

Abstract
Objective
Clinical, neuroimaging, and genetic characterization of 3 patients with LINS1-associated developmental regression, intellectual disability, dysmorphism, and further neurologic deficits.
Methods
Three affected brothers from a consanguineous family from Afghanistan, their 2 healthy siblings, and both parents were all assessed in the clinic. General and neurologic examination, expert dysmorphology examination, and 3T brain MRI were performed. Whole-exome sequencing was performed for the 3 affected brothers, followed by Sanger sequencing in all available family members.
Results
The index patient and his 2 affected brothers presented a complex neurologic syndrome with similar features but marked intrafamilial phenotypical variability, including varying degrees of cognitive impairment, speech impairment, dystonia, abnormal eye movements, and dysmorphic features. All 3 affected brothers are homozygous for a novel, pathogenic frameshift mutation in LINS1, c.1672_1679del, and p.Gly558Profs*22, whereas both parents and healthy siblings are heterozygous for the mutation. No major brain malformations were evident in 3T brain MRI of the affected brothers.
Conclusion
This consanguineous family with a novel mutation expands the spectrum of LINS1-associated disorder to include developmental regression, oculomotor signs, and dystonia, previously not described in the published 9 cases of this rare disorder. The 3T-MRI data from our 3 patients and review of the neuroimaging data in the literature showed unspecific brain MRI changes. LINS1 protein is a known modulating factor of the Wnt signaling pathway, with important roles in organogenesis including of the cerebral cortex. More research is warranted to disentangle the underlying pathophysiologic mechanisms, leading to cognitive impairment and the complex phenotype of LINS1-associated disorder.
Pathogenic mutations in LINS1 (Online Mendelian Inheritance in Man [OMIM] *610350) have first been described in 2011 in 4 siblings with intellectual disability of one consanguineous family.1 Two affected children of another consanguineous family with LINS1 mutations showed in addition stereotypies and facial dysmorphism.2 In total, only 9 patients from 4 families have been reported in the literature1–4 (table 1).
Table 1.
Clinical and genetic features of LINS1-associated neurodevelopmental disorder in the literature and in this family

The gene product, LINS1, is a modulating factor of the Wnt signaling pathway,5 which plays a role in development and growth, especially during embryogenesis.6,7 In a recent genome-wide association meta-analysis of brain MRI data, genetic variants influencing the structure of the human cerebral cortex were found to cluster near genes involved in the Wnt signaling pathway.8 Wnt proteins are known to regulate neural progenitor fate decisions.9 Of interest, mutations in Cip1-interacting zinc finger protein 1 have been shown to lead to primary cervical dystonia by influencing the Wnt signaling pathway.10
Methods
Patient consent
Genetic studies were performed in a diagnostic context after written informed consent or assent from legal guardians.
Patients and study design
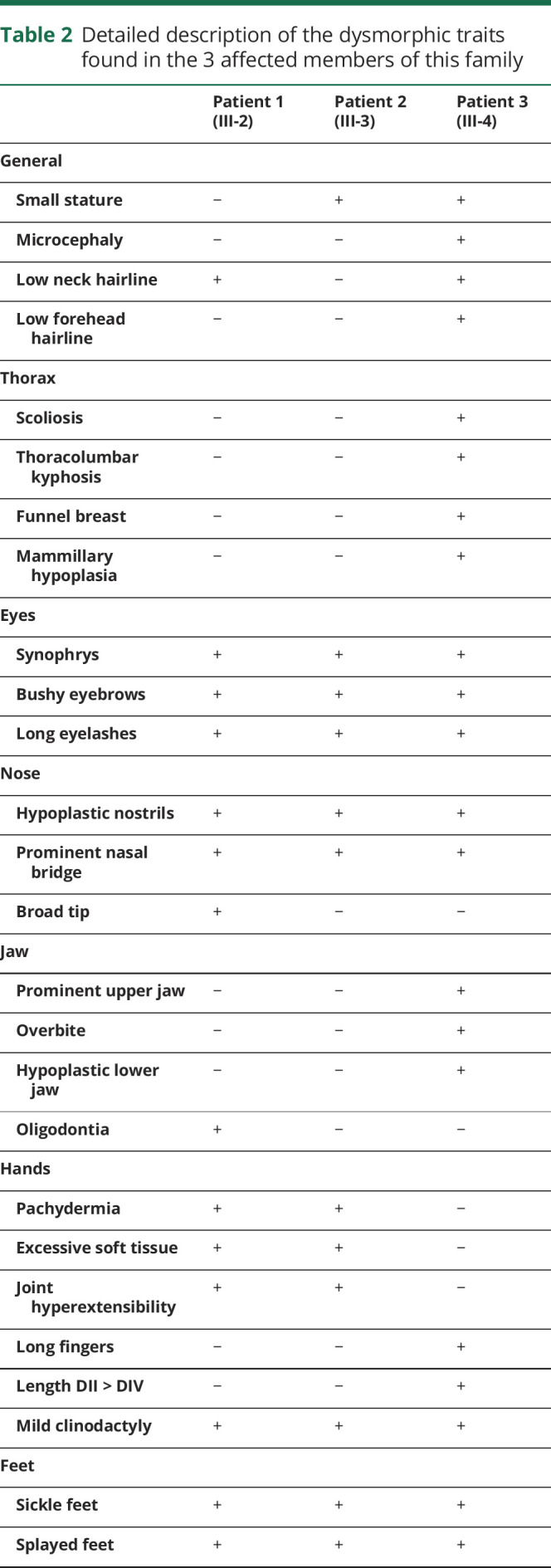
The 19-year-old index patient from a consanguineous family from Afghanistan (figure) was referred to the clinic with dystonia. We also assessed his 4 siblings, 2 of whom also presented with severe neurologic signs, and both parents, including neurologic and general physical examination (table 2). Formal cognitive testing was not possible because of the language barrier and the lack of cooperation. A dysmorphology expert assessed all 3 affected siblings.
Figure. Neuroradiologic findings, dysmorphic features, and pedigree.

(A) 3T brain MRI scans of patient 1 showed normal cortex and gray-white differentiation in 3D T1w images and no signs of microbleeds or hemorrhage in T2*. There was a slight bilateral widening of the postcentral and intraparietal sulci. FLAIR images showed multiple small, unspecific white matter lesions in the left parietal and bilateral frontal white matter. There was a minimal asymmetry with wider temporal horn of the left lateral ventricle. (B and C) Because of movement artifacts, the only scans of diagnostic quality from patients 2 and 3 were fast T2w Propeller acquisitions, repeated several times in multiple orientations. They showed no obvious brain malformations and no signs of leukoencephalopathy. Slice orientation is asymmetric because of the movement of patient 3 (C). The 3 affected brothers (patient 1: A, E, and I; patient 2: B, F, and J; patient 3: C, G, H, and K) presented with varying degrees of several dysmorphic features, including dysmorphism of the face (E–G), fingers (I–K), and feet (H). All had clinodactyly of digit V, and patient 3 showed long and thin fingers, with digit II longer than digit IV (K). Pedigree of this consanguineous family (D) showing the 3 affected siblings (III-2: patient 1, index; III-3: patient 2; and III-4: patient 3). The 2 healthy siblings and both parents were also genetically tested and were found to be heterozygous for the novel LINS1 mutation.
Table 2.
Detailed description of the dysmorphic traits found in the 3 affected members of this family
Next-generation sequencing workflow and sequencing analyses
Whole-exome sequencing (WES) was performed for the 3 patients. Sanger sequencing was performed to confirm the LINS1 mutation in the affected siblings and to test the carrier status in the parents and healthy siblings. After enrichment of exonic DNA fragments with a SureSelect Human All Exon Kit (Agilent, 50 Mb V5), sequencing was performed on a HiSeq2500 system (Illumina). Assuming an autosomal recessive mode of inheritance, the exome data were searched for homozygous or compound heterozygous variants with an allele frequency of less than 1%. The prioritized mutations were evaluated for known associations with inherited disease in the literature and OMIM database.
Neuroimaging studies
A 3T MRI of the brain was performed for the 3 affected siblings and evaluated by an experienced neuroradiologist. MRI was acquired on a General Electric Signa HDxt 3T scanner. Acquisition parameters are described in detail in Supplemental Data (e-Methods, links.lww.com/NXG/A310).
Data availability
Any data from this work not published within this article will be shared on reasonable request from any qualified investigator. Individual participant data will only be shared in a deidentified form. The data are not publicly available because of the information that could compromise the privacy of the participants.
Results
Clinical reports
Index patient 1 (III-2)
This 19-year-old male patient, second of 5 siblings of a consanguineous family from Afghanistan, was referred to our clinic for episodes of hand “cramps” (figure, A, E, I). After normal pregnancy, normal delivery at term and normal development during the first 3 years of life, regression of psychomotor development became apparent at age 3 years, followed by learning disability. From age 8 years on, stereotypical movement of the hands was noted. Episodes of cramps of the hands were reported, triggered by stress or repetitive hand movements, compatible with dystonia. General examination showed facial dysmorphism. Neurologic examination revealed stuttering, hypometric vertical saccades, transiently restricted abduction and impaired conjugation of the eyes, dysdiadochokinesis, impaired fine motor skills, and mild postural tremor of the hands. He works in a sheltered workshop and has learned some German. Formal cognitive testing was not possible because of language difficulties and short attention span.
Patient 2 (III-3)
This 18-year-old male sibling was born at term after unremarkable pregnancy and birth (figure, B, F, J). He had normal behavior, speech, and psychomotor development until the age of 4 years. Behavioral changes started at 4 years of age, with challenging behavior, increasingly restless, and defiant. He also developed urinary incontinence and stuttering. From age 14 years, slowly progressive cognitive impairment was noted, as were episodes of challenging behavior, some of which required inpatient psychiatric assessment and antipsychotic therapy. General examination was remarkable for obesity and facial dysmorphism. Neurologic examination showed divergent strabismus, unconjugated eye movements, and mild dystonic gait in a friendly and cooperative but restless and inattentive patient, who obeyed simple commands and tried to communicate but only in Farsi.
Patient 3 (III-4)
This 17-year-old brother was the most severely affected (figure G, H, K). After unremarkable pregnancy and delivery at term, early motor development was normal. He first crawled at 6 months but started walking only at 2 years. He never learned to talk but for a short period could vocalize the syllables “nam” and “mam”. At age 2 years, he started having recurrent episodes of vomiting. He started swallowing objects (pica eating disorder) and putting objects in his ears, necessitating constant supervision, resulting in several gastrointestinal operations (due to gastric hemorrhages and intestine perforations) and ENT procedures for tympanic reconstruction and chronic otitis. He has bladder and bowel incontinence. Short stature (151 cm), cachectic appearance (27 kg), microcephaly, and dysmorphism were evident on general examination. Neurologic examination was remarkable for mutism, although the patient was partly cooperative and could interact nonverbally and follow very simple commands in Farsi. He smiled and reacted to eye contact and direct physical contact. He was restless, exhibiting frequent stereotypic movements or behaviors, for example, rumination, fumbling, or swaying movements of the upper body. He had unconjugated eye movements and a dystonic gait but could walk alone for a short distance.
Genetic studies
Through WES, we identified rare variants in 61 genes associated with an autosomal recessive mode of inheritance, 8 of which were listed in OMIM: CYP2C19, LINS1, IFT140, ABCA4, TSPEAR, ALMS1, C1GALT1C1, and G6PD. Of those, only LINS1 had been previously associated with phenotypic features found in our patients (table 1). Regions of homozygosity found for the three patients are listed in Supplemental Data table e1 (links.lww.com/NXG/A311).
All affected brothers carried the novel homozygous frameshift mutation in LINS1, c.1672_1679del/p.Gly558Profs*22 (NM_001040616.2), which was later confirmed by Sanger sequencing. Both healthy parents and healthy siblings are heterozygous carriers of the LINS1 mutation, as shown by Sanger sequencing. The genetic variant was not found in exomes from about 8,000 individuals from an in-house database and about 120,000 alleles from the Exome Aggregation Consortium. This frameshift mutation predicts a premature stop of the translation of the LINS1 protein.
Neuroimaging findings
The 3T brain MRI of the index patient showed multiple small, unspecific white matter lesions in the left parietal and bilateral frontal white matter, and a slight bilateral widening of the postcentral and intraparietal sulci (figure, A). Also, for patients 2 and 3, no obvious brain malformation was detected and no signs of leukoencephalopathy (figure, B, C), but image quality was limited by movement artifacts.
Dysmorphology assessment
All 3 affected brothers had dysmorphism, but, interestingly, their physical appearance was very diverse (table 2). Common features included prominent nasal bridge with hypoplastic nostrils, bushy eyebrows with synophrys and long eyelashes (figure, E–G), and mild bilateral radial clinodactyly of the 5th digit (figure, I–K). Dysmorphism was most pronounced in patient 3, who had microcephaly with low forehead and neck hairline, prominent upper and hypoplastic lower jaw with pronounced overbite (figure, G), thoracic dysplasia with funnel thorax, mammillary hypoplasia, scoliosis, and sickle feet (figure, H).
Discussion
Through WES in a consanguineous family with 3 affected siblings, we identified the novel 8-bp deletion c.1672_1679del (p.Gly558Profs*22) in LINS1 causing a complex neurologic syndrome and dysmorphism, with a remarkable broad intrafamilial severity spectrum.
In the literature so far, only 9 patients from 4 families had been described with LINS1-associated syndromes. After initially normal development, our patients showed developmental regression (from 3 years, 4 years and 6 months of life, respectively), leading to intellectual disability in varying degrees, with delayed or even absent speech development. We found stereotypic movements, and dysmorphism, as already described in the literature. Here, we also describe novel phenotypical features, including behavioral abnormalities, pica eating disorder, oculomotor disorder, and dystonia. Importantly, we provide a thorough description of the dysmorphic features of the head, face, and extremities, expanding the spectrum of LINS1-associated disease and highlighting its complex phenotype.
We found no other genetic variants, which may contribute to the phenotype of the 3 patients, but cannot exclude entirely the possibility of other genetic variants contributing to the phenotype.
The mechanisms linking LINS1 to cognition remain mostly unclear. The gene product, LINS1, is a regulating factor of the Wnt signaling pathway,5 which plays a role in embryogenesis, development, and growth, and has links to neurodegenerative disease.6,7 Further studies are warranted to better understand the pathophysiologic mechanisms underlying the complex clinical presentation caused by LINS1 genetic variation.
Acknowledgment
The authors are grateful to the patients and the family members, who agreed to be a part of this study. T. Klopstock is a member of the European Reference Network for Rare Neurological Diseases (ERN-RND).
Glossary
- OMIM
Online Mendelian Inheritance in Man
- WES
whole-exome sequencing
Appendix. Authors
Study funding
No funders.
Disclosure
C. Neuhofer reports no disclosures relevant to the manuscript. C.B. Catarino reports no disclosures relevant to the manuscript. H. Schmidt reports no disclosures relevant to the manuscript. K. Seelos reports no disclosures relevant to the manuscript. B. Alhaddad reports no disclosures relevant to the manuscript. T.B. Haack reports no disclosures relevant to the manuscript. T. Klopstock reports no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011;478:57–63. [DOI] [PubMed] [Google Scholar]
- 2.Akawi NA, Al-Jasmi F, Al-Shamsi AM, Ali BR, Al-Gazali L. LINS, a modulator of the WNT signaling pathway, is involved in human cognition. Orphanet J Rare Dis 2013;8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheth J, Ranjan G, Shah K, Bhavsar R, Sheth F. Novel LINS1 missense mutation in a family with non-syndromic intellectual disability. Am J Med Genet A 2017;173:1041–1046. [DOI] [PubMed] [Google Scholar]
- 4.McMillan HJ, Holahan AL, Richer J. Worster-Drought syndrome associated with LINS mutations. Child Neurol Open 2018;5:2329048X18791083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatini V, Bokor P, Goto-Mandeville R, DiNardo S. Tissue- and stage-specific modulation of Wingless signaling by the segment polarity gene lines. Genes Dev 2000;14:1364–1376. [PMC free article] [PubMed] [Google Scholar]
- 6.Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci 2010;11:77–86. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasby KL, Jahanshad N, Painter JN, et al. The genetic architecture of the human cerebral cortex. Science 2020:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 2011;31:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao J, Uitti RJ, Zhao Y, et al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann Neurol 2012;71:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data from this work not published within this article will be shared on reasonable request from any qualified investigator. Individual participant data will only be shared in a deidentified form. The data are not publicly available because of the information that could compromise the privacy of the participants.