1H), 7.02 – 6.94 (m, 2H), 6.88 (q, J = 6.9 Hz, 1H), 4.21 (dq, J = 13.4, 6.6 Hz, 1H), 3.14 (s, 1H), 1.22 (dd, J = 13.5, 7.2 Hz, 9H); 13C NMR (75 MHz, CDCl3): δ 162.83 (s), 161.78 (s), 158.43 (s), 138.78 (s), 134.98 (s), 134.69 (s), 133.85 (s), 133.35 (s), 131.85 (s), 131.74 (s), 131.35 (s), 129.73 (s), 128.83 (d, JC-F = 12.7 Hz), 124.43 (d, JC-F = 3.3 Hz), 122.08 (s), 121.36 (s), 116.04 (d, JC-F = 21.6 Hz), 81.53 (s), 79.62 (s), 49.87 (s), 40.69 (s), 22.93 (s), 22.85 (s), 15.00 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H22FN4O 401.1772; found 401.1776.
(S)-N-(tert-Butyl)-8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carbox- amide (GL-I-41, 8e)
The t-butyl amide 8e was prepared from the carboxylic acid 6 following the general procedure for esters/thioester/amides with dry t-butylamine as the nucleophile. The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 1:1) to yield the pure t-butyl amide 8e as a white powder (0.19 g, 83%): mp 136–137 °C; [α]D25 = +8.7 (c 0.23, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.80 (s, 1H), 7.65 (dd, J = 15.1, 7.7 Hz, 2H), 7.51 (d, J = 8.3 Hz, 1H), 7.44 (dd, J = 10.1, 7.6 Hz, 2H), 7.24 (t, J = 7.5 Hz, 1H), 7.02 (dd, J = 15.8, 5.8 Hz, 2H), 6.87 (q, J = 7.1 Hz, 1H), 3.15 (s, 1H), 1.46 (s, 9H), 1.27 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 162.84 (s), 162.11 (s), 160.15 (d, JC-F = 250.3 Hz), 138.60 (s), 134.97 (s), 134.76 (s), 133.85 (s), 133.02 (s), 132.59 (s), 131.84 (d, JC-F = 7.8 Hz), 131.39 (d, JC-F = 2.4 Hz), 129.81 (s), 128.79 (d, JC-F = 11.8 Hz), 124.45 (d, JC-F = 3.4 Hz), 122.07 (s), 121.37 (s), 116.07 (d, JC-F = 21.5 Hz), 81.56 (s), 79.49 (s), 50.91 (s), 49.90 (s), 28.98 (s), 14.96 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C25H24FN4O 415.1926; found 415.1926.
(S)-8-Ethynyl-6-(2-fluorophenyl)-N,N,4-trimethyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxamide (GL-I-54, 8f)
The dimethyl amide 8f was prepared from 6 following the general procedure with dry dimethylamine as the nucleophile. The crude residue was purified by a column chromatography (silica gel, EtOAc and 1% MeOH) to yield pure dimethyl amide 8f as a light yellow powder (0.15 g, 71%): mp 142–144 °C; [α]D25 = −16.0 (c 0.75, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.94 (s, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.52 (d, J = 8.2 Hz, 1H), 7.48 – 7.41 (m, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.04 (t, J = 9.3 Hz, 1H), 4.33 (q, J = 6.0 Hz, 1H), 3.16 (s, 1H), 3.14 (s, 3H), 3.01 (s, 3H), 1.94 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 166.14 (s), 162.60 (s), 160.25 (d, JC-F = 252.3 Hz), 135.31 (s), 134.73 (d, JC-F = 7.2 Hz), 133.83 (s), 133.73 (s), 132.79 (d, JC-F = 12.7 Hz), 132.05 (d, JC-F = 9.4 Hz), 131.78 (s), 131.28 (s), 129.25 (s), 127.69 (d, JC-F = 10.8 Hz), 124.51 (d, JC-F = 3.4 Hz), 122.83 (s), 121.50 (s), 116.17 (d, JC-F = 21.7 Hz), 81.57 (s), 79.46 (s), 52.21 (s), 39.09 (s), 35.03 (s), 18.50 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C23H20FN4O 387.1616; found 387.1624.
General Procedure for the Preparation of Oxadiazoles (9a-9c)
The ethyl ester 2 (200 mg, 0.52 mmol) was dissolved in dry THF (20 mL) at rt under argon. In a separate flask which contained 3Å molecular sieves, the corresponding oxime (2.08 mmol) was dissolved in dry THF (30 mL) under argon and treated with sodium hydride (60% dispersion in mineral oil, 0.57 mmol). The mixture, which resulted, was stirred for 15 min at which point the solution containing the ethyl ester was added. The reaction mixture, which resulted, was stirred at rt for 2 h until the starting material was consumed as indicated on analysis by TLC (silica gel). The reaction mixture was quenched with a saturated aq NaHCO3 solution (50 mL). Water (50 mL) was then added and the product was extracted with EtOAc (3 × 100 mL). The organic layers were combined, washed with brine (30 mL) and dried (Na2SO4). The solvent was removed under reduced pressure. The solid, which resulted, was purified by flash column chromatography (silica gel) to afford the pure corresponding oxadiazole.
(S)-5-(8-Ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)-3-methyl-1,2,4-oxadiazole (GL-I-65, 9a)
The methyl oxadiazole 9a was prepared from 2 following the general procedure for oxadiazoles with the methyl oxime (0.154 g, 2.08 mmol). The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 3:2) to yield pure methyl oxadiazole 9a as a white powder (0.174 g, 85%): mp 225–226 °C; [α]D25 = +32.52 (c 2.86, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.08 (s, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 7.48 (dd, J = 18.1, 11.6 Hz, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.06 (t, J = 9.2 Hz, 1H), 6.75 (q, J = 7.1 Hz, 1H), 3.18 (s, 1H), 2.47 (s, 3H), 1.35 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 170.75 (s), 167.42 (s), 163.40 (s), 160.09 (d, JC-F = 250.5 Hz), 139.26 (s), 136.32 (s), 135.34 (s), 134.17 (s), 134.05 (s), 132.06 (d, JC-F = 8.5 Hz), 131.20 (s), 129.63 (s), 128.55 (d, JC-F = 12.3 Hz), 124.83 (s), 124.50 (d, JC-F = 3.2 Hz), 122.21 (s), 121.88 (s), 116.21 (d, JC-F = 21.4 Hz), 81.34 (s), 79.95 (s), 50.16 (s), 14.94 (s), 11.66 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C23H17FN5O 398.1412; found 398.1419.
(S)-3-Ethyl-5-(8-ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)-1,2,4-oxadiazole (GL-I-66, 9b)
The ethyl oxadiazole 9b was prepared from 2 following the general procedure for oxadiazoles with the ethyl oxime (0.183 g, 2.08 mmol). The crude residue was purified by flash column chromatography (silica gel, EtOAc/Hexane 3:2) to yield pure ethyl oxadiazole 9b as a white powder (0.168 g, 79%): mp 199–200 °C; [α]D25 = +50.00 (c 0.22, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.08 (s, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 8.3 Hz, 2H), 7.54 – 7.40 (m, 2H), 7.27 (t, J = 7.4 Hz, 1H), 7.06 (t, J = 9.2 Hz, 1H), 6.75 (q, J = 14.4, 7.2 Hz, 1H), 3.18 (s, 1H), 2.84 (q, J = 7.6 Hz, 2H), 1.40 (d, J = 7.6 Hz, 3H), 1.36 (t, J = 3.6 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 171.88 (s), 170.70 (s), 163.41 (s), 160.10 (d, JC-F = 251.4 Hz), 139.22 (s), 136.26 (s), 135.33 (s), 134.22 (s), 134.07 (s), 132.04 (d, JC-F = 8.7 Hz), 131.15 (s), 129.58 (d, JC-F = 6.0 Hz), 128.58 (d, JC-F = 11.9 Hz), 124.99 (s), 124.51 (d, JC-F = 3.2 Hz), 122.18 (s), 121.88 (s), 116.22 (d, JC-F = 21.5 Hz), 81.34 (s), 79.90 (s), 50.23 (s), 19.74 (s), 14.97 (s), 11.51 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C24H19FN5O 412.1568; found 412.1569.
(S)-5-(8-Ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)-3-isopropyl-1,2,4-oxadiazole (GL-I-81, 9c)
The isopropyl oxadiazole 9c was prepared from 2 following the general procedure for oxadiazoles with the isopropyl oxime (0.212 g, 2.08 mmol). The crude residue was purified by flash column chromatography (silica gel, EtOAc/hexane 3:2) to yield pure isopropyl oxadiazole 9c as a white powder (0.180 g, 82%): mp 205–206 °C; [α]D25 = +22.37 (c 0.76, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.06 (s, 1H), 7.66 (d, J = 8.3 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 7.44 – 7.30 (m, 2H), 7.17 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 9.2 Hz, 1H), 6.67 (q, J = 7.0 Hz, 1H), 3.14 (s, 1H), 3.09 (q, J = 7.0 Hz, 1H), 1.32 (d, J = 7.0 Hz, 6H), 1.28 (d, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 175.24 (s), 170.61 (s), 163.38 (s), 160.03 (d, JC-F = 251.5 Hz), 139.17 (s), 136.34 (s), 135.36 (s), 134.17 (s), 133.98 (s), 132.01 (d, JC-F = 8.3 Hz), 131.10 (s), 129.48 (s), 128.58 (d, JC-F = 12.3 Hz), 124.96 (s), 124.48 (d, JC-F = 3.1 Hz), 122.28 (s), 121.82 (s), 116.16 (d, JC-F = 21.4 Hz), 81.30 (s), 80.03 (s), 50.22 (s), 26.70 (s), 20.57 (s), 20.51 (s), 14.94 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C25H21FN5O 426.1725; found 426.1728.
(S)-8-Ethynyl-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carbonitrile (MP-III-018.A, 10)
The ethyl ester SH-053–2’F-S-CH3 2 (0.5 g, 1.40 mmol) was stirred in dry xylene (35 mL) at rt. Using a glass syringe and a metal needle, dimethylaluminum amine (0.67 M, 12.5 mL, 8.42 mmol) was carefully added to the solution of starting material 2. The reaction was then heated to 80 ˚C in an oil bath and was monitored on analysis by TLC (silica gel) until the starting material had been consumed in 2 h. Once the reaction was complete, the mixture was cooled to rt and then quenched with cold water (15 mL). The product was extracted with EtOAc (5 × 50 mL) and the organic layers were combined, washed with brine and dried (Na2SO4). The solvent was removed under reduced pressure. The solid, which resulted, was purified by flash column chromatography (silica gel, EtOAc/hexane 1:1) which afforded the nitrile 10 as a white solid (0.331 g, 75.4%): mp 131–132 °C; [α]D25 = −165.16 (c 1.55, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.98 (s, 1H), 7.78 (d, J = 7.1 Hz, 1H), 7.67 (s, 1H), 7.62 – 7.53 (m, 1H), 7.49 (s, 2H), 7.28 (s, 1H), 7.04 (t, J = 8.7 Hz, 1H), 4.36 (d, J = 4.8 Hz, 1H), 3.20 (s, 1H), 2.18 (d, J = 4.6 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 163.64 (s), 161.94 (d, JC-F = 253.4 Hz), 143.66 (s), 135.75 (s), 135.69 (s), 133.97 (s), 133.35 (s), 132.89 (d, JC-F = 9.7 Hz),131.37 (s), 129.22 (s), 126.85 (d, JC-F = 13.0 Hz), 124.73 (d, JC-F = 6.5 Hz), 122.88 (s), 122.64 (s), 116.50(d, JC-F = 19.7 Hz), 114.54 (s), 110.74 (s), 81.13 (s), 80.33 (s), 51.68 (s), 18.09 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C21H14FN4 341.1137; found 341.1142.
Preparation of the 0.67 M dimethylaluminum amine solution
The ammonia gas was bubbled into dry DCM (20 mL) at 0 °C until the solution was saturated. This took about 10–15 min. The trimethylaluminum (10 mL, 2.0 M in toluene) was then added to the solution. The solution was stirred at rt for 5 min and transferred directly for use in the reaction above using a glass syringe and metal needle.
Supplementary Material
Acknowledgements
We wish to acknowledge the NIH for generous financial support (R01MH096463, R01NS076517, R01HL118561). We also thank the Milwaukee Institute for Drug Discovery and University of Wisconsin-Milwaukee’s Shimadzu Laboratory for Advanced and Applied Analytical Chemistry for help with spectroscopy.
References
- (1).Frangou S Medicine 2008, 36, 405. [Google Scholar]
- (2).Kapur S; Mamo D Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 1081. [DOI] [PubMed] [Google Scholar]
- (3).Leucht S; Corves C; Arbter D; Engel RR; Li C; Davis JM The Lancet 2009, 373, 31. [DOI] [PubMed] [Google Scholar]
- (4).Longo LP; Johnson B Am. Fam. Physician 2000, 61, 2121. [PubMed] [Google Scholar]
- (5).Wassef A; Baker J; Kochan LD J. Clin. Psychopharmacol. 2003, 23, 601. [DOI] [PubMed] [Google Scholar]
- (6).Fee C; Banasr M; Sibille E Biol. Psychiatry 2017, 82, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Richter L; de Graaf C; Sieghart W; Varagic Z; Morzinger M; de Esch IJ; Ecker GF; Ernst M Nat. Chem. Biol. 2012, 8, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Olsen RW; Sieghart W Neuropharmacology 2009, 56, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cossart R; Bernard C; Ben-Ari Y Trends Neurosci. 2005, 28, 108. [DOI] [PubMed] [Google Scholar]
- (10).Connolly CN; Krishek BJ; McDonald BJ; Smart TG; Moss SJ J. Biol. Chem. 1996, 271, 89. [DOI] [PubMed] [Google Scholar]
- (11).Sieghart W Pharmacol. Rev. 1995, 47, 181. [PubMed] [Google Scholar]
- (12).Gielen MC; Lumb MJ; Smart TG J. Neurosci. 2012, 32, 5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rudolph U; Mohler H Curr. Opin. Pharmacol. 2006, 6, 18. [DOI] [PubMed] [Google Scholar]
- (14).Rudolph U; Crestani F; Benke D; Brunig I; Benson JA; Fritschy JM; Martin JR; Bluethmann H; Mohler H Nature 1999, 401, 796. [DOI] [PubMed] [Google Scholar]
- (15).Low K; Crestani F; Keist R; Benke D; Brunig I; Benson JA; Fritschy JM; Rulicke T; Bluethmann H; Mohler H; Rudolph U Science 2000, 290, 131. [DOI] [PubMed] [Google Scholar]
- (16).Crestani F; Keist R; Fritschy JM; Benke D; Vogt K; Prut L; Bluthmann H; Mohler H; Rudolph U Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Collinson N; Kuenzi FM; Jarolimek W; Maubach KA; Cothliff R; Sur C; Smith A; Otu FM; Howell O; Atack JR; McKernan RM; Seabrook GR; Dawson GR; Whiting PJ; Rosahl TW J. Neurosci. 2002, 22, 5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cook JM; Zhou H; Huang S; Sarma PVVS; Zhang C US Patent 7,618,958 B2, Nov. 17, 2009.
- (19).Savic MM; Clayton T; Furtmuller R; Gavrilovic I; Samardzic J; Savic S; Huck S; Sieghart W; Cook JM Brain Res. 2008, 1208, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Landoni MF; Soraci A Curr. Drug. Metab. 2001, 2, 37. [DOI] [PubMed] [Google Scholar]
- (21).Patocka J; Ales DJ Appl. Biomed. 2004, 2, 95. [Google Scholar]
- (22).Mellin GW; Katzenstein MN Engl. J. Med. 1962, 267, 1184. [DOI] [PubMed] [Google Scholar]
- (23).Clayton T; Chen JL; Ernst M; Richter L; Cromer BA; Morton CJ; Ng H; Kaczorowski CC; Helmstetter FJ; Furtmuller R; Ecker G; Parker MW; Sieghart W; Cook JM Curr. Med. Chem. 2007, 14, 2755. [DOI] [PubMed] [Google Scholar]
- (24).Gill KM; Cook JM; Poe MM; Grace AA Schizophr. Bull. 2014, 40, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gill KM; Lodge DJ; Cook JM; Aras S; Grace AA Neuropsychopharmacology 2011, 36, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Piantadosi SC; French BJ; Poe MM; Timic T; Markovic BD; Pabba M; Seney ML; Oh H; Orser BA; Savic MM; Cook JM; Sibille E Front. Pharmacol. 2016, 7, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Batinic B; Santrac A; Jancic I; Li G; Vidojevic A; Markovic B; Cook JM; Savic MM Int. J. Dev. Neurosci. 2017, 61, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Forkuo GS; Nieman AN; Yuan NY; Kodali R; Yu OB; Zahn NM; Jahan R; Li G; Stephen MR; Guthrie ML; Poe MM; Hartzler BD; Harris TW; Yocum GT; Emala CW; Steeber DA; Stafford DC; Cook JM; Arnold LA Mol. Pharm. 2017, 14, 2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mizuta K; Xu D; Pan Y; Comas G; Sonett JR; Zhang Y; Panettieri RA Jr.; Yang J; Emala CW Sr. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gallos G; Yocum GT; Siviski ME; Yim PD; Fu XW; Poe MM; Cook JM; Harrison N; Perez-Zoghbi J; Emala CW Sr. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fischer BD; Licata SC; Edwankar RV; Wang Z-J; Huang S; He X; Yu J; Zhou H; Johnson EM; Cook JM; Furtmüller R; Ramerstorfer J; Sieghart W; Roth BL; Majumder S; Rowlett JK Neuropharmacology 2010, 59, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Savić MM; Majumder S; Huang S; Edwankar RV; Furtmüller R; Joksimović S; Clayton T; Ramerstorfer J; Milinković MM; Roth BL; Sieghart W; Cook JM Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Richetto J; Labouesse MA; Poe MM; Cook JM; Grace AA; Riva MA; Meyer U Int. J. Neuropsychopharmacol. 2015, 18, pyu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Takai S; Matsuda A; Usami Y; Adachi T; Sugiyama T; Katagiri Y; Tatematsu M; Hirano K Biol. Pharm. Bull. 1997, 20, 869. [DOI] [PubMed] [Google Scholar]
- (35).Clayton T; Poe MM; Rallapalli S; Biawat P; Savic MM; Rowlett JK; Gallos G; Emala CW; Kaczorowski CC; Stafford DC; Arnold LA; Cook JM Int. J. Med. Chem. 2015, 2015, 430248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Batinić B; Santrač A; Jančić I; Li G; Vidojević A; Marković B; Cook JM; Savić MM Int. J. Dev. Neurosci. 2017, 61, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Poe MM; Methuku KR; Li G; Verma AR; Teske KA; Stafford DC; Arnold LA; Cramer JW; Jones TM; Cerne R; Krambis MJ; Witkin JM; Jambrina E; Rehman S; Ernst M; Cook JM; Schkeryantz JM J. Med. Chem. 2016, 59, 10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fischer BD; Schlitt RJ; Hamade BZ; Rehman S; Ernst M; Poe MM; Li G; Kodali R; Arnold LA; Cook JM Brain Res. Bull. 2017, 131, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Witkin JM; Cerne R; Wakulchik M; S; Gleason; Jones; Li; Arnold; Li; Schkeryantz; Methuku; Cook; Poe Pharmacol. Biochem. Behav. 2017, 157, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wallace DJ; Chen CY Tetrahedron Lett. 2002, 43, 6987. [Google Scholar]
- (41).Otera J Chem. Rev. 1993, 93, 1449. [Google Scholar]
- (42).Cook JM; Clayton TS; Jain HD; Johnson YT; Yang J; Rallipalli SK; Wang ZJ; Namjoshi OA; Poe MM US Patent 9,006,233 B2, Apr. 14, 2015.
- (43).Patani GA; LaVoie EJ Chem. Rev. 1996, 96, 3147. [DOI] [PubMed] [Google Scholar]
- (44).Namjoshi OA; Wang Z.-j.; Rallapalli SK; Johnson EM; Johnson Y-T; Ng H; Ramerstorfer J; Varagic Z; Sieghart W; Majumder S; Roth BL; Rowlett JK; Cook JM Bioorganic Med. Chem. 2013, 21, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Fleming FF; Yao L; Ravikumar PC; Funk L; Shook BC J. Med. Chem. 2010, 53, 7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wood JL; Khatri NA; Weinreb SM Tetrahedron Lett. 1979, 20, 4907. [Google Scholar]
- (47).Sanberg PR; Bunsey MD; Giordano M; Norman AB Behav. Neurosci. 1988, 102, 748. [DOI] [PubMed] [Google Scholar]
- (48).Bakshi VP; Geyer MA Psychopharmacology (Berl.) 1995, 122, 198. [DOI] [PubMed] [Google Scholar]
- (49).Geng Y; Bush M; Mosyak L; Wang F; Fan QR Nature 2013, 504, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Prevot T; Li G; Vidojevic A; Santrac A; Misquitta K; Fee CJ; Knutson D; Stephen MR; Kodali R; Zahn N; Arnold A; Scholze P; Fisher J; Banasr M; Cook JM; Savic M; Sibille E, manuscript in preparation. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.