

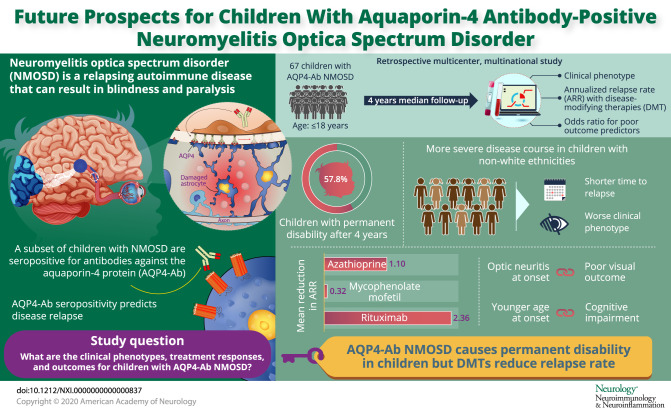
Abstract
Objective
To describe the clinical phenotypes, treatment response, and outcome of children with antibodies against aquaporin-4 (AQP4-Ab) neuromyelitis optica spectrum disorder (NMOSD).
Methods
Retrospective, multicenter, and multinational study of patients with AQP4-Ab NMOSD aged <18 years at disease onset from a center in Brazil and 13 European centers. Data on demographics, clinical findings, and laboratory results were analyzed; calculation of annualized relapse rates (ARRs) pre- and on-treatment with disease-modifying therapies (DMTs) and of ORs for predictors of poor outcome was performed.
Results
A total of 67 children were identified. At last follow-up (median 4 years, interquartile range 2–10 years), 37/67(57.8%) were found to have permanent disability. A more severe disease course was seen in the non-White ethnicity with both a shorter time to first relapse (p = 0.049) and a worse Expanded Disability Status Scale score at last follow-up (p = 0.008). The median ARR on treatment was 0.18 on azathioprine (n = 39, range 0–4), 0 on mycophenolate mofetil (n = 18, range 0–3), and 0 on rituximab (n = 29, range 0–2). No patient treated with rituximab as first-line therapy relapsed. Optic neuritis at onset was associated with a poor visual outcome below 20/200 (OR 8.669, 95% CI 1.764–42.616, p = 0.008), and a younger age at onset was associated with cognitive impairment (OR 0.786, 95% CI 0.644–0.959, p = 0.018).
Conclusions
AQP4-Ab NMOSD in children is an aggressive disease with permanent disabilities observed in over half the cohort. All DMTs were associated with a reduction of ARR. First-line rituximab prevented further clinical relapses. International consensus on treatment protocols for children is required to reduce heterogeneity of treatment regimens used worldwide.
Classification of evidence
This study provides Class IV evidence that for children with AQP4-Ab NMOSD, all DMTs, particularly first-line rituximab, reduced the ARR and prevented further clinical relapses.
Antibodies against aquaporin-4 (AQP4-Ab) were first described in 2004 in patients with neuromyelitis optica (NMO)1 allowing the expansion of the phenotype.2 The most recent criteria for the diagnosis of NMO spectrum disorder (NMOSD) stratify patients by the presence/absence of AQP4-Ab.3 AQP4-Ab seropositivity is associated with relapsing disease.4,5 This led to the use of B-cell targeting therapies, which clearly reduce relapse rates.6 This reduction is not seen when therapies known to be effective for MS7 are used in NMOSD.
The clinical features and MRI abnormalities in children with AQP4-Ab NMOSD are similar to the adult phenotype.8–11 The prevalence of AQP4-Ab was reported in 0.7% (2/279)12 to 4.5% (3/64)13 of children presenting with a first presentation of acquired demyelinating syndrome (ADS) and 8/102 (7.8%) of children with relapsing syndromes.14 Children are reported to have a less severe disease course and may take longer to reach disability than adults.15 Children are at a higher risk of visual impairment compared with adults but are less likely to acquire motor deficits.16 Previous pediatric publications highlighted that AQP4-Ab NMOSD in Europe is rare,9 whereas the prevalence in South America8 is higher.
With the rarity of pediatric presentation, treatment is derived from adult guidelines and can be influenced by medication availability and cost. Current available treatments used, such as azathioprine (AZA), mycophenolate mofetil (MMF), and rituximab, have not received regulatory approval for NMOSD. In this retrospective, multicenter, and multinational study, patients' demographics, first attack features, paraclinical characteristics, and disease course are described to ultimately evaluate responses to different treatment strategies in children with AQP4-Ab NMOSD.
Methods
Participants
In this multicenter, multinational study, we collected demographic, clinical, and radiologic data of 67 patients from a single center in Brazil (São Paulo, n = 20) and from 13 centers in 7 countries as part of the EU Paediatric Demyelinating Disease Consortium (United Kingdom [n = 18], France [n = 11], Spain [n = 6], Germany/Austria [n = 5], the Netherlands/Belgium [n = 4], Italy [n = 2], and Ukraine [n = 1]). This consortium was initiated to study children with ADS, as part of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Disease.
We retrospectively identified participants who were recruited into the respective centers or national demyelination programs and fulfilled the following inclusion criteria: (1) NMOSD, fulfilling the 2015 International Panel for NMO diagnosis criteria,3 (2) AQP4-Abs detected at onset or at the time of a clinical relapse, using live cell-based assays in the local laboratories, and (3) age <18 years at first presentation.
Standard protocol approvals, registrations, and patient consents
Patients included in this study had been enrolled in national programs with respective review board/ethical committee approvals (Brazil [Hospital das Clínicas, Faculty of Medicine, University of São Paulo, São Paulo], France [Hôpital Bicêtre, Paris], the Netherlands [Medisch Ethische Toetsingscommissie Erasmus Medical Centre, Rotterdam], United Kingdom [West Midlands-South Birmingham Research Ethics Committee], Germany and Austria [University of Innsbruck Ethics Committee], and Spain [Hospital Clinic and by Sant Joan de Déu Children's Hospital] or provided verbal and/or written informed consent to the respective referring physician (Italy, Ukraine). All data were deidentified.
Procedure
Clinical data already collected as part of national demyelination programs were deidentified and entered by each participating investigator onto a unified case reporting form (CRF), detailing selected demographics, clinical findings and laboratory results (AQP4-Abs, CSF cell count, protein, and oligoclonal bands), first and subsequent attack characteristics, and treatment information. Demyelinating phenotype at onset and relapses were clinically determined according to established criteria18 as being optic neuritis (ON), transverse myelitis (TM), brainstem and/or cerebellar and/or hemispheric syndromes (associated with encephalopathy or without encephalopathy).
Brain and spinal cord MRI were performed in 61 and 50 patients, respectively, at disease onset. All patients had undergone brain and spinal cord imaging according to local MRI protocols (not routinely including orbits). Gadolinium-enhanced imaging was performed in 54 patients. Lumbar puncture for CSF analysis was performed in 57/67 (85.0%) patients.
The acute treatment for each of these patients at presentation and subsequent episodes of relapses was decided by the treating pediatricians, on the basis of protocols influenced by their regional and/or national reference center for CNS demyelination, guided by severity and persistence of symptoms. Disease-modifying therapies (DMTs) referred to all forms of maintenance immunomodulation or immunosuppression therapies.
Annualized relapse rates (ARRs) on retrospective data were calculated as number of relapses per year and only included patients with at least 6 months of follow-up after initiation of treatment. If time to treatment was less than 6 months, the pretreatment ARR was calculated over a 6-month period. The outcomes, as measured by a range of difficulties the patients were experiencing (cognitive, visual, and motor), at last follow-up were retrieved from the patient's medical records to represent the most contemporary assessment of disability. If unavailable, this assessment was obtained directly from the patient's primary treating physician. Patients were considered to have motor disability if they were a wheelchair user, needed crutches (or a cane) to walk, or could walk for less than 500 m without support. Cognitive performance was estimated by looking at their school performance at last follow-up: patients were considered to have cognitive difficulties if there was significant school support, grade repetition, or needing of special education schools. Patients were considered to have visual sequelae if the logMAR scale was <0.3 (Snellen <20/40) or persistent visual field defects. The Expanded Disability Status Scale (EDSS) scores were documented at first attack nadir and at final follow-up (at point of disease stability at least 3 months from acute or relapsing events).
All CRFs were initially reviewed by the respective national leads and subsequently analyzed by 3 investigators (R.B.P., Y.H., and K.D.).
AQP4-Ab testing
Within 1 month of an acute event (either onset or relapse), clinically symptomatic children underwent testing for serum AQP4-Ab, using live cell-based assays in the respective reference laboratories of the following referring countries (France,18 Brazil,19 United Kingdom,20 Spain,21 the Netherlands,22,23 in Germany/Austria in the Clinical Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria and in Italy and Ukraine anti-AQP4 antibodies testing were performed with commercial IFA [Euroimmun]). Routine assessments of antibody testing in the CSF were not performed.
Statistical analysis
We tested the hypothesis that patients treated with AZA, MMF, and/or rituximab experienced a reduction in ARR providing Class IV evidence for therapeutic decision.
Descriptive data of demographic, clinical, radiologic, and serologic characteristics were reported for children with AQP4-Ab NMOSD. To compare between the different ethnicities, parametric or nonparametric statistical tests (Mann-Whitney U and Kruskal-Wallis tests) were used for continuous distributions, as appropriate given normality, and χ2 or Fisher exact tests for nominal data. In particular, comparisons between White and non-White ethnicities and stratified EDSS score at last follow-up were performed. Logarithm transformation was performed for nonparametric data. Effect sizes were calculated by OR, Cohen d, and eta-squared (η2) measures.
In addition, parametric or nonparametric statistical tests were used for continuous distributions, as appropriate, and χ2 or Fisher exact tests for nominal data to compare patients who were treated with AZA, MMF, and rituximab as first-line treatment. A paired 2-tailed t test was used to compare ARRs before and during treatment.
Univariate logistic regression was used to evaluate age at onset, sex, ethnicity, demyelinating phenotype at onset, abnormal MRI at onset, time to treatment, number of relapses before treatment, and follow-up duration as potential predictors of motor (wheelchair or walking aid), visual (blindness), and cognitive outcomes at last follow-up. Multivariable analyses were modeled for the same outcomes, including as covariates those variables significant at the (p < 0.05) level in the univariate analysis. Results were presented as ORs and 95% CIs. A 2-sided p < 0.05 was considered significant. Data were analyzed using SPSS 20 software and GraphPad Prism 5.
Data availability
Anonymized data will be shared by request from qualified investigators.
Results
Patients
A total of 67 children with AQP4-Ab NMOSD were included in this study (table 1). Mean age at onset was 10.2 ± 3.6 years (range 2–16 years). The female/male ratio was 4.1:1. Of the 67 children, 29 (43.3%) were White, 14 (20.9%) Black, 13 (19.4%) Brazilian mixed ethnicity, and 11 (16.4%) other ethnicities. Of the 67 children, 29 (43.3%) were White, 14 (20.9%) Black, 13 (19.4%) Brazilian mixed ethnicity and 11 (16.4%) other ethnicities (Asian, Indians, Nepali, Afro-Caribbean, Omani). Non-White ethnicity was more common in both European (25/47) and Brazilian (13/20) populations. Family history of autoimmune diseases was reported in 9 (13.7%) patients, personal history of inflammatory diseases in 7 (10.4%), and 8 (11.9%) had a preceding infection. Patients' demographic, clinical, and paraclinical features and EDSS scores, stratified to ethnicities, are summarized in table 1. The groups were similar except for shorter time to first relapse (η2 = 0.071, p = 0.049) and higher EDSS score at last follow-up (η2 = 0.056, p = 0.008) in the non-White population.
Table 1.
Clinical/paraclinical features and outcome in children with AQP4-Ab NMOSD stratified to the different ethnicities

First attack features
The most frequent phenotype at onset was isolated ON (n = 20, 29%) followed by isolated TM (n = 15, 22%) and isolated area postrema syndrome (n = 11, 16.4%). Of the children presenting with ON, 9/20 (45%) had bilateral ON. Of the 15 children presenting with isolated TM, 10 (66.7%) had longitudinally extensive TM. Six (9%) patients had simultaneous ON and TM. Six (9%) patients presented with ADEM, 1 (1.5%) patient with isolated diencephalic syndrome, 3 (4.5%) with isolated brainstem syndrome, and 5 (7.5%) patients with multifocal syndromes involving the spinal cord, optic nerve, and brain (brainstem, area postrema, or diencephalic syndromes).
At onset, 44/61 (72.1%) patients had abnormal brain MRI, 32/50 (64%) patients had abnormal spinal cord MRI, and 16/54 (29.6%) had contrast-enhancing lesions. Abnormal CSF was reported in 37/57 (64.9%) children, with oligoclonal bands detected in 7/57 (12.2%). CSF pleocytosis was present in 35/50 (70%) examinations, with a median of 10 cells (interquartile range [IQR] 2.5–38.5 cells). Elevated CSF protein was found in 13/27 (48%) examinations with a median of 0.030 g/dL (IQR 0.002–0.850 g/dL). Myelin oligodendrocyte glycoprotein antibodies (MOG-Abs) were negative in the 60 patients who were tested.
Disease course and outcome
A total of 297 attacks were reported in the cohort. The median time to first relapse was 4 months (range 1.7–100.5 months). At last follow-up (median disease duration of 4 years, IQR 2–10 years), 58 (86.6%) patients had a second clinical event with a median ARR of 1.05 relapses per year and median (IQR) EDSS score = 2.0 (1.0–3.5). Moderate disability with EDSS score ≥3 was reported in 29/67 (43.2%). Visual impairment was seen in 32 (47.8%) patients, of which 20 were registered blind (visual acuity <20/200; logMAR >1), motor deficits were found in 14 (21.2%, of which 5 were wheelchair users and 2 used a walker), and cognitive impairment was detected in 17 (25.4%) patients.
A more severe disease course was seen in the non-White ethnicity with worse EDSS score at last follow-up (p = 0.008) and a shorter time to first relapse (p = 0.049).
Response to immunotherapy
DMTs were given in 63/67 (95%) children. A total of 41 patients were treated with 1 DMT, 12 with 2, 7 with 3 DMTs, 2 with 4 DMTs, and 1 with 5 sequential DMTs. Patients were commenced on DMTs at a median of 6 months from symptom onset (IQR 2–13.5 months) and a median of 2 clinical attacks (IQR 1–3 clinical attacks). Of the 4 untreated patients, at a median follow-up of 5 years (range 2–10 years), only 1 patient relapsed despite persistent antibody positivity.
A total number of 139 relapses on treatment were observed in the cohort. Relapses occurred on all treatments, except the 2 patients treated with ofatumumab and the 1 patient who underwent hematopoietic stem cell transplantation (HSCT). The clinical course and disease activity in patients who underwent therapy with maintenance treatment are illustrated in figure 1. Most frequently used treatments were AZA (total treatment duration 209.5 patient years, relapses = 92; 0.43 relapses/treated year), rituximab (total treatment duration 88 years, relapses = 13; 0.15 relapses/treated year), and MMF (total treatment duration 46.2 years, relapses = 23; 0.49 relapse/treated year). Patient characteristic stratified to first-line treatments is summarized in table 2 and table e-4, links.lww.com/NXI/A287.
Figure 1. Disease course in relation to respective therapies.

Patient relapsed on all treatments with a total of 139 relapses on treatment reported in the cohort. Forty-three (69.3%) remained relapse free on treatment; 10/35 (28.6%) treated with azathioprine (AZA), 10/17 (58.8%) treated with mycophenolate mofetil (MMF); and 23/29 (79.3%) treated with rituximab. One patient who relapsed AZA and cyclosporine underwent hematopoietic stem cell transplantation (HSCT) and has not relapsed (follow-up 2 years).
Table 2.
Patients characteristic stratified to first-line treatment

Thirty-nine children were treated with AZA, 35 (89.7%) as first-line therapy, and 4 as second-line treatment. AZA treatment was associated with a mean reduction in the ARR of 1.10 (mean ARR pretreatment 1.69 vs 0.59 on treatment, mean difference 1.10, 95% CI 0.54–1.66, t = 4.01, Cohen d = 0.63, p < 0.001). Twenty-five of 39 (64.1%) patients relapsed on treatment. Of these, treatment was escalated in 16 patients to MMF (n = 6), rituximab (n = 8), cyclosporine (n = 1), and glatiramer acetate (n = 1). Of the 4 who were treated with AZA as second line, 3 previously failed treatment with methotrexate, glatiramer acetate, and cyclophosphamide, and in 1, treatment was de-escalated after being relapse free on rituximab.
Twenty-nine children were treated with rituximab, 14 (48.3%) first line, 9 (31.0%) second line, 5 (17.2%) third line, and 1 (3.4%) fourth line. The treatment with rituximab was associated with a mean reduction in the ARR of 2.36 (mean ARR pretreatment 2.50 vs 0.14 on treatment, mean difference 2.36, 95% CI 1.57–3.15, t = 6.13, Cohen d = 1.14, p < 0.001). All 14 patients treated with rituximab as first line did not have further relapses. Of these, 1 patient had prolonged oral steroids (for 1 year) before commencing rituximab, and a second patient had additional intravenous immunoglobulin for the first 6 months. Seven of 29 patients (24.1%) treated with rituximab relapsed on treatment, and these patients were previously treated with AZA (n = 5) and MMF (n = 2). In the 4 children in whom the CD19 count was measured acutely, 1 relapsed with absent B cells and 3 during B cell reconstitution.
Eighteen children were treated with MMF, 10 (55.6%) first line, 6 (33.3%) second line, 1 (5.6%) third line, and 1 fifth line (5.6%). MMF treatment was associated with a mean reduction in the ARR of 0.32 (mean ARR pretreatment 1.04 vs 0.72 on treatment, mean difference 0.32, 95% CI −0.57 to 1.22, t = 0.77, Cohen d = 0.18, p = 0.452). Ten patients remained relapse free on treatment. Of the 8 who relapsed on treatment, 5 were changed to rituximab.
Tocilizumab was used in 2 patients as third-line DMT, and both relapsed and satralizumab in 2 patients (who were enrolled in the open-label SAkuraSky trial as an add-on treatment to MMF), who have not relapsed. Two patients were treated with ofatumumab following infusion reaction with rituximab and have not relapsed. Three patients treated with cyclophosphamide, 2 with methotrexate and 1 with cyclosporine; all continued to relapse with worsening ARR. Two patients were treated with glatiramer acetate for a presume diagnosis of MS, and both relapsed on treatment. ARRs before and after treatment initiation are shown in figure 2.
Figure 2. The efficacy of various disease-modifying therapies in children with AQP4-Ab NMOSD.

Annualized relapse rates (ARRs) before and after treatment initiation with the most frequently used medications. (A) Azathioprine (AZA). (B) Mycophenolate mofetil (MMF). (C) Rituximab. (D) After escalation from treatment with AZA or MMF to rituximab.
Adverse events
Of the 37 patients treated with AZA, 5 (13.5%) developed lymphopenia (<0.5 × 109/L) without infectious complications, 3 developed infections (viral meningitis, pneumonia, and varicella), and 3 did not tolerate the treatment in view of gastrointestinal symptoms or raised liver function test. One child developed lymphopenia (<0.5 × 109/L) on MMF. Infusion reactions with rituximab occurred in 3 of 29 (10.3%) children, of which 2 were switched to ofatumumab due to the side effects. One child developed persisted neutropenia on rituximab and was changed to AZA.
Predictors of poor outcome
ORs to estimate the effects of parameters at onset on the occurrence of disability (EDSS score ≥3) are illustrated in table 2. Children with an EDSS score ≥3 (n = 29, 43.2%) were younger, presented with ON, and had a higher EDSS score at nadir. Patients with a better outcome at last follow-up (EDSS score <3.0) were more commonly girls and presented with cerebral syndrome (table 3). Logistic regression looking at age at onset, ethnicity, demyelinating phenotype at onset, abnormal MRI at onset, time to treatment, and number of relapses before treatment were evaluated as predictors of motor, visual, and cognitive outcomes (table e-1, links.lww.com/NXI/A284, table e-2, links.lww.com/NXI/A285, and table e-3, links.lww.com/NXI/A286). Multivariable analysis did not identify any predictors for motor disabilities (wheelchair or walking aid). ON at onset was associated with worse visual outcome below 20/200 (p = 0.008, OR 8.669, 95% CI 1.764–42.616), and younger age at onset was associated with worse cognitive impairment (p = 0.018, OR 0.786, 95% CI 0.644–0.959).
Table 3.
Univariable analysis and OR for parameters associated with the EDSS score (EDSS ≥3) at final follow-up

Discussion
AQP4-Ab NMOSD is now well recognized in children. Natural history studies suggest an attack-related stepwise accumulation of disabilities. Therefore, attack prevention strategies are used as maintenance treatment. In this multinational cohort of children with AQP4-Ab NMOSD, the phenotype observed was overall similar to that reported previously in children or adults.3 The time to first relapse was short, and the ARR pretreatment was high, indicating a high risk of relapses and permanent disability in children with AQP4-Ab NMOSD.
We detected cognitive impairment in 25.4% of the cohort. Abnormal brain MRI was not a risk factor, but younger age at onset and brainstem/cerebral attacks were associated with cognitive deficits. Cognitive impairment had not been systematically addressed in pediatric NMOSD, and our results highlight its importance. Although no objective cognitive measure was used in this study, we selected measures that are consistent across the diverse educational systems that reflect clear difficulties and thus would be a reasonable surrogate for cognitive performance. ON at onset was a risk for visual impairment and blindness in keeping with previous report that young children do not recover well following ON.16 Of interest, time to starting treatment and number of relapses before starting treatment were not associated with worst outcome. As previously reported,24 non-White ethnicity was associated with a more aggressive disease course but was not predictive of moderate disabilities at last follow-up.
Patients were treated according to each center experience, and the choices of treatment were additionally influenced by the access to treatments in each country and their related costs. Almost all of the Brazilian children were commenced on AZA, and none were given rituximab as first-line treatment. In contrast, the European children were frequently commenced on rituximab and MMF (table 3). A previous pediatric cohort10 (only 20/58 patients were treated with DMTs) reported residual disability in 43/48 (90%), with 26/48 (54%) having visual impairment and 21/48 (44%) with motor deficits. Comparing to this cohort, the visual but not the motor outcome is similar, which might be related to treatment.
A key observation from our study was that all treatments known to reduce relapses in adults (AZA, MMF, and rituximab) were associated with a reduction in the ARR. Moreover, rituximab treatment resulted in the lowest ARR, and it is remarkable that all 14 patients who started on rituximab as first line did not have any further relapses. Treatment escalation from AZA and MMF to rituximab was beneficial in 12/13, with only 1 child maintaining the same ARR (figure 2). The benefit of early treatment with rituximab was previously demonstrated in a pediatric cohort with a range of CNS disorders including NMOSD.25 This apparent superiority could be partially explained by the early treatment effect of rituximab. In addition, in our cohort, patients were also initiated with rituximab sooner compared with AZA/MMF (1 month vs 6–10 months) after the attack. CD19 count was not performed in all 7 patients who relapsed with rituximab. Monitoring B-cell repopulation and redosing rituximab might prevent relapses as demonstrated in a pediatric study.26 Three patients treated with rituximab changed therapy due to adverse effect, and none had severe infection. Prospective follow-up was not performed to check on hypogammaglobulinemia.27
Although we did not perform a direct comparison, the treatment effect observed in this pediatric cohort appeared greater than reported in adults with AQP4-Ab NMOSD.6,28–30 The greater reduction in the ARR following initiation of DMTs is also reported in children with MS compared with adults and likely reflects the more inflammatory disease seen in the younger patients.31 Of interest, in comparison to a cohort of children with MOG-Ab–associated disease,32 despite the higher ARR before treatment, the ARR on treatment was lower across all treatments in the AQP4-Ab NMOSD group.
Our cohort was not optimal for a direct evaluation of an individual treatment or a sequence of treatment effect, which is better suited to a study design where the sequence of DMTs, the lag phase of efficacy or washout period of specific therapies are prospectively controlled. One particular treatment that deserves specific attention would be the cumulative use of corticosteroids, often used in conjunction with DMTs and at low doses, but also during acute attacks. Time to initiation of acute treatment, the choice of treatment, and treatment escalation are major predictors of long-term outcome in AQP4-Ab NMOSD.33–35 As many of the children presented initially to district general hospital, timing, duration, and sequence of acute treatment were not included in the analysis.
A major limitation of this study is that not all patients were systematically managed, with possible biases in treatment initiation and/or escalation. Nevertheless, these real-world clinical data from multiple countries allowed us to make important observations about the treatment responsiveness of children with AQP4-Ab NMOSD. The important question our study raises is whether rituximab should be first-line treatment for children. Moreover, should children who are already stable on AZA and MMF be changed to the more efficacious therapies with a higher risk profile? Furthermore, with new monoclonal antibodies (satralizumab,36 inebilizumab,37 and eculizumab38) likely to become available, treatment algorithms that accommodate risk stratification of poorer prognosis (such as the patients age and severity of the attack) and grouping of treatments with similar efficacy within the sequence and choice of treatments in pediatric AQP4-Ab NMOSD are urgently required.
Acknowledgment
The authors thank all collaborators who worked in this study in the collection of data: Austria and Germany: M. Reindl, Clinical Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria. Brazil: Dr. Dagoberto Callegaro (Department of Neurology, Hospital das Clínicas, Faculty of Medicine, São Paulo, Brazil). Laboratory of Brain Institute of Rio Grande do Sul (Brains), Pontifical Catholic University of Rio Grande Do Sul (PUCRS), Porto Alegre, Brazil. France: Dr. Maurey, Assistance Publique-Hôpitaux de Paris, University Hospitals Paris South, Bicêtre Hospital, Pediatric Neurology Department, Le Kremlin Bicêtre, Dr. Pascale Chrétien, University Hospitals Paris South, Bicêtre Hospital, Immunology Department, Le Kremlin Bicêtre, Dr. Aubart, Assistance Publique-Hôpitaux de Paris, Necker Hospital, Paris, Dr. Cheuret, Hôpital des Enfants, Pediatric Neurology Department, Toulouse, Dr. Lepine, Centre Hopitalo-Universitaire de Marseille, Marseille, Pr Laugel, CHU de Strasbourg - Hôpital de Hautepierre, Pediatric Neurology Department, Strasbourg. Spain: Dr. Maria Sepulveda (Neuroimmunology Program, Hospital Clinic-IDIBAPS, University of Barcelona, Spain), Dr. Victor Soto-Insuga (Pediatric Neurology Section, Pediatric Service, Fundacion Jimenez Díaz, Madrid, Spain), Maria C. Miranda-Herrero (Pediatric Neurology Section, Pediatric Service, Hospital Gregorio Marañon, Madrid, Spain), Montserrat Garcia-Puig (Pediatric Neurology Section, Pediatric Service, Hospital Parc Taulí, Sabadell, Spain). The Netherlands: Prof. Rogier Hintzen, MD, PhD, was involved in this study before his sudden death. Dörte Hamann and Kyra Gelderman from the Department of Immunopathology and Blood Coagulation, Sanquin Diagnostic Services, Amsterdam, the Netherlands. AQP4 antibodies were routinely assayed in a cell-based assay using a fluorescence-activated cell sorter (FACS, LSRII, and DIVA software, Becton Dickinson, San Jose). United Kingdom: UK Childhood Inflammatory Disorders (UK-CID) Study Group: Antonia Clarke, MD (Department of Paediatric Neurology, St George's Hospital, London), Katherine Forrest, MD (Department of Paediatric Neurology, Southampton Children's Hospital, Southampton), Andrex Lux, MD (Department of Paediatric Neurology, University Hospitals Bristol, Bristol), Saleel Chandratre, MD (Department of Paediatric Neurology, John Radcliffe Hospital, Oxford), Kshitij Mankad, MD, and Kling Chong, MD (Paediatric Neuroradiology, Great Ormond Street Hospital, London). Laboratory: Paddy Water, PhD, Mark Woodhall, PhD, and Jackie Palace, MD, PhD (Oxford Autoimmune Neurology Group and National Health Service National Specialised Commissioning Group for Neuromyelitis Optica, United Kingdom).
Glossary
- ADS
acquired demyelinating syndrome
- AQP4-Ab
antibodies against aquaporin-4
- ARR
annualized relapse rate
- AZA
azathioprine
- CRF
case reporting form
- DMT
disease-modifying therapy
- EDSS
Expanded Disability Status Scale
- IQR
interquartile range
- MMF
mycophenolate mofetil
- MOG-Ab
myelin oligodendrocyte glycoprotein antibody
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- TM
transverse myelitis
Appendix. Authors
Study funding
This study was supported in part by the Marato TV3 Foundation (20141830 to T.A. and A.S.), Red Española de Esclerosis Múltiple (REEM) (RD16/0015/0002; RD16/0015/0003 to A.S.), and Torrons Vicens Foundation (PFNR0144 to TA).
Disclosure
R.B. Paolilo received support for participating in scientific meetings from Roche and Merck. Y. Hacohen and E. Yazbeck report no disclosures. T. Armangue received speaking honoraria from Novartis and travel expenses for participating in scientific meetings from Roche. A. Bruijstens and C. Lechner report no disclosures. S.L. Apostolos-Pereira served as board advisory for Novartis and Roche and received support for participating in scientific meetings from Genzyme, Roche, and Novartis. Y. Martynenko, M. Breu, C. de Medeiros Rimkus, E. Wassmer, M. Baumann, and L. Papetti report no disclosures. M. Capobianco received personal honoraria for speaking at meeting or participating in advisory boards from Biogen, Merck, Novartis, Roche, Sanofi, and Teva. B. Kornek, K. Rostásy, and J.A. da Paz report no disclosures. O. Ciccarelli receives research funding from the UK MS Society, Rosetrees Trust, and NIHR UCLH BRC and serves as a consultant for Biogen, Novartis, Roche, and GE Healthcare; she is on the editorial board of Neurology. M. Lim received consultation fees from CSL Behring; received travel grants from Merck Serono; and was awarded educational grants to organize meetings by Novartis, Biogen Idec, Merck Serono, and Bayer. A. Saiz reports no disclosures. R. Neuteboom received consultation fees from Novartis and Zogenix. R. Marignier serves on the scientific advisory board for Novartis and MedImmune and received speaker honoraria and travel funding from Novartis, Biogen, Teva, Sanofi-Aventis/Genzyme, and Merck. C. Hemingway received consulting and educational grants from Novartis; served on the Global Paediatric MS Steering committee and received educational grants from Biogen; and received consultation fees from Alexion. D.K. Sato received research support from CAPES/Brasil, CNPq/Brasil, FAPERGS/Brasil, and Japan Society of Science and received speaker honoraria from Biogen, Novartis, Genzyme, Merck Sereno, Quest/Athena Diagnosis, Shire, and Teva. K. Deiva received speaking honoraria at meetings and for participation in advisory boards from Novartis, Sanofi, and Servier. Go to Neurology.org/NN for full disclosures.
References
- 1.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. . A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 2.Sato DK, Nakashima I, Takahashi T, et al. . Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology 2013;80:2210–2216. [DOI] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Banwell B, Bennett JL, et al. . International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rostasy K, Reindl M. Role of autoantibodies in acquired inflammatory demyelinating diseases of the central nervous system in children. Neuropediatrics 2013;44:297–301. [DOI] [PubMed] [Google Scholar]
- 5.Tillema JM, McKeon A. The spectrum of neuromyelitis optica (NMO) in childhood. J Child Neurol 2012;27:1437–1447. [DOI] [PubMed] [Google Scholar]
- 6.Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol 2014;71:324–330. [DOI] [PubMed] [Google Scholar]
- 7.Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010;67:1016–1017. [DOI] [PubMed] [Google Scholar]
- 8.Banwell B, Tenembaum S, Lennon VA, et al. . Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology 2008;70:344–352. [DOI] [PubMed] [Google Scholar]
- 9.Huppke P, Bluthner M, Bauer O, et al. . Neuromyelitis optica and NMO-IgG in European pediatric patients. Neurology 2010;75:1740–1744. [DOI] [PubMed] [Google Scholar]
- 10.McKeon A, Lennon VA, Lotze T, et al. . CNS aquaporin-4 autoimmunity in children. Neurology 2008;71:93–100. [DOI] [PubMed] [Google Scholar]
- 11.Chitnis T, Ness J, Krupp L, et al. . Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology 2016;86:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banwell B, Bar-Or A, Arnold DL, et al. . Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 2011;10:436–445. [DOI] [PubMed] [Google Scholar]
- 13.Hacohen Y, Absoud M, Woodhall M, et al. . Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry 2014;85:456–461. [DOI] [PubMed] [Google Scholar]
- 14.Hacohen Y, Mankad K, Chong WK, et al. . Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology 2017;89:269–278. [DOI] [PubMed] [Google Scholar]
- 15.Collongues N, Marignier R, Zephir H, et al. . Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology 2010;75:1084–1088. [DOI] [PubMed] [Google Scholar]
- 16.Kitley J, Leite MI, Nakashima I, et al. . Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012;135:1834–1849. [DOI] [PubMed] [Google Scholar]
- 17.Krupp LB, Tardieu M, Amato MP, et al. . International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 2013;19:1261–1267. [DOI] [PubMed] [Google Scholar]
- 18.Marignier R, Bernard-Valnet R, Giraudon P. Aquaporin-4 antibody-negative neuromyelitis optica: distinct assay sensitivity-dependent entity. Neurology 2013;80:2194–2200. [DOI] [PubMed] [Google Scholar]
- 19.Sato DK, Callegaro D, Lana- Peixoto MA, et al. . Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol 2014;5:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Höftberger R, Sepulveda M, Armangue T. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler 2015;21:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Pelt ED, Wong YY, Ketelslegers IA. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol 2015;23:580–587. [DOI] [PubMed] [Google Scholar]
- 23.Ketelslegers IA, Modderman PW, Vennegoor A. Antibodies against aquaporin-4 in neuromyelitis optica: distinction between recurrent and monophasic patients. Mult Scler 2011;17:1527–1530. [DOI] [PubMed] [Google Scholar]
- 24.Kim SH, Mealy MA, Levy M, et al. . Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018;91:e2089–e2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dale RC, Brilot F, Duffy LV, et al. . Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 2014;83:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nosadini M, Alper G, Riney CJ, et al. . Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 2016;3:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcinno A, Marnetto F, Valentino P, et al. . Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 2018;5:e498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costanzi C, Matiello M, Lucchinetti CF, et al. . Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 2011;77:659–666. [DOI] [PubMed] [Google Scholar]
- 29.Huh SY, Kim SH, Hyun JW, et al. . Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol 2014;71:1372–1378. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Jeong IH, Hyun JW, et al. . Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol 2015;72:989–995. [DOI] [PubMed] [Google Scholar]
- 31.Gorman MP, Healy BC, Polgar-Turcsanyi M, Chitnis T. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol 2009;66:54–59. [DOI] [PubMed] [Google Scholar]
- 32.Hacohen Y, Wong YY, Lechner C, et al. . Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 2018;75:478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleiter I, Gahlen A, Borisow N, et al. . Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 2016;79:206–216. [DOI] [PubMed] [Google Scholar]
- 34.Kleiter I, Gahlen A, Borisow N, et al. . Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 2018;5:e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnan M, Valentino R, Debeugny S, et al. . Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry 2018;89:346–351. [DOI] [PubMed] [Google Scholar]
- 36.Yamamura T, Kleiter I, Fujihara K, et al. . Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med 2019;381:2114–2124. [DOI] [PubMed] [Google Scholar]
- 37.Cree BAC, Bennett JL, Kim HJ, et al. . Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019;394:1352–1363. [DOI] [PubMed] [Google Scholar]
- 38.Pittock SJ, Berthele A, Fujihara K, et al. . Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 2019;381:614–625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from qualified investigators.