

Abstract
The use of single-domain antibody fragments, or nanobodies, has gained popularity in recent years as an alternative to traditional monoclonal antibody-based approaches. Relatively little is known, however, about the utility of nanobodies as targeting agents for delivery of therapeutic cargoes, particularly to vascular epitopes or in the setting of acute inflammatory conditions. We used a nanobody (VCAMelid) directed against mouse vascular cell adhesion molecule 1 (VCAM-1) and techniques for site-specific radiolabeling and bioconjugation to measure targeting to sites of constitutive and inducible antigen expression and investigate the impact of various characteristics (affinity, valence, circulation time) on nanobody biodistribution and pharmacokinetics. Engineering of VCAMelid for bivalent binding (BiVCAMelid) increased affinity by an order of magnitude and provided 2.8- and 3.6-fold enhancements in splenic and brain targeting in naive mice, with a further 2.6-fold increase in brain uptake in the setting of focal CNS inflammation. In contrast, introduction of an albumin-binding arm (VCAM/ALB8) did not affect binding affinity, but its prolonged circulation time resulted in 3.5-fold and 17.4-fold increases in splenic and brain uptake at 20 min post-dose and remarkable 40-, 25-, and 15-fold enhancements in overall exposure of blood, spleen, and brain, respectively, relative to both VCAMelid and BiVCAMelid. Both therapeutic protein (superoxide dismutase, SOD-1) and nanocarrier (liposome) delivery were enhanced by conjugation to VCAM-1 targeted nanobodies. The bispecific VCAM/ALB8 maintained its superiority over VCAMelid in enhancing both circulation time and organ targeting of SOD-1, but its advantages were largely blunted by conjugation to liposomes.
Graphical Abstract
INTRODUCTION
Targeted drug delivery to sites of vascular injury, inflammation, or disease can be accomplished using a variety of immunologic affinity moieties, including monoclonal antibodies (mAb) and recombinant single chain antigen-binding fragments (scFv).1,2 Recently, a new class of recombinant affinity ligands, derived from camelid heavy chain antibodies,3 has garnered attention as a promising alternative to traditional immunoglobulins in a wide array of biomedical applications.4–6 These agents, often termed single domain antibodies (sdAb) or nanobodies, consist solely of a variable heavy chain fragment and represent the smallest binding region derived from a functional immunoglobulin, with an average molecular weight of ~15 kDa.4 Nanobodies have several characteristics which distinguish them from traditional mAb and scFv, including smaller size, allowing potential access to sterically obscured or otherwise cryptic epitopes, high solubility, and remarkable stability to variations in pH, temperature, and other physical stressors.4 To date, nanobodies have been utilized in vivo primarily as agents for molecular imaging,6–8 although the recent clinical success and approvals in Europe and the USA of an anti-von Willebrand Factor sdAb, caplacizumab, has spurred on the investigation of many other preclinical applications.9–11 A few reports have investigated the use of nanobodies as targeting molecules,12–19 i.e., affinity ligands for the delivery of radionuclides, biotherapeutic cargo, or even macromolecular drug carriers, but the molecular properties which may make them more or less advantageous for these applications remain poorly defined. Likewise, little has been done to quantify delivery of nanobody-targeted cargoes to the vascular endothelium in either naive or inflammatory conditions.
Several factors may limit the utility of nanobodies as targeting molecules. Their small size results in rapid elimination from the circulation,20 limiting the plasma concentration needed to drive binding and uptake. In addition, the nanobody has just a single binding arm and engages targets in a monovalent fashion, often manifesting in lower affinities and more rapid dissociation kinetics than traditional antibodies. These issues are potential liabilities not only for drug delivery, but also antigen capture and receptor blockade, applications which have progressed to industrial development and clinical trials. As such, it is not surprising that a number of strategies have been developed to address both the pharmacokinetic (PK) profile and monovalent binding of sdAbs. Molecular modifications include conjugation to branched or linear polyethylene glycol (PEG),21 fusion with albumin-binding domains,22–25 fusion with Fc fragments of IgG,26,27 and generation of multivalent nanobody fusions.24,28,29 While these approaches have found utility in other applications—caplacizumab, for example, is a bivalent nanobody—their impact on nanobodies as a recombinant affinity ligand for targeted delivery of therapeutic cargoes has not been extensively studied.
In the present work, we provide a quantitative evaluation of the biodistribution of a nanobody against mouse vascular cell adhesion molecule 1 (mVCAM-1), both in naive animals and in models of acute vascular inflammation. Furthermore, we take advantage of several recently reported molecular modifications to investigate the impact of nanobody binding affinity, avidity, and pharmacokinetics on vascular targeting of organs with high constitutive VCAM-1 expression (spleen) and sites of VCAM-1 induction following focal inflammatory insult (brain). Finally, we explore the use of anti-VCAM-1 nanobody—and several of its molecularly engineered derivatives—as targeting molecules for the delivery of therapeutic protein (superoxide dismutase-1) and translational nanoparticles (liposomes) to healthy and injured tissues.
RESULTS
Monovalent Nanobody Characterization.
One of several reported anti-VCAM-nanobody clones (hereafter referred to as “VCAMelid”)30 was synthesized in a “sortagged” construct described previously.31 VCAMelid, also known as cAbVCAM1–5,30 is a high-affinity mouse/human cross-reactive nanobody. The C-terminal sortag enables transpeptidation by the bacterial enzyme Sortase A (SrtA) and, through the use of short, functionalized peptides, site-specific addition of fluorophores, radiolabels, or reactive chemical groups. These in turn facilitate the measurement of binding or biodistribution (i.e., via radiotracing) and the conjugation of the nanobody to cargo, without compromising affinity for the target antigen.31–33 This is particularly important for small protein affinity ligands like nanobodies, which are more likely to be affected by nonspecific amine- and thiol-based conjugation chemistry.31 Sortase reaction efficiency for nanobodies was similar to that previously reported for scFv.31,34 As shown in Figure S1a, the purity of all sortagged nanobodies was confirmed to be >90% via SEC-HPLC. Specific binding of VCAMelid to mouse VCAM-1 expressing REN cells was confirmed using flow cytometry, and the equilibrium dissociation constant (KD) was estimated to be 58.3 ± 5.8 (mean ± SEM) nM (Figure 1a). In all experiments, the nontargeted control was a previously described anti-human intercellular adhesion molecule 1 (hICAM-1) nanobody,35 which bound with high affinity to hICAM-1-expressing cells (Figure S2). The importance of site-specific modification was demonstrated by comparing the measured binding affinity for C-terminal-modified (SrtA transpeptidation) and amine-modified (NHS-ester) VCAMelid. Random, amine coupling of fluorophore led to an ~10-fold reduction in binding affinity to VCAM-1 expressing REN cells compared to C-terminal modification (509 ± 200 nM vs 58.3 ± 5.8 nM) (Figure S3).
Figure 1.
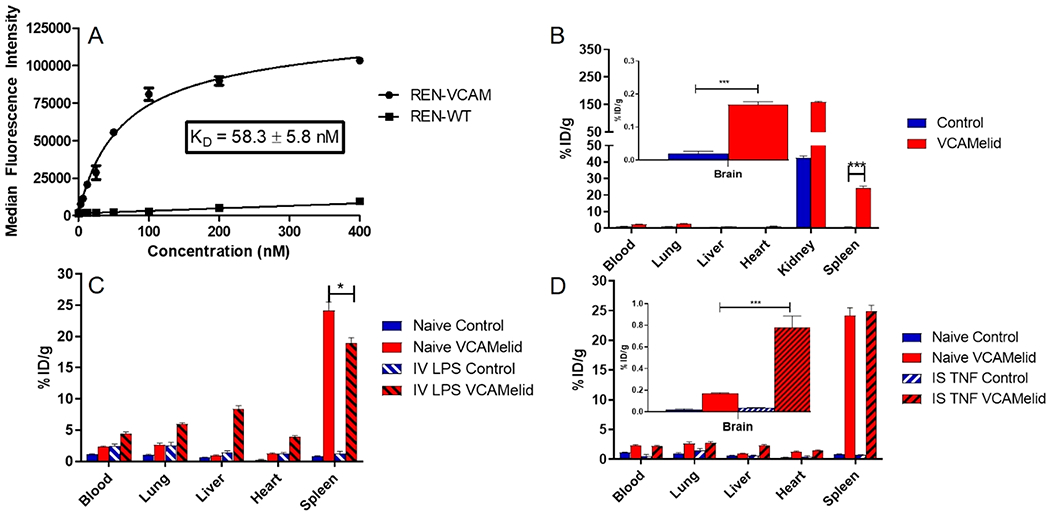
Characterization of VCAMelid targeting. (a) Specific binding of VCAMelid to VCAM-expressing cells. (b) Whole-body biodistribution of VCAMelid and untargeted control in naive, (c) LPS-injected animals, and (d) intrastriatal (IS) TNF-injected animals. Comparisons were made by one-way ANOVA with Tukey’s post-hoc test, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). All biodistributions were performed 20 min post-injection of 5 μg of nanobody.
Figure 1b shows the biodistribution of radiolabeled VCAMelid and control nanobody in naive animals. Both were rapidly eliminated from the circulation, with 2.36 ± 0.10%ID/g and 1.13 ± 0.07%ID/g remaining in the blood at 20 min post-injection, respectively. Consistent with previous reports, high uptake was seen in the kidney, likely due to efficient renal filtration.30,36,37 In naive mice, VCAMelid was found to specifically target the spleen compared to control (24.2 ± 1.3%ID/g vs 0.836 ± 0.088%ID/g, p < 0.0001 by unpaired t test), consistent with high basal expression of VCAM-1 on splenic endothelial cells, myeloid cells, and follicular dendritic cells.38 VCAMelid also showed specific uptake in the brain (0.167 ± 0.009%ID/g vs 0.0205 ± 0.0068%ID/g, p = 0.0002), consistent with the high basal expression of its ligand on brain endothelial cells. Small but significant differences were seen in the uptake of the VCAMelid vs control in the lung, liver, and heart, and since the animal was perfused prior to organ harvest, these appear to represent VCAM-1 binding, rather than residual blood content.
The biodistribution of both VCAMelid and control were significantly affected by the induction of systemic inflammation (intravenous endotoxin, Figure 1c). Blood concentrations of both nanobodies increased to 4.45 ± 0.26%ID/g (p = 0.0016 vs naive) and 2.50 ± 0.28%ID/g (p = 0.0092 vs naive), respectively. VCAMelid uptake in lung, liver, and heart also increased, reflecting greater expression or accessibility of VCAM-1 in the presence of inflammation. Notably, control uptake also increased in these organs (Figure 1c), indicating some contribution of non-ligand-mediated extravasation of these relatively small proteins following endotoxin exposure. Additionally, increased blood and tissue concentrations in the context of systemic inflammation may be due to reductions in cardiac output and renal function. In contrast to other organs, splenic uptake of VCAMelid was slightly reduced (24.2 ± 1.3% ID/g vs 18.9 ± 0.9%ID/g, p = 0.0286).
Finally, we examined the biodistribution of the nanobodies in the presence of focal CNS inflammation (Figure 1d). In contrast to systemic endotoxinemia, focal brain insult had no effect on the blood concentration of either nanobody (2.36 ± 0.10%ID/g vs 2.26 ± 0.11%ID/g for the VCAMelid, p = 0.5533) or VCAMelid targeting to spleen (24.2 ± 1.3 vs 25.0 ± 0.9%ID/g, p = 0.6416). Brain uptake (Figure 1d), however, was increased 4.7-fold (0.167 ± 0.009 vs 0.781 ± 0.106%ID/g, p = 0.0046).
Bivalent Nanobody Characterization.
One strategy that has been reported for improving the in vivo performance of nanobodies is the engineering of recombinant bivalent molecules, with the goal of increasing avidity and binding affinity, and slowing renal clearance by increasing molecular size. To study the impact of these alterations on the quantitative biodistribution of the VCAMelid, we synthesized a “BiVCAMelid” using the upper hinge of llama IgG2 as a linker, a strategy previously reported to promote proper folding and optimal orientation of the two nanobody arms.28 Protein purity obtained via SEC HPLC is shown in Figure S1b. Inclusion of a second binding site enhanced the affinity for mVCAM-1 expressing cells by 8.6-fold (KD = 6.80 ± 0.46 nM) (Figure 2a), as compared to the monovalent nanobody.
Figure 2.
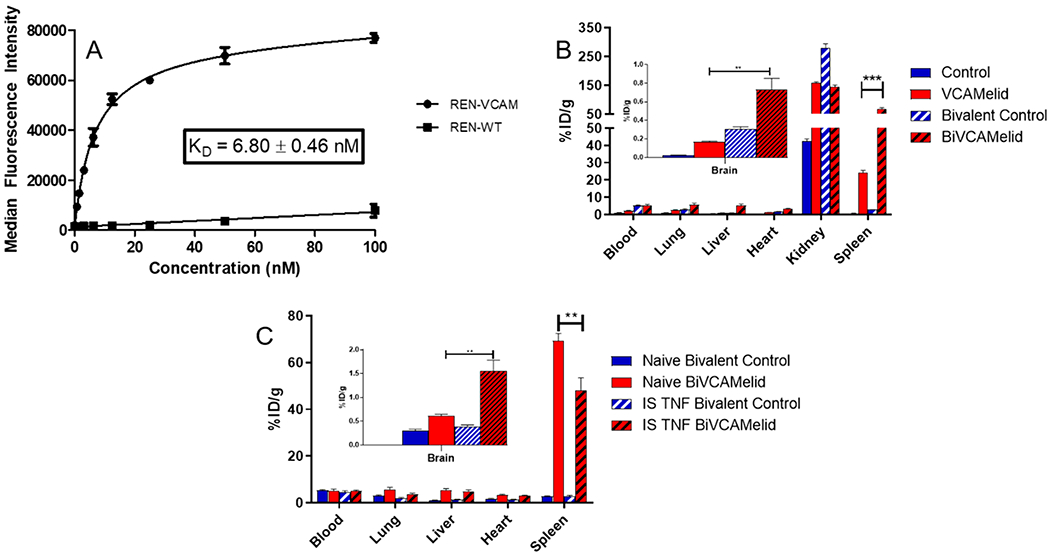
Characterization of BiVCAMelid targeting. (a) Specific binding of BiVCAMelid to VCAM-expressing cells, (b) whole-body biodistribution of VCAMelid and BiVCAMelid in naive animals, and (c) whole-body biodistribution of BiVCAMelid and untargeted control in naive and injured animals. Comparisons were made by one-way ANOVA with Tukey’s post-hoc test, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). All biodistributions were performed 20 min post-injection of 10 μg of bivalent nanobody.
Figure 2b shows the biodistribution of BiVCAMelid, as compared to its monovalent form. While elimination from the blood remained rapid, concentration was increased at 20 min post-injection (Figure 2b, 5.22 ± 0.71%ID/g vs 2.36 ± 0.10% ID/g, p = 0.016). This effect appears to be related to molecular size, as a similar increase was observed for bivalent control (5.27 ± 0.39%ID/g vs 1.13 ± 0.07%ID/g for control, p = 0.0005). Apart from the kidneys, uptake of the BiVCAMelid was increased in all other organs, relative to VCAMelid (Figure 2b), with particularly large effects in the spleen (69.2 ± 3.3% ID/g vs 24.2 ± 1.3%ID/g, p = 0.0002) and brain (0.607 ± 0.041%ID/g vs 0.167 ± 0.009, p = 0.0181). Some component of this increase in uptake may not be antigen-specific, however, as a number of organs (e.g., lung, heart, spleen) also demonstrated increased uptake of bivalent control, as compared to its monovalent form (Figure 2b).
We next assessed the performance of the BiVCAMelid in the focal brain inflammation model (Figure 2c). As with monovalent VCAMelid, focal injury had little effect on blood concentration or uptake by lung, liver, or heart. Splenic targeting was reduced (69.2 ± 3.3%ID/g vs 48.0 ± 5.5%ID/g, p = 0.0303). In contrast, targeting to the brain was markedly increased following TNF-α injection (0.607 ± 0.041%ID/g vs 1.56 ± 0.22%ID/g in naive vs injured animals, p = 0.0137) (Figure 2c,d). No statistically significant changes were seen for the bivalent control (Figure 2c).
Bispecific Nanobody Characterization.
We next produced a genetic fusion of VCAMelid and a species cross-reactive (mouse, monkey, and human) albumin-binding nanobody (ALB8),22,23 using the same llama IgG2 linker. Binding to serum albumin is a widely reported strategy for extending the pharmacokinetics of small proteins and peptides,39 thought to extend circulation time by decreasing renal filtration and taking advantage of the recycling of albumin by the neonatal Fc receptor (FcRn).40,41 The expression and purity of the bispecific VCAM/ALB8 were similar to the other engineered forms of the nanobody (Figure S1c) and the protein was readily modified by SrtA. Binding to mouse VCAM-1 expressing cells was shown to be preserved in the presence of the albumin-binding arm, with KD (32.1 ± 3.1 nM) similar to that of the VCAMelid (Figure 3a). We also confirmed binding to human serum albumin (HSA) (KD = 5.83 ± 1.32 nM, Figure 3b) and the ability of the bispecific VCAM/ALB8 to simultaneously engage mVCAM-1 and HSA (Figure 3c).
Figure 3.
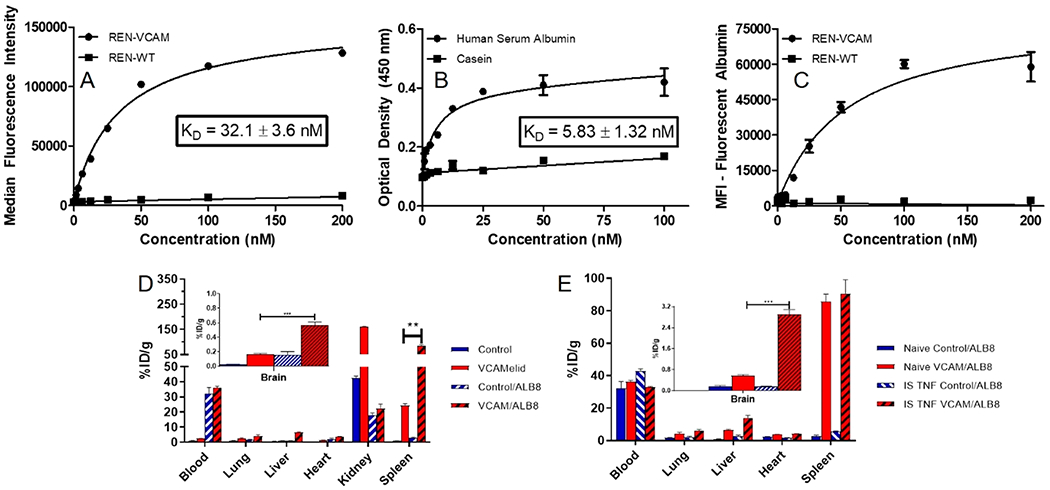
Characterization of VCAM/ALB8 bispecific nanobody. (a) Binding of VCAM/ALB8 to VCAM-expressing cells, (b) binding of VCAM/ALB8 to human serum albumin, measured using plate-based ELISA, (c) simultaneous binding of VCAM/ALB8 to VCAM-expressing cells and to human serum albumin, measured using fluorescently labeled HSA, mouse VCAM-1 expressing cells (or controls), and varying concentrations of VCAM/ALB8, (d) whole-body biodistribution of VCAM/ALB8 and untargeted control, as compared to monovalent forms, and (e) distribution of VCAM/ALB8 and untargeted control in naive and injured animals. Comparisons were made by one-way ANOVA with Tukey’s post-hoc test, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). All biodistributions were performed 20 min post-injection of 10 μg of bispecific nanobody.
The impact of binding to serum albumin is evident in Figure 3d, which shows a large percentage of the dose remaining in the blood 20 min post-injection for both bispecific VCAM/ALB8 (36.3 ± 0.8%ID/g vs 1.13 ± 0.07%ID/g, p < 0.0001, relative to VCAMelid) and control/ALB8 (32.3 ± 4.1%ID/g vs 2.36 ± 0.10%ID/g, p = 0.0016, relative to control). VCAM/ALB8 also had significantly increased uptake in all organs except for the kidney, as compared to VCAMelid, with particularly large effects in the spleen (85.5 ± 4.6% ID/g vs 24.2 ± 1.3%ID/g, p = 0.0002) and brain (0.570 ± 0.039%ID/g vs 0.167 ± 0.009, p = 0.0006). Interestingly, splenic uptake of VCAM/ALB8 in naive mice was significantly greater than that of BiVCAMelid (85.5 ± 4.6 vs 69.2 ± 3.3%ID/g, p < 0.05), whereas brain uptake was nearly identical (0.570 ± 0.039 vs 0.607 ± 0.041%ID/g, p = 0.3427), indicating subtle differences in the relative importance of prolonged circulation time and bivalent binding in targeting VCAM-1 in these organs.
Figure 3e shows the biodistribution of VCAM/ALB8 in mice with focal brain inflammation. While splenic uptake was unchanged (85.5 ± 4.6%ID/g vs 90.5 ± 8.6%ID/g, p = 0.6349) (Figure 3e), brain targeting was enhanced 5.1-fold (0.570 ± 0.039%ID/g vs 2.90 ± 0.18%ID/g, p = 0.0002). This was entirely antigen specific, as no induction of targeting was seen for control/ALB8 (0.156 ± 0.046%ID/g vs 0.157 ± 0.057%ID/g, p = 0.9756) (Figure 3e). Notably, brain targeting by VCAM/ALB8 in injured mice was significantly greater than both VCAMelid (2.90 ± 0.18 vs 0.781 ± 0.106%ID/g, p < 0.001) and BiVCAMelid (1.56 ± 0.22%ID/g, p < 0.01, by one-way ANOVA with Tukey’s test).
Nanobody Blood and Tissue Pharmacokinetics.
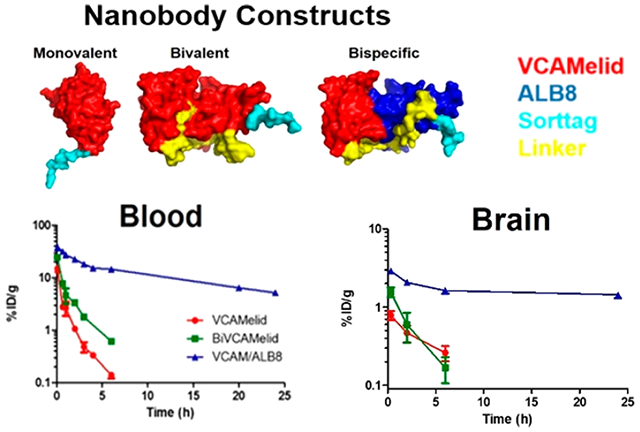
Having fully characterized the biodistribution of VCAMelid, BiVCAMelid, and VCAM/ALB8 in naive and injured mice at a single time point, we next aimed to fully characterize the blood (Figure 4a) and tissue (Figure 4b,c, Table S1) pharmacokinetics of these recombinant affinity ligands. This seemed particularly important given the large difference in blood concentrations at 20 min post-dose. We focused on animals with focal brain inflammation, in which targeted therapeutic delivery would be of particular interest. As shown in Figure 4, the blood (Figure 4a), spleen (Figure 4b), and brain (Figure 4c) concentrations of VCAM/ALB8 were significantly enhanced, as compared to VCAMelid and BiVCAMelid, persisting at fairly stable levels for at least 24 h. Non-compartmental analysis (NCA) was used to quantify the differences in PK of the various engineered forms of the nanobody (Table 1). The area under the blood concentration vs time curve (AUCinf) was increased for VCAM/ALB8 relative to both VCAMelid and BiVCAMelid (p < 0.0001 by one-way ANOVA with Tukey’s post-hoc test), while no significant changes were found between VCAMelid and BiVCAMelid. Likewise, the terminal half-life (t1/2,β) was unchanged by engineering of bivalent binding, but was significantly enhanced via albumin binding (p = 0.0002 by one-way ANOVA with Tukey’s post-hoc test).
Figure 4.
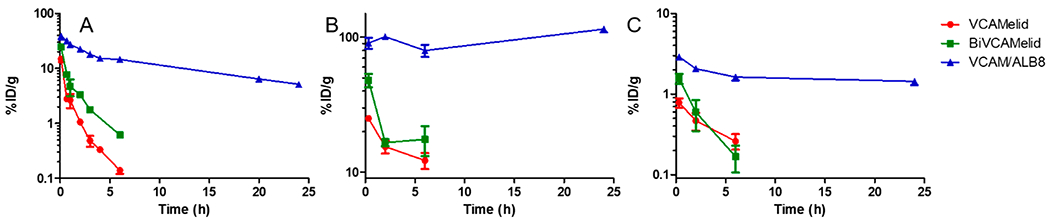
Pharmacokinetics of VCAMelid, BiVCAMelid, and VCAM/ALB8 in TNF-α injected mice. (a) Blood, (b) Spleen, and (c) Brain concentration vs time profile. PK studies were performed after injection of equimolar doses of monovalent (VCAMelid, 5 μg), bivalent (BiVCAMelid, 10 μg) and bispecific (VCAM/ALB8, 10 μg) nanobodies.
Table 1.
Pharmacokinetic Parameters for VCAMelid, BiVCAMelid, and VCAM/ALB8
| Parametera | VCAMelid | BiVCAMelid | VCAM/ALB8 |
|---|---|---|---|
| AUCinf (%ID/g h) | 9.56 (1.40)### | 19.8 (7.6)### | 389 (40) |
| CL (mL/h) | 10.6 (1.6)*,## | 5.79 (2.87)# | 0.259 (0.025) |
| t1/2,β (h) | 1.59 (0.17)### | 1.97 (0.05)### | 13.2 (2.8) |
| Vss (mL) | 16.9 (7.0) | 7.87 (0.48) | 9.00 (0.21) |
Parameters are displayed as Mean (SEM). Comparisons were made using one-way ANOVA with Tukey’s post-hoc test
(p < 0.05)
(p < 0.01)
(p < 0.001) vs BiVCAMelid
(p < 0.05)
(p < 0.01)
(p < 0.001) vs VCAM/ALB8.
Conjugation to Superoxide Dismutase 1 (SOD-1).
We next sought to assess the utility of the VCAMelid nanobody and its bispecific, albumin-binding form as targeting ligands for the delivery of a model therapeutic payload, selecting Superoxide Dismutase-1 (SOD-1) for proof-of-concept studies (Figure 5a). Studies in transgenic42 and knockout43 mice indicate that endothelial SOD-1 plays a significant role in a variety of cerebrovascular functions, including endothelial-dependent vasodilation, and targeted delivery to areas of vascular inflammation has demonstrated therapeutic effect in a number of animal models.44–47 Moreover, untargeted SOD-1 is rapidly eliminated from the circulation (4.54 ± 0.17%ID/g 20 min post-dose) and neither accumulates in the spleen (0.919 ± 0.136%ID/g) or the injured brain (0.055 ± 0.003% ID/g), suggesting that drug targeting could have a meaningful difference in uptake at sites of VCAM expression (Figure S4).
Figure 5.
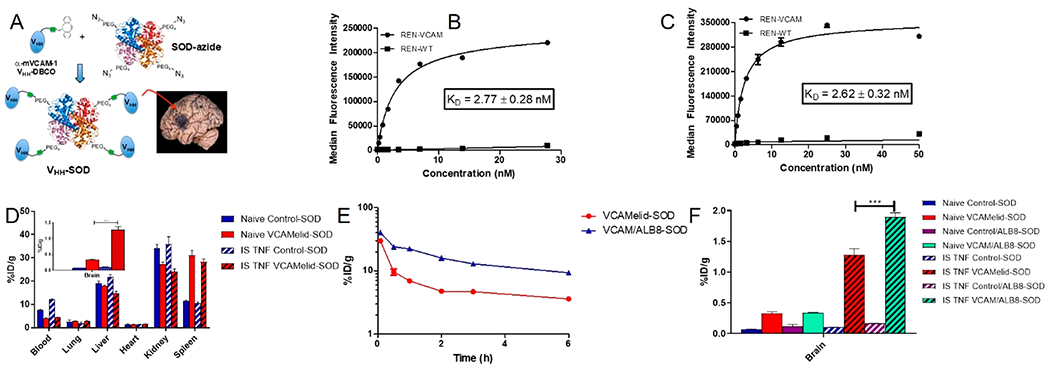
Characterization of nanobodies as drug targeting agents. (a) Depiction of the hypothesized conjugate structure. Binding affinity of (b) VCAMelid-SOD conjugates and (c) VCAM/ALB8-SOD conjugates. (d) Whole-body biodistribution of VCAMelid-SOD conjugates, (e) blood pharmacokinetics of VCAMelid-SOD and VCAM/ALB8-SOD conjugates in TNF-a injected mice, and (f) biodistribution of VCAM-SOD and VCAM/ALB8-SOD conjugates in brain. All biodistributions were performed 20 min post-injection of equimolar doses of VCAMelid-SOD (10 μg) or VCAM/ALB8-SOD (16 μg) conjugates.
Reactions of azide-modified SOD-1 with DBCO-modified VCAMelid were carried out at several ratios, all of which showed a conjugation efficiency of ~80% (Figure S5a). The conjugate that was selected for in vivo studies was reacted at a 4:1 excess of nanobody, giving an average nanobody/SOD-1 ratio of 3.2. A similar degree of conjugation efficiency was confirmed for VCAM/ALB8-SOD (Figure S5b). In vitro binding affinity, measured on REN-VCAM cells, was 2.77 ± 0.27 nM for VCAM-SOD conjugates (Figure 5b) and 2.62 ± 0.32 nM for VCAM/ALB8-SOD conjugates (Figure 5c). The enhanced affinity relative to the monomeric, unconjugated affinity ligands is likely due to the increased avidity of the SOD conjugates.
In Vivo Behavior of Nanobody-SOD-1 Conjugates.
Like unconjugated SOD-1, VCAMelid-SOD conjugates were cleared quickly from the blood (4.12 ± 0.13%ID/g, 20 min post-dose) in both naive and TNF-a injected mice. Slightly higher blood concentrations were seen for the untargeted conjugate, control-SOD (Figure 5d). Specific targeting of VCAMelid-SOD, as compared to unconjugated SOD-1 and control-SOD, was observed in the spleen (31.1 ± 2.1%ID/g vs 0.919 ± 0.136%ID/g vs 11.4 ± 0.5%ID/g, p < 0.0001), and the brain (0.328 ± 0.029%ID/g vs 0.0303 ± 0.0156%ID/g vs 0.0714 ± 0.0038%ID/g, p < 0.0001), with a significant increase in brain targeting following focal inflammatory insult (1.28 ± 0.10%ID/g vs 0.0547 ± 0.0029%ID/g vs 0.102 ± 0.005%ID/g, p < 0.0001) (Figure 5d).
We next compared the in vivo behavior of VCAM/ALB8 and VCAMelid-SOD conjugates. A blood PK study in TNF-α-injured mice showed that the albumin-binding bispecific nanobody conferred a pharmacokinetic advantage, with a 2.2-fold enhancement in AUCinf over VCAMelid-SOD (Figure 5e), demonstrating that conjugation to a therapeutic protein did not ablate the effect of albumin binding. Furthermore, the improvement in circulation time translated into a significant increase in splenic uptake (40.3 ± 2.2%ID/g vs 31.1 ± 2.1% ID/g, p = 0.0302), albeit modest in comparison to the effect seen in the absence of the enzymatic cargo (Figure 5e). Likewise, both VCAMelid-SOD and VCAM/ALB8-SOD demonstrated significant targeting to the cerebrovasculature, but no difference was seen in their brain uptake in naive mice (0.341 ± 0.005%ID/g vs 0.328 ± 0.029%ID/g, p = 0.7203), despite the longer circulation time of the latter. The bispecific nanobody conjugates did show an advantage over VCAMelid-SOD in targeting the brain of injured mice (1.90 ± 0.07 vs 1.28 ± 0.10%ID/g, p = 0.0005) (Figure 5f). Therefore, while the performance of the albumin-binding bispecific affinity ligand was somewhat blunted by conjugation to protein cargo (Figure 3d), its capacity for therapeutic delivery to the primary target site was superior to that of the unmodified nanobody.
Nanobody-Targeted Liposomes.
Finally, we evaluated the utility of VCAMelid as a targeting ligand for liposomes, an extensively studied nanocarrier and one capable of delivery of a wide variety of therapeutic agents. Our group recently described a strategy for oriented conjugation of recombinant affinity ligands to the surface of PEGylated liposomes,34 which we applied here for nanobodies. The biodistribution of VCAMelid liposomes, as compared to untargeted control liposomes, is shown in Figure 6a. Blood concentrations at 20 min post-injection were low, although somewhat higher than those of the SOD conjugates, and unaffected by VCAM-1 targeting. Likewise, splenic uptake was similar for targeted and untargeted liposomes, presumably due to the high baseline uptake of immunoliposomes by the spleen. VCAMelid-coated liposomes also failed to show specific accumulation in the brain of naive animals, although significant targeting was seen in the inflamed brain (1.04 ± 0.25 VCAMelid/injured vs 0.132 ± 0.011 VCAMelid/naive vs 0.427 ± 0.049 Untargeted/injured vs 0.264 ± 0.013%ID/g Untargeted, p = 0.0082) (Figure 6a). Finally, we generated VCAM/ALB8-coated liposomes and measured their biodistribution in naive and injured animals. As shown in Figure 6b, the VCAM/ALB8-liposomes behaved quite similarly to VCAMelid-liposomes. No difference was seen in blood levels or splenic uptake, and while targeting was seen to the injured brain, the albumin-binding bispecific nanobody provided no benefit relative to VCAMelid-coated liposomes (1.16 ± 0.23 vs 1.04 ± 0.25%ID/g in injured brain, p = 0.7556).
Figure 6.
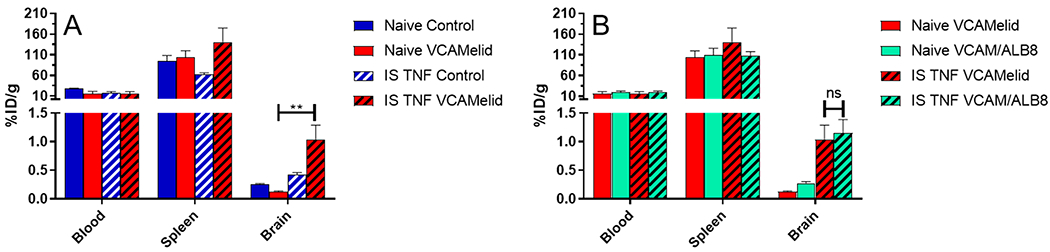
Nanobody-targeted liposomes. Biodistribution of (a) VCAMelid and untargeted control-coated liposomes and (b) VCAMelid and VCAM/ALB8-coated liposomes in select tissues of naive and injured mice. All biodistributions were performed 20 min post-injection of 50 μL of liposomes.
DISCUSSION
Targeted drug delivery of small molecules, proteins, and nanocarriers via attachment of affinity ligands remains a relatively underdeveloped field of pharmacology, despite clear potential for improvements in PK, potency, and therapeutic index. Notwithstanding a steady stream of “proof-of-concept” reports, only a small number of targeted therapeutics have demonstrated convincing efficacy in human disease and achieved success in the clinical domain. One reason for the slow progress and limited bench-to-bedside translation may be the relative lack of information regarding the optimal characteristics of targeting ligands or even general guidelines regarding which attributes are of particular value for specific applications. Even for monoclonal antibodies, where the molecular properties of the affinity ligand have been exhaustively studied, little is known about the relevance of these factors following bioconjugation to different cargoes. The Fc fragment is an example—despite a marked effect on the plasma half-life of isolated antibodies (i.e., via FcRn recycling), the effect on the circulation time of protein or nanoparticle cargoes remains unclear and, indeed, surface conjugation of full-length antibodies may accelerate clearance by promoting opsonization or interaction with cells of the reticuloendothelial system.
Nanobodies are a relatively new entry into the world of drug targeting and have attracted intense interest due to their relatively high affinity, molecular stability, small size (a potential advantage for sterically obscured or extravascular targets), and lack of Fc fragment. Despite industrial development for other biomedical applications (e.g., antigen capture and molecular imaging) and a number of reports promoting their use for drug delivery, relatively little work has been done to understand their relative advantages and disadvantages. The VCAM-targeting nanobody utilized in this study, for example, has been extensively characterized as a molecular imaging agent for atherosclerosis in several formats,30,36,37,48 but, to the best our knowledge, the current manuscript is the first to rigorously characterize its biodistribution in the presence of acute focal or systemic inflammatory insults or to evaluate its potential for delivery of protein and nanoparticle cargo to sites of endothelial activation.
Our current results are focused in two areas: (l) understanding the impact of changes in binding avidity and circulation time on the in vivo performance of an affinity ligand, and (2) determining the effects of these properties on delivery of potential therapeutic cargoes. Due to their compact molecular folding and relative ease of expression, nanobodies are in many ways an ideal substrate for this work—particularly given established techniques for production of bivalent and bispecific molecular variants.28 Pairing these engineered forms with our previously reported method of site-specific modification31‘34 (which allows radiolabeling and bioconjugation without appreciable loss of binding affinity), we believe we have developed a useful platform for systematic study of affinity ligand attributes and their impact on biodistribution, pharmacokinetics, and drug delivery. It is worth noting the role of target accessibility in this work. In contrast to the relatively large body of research on nonvascular neoplastic epitopes, drug targeting to endothelial surface antigens benefits from a lack of barriers to target access, allowing straightforward conclusions to be drawn regarding the impact of molecular engineering on the in vivo behavior of affinity ligands. Thus, while improved drug delivery to solid tumors and metastases is a medical priority, vascular accessible antigens may provide additional insights into factors controlling biodistribution and drug delivery.
Through head-to-head comparison of various engineered forms of the same nanobody, the current work arrives at several conclusions. First, modulation of both affinity/avidity (BiVCAMelid vs VCAMelid) and circulation time (VCAM/ALB8 vs VCAMelid) results in significant improvement in uptake by the spleen (Figures 2b, 3d), a noninducible source of VCAM-1, and the brain (Figures 2c, 3e), a largely inducible source of endothelial VCAM-1. Indeed, our results indicate that these modifications have nearly identical impact at the earliest time point (20 min post-injection), although a rapid decline in the blood concentration of the BiVCAMelid eliminates any benefit over the monovalent nanobody at later time points (2 and 6 h). As blood levels drop, the binding equilibrium presumably shifts to a state favoring dissociation, and in spite of the higher affinity, the end result is a rather paltry 1.4- and 1.3-fold increase in spleen and brain AUC, respectively. In contrast, the prolonged circulation time (40fold enhancement in blood AUC) conferred by the albumin-binding arm of VCAM/ALB8 results in 25.5- and 15.6-fold improvements in spleen and brain AUC. This analysis makes it clear that circulation time outweighs avidity under these specific circumstances, although similar empiric testing is likely necessary to determine the relative importance of these attributes for other targets, epitopes, or types of affinity ligand. For example, a ligand–target interaction which induces rapid internalization or one with an extremely slow dissociation rate constant (e.g., half-life on the order of several hours) might achieve prolonged delivery to target sites without the need for half-life extension. In fact, one intriguing area of future investigation is that of affinity engineering (rather than the molecular engineering approach taken in this manuscript), in which site-directed mutagenesis or display-based techniques could be used to produce nanobodies with varying binding kinetics. An ultra-slow dissociating nanobody, in particular, could have great utility in certain drug delivery or imaging applications where both rapid blood clearance and prolonged target engagement are desired.
Our group recently reported quantitative assessment of VCAM-1 as a target for selective delivery of therapeutics to activated/injured cerebrovascular endothelium, using unilateral striatal injection of TNF-α as a simple model of lateralizing brain inflammation.49 A key finding of this publication was that a monoclonal antibody (mAb) directed against mouse VCAM-1 (clone M/K-2.7) demonstrated not only high, specific uptake in naive brain (~1.7% ID/g), but a 10-fold increase in distribution (~17% ID/g) to the TNF-α injected hemisphere. Based on these results and the potential biomedical significance of this application, we were interested in determining the biodistribution of the VCAMelid and its modified forms in this model. Our results demonstrate that prolonged circulation again outweighs bivalent binding in determining brain uptake—in fact, in the presence of focal inflammation, VCAM/ALB8 is superior to BiVCAMelid or VCAMelid even at the earliest time point. It is also interesting to note the relative inferiority of VCAM/ALB8 relative to the M/K-2.7 mAb, which has ~3-fold higher brain uptake in naive animals and ~7-fold higher uptake in TNF-α injected hemispheres. It is tempting to speculate that this might be due to M/K-2.7’s combination of long circulation time and bivalent binding, but it is impossible to make direct comparisons, as the two ligands almost certainly engage distinct epitopes on VCAM-1. Engineering the VCAMelid into a more “IgG-like” molecule, which combines bivalent binding and albumin- or FcRn-binding would be one way to directly assess this and may be an avenue for future investigation.
Having characterized the in vivo behavior of the nanobody—and its engineered forms—in both normal and TNF-α challenged mice, we next assessed its performance as a targeting ligand and sought to determine the effects of its molecular engineering on the circulation and biodistribution of various cargoes. Azide-modified SOD-1 was used as a model protein, while irrelevant nanobody-functionalized PEGylated liposomes were used as representative nanoparticle drug carriers. Conjugation of the VCAMelid itself had variable impact on these two entities, causing a marked shift in the biodistribution of SOD-1, while having relatively little impact on liposomes in naive animals. This finding was somewhat surprising, in light of our recently published studies, which showed a marked increase in brain uptake of immunoliposomes targeted with the M/K-2.7 monoclonal antibody (~20-fold greater than IgG-control in naive mice and >100-fold increase in TNF-α injected hemispheres), and it underscores the difficulty predicting a priori the utility of a given affinity ligand for drug delivery. The limited circulation time of the VCAMelid seems unlikely to be the culprit, as both M/K-2.7 liposomes and VCAM/ALB8-conjugated particles demonstrate similar blood concentrations at early time points post-injection. Likewise, conjugation of the bispecific nanobody offered no improvement in tissue uptake over the VCAMelid-liposomes, suggesting that other factors (e.g., epitope) are likely responsible for the much higher brain biodistribution of M/K-2.7 liposomes.
Unlike liposomes, targeting of SOD-1 was quite effective for both VCAMelid and VCAM/ALB8, with each demonstrating 20–40-fold increases in splenic and brain uptake vs untargeted controls (e.g., control-SOD). By comparing VCAMelid to the bispecific nanobody, the latter had a significant effect on the blood PK of the SOD conjugates (2.2-fold enhancement in blood AUC), but clearly far more modest than was seen for the affinity ligands alone. This translated into small, albeit significant, 1.3- and 1.5-fold differences in spleen and brain uptake, the latter being in TNF-α injected animals. Since each SOD-1 molecule had an average of 3.2 surface conjugated affinity ligands, it is possible that the avidity of the conjugates is responsible for the relatively small differences in targeting at the early time point. After all, a similar relationship was noted in the biodistribution of the BiVCAMelid vs VCAM/ALB8, i.e., the two were nearly identical at 20 min post-injection, despite large differences in blood concentrations, but then diverged at later time points. Future experiments could directly assess this through synthesis and purification of conjugates with varying levels of avidity.
In conclusion, we have utilized a toolbox of protein engineering strategies to systematically evaluate the impact of binding affinity and circulation time on the biodistribution of a VCAM-1 specific nanobody affinity ligand and its capacity for delivery of protein and nanoparticle therapeutics (Figure 7). These variables were assessed under both basal and inflammatory conditions and at sites of noninducible (spleen) and largely inducible (brain) VCAM-1 expression. Our results show that for this ligand, target, and epitope, engineering for enhanced pharmacokinetics has greater impact than improved avidity and that this held true for different organs, expression levels, and physiologic states. At the same time, our results clearly indicate that much of the benefit of affinity ligand engineering may be “drowned out” by the characteristics of a given therapeutic cargo. Ultimately, the most significant impact of the work may be demonstrating how little we currently understand about the determinants of in vivo behavior and the critical need for systematic study as a means of establishing future mathematical models or at least guidelines for the design of novel affinity targeted interventions.
Figure 7.
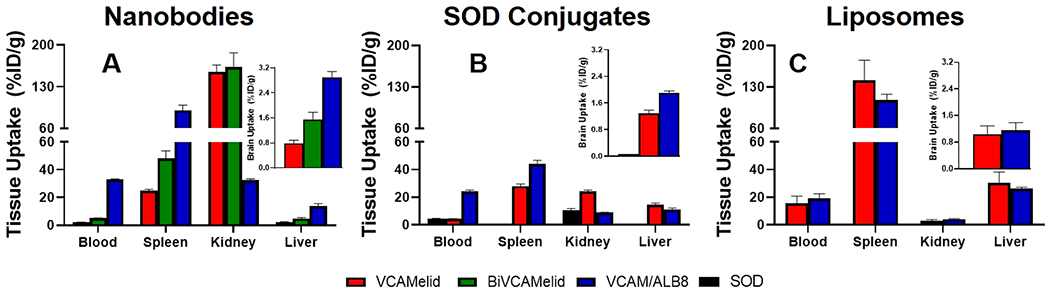
Comparison of all tested constructs. Biodistribution data for (a) nanobodies, (b) nanobody–SOD conjugates, and (c) nanobody-targeted liposomes are shown for blood, target organs (spleen and brain), and elimination organs (liver and kidney) 20 min post-IV injection in mice that had previously received local brain injury (IS TNF-α). Brain data is shown as an inset on all plots. Blood, spleen, and brain data are reproduced from Figures 1, 2, 3, 5, and 6. The same y-axis scale was used across all plots for ease of comparison.
EXPERIMENTAL PROCEDURES
Materials.
Restriction enzymes were purchased from New England BioLabs (Ipswich, MA). Custom peptides were synthesized by Thermo Fisher (Carlsbad, CA). Lipids and cholesterol were obtained from Avanti Polar Lipids (Alabaster, AL). All chemicals and other reagents were purchased from SigmaAldrich (St. Louis, MO), unless otherwise noted in the text.
Protein Expression and Purification.
DNA sequences encoding single-domain antibodies directed against mouse vascular cell adhesion molecule 1 (mVCAM-1),30 nontargeted control,35 and serum albumin50 were obtained from the literature. The nontargeted control is a previously reported anti-human ICAM-1 nanobody. Bivalent or bispecific nanobodies were produced as genetic fusions, with nanobodies linked by the upper hinge region of llama IgG228 (Figure S6). Linear DNA fragments encoding the molecules were ordered as gBlocks (IDT, Coralville, IA) with 5′ NcoI and 3′ NheI restriction enzyme sites to allow insertion into the vector pBAD/LPET. This plasmid was derived from the commercially available pBAD/gIII vector (ThermoFisher, Carlsbad, CA) by addition of a sequence encoding a C-terminal Sortase A recognition motif (LPETG), triple FLAG tag, and stop codon.31 Following ligation into pBAD/LPET, DNA was transformed into Top10 E. coli (ThermoFisher, Carlsbad, CA) to allow for periplasmic expression of proteins.
Expression and purification of nanobodies were performed using a periplasmic expression system. Briefly, E. coli were grown up to an optical density of 0.5–0.8 at 600 nm in Terrific Broth containing 100 μg/mL ampicillin at 37 °C. Protein expression was induced using 0.02%w/v arabinose and allowed to proceed overnight at 18 °C. Cell pellets were collected via centrifugation for 20 min at 8000×g. Periplasmically expressed proteins were extracted from cell pellets using a standard osmotic shock protocol. Briefly, cell pellets were resuspended in a buffer consisting of 30 mM Tris-HCl, 20% w/v sucrose, and 1 mM EDTA, pH 8.0, following by centrifugation as described above. Release of periplasmic proteins was achieved by resuspending in 5 mM magnesium sulfate, and cell debris was removed by centrifugation. Nanobodies were purified from the shock fluid using L5-agarose (anti-FLAG) affinity chromatography (BioLegend, San Diego, CA). Protein purity was assessed using a size exclusion (SEC) high-performance liquid chromatography (HPLC) (Waters, Milford, MA), with separation being performed on a Yarra 2000-GFC column (Phenomenex, Torrance, CA).
Sortase A Modification of Proteins.
Sortase A (SrtA) was produced in T7 Shuffle E. coli, as previously described,31 with a C-terminal hexahistidine tag to allow purification via Ni-NTA chromatography. In a typical reaction, sortagged nanobody proteins (40 μM) were incubated with equimolar SrtA (40 μM), an excess of H2N-GGG-containing peptide (100 μM), and Ca2+ (1 mM) in Tris-buffered saline (TBS), pH 7.4, overnight at room temperature. SrtA was removed from the reaction mixture using Ni-NTA-agarose beads and unreacted peptide was removed via centrifugation over a 10 kDa cutoff filter (Amicon). Reacted proteins were stored in PBS containing 5 mM EDTA to quench the activity of any residual SrtA.
NHS-Ester Modification of Nanobodies.
Nanobodies were labeled with AlexaFluor488 via amine coupling chemistry. Briefly, AlexaFluor488-NHS ester was reacted with nanobodies at a 20:1 molar ratio for 1 h at room temperature. Unreacted dye was removed via centrifugation over a 10 kDa cutoff filter.
VCAM-1 Binding Affinity.
Binding between nanobodies and mVCAM was measured by flow cytometry using a BD Accuri C6 flow cytometer. Briefly, nanobodies were conjugated to a 5-carboxyfluorescein (FAM)-containing peptide (GGGK-[5-FAM]GGGSK[azide]) via SrtA-mediated transpeptidation were incubated with formaldehyde-fixed cells transfected to express mouse VCAM-1 (REN-VCAM)49 or wild-type cells (REN-WT) at a range of concentrations for 1 h on ice in PBS containing 3% v/v fetal bovine serum (FBS). Following a wash with PBS, cells were resuspended in PBS and binding was measured on the cytometer. Binding data was fit to a typical binding equation in order to obtain estimates of the equilibrium dissociation constant (KD).
Albumin Binding Affinity.
Binding of nanobodies to human serum albumin (HSA) was measured using an enzyme-linked immunosorbent assay (ELISA). Nunc MaxiSorp 96 well plates were coated overnight at 4 °C with 0.5 mg/mL of HSA or casein in 0.1 M sodium phosphate, monobasic. Plates were then blocked for 2 h at 37 °C with 1% w/v casein in PBS, followed by washing with PBS. Varying concentrations (0–400 nM) of albumin-binding nanobody were added to the wells in PBS and incubated for 2 h at 37 °C, followed by PBS washes. Secondary anti-FLAG (M2) monoclonal antibody conjugated to horseradish peroxidase (HRP) was added to the wells at a 1:15,000 dilution in PBS for 1 h at 37 °C, followed by washes as described above. TMB substrate (BioLegend, San Diego, CA) was then added (200 μL/well) and allowed to develop in the dark for 15 min and the reaction was stopped by addition of 50 μL of 10% v/v H2SO4. The plates were then read using a SpectraMax M2 plate reader (Molecular Devices, San Jose, CA) at 450 nm to assess binding to HSA.
Simultaneous Binding to VCAM-1 and HSA.
Flow cytometry was used to assess the ability of bispecific nanobodies to simultaneously engage both VCAM-1 and albumin. Briefly, unmodified nanobody was added to either REN-VCAM or REN-WT cells at a range of concentrations, as described above. Detection was performed by addition of 50 nM of AlexaFluor647-modified HSA, and incubation on ice for 1 h, followed by washing with PBS. The degree of binding was measured using flow cytometry, as described above.
Camelid Radiolabeling.
Nanobodies were site-specifically radiolabeled with 111In using SrtA-mediated transpeptidation (Figure S7). Briefly, 111In (Nuclear Diagnostics) was resuspended in metal-free 0.5 M tetramethylammonium acetate (TMAA), pH 4.5, and incubated with a peptide containing a radiometal chelating group (H2N-GGGK-DOTA, Click Chemistry Reagents, San Diego, CA) for 1 h at 37 °C. The reaction was quenched by neutralization with 2 M Tris and degree of radiometal incorporation was confirmed to be >95% using thin-layer chromatography (TLC). The radiolabeled peptide was conjugated to nanobodies using SrtA-mediated transpeptidation, as described above. The reaction mixture was then purified first by removing SrtA using Ni-NTA agarose beads, followed by desalting using a 10DG column to remove unreacted peptide.
Conjugation to Superoxide Dismutase.
Superoxide dismutase (SOD-1), obtained from bovine erythrocytes, (Sigma-Aldrich) was modified by mixing 2 mg/mL SOD-1 with a 60-fold excess of azido-PEG4-NHS ester (Click Chemistry Tools) in PBS containing 0.1 M sodium bicarbonate, pH 8.3, and incubating for 1 h at room temperature. Unreacted NHS-ester was removed by repeated spins through a 10 kDa cutoff filter. Conjugation with nanobodies was carried out using strain-promoted alkyne–azide cycloaddition (SPAAC) (“click chemistry”) (Figure S8). As described previously,34 a peptide containing a free C-terminal thiol (GGGK[5-FAM]GGSC) was modified with dibenzylocyclooctyne (DBCO) through maleimide chemistry, by reaction with DBCO-PEG4-maleimide (Click Chemistry Tools, Scottsdale, AZ). This peptide was then site-specifically reacted with nanobodies using SrtA-mediated transpeptidation, as described above. Conjugation was carried out at varying ratios of nanobody:SOD-1, ranging from 1:1 to 4:1, in PBS, pH 7.4, and the degree of conjugation was assessed using SEC HPLC. For further evaluation, unreacted nanobody and SOD were removed by centrifugation through appropriate molecular weight cutoff filters.
SOD-1 Radiolabeling.
SOD-1-azide was directly radiolabeled with [125]NaI (PerkinElmer, Waltham, MA) using iodination beads and purified using Zeba spin columns. Radiochemical purity was confirmed to be >95% via TLC.
Liposome Production, Radiolabeling, and Conjugation.
Liposomes were produced as described previously.34 Briefly, 16:0 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene glycol)-2000] (DSPE-PEG(2000)-azide), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid (PE-DTPA) were mixed in a ratio of 69:25:1:0.1 and chloroform was evaporated to form lipid films. Films were rehydrated in 0.2 M sodium citrate, pH 4.5, and extruded using a 200 nm filter at 50 °C. Liposomes were radiolabeled with 111In by incubating at 37 °C for 1 h. The degree of radiometal incorporation was assessed using TLC and any residual free radiometal was removed by use of a 10 kDa MWCO Amicon filter unit (Millipore). Liposomes were then buffer-exchanged into PBS, pH 7.4, to allow for SPAAC-mediated conjugation of DBCO-modified nanobodies. Nanobodies were added to liposomes and reacted overnight at 37 °C (Figure S8). At all steps, the size and polydispersity index (PDI) of liposomes were measured using dynamic light scattering (DLS) on a Malvern Zetasizer (Malvern, UK).
Liposomes were buffer-exchanged into PBS, pH 7.4, and DBCO-modified nanobodies were added to azide-liposomes at a ratio of 400 affinity ligands/particle. Following overnight reaction at 37 °C, liposomes were purified using a Sepharose CL-4B column and reaction efficiency was quantified via fluorescence plate assay (Figure S9).
Animal Use.
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
In Vivo Studies.
Systemic inflammation was induced by intravenous (IV) injection of 2 mg/kg lipopolysaccharide (LPS) B5. Biodistribution studies were carried out 5 h post-LPS injection. Local, neurovascular inflammation was induced as described previously.51 Briefly, 0.5 μg of human tumor necrosis factor-α (hTNF-α) (BioLegend, San Diego, CA) was injected into the right striatum of 6–8-week-old male C57BL/6 mice using a stereotactic frame 16–18 h prior to in vivo studies.
All formulations were injected intravenously via the retro-orbital plexus in a volume of 100 μL or less. Blood and organs (lung, liver, kidney, heart, spleen, and brain) were collected at 20 min, 2 h, 6 h, or 24 h for whole-body biodistribution studies. Prior to organ collection, all animals were perfused with 20 mL of ice-cold PBS via the right ventricle to remove residual blood from tissues. For investigation of blood pharmacokinetics (PK), blood samples were collected from the retro-orbital plexus using heparinized capillary tubes at various time points following injection. Radioactivity in blood and tissue samples was counted using a gamma counter.
Statistics.
All statistical and regression analyses were performed using GraphPad Prism 5.0. Comparisons between 2 groups were performed using unpaired t tests, while comparisons of 3 or more groups were performed using one-way ANOVA with Tukey’s post-hoc test. Statistical significance is represented as * (p < 0.05), ** (p < 0.01), *** (p < 0.001), **** (p < 0.0001).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant numbers: T32HL007954 (P.M.G.), T32HL007971 (P.M.G.), R01HL12687401A1 (V.R.M.), 1R01HL12839801 (V.R.M.), 1RO1HL1254621A1 (V.R.M.), and K08HL130430 (C.F.G.).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.0c00003
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.0c00003.
Characterization of protein purity and binding affinity and full tissue distribution data (PDF)
Amino acid sequences of recombinant protein constructs (PDF)
The authors declare no competing financial interest.
Contributor Information
Patrick M. Glassman, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Landis R. Walsh, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Carlos H. Villa, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Oscar A. Marcos-Contreras, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Elizabeth D. Hood, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Vladimir R. Muzykantov, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Colin F. Greineder, Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States; Department of Emergency Medicine and Department of Pharmacology, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Muzykantov VR (2005) Biomedical aspects of targeted delivery of drugs to pulmonary endothelium. Expert Opin. Drug Delivery 2 (5), 909–26. [DOI] [PubMed] [Google Scholar]
- (2).Koren E, and Torchilin VP (2011) Drug carriers for vascular drug delivery. IUBMB Life 63 (8), 586–95. [DOI] [PubMed] [Google Scholar]
- (3).Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, and Hamers R (1993) Naturally occurring antibodies devoid of light chains. Nature 363 (6428), 446–8. [DOI] [PubMed] [Google Scholar]
- (4).Hu Y, Liu C, and Muyldermans S (2017) Nanobody-Based Delivery Systems for Diagnosis and Targeted Tumor Therapy. Front. Immunol 8, 1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Beghein E, and Gettemans J (2017) Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Inter-action Analysis, and Protein Function Exploration. Front. Immunol 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chakravarty R, Goel S, and Cai W (2014) Nanobody: the “magic bullet” for molecular imaging? Theranostics 4 (4), 386–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Senders ML, Hernot S, Carlucci G, van de Voort JC, Fay F, Calcagno C, Tang J, Alaarg A, Zhao Y, Ishino S, et al. (2019) Nanobody-Facilitated Multiparametric PET/MRI Phenotyping of Atherosclerosis. JACC Cardiovasc Imaging 12 (10), 2015–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fumey W, Koenigsdorf J, Kunick V, Menzel S, Schutze K, Unger M, Schriewer L, Haag F, Adam G, Oberle A, et al. (2017) Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38(+) tumors in mouse models in vivo. Sci Rep. 7 (1), 14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, et al. (2019) Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med 380 (4), 335–346. [DOI] [PubMed] [Google Scholar]
- (10).Duggan S (2018) Caplacizumab: First Global Approval. Drugs 78 (15), 1639–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Peyvandi F, Scully M, Kremer Hovinga JA, Knobl P, Cataland S, De Beuf K, Callewaert F, De Winter H, and Zeldin RK (2017) Caplacizumab reduces the frequency of major throm-boembolic events, exacerbations and death in patients with acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemostasis 15 (7), 1448–1452. [DOI] [PubMed] [Google Scholar]
- (12).Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, and Revets H (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 64 (8), 2853–7. [DOI] [PubMed] [Google Scholar]
- (13).Oliveira S, Schiffelers RM, van der Veeken J, van der Meel R, Vongpromek R, van Bergen En Henegouwen PM, Storm G, and Roovers RC (2010) Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J. Controlled Release 145 (2), 165–75. [DOI] [PubMed] [Google Scholar]
- (14).Arias JL, Unciti-Broceta JD, Maceira J, Del Castillo T, Hernandez-Quero J, Magez S, Soriano M, and Garcia-Salcedo JA (2015) Nanobody conjugated PLGA nanoparticles for active targeting of African Trypanosomiasis. J. Controlled Release 197, 190–8. [DOI] [PubMed] [Google Scholar]
- (15).van Driel P, Boonstra MC, Slooter MD, Heukers R, Stammes MA, Snoeks TJA, de Bruijn HS, van Diest PJ, Vahrmeijer AL, van Bergen En Henegouwen PMP, et al. (2016) EGFR targeted nanobody-photosensitizer conjugates for photo-dynamic therapy in a pre-clinical model of head and neck cancer. J. Controlled Release 229, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yu Y, Li J, Zhu X, Tang X, Bao Y, Sun X, Huang Y, Tian F, Liu X, and Yang L (2017) Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lympho-blastic leukemia potential. Int. J. Nanomed 12, 1969–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pruszynski M, D’Huyvetter M, Bruchertseifer F, Morgenstern A, and Lahoutte T (2018) Evaluation of an Anti-HER2 Nanobody Labeled with (225)Ac for Targeted alpha-Particle Therapy of Cancer. Mol. Pharmaceutics 15 (4), 1457–1466. [DOI] [PubMed] [Google Scholar]
- (18).Fang T, Duarte JN, Ling J, Li Z, Guzman JS, and Ploegh HL (2016) Structurally Defined alphaMHC-II Nanobody-Drug Conjugates: A Therapeutic and Imaging System for B-Cell Lymphoma. Angew. Chem., Int. Ed 55 (7), 2416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rotman M, Welling MM, Bunschoten A, de Backer ME, Rip J, Nabuurs RJ, Gaillard PJ, van Buchem MA, van der Maarel SM, and van der Weerd L (2015) Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J. Controlled Release 203, 40–50. [DOI] [PubMed] [Google Scholar]
- (20).Cortez-Retamozo V, Lauwereys M, Hassanzadeh Gh G, Gobert M, Conrath K, Muyldermans S, De Baetselier P, and Revets H (2002) Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 98 (3), 456–62. [DOI] [PubMed] [Google Scholar]
- (21).Vugmeyster Y, Entrican CA, Joyce AP, Lawrence-Henderson RF, Leary BA, Mahoney CS, Patel HK, Raso SW, Olland SH, Hegen M, et al. (2012) Pharmacokinetic, biodistribution, and biophysical profiles of TNF nanobodies conjugated to linear or branched poly(ethylene glycol). Bioconjugate Chem. 23 (7), 1452–62. [DOI] [PubMed] [Google Scholar]
- (22).Tijink BM, Laeremans T, Budde M, Stigter-Van Walsum M, Dreier T, de Haard HJ, Leemans CR, and van Dongen GA (2008) Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol. Cancer Ther 7 (8), 2288–97. [DOI] [PubMed] [Google Scholar]
- (23).Vosjan MJ, Vercammen J, Kolkman JA, Stigter-Van Walsum M, Revets H, and van Dongen GA (2012) Nanobodies targeting the hepatocyte growth factor: potential new drugs for molecular cancer therapy. Mol. Cancer Ther 11 (4), 1017–25. [DOI] [PubMed] [Google Scholar]
- (24).Krasniqi A, Bialkowska M, Xavier C, Van der Jeught K, Muyldermans S, Devoogdt N, and D’Huyvetter M (2018) Pharmacokinetics of radiolabeled dimeric sdAbs constructs targeting human CD20. New Biotechnol 45, 69–79. [DOI] [PubMed] [Google Scholar]
- (25).Li Q, Barrett A, Vijayakrishnan B, Tiberghien A, Beard R, Rickert KW, Allen KL, Christie RJ, Marelli M, Harper J, et al. (2019) Improved Inhibition of Tumor Growth by Diabody-Drug Conjugates via Half-Life Extension. Bioconjugate Chem. 30 (4), 1232–1243. [DOI] [PubMed] [Google Scholar]
- (26).Rotman M, Welling MM, van den Boogaard ML, Moursel LG, van der Graaf LM, van Buchem MA, van der Maarel SM, and van der Weerd L (2015) Fusion of hIgG1-Fc to 111In-anti-amyloid single domain antibody fragment VHH-pa2H prolongs blood residential time in APP/PS1 mice but does not increase brain uptake. Nucl. Med. Biol 42 (8), 695–702. [DOI] [PubMed] [Google Scholar]
- (27).Farrington GK, Caram-Salas N, Haqqani AS, Brunette E, Eldredge J, Pepinsky B, Antognetti G, Baumann E, Ding W, Garber E, et al. (2014) A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 28 (11), 4764–78. [DOI] [PubMed] [Google Scholar]
- (28).Els Conrath K, Lauwereys M, Wyns L, and Muyldermans S (2001) Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem 276 (10), 7346–50. [DOI] [PubMed] [Google Scholar]
- (29).Iqbal U, Trojahn U, Albaghdadi H, Zhang J, O’Connor-McCourt M, Stanimirovic D, Tomanek B, Sutherland G, and Abulrob A (2010) Kinetic analysis of novel mono- and multivalent VHH-fragments and their application for molecular imaging of brain tumours. Br. J. Pharmacol 160 (4), 1016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Broisat A, Hernot S, Toczek J, De Vos J, Riou LM, Martin S, Ahmadi M, Thielens N, Wernery U, Caveliers V, et al. (2012) Nanobodies targeting mouse/human VCAM1 for the nuclear imaging of atherosclerotic lesions. Circ. Res 110 (7), 927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Greineder CF, Villa CH, Walsh LR, Kiseleva RY, Hood ED, Khoshnejad M, Warden-Rothman R, Tsourkas A, and Muzykantov VR (2018) Site-Specific Modification of Single-Chain Antibody Fragments for Bioconjugation and Vascular Immunotargeting. Bioconjugate Chem. 29 (1), 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).van Lith SA, van Duijnhoven SM, Navis AC, Leenders WP, Dolk E, Wennink JW, van Nostrum CF, and van Hest JC (2017) Legomedicine-A Versatile Chemo-Enzymatic Approach for the Preparation of Targeted Dual-Labeled Llama Antibody-Nano-particle Conjugates. Bioconjugate Chem. 28 (2), 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Massa S, Vikani N, Betti C, Ballet S, Vanderhaegen S, Steyaert J, Descamps B, Vanhove C, Bunschoten A, van Leeuwen FW, et al. (2016) Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: a versatile strategy for multiple molecular imaging modalities. Contrast Media Mol. Imaging 11 (5), 328–339. [DOI] [PubMed] [Google Scholar]
- (34).Hood ED, Greineder CF, Shuvaeva T, Walsh L, Villa CH, and Muzykantov VR (2018) Vascular Targeting of Radiolabeled Liposomes with Bio-Orthogonally Conjugated Ligands: Single Chain Fragments Provide Higher Specificity than Antibodies. Bioconjugate Chem. 29 (11), 3626–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Abulrob A, Arbabi-Gahroudi M, and Stanimirovic D (2011) Anti-ICAM-1 Single Domain Antibody and Uses Thereof. US 8623369 B2.
- (36).Bala G, Crauwels M, Blykers A, Remory I, Marschall ALJ, Dubel S, Dumas L, Broisat A, Martin C, Ballet S, et al. (2019) Radiometal-labeled anti-VCAM-1 nanobodies as molecular tracers for atherosclerosis - impact of radiochemistry on pharmacokinetics. Biol. Chem 400 (3), 323–332. [DOI] [PubMed] [Google Scholar]
- (37).Bala G, Blykers A, Xavier C, Descamps B, Broisat A, Ghezzi C, Fagret D, Van Camp G, Caveliers V, Vanhove C, et al. (2016) Targeting of vascular cell adhesion molecule-1 by 18F-labelled nanobodies for PET/CT imaging of inflamed atherosclerotic plaques. Eur. Heart J. Cardiovasc Imaging 17 (9), 1001–8. [DOI] [PubMed] [Google Scholar]
- (38).Ulyanova T, Scott LM, Priestley GV, Jiang Y, Nakamoto B, Koni PA, and Papayannopoulou T (2005) VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. Blood 106 (1), 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, and Damico LA (2002) Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem 277 (38), 35035–43. [DOI] [PubMed] [Google Scholar]
- (40).Kim J, Bronson CL, Hayton WL, Radmacher MD, Roopenian DC, Robinson JM, and Anderson CL (2006) Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am. J. Physiol Gastrointest Liver Physiol 290 (2), G352–60. [DOI] [PubMed] [Google Scholar]
- (41).Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, and Anderson CL (2003) The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med 197 (3), 315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, and Carlson GA (1999) SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci 2 (2), 157–61. [DOI] [PubMed] [Google Scholar]
- (43).Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, and Lin MT (2009) Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 23 (8), 2459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shuvaev VV, Han J, Tliba S, Arguiri E, Christofidou-Solomidou M, Ramirez SH, Dykstra H, Persidsky Y, Atochin DN, Huang PL, et al. (2013) Anti-inflammatory effect of targeted delivery of SOD to endothelium: mechanism, synergism with NO donors and protective effects in vitro and in vivo. PLoS One 8 (10), No. e77002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shuvaev VV, Kiseleva RY, Arguiri E, Villa CH, Muro S, Christofidou-Solomidou M, Stan RV, and Muzykantov VR (2018) Targeting superoxide dismutase to endothelial caveolae profoundly alleviates inflammation caused by endotoxin. J. Controlled Release 272, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Shuvaev VV, Muro S, Arguiri E, Khoshnejad M, Tliba S, Christofidou-Solomidou M, and Muzykantov VR (2016) Size and targeting to PECAM vs ICAM control endothelial delivery, internalization and protective effect of multimolecular SOD conjugates. J. Controlled Release 234, 115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Han J, Shuvaev VV, and Muzykantov VR (2011) Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor. J. Pharmacol. Exp. Ther 338 (1), 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Punjabi M, Xu L, Ochoa-Espinosa A, Kosareva A, Wolff T, Murtaja A, Broisat A, Devoogdt N, and Kaufmann BA (2019) Ultrasound Molecular Imaging of Atherosclerosis With Nanobodies: Translatable Microbubble Targeting Murine and Human VCAM (Vascular Cell Adhesion Molecule) 1. Arterioscler., Thromb., Vasc. Biol 39 (12), 2520–2530. [DOI] [PubMed] [Google Scholar]
- (49).Marcos-Contreras OA, Greineder CF, Kiseleva RY, Parhiz H, Walsh LR, Zuluaga-Ramirez V, Myerson JW, Hood ED, Villa CH, Tombacz I, et al. (2020) Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood-brain barrier. Proc. Natl. Acad. Sci U. S. A 117 (7), 3405–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Beirnaert E, Revets HAP, Hoogenboom HRJM, Jonckheere HMF, and Dreier T (2007) Serum albumin binding proteins with long half-lives. US 2007/0269422 A1.
- (51).Montagne A, Gauberti M, Macrez R, Jullienne A, Briens A, Raynaud JS, Louin G, Buisson A, Haelewyn B, Docagne F, et al. (2012) Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage 63 (2), 760–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.