

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Platelets prevent plasma leakage during neutrophil diapedesis by binding to endothelial VWF and releasing Angpt1.
Platelet-released Angpt1 stimulates endothelial Tie-2, thereby activating the GEF FGD5, which strengthens cortical actin bundles.
Abstract
Neutrophil extravasation requires opening of the endothelial barrier but does not necessarily cause plasma leakage. Leaks are prevented by contractile actin filaments surrounding the diapedesis pore, keeping this opening tightly closed around the transmigrating neutrophils. We have identified the receptor system that is responsible for this. We show that silencing, or gene inactivation, of endothelial Tie-2 results in leak formation in postcapillary venules of the inflamed cremaster muscle at sites of neutrophil extravasation, as visualized by fluorescent microspheres. Leakage was dependent on neutrophil extravasation, because it was absent upon neutrophil depletion. We identified the Cdc42 GTPase exchange factor FGD5 as a downstream target of Tie-2 that is essential for leakage prevention during neutrophil extravasation. Looking for the Tie-2 agonist and its source, we found that platelet-derived angiopoietin-1 (Angpt1) was required to prevent neutrophil-induced leaks. Intriguingly, blocking von Willebrand factor (VWF) resulted in vascular leaks during transmigration, indicating that platelets interacting with endothelial VWF activate Tie-2 by secreting Angpt1, thereby preventing diapedesis-induced leakiness.
Visual Abstract

Introduction
Leukocyte extravasation and enhanced vascular permeability are hallmarks of inflammation. Yet, whether the transmigration of leukocytes through the endothelial barrier would automatically lead to increased vascular leakage of plasma components or whether leaks are induced separately has been debated for decades. Evidence for a link of both processes was based on findings that blocking leukocyte adhesion to, and extravasation through, endothelium could reduce the increase in vascular permeability in certain inflammatory settings, such as postischemic skeletal muscle, platelet-activating factor–stimulated mesentery, and tumor necrosis factor-α (TNF-α)–activated cremaster muscle.1-4 This clearly established a possible link between the 2 processes but left open whether leaks were caused directly by the transmigration process per se or by factors released from the adhering neutrophils5 in response to the cocktail of proinflammatory mediators present in certain inflammatory settings.
There is strong evidence that the transmigration itself does not automatically cause leaks. Investigating an allergen-induced inflammation model in the trachea, it was shown that leaks occurred in postcapillary venules, whereas the exit of leukocytes was observed in venules.6 Furthermore, the 2 processes were triggered with different kinetics in a sterile cutaneous wound model.7 Finally, we have recently shown in vivo that leukocyte transmigration, on the 1 hand, and permeability induction, on the other hand, destabilize endothelial junctions by affecting the phosphorylation of different tyrosine residues on VE-cadherin.8 Thus, both processes address the opening of endothelial junctions in different ways. Collectively, these observations raise the question of how the endothelial diapedesis pore can be kept sealed for plasma content while a leukocyte is moving through. A mechanistic basis for this was proposed recently: during leukocyte diapedesis, leakage is prevented by the contraction of endothelial circumferential actin, which confines the diapedesis pore around the transmigrating leukocyte; a process accompanied by local RhoA signaling.9 However, it is still unknown what stimulates the contraction of the actomyosin system, which keeps the diapedesis pore tight.
The receptor tyrosine kinase Tie-2 is an important regulator of angiogenesis and vessel remodeling,10,11 which is also of crucial importance for the regulation of vascular quiescence and the maintenance of endothelial barrier function. The Tie-2 agonist angiopoietin-1 (Angpt1) promotes the stability of endothelial junctions and, thereby, the barrier function of endothelium.12,13 Accordingly, Angpt1 treatment was shown to protect against the permeability-enhancing effects of various inflammatory mediators in vitro and in vivo.14-18 Tie-2 is a substrate of the endothelial phosphatase VE-PTP and, as such, is the functional target by which a highly specific inhibitor of VE-PTP counteracts vascular permeability induction in diabetic retinopathy and other inflammation models.19,20 Mechanistically, VE-PTP inhibition and Tie-2 activation were shown to induce rearrangements of the actin cytoskeleton, leading to dampening of actin stress fibers and reinforcement of junctional actin. This process required activation of the small GTPases Rap1 and Rac1 and the GTPase exchange factor (GEF) FGD5.20,21 Collectively, these studies show the great potential of Tie-2 in stabilizing endothelial junctions; however, much less is known about the physiological processes that make use of endogenously provided Angpt1 for junction stabilization.
Platelets play a fundamental role in hemostasis but have also become more and more recognized for their contributions to the inflammatory process.22-24 They are potent supporters of leukocyte extravasation in various tissues and inflammatory settings.25-28 Platelets can bind to von Willebrand factor (VWF) under flow conditions,29,30 and we and other investigators have shown in vivo that blocking this interaction on the endothelial cell surface interfered with the extravasation of neutrophils in peritonitis and inflamed cremaster muscle.27,31 In different inflammation models, it was shown that platelet factors, such as thromboxane A2 or serotonin, enhanced vascular permeability in inflammation and supported neutrophil extravasation.25,32 On the other hand, platelets can also protect vascular integrity. In skin and lung, platelet depletion leads to inflammatory bleeding.33-35 Platelets prevent this bleeding via coagulation-unrelated mechanisms33 and independent of secreted junction-stabilizing factors.36 Instead, plugging of vascular exit sites subsequent to the diapedesis of a neutrophil may prevent the passive exit of erythrocytes.37 Platelet granules contain various junction-stabilizing factors, such as sphingosine-1 phosphate, serotonin, and Angpt1. The latter 2 were reported to prevent intratumor hemorrhage38; furthermore, Angpt1 was required for vascular integrity in a mouse model of cerebral malaria.39
Here, we have analyzed what triggers the sealing of the diapedesis pore for macromolecules during leukocyte diapedesis. Using the accumulation of 20-nm fluorescent microspheres at sites of leukocyte extravasation as a read-out for leak formation, we found that Tie-2 and its downstream target FGD5 were essential to prevent leaks during diapedesis. The stimulation of Tie-2 was achieved by platelets that provided Angpt1 by interacting with VWF at the endothelial surface.
Methods
Mice
All mice used in this study were on the C57BL/6 genetic background. Tektm1a(EUCOMM)Hmgu embryonic stem cells were purchased from EUCOMM and injected into C57BL/6 blastocysts, generating transgenic mice with an inactivated Tek gene mediated by a LacZ trapping cassette. By mating these mice with mice expressing Flp recombinase, the FRT-flanked lacZ trapping cassette was excised, producing a conditional knockout (KO) allele in which exon 8 is flanked by loxP sites (Teklox/lox). To inactivate Tek postnatally in endothelial cells, Teklox/lox mice were bred to tamoxifen-inducible Pdgfb-iCre transgenic mice,40 resulting in TekiECKO mice. Tek_fw (5′-CCTGGGTGGTGTGTTGTCA-3′) and Tek_rev (5′-GTCTTTGCCATGACAGTACAGT-3′) primers were used for genotyping, generating a 344-bp polymerase chain reaction (PCR) product in wild-type (WT) mice and a 299-bp product in Teklox/lox mice. Angpt1lox/lox mice41 (provided by Sean Morrison, University of Texas Southwestern, Dallas, TX) and bred with PF4-Cre mice42 to obtain Angpt1PtlKO mice. Angpt1_fl_F (5′-GGACTCAACTTCCCTGGGTAAGC-3′) and Angpt1_fl_R (5′-GGCTTTGACAGTCAAAATGCC-3′) primers were used for genotyping, generating a 150-bp product for the WT allele and a 250-bp product for the Angpt1lox/lox allele. C57BL/6 WT mice43 were used for in vivo small interfering RNA (siRNA) experiments and LysM-eGFP mice were used for intravital imaging. Animals were kept in a barrier facility under special pathogen-free conditions. All experiments were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen.
Antibodies
Polyclonal antibodies VD68 against FGD5,21 VE-PTP-C against VE-PTP,44 and VE-42 against VE-cadherin,45 as well as monoclonal antibodies 1G5.1 and 5D2.6 against mPECAM-1,46 11D4.1 against mVE-cadherin,47 and 3G1 against Tie-2,48 have been described previously. The following antibodies were purchased: monoclonal antibodies against Ly-6G/Gr-1 (RB6-8C5; BD Bioscience) and an isotype-matched control (eB149/10H5; eBioscience), a mixture of monoclonal antibodies against mGPIbα (R300) and an isotype-matched control (C301; both from emfret ANALYTICS), mGPIbβ (X649; emfret ANALYTICS), mPECAM-1 (390; eBioscience), α-tubulin (B-5-1-2; Sigma-Aldrich), and polyclonal antibodies against VWF (A0082; Dako) and mMRP14 (R&D Systems). Horseradish peroxidase–coupled secondary antibodies were purchased from Dianova, and goat anti-Mmouse Alexa Fluor–coupled antibodies were purchased from Invitrogen.
Cell culture
Human umbilical vascular endothelial cells (HUVECs) were cultured in EBM-2 medium supplemented with SingleQuots (Lonza). Human polymorphonuclear leukocytes (PMNs) were isolated from peripheral blood by density gradient centrifugation using Histopaque 1077 and 1119 (Sigma-Aldrich) The granulocyte-containing phase was collected and washed twice in Hanks balanced salt solution, 25 mM HEPES (pH 7.3), and 10% fetal calf serum. Erythrocytes were lysed by incubation with 0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA for 4 minutes at room temperature. PMNs were immediately used for transmigration assays.
RNA interference
For in vitro RNA interference with expression of FGD5, the following siRNAs were used: Hs_FGD5_6, 5′-GCGCUGACACUUCAGACUA-3′ or Hs_FGD5_2, 5′-GGAUGACAUGGACCAUGAA-3′. siRNA not targeting any known mammalian gene (5′-UUCUCCGAACGUGUCACGU-3′; QIAGEN) was used as a negative control. Routinely, 106 HUVECs were transfected with 40 ng of siRNA using INTERFERin (Polyplus Transfection), according to the manufacturer’s instructions. Cells were analyzed 72 hours after transfection. For RNA interference with FGD5 or Tie-2 expression in vivo, siRNA sequences 5′-CAGUCGGUGUAUAGUUAGUAA-3′ (FGD5) and 5′-CAGGAAGGTATGAGTACAAAT-3′ (Tie-2) were used, as described previously.20,21 As a negative control, siRNA not targeting any known mammalian gene (5′-TAGCGACTAAACACATCAA-3′; siSTABLE; GE Healthcare) was used. Mice were injected intrascrotally with 60 μg of siRNA using in vivo-jetPEI as transfer reagent (Polyplus Transfection), according to the manufacturer’s instructions, and analyzed 48 hours later.
In vitro transmigration assay
To analyze permeability induction during PMN transmigration, 2 × 104 HUVECs were seeded on fibronectin-coated Transwell filters (6.5 mm, 5-µm pore size; Corning), grown to confluence, and stimulated with 5 nM recombinant human TNF-α (PeproTech) 16 hours prior to the assay. PMNs (2 × 105) were allowed to transmigrate toward the chemokine interleukin-8 (IL-8; R&D Systems) for 40 minutes in the presence of 0.25 mg/mL fluorescein isothiocyanate (FITC)-dextran (40 kilodalton [kDa]; Sigma-Aldrich). Fluorescence in the lower chamber was measured on a plate reader (Synergy 2; Lab-Tek), and transmigrated PMNs were counted using a CASY Cell Counter TT+ (Roche).
Immunoblot analysis
Cells were lysed in lysis buffer containing 20 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 2 mM CaCl2, 1.5 mM MgCl2, 1 mM Na3VO4, 1% Triton X‐100, 0.04% NaN3, and 1× cOmplete, EDTA‐free Proteinase Inhibitor Cocktail (Roche). Lysates were centrifuged at 20 000g and 4°C for 30 minutes before aliquots for immunoblot analysis were taken. Murine lungs were homogenized using an ULTRA-TURRAX (IKA-Werke) or a Precellys Evolution (Bertin Instruments) in radioimmunoprecipitation assay buffer containing 1% NP-40, 1% sodium deoxycholate, 0.01 M NaPi, 150 mM NaCl, 2 mM EDTA, 1 mM Na3VO4, and 2× cOmplete. Total cell or organ lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose (Schleicher & Schuell) by wet blotting. Blots were analyzed as described previously.49
Immunofluorescence of cremaster whole mounts
Mice received intraperitoneal injections of 100 µg anti-Ly6G/Gr-1 antibodies to deplete neutrophils or anti-rat control immunoglobulin G (IgG) 24 hours before whole mount stainings of the cremaster muscle were prepared. Local inflammation was induced by intrascrotal injection of 50 ng of IL-1β (Biomol). Teklox/lox mice, TekiECKO mice, or in vivo siRNA-transfected mice were stimulated for 3 hours, Angpt1lox/lox mice, Angpt1PltKO mice, or platelet-depleted mice were stimulated for 4 hours. Platelet depletion was achieved by IV injection of anti-GPIbα antibodies (R300) at 2 µg/g body weight within 1 hour. Then, mice were injected IV with 0.02 µm FluoSpheres fluorescent microspheres (crimson-labeled; Invitrogen) and sacrificed 5 minutes later. To analyze the blockade of VWF, C57BL/6 mice received 50 µg of anti-VWF or control rabbit IgG antibodies IV 5 minutes prior to IL-1β injection. The cremaster muscle was dissected and left in situ for initial fixation for 10 minutes with 4% paraformaldehyde solution in phosphate-buffered saline (PBS) before it was prepared and postfixed for 1 hour in 4% paraformaldehyde solution in PBS, blocked, permeabilized in 2% ovalbumin and 0.5% Triton X-100-PBS, and incubated with primary antibodies against MRP14 and Alexa Fluor 555–coupled anti–PECAM-1 antibodies. Donkey anti-goat Alexa Fluor 488 secondary antibodies were used to detect primary antibodies. Tissue was mounted in Dako fluorescent mounting medium, and Z-stack projections were acquired using a Zeiss LSM880 confocal microscope and analyzed with ImageJ and Imaris (Bitplane).
Statistical analysis
Data sets were tested for normality (Shapiro-Wilk) and equal variance. Statistical significance was analyzed using a 2-tailed Student t test, a Mann-Whitney rank test, 1-way analysis of variance (ANOVA) or 2-way ANOVA for independent samples. Tukey’s test was applied to correct for multiple comparisons. The ROUT method was used to identify outliers, with a maximum false discovery rate of 0.1%. GraphPad Prism 7 software was used for this analysis. Data are shown as mean ± standard error of the mean (SEM). Immunoblot signals were quantified using ImageJ.
Results
Interference with FGD5 expression induces leakiness during the extravasation of neutrophils in vitro and in vivo
We have previously demonstrated that the GEF FGD5, although not necessary for barrier integrity under basal conditions, is required for the mechanism by which VE-PTP inhibition/Tie-2 activation protect endothelial junction stability against the challenge of inflammatory mediators in vivo. In this setup, FGD5 was necessary for the dissolution of radial stress fibers, as well as the reinforcement of cortical actin.21 Since Heemskerk et al showed that during the transmigration of neutrophils, endothelial cells are protected from permeability induction by a contractile actin ring,9 we wanted to know whether FGD5 was required for this process. To this end, we compared simultaneously measured neutrophil transmigration and 40 kDa FITC-dextran leakage across TNF-α–stimulated HUVEC monolayers treated with control or FGD5 siRNA. We found that neutrophil transmigration did not affect permeability of control siRNA-treated cell layers but enhanced leakiness of FGD5-depleted monolayers by 50%, while increasing transmigration by only 18% (Figure 1A-B). Inhibition of FGD5 expression was confirmed by immunoblot analysis (Figure 1C).
Figure 1.
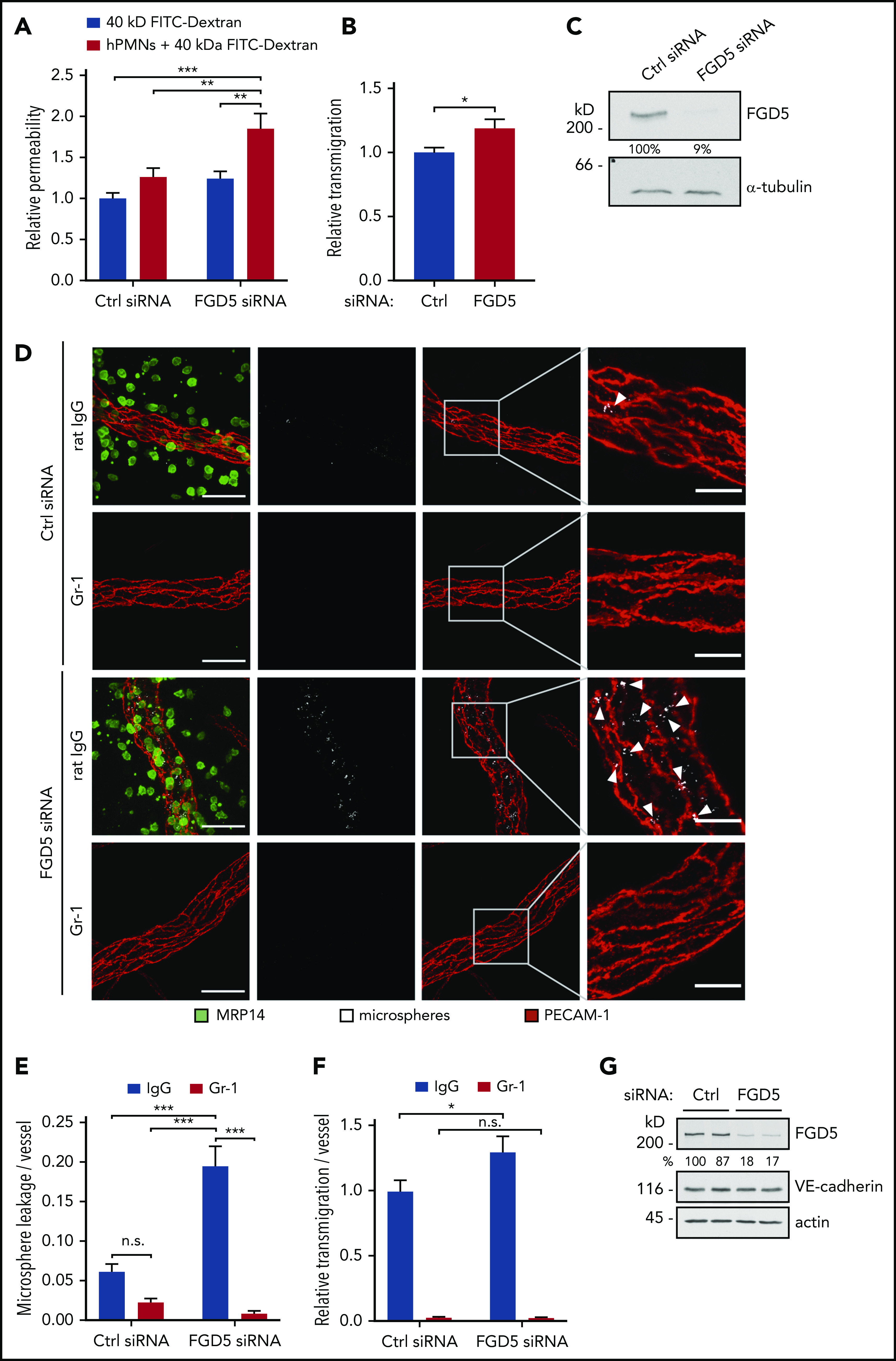
FGD5 is necessary to prevent neutrophil-induced leakiness in vitro and in vivo. (A) Paracellular permeability for 40 kDa FITC-dextran was determined for TNF-α–stimulated HUVEC that were pretreated with control (Ctrl) or FGD5 siRNA in the presence or absence of human neutrophils freshly isolated from whole blood (as indicated). Values were normalized to Ctrl siRNA monolayers without PMNs. (B) Relative transmigration of human PMNs across HUVEC monolayers treated with Ctrl or FGD5 siRNA. (C) HUVEC transfected with Ctrl or FGD5-targeting siRNA were immunoblotted for FGD5 and α-tubulin. (D) Mice were injected intrascrotally with FGD5 or Ctrl siRNA and received anti–Gr-1 antibodies or control IgG intraperitoneally the following day to deplete neutrophils. Twenty-four hours later, mice were stimulated with IL-1β for 3 hours before fluorescent microspheres were injected IV, and mice were euthanized 5 minutes later. Whole mounts of the cremaster muscle were stained with antibodies against PECAM-1 and MRP14. Arrowheads indicate microsphere leakage. Scale bars, 40 µm (left panels), 15 µm (right panels). Quantification of microspheres (E) and extravasated neutrophils (F) per vessel area. (G) Total lung lysates of mice treated with Ctrl siRNA or FGD5 siRNA were immunoblotted for the indicated antigens. Data are mean ± SEM. Results are representative of (C,D,G) or pooled from 4 independent experiments (A-B) or pooled from 4 independent experiments, with a total of 35 to 40 vessels analyzed per condition (E-F). *P < .05, **P < .01, ***P < .001, Student t test (B), 2-way ANOVA (A,E-F). n.s., not significant.
To validate these findings in vivo, we treated C57BL/6 mice with control or FGD5 siRNA complexed with a polyethylenimine-based transfection reagent for 48 hours, which resulted in efficient FGD5 knockdown in total lung lysates (Figure 1G) and the endothelium of venules in the cremaster muscle, as shown by whole mount staining (supplemental Figure 1, available on the Blood Web site). Local inflammation in the cremaster was induced by IL-1β stimulation for 3 hours, and fluorescent microspheres were injected IV to mark sites of vascular leakiness. To specifically quantify the leakiness induced by extravasating neutrophils and to distinguish it from the effect of IL-1β stimulation, a control condition was included, in which neutrophils were depleted from the circulation by injection of anti–Gr-1 antibodies. Whole mounts of the cremaster muscle were stained with antibodies against PECAM-1 and MRP14 to visualize endothelial cells and neutrophils, respectively. In agreement with the in vitro results, only minor leakage of microspheres was observed in the cremaster of control siRNA–treated mice. In sharp contrast to this, interference with FGD5 expression induced microsphere leakage along vessels at numerous sites. Depletion of neutrophils revealed that this leakiness was indeed induced during the extravasation process, because microsphere leakage was not observed in Gr-1–injected mice (Figure 1D). Quantification demonstrated that FGD5 depletion resulted in a threefold increase in neutrophil-induced leakiness compared with control siRNA (Figure 1E), whereas counts of extravasated neutrophils in the analyzed images revealed only a slight increase (20%) upon FGD5 siRNA treatment (Figure 1F). Importantly, depletion of neutrophils by anti–Gr-1 injection blocked the permeability effects (Figure 1E), demonstrating that the leakage observed in FGD5 siRNA–treated mice is induced by transmigrating neutrophils.
Tie-2 mediates the protection against neutrophil-induced leakiness in vivo
Next, we sought to identify the receptor acting upstream of FGD5 to prevent neutrophil-induced leakiness during inflammation. We have previously shown that FGD5 is an essential downstream target by which Tie-2 counterregulates vascular permeability induced by inflammatory mediators, such as histamine or thrombin.21 Hence, we decided to investigate the role of Tie-2 in preventing neutrophil-induced leakage.
To this end, we first inhibited Tie-2 expression in vivo using the in vivo–silencing approach with siRNA established previously20 which resulted in an 80% decrease in Tie-2 protein expression in total lung lysates (Figure 2D). Upon intrascrotal injection of IL-1β, IV-administered fluorescent microspheres accumulated around inflamed vessels in Tie-2 siRNA–treated mice, but not in control siRNA–treated mice, marking the sites of fluid leakage (Figure 2A). Depletion of neutrophils from the circulation by anti–Gr-1 antibodies completely blocked the accumulation of microspheres, revealing that leakiness was induced by neutrophils during the transmigration process (Figure 2A-B). In contrast to microsphere leakage, which was increased threefold in Tie-2 siRNA–treated mice vs control siRNA–treated mice (Figure 2B), relative transmigration of neutrophils per vessel was only slightly and insignificantly different between the 2 groups (Figure 2C). However, we did observe some variance in the severity of microsphere leakiness within Tie-2 siRNA–treated mice and even between different vessels in the same animal, which is likely caused by unequal targeting of the vessels by Tie-2 siRNA.
Figure 2.
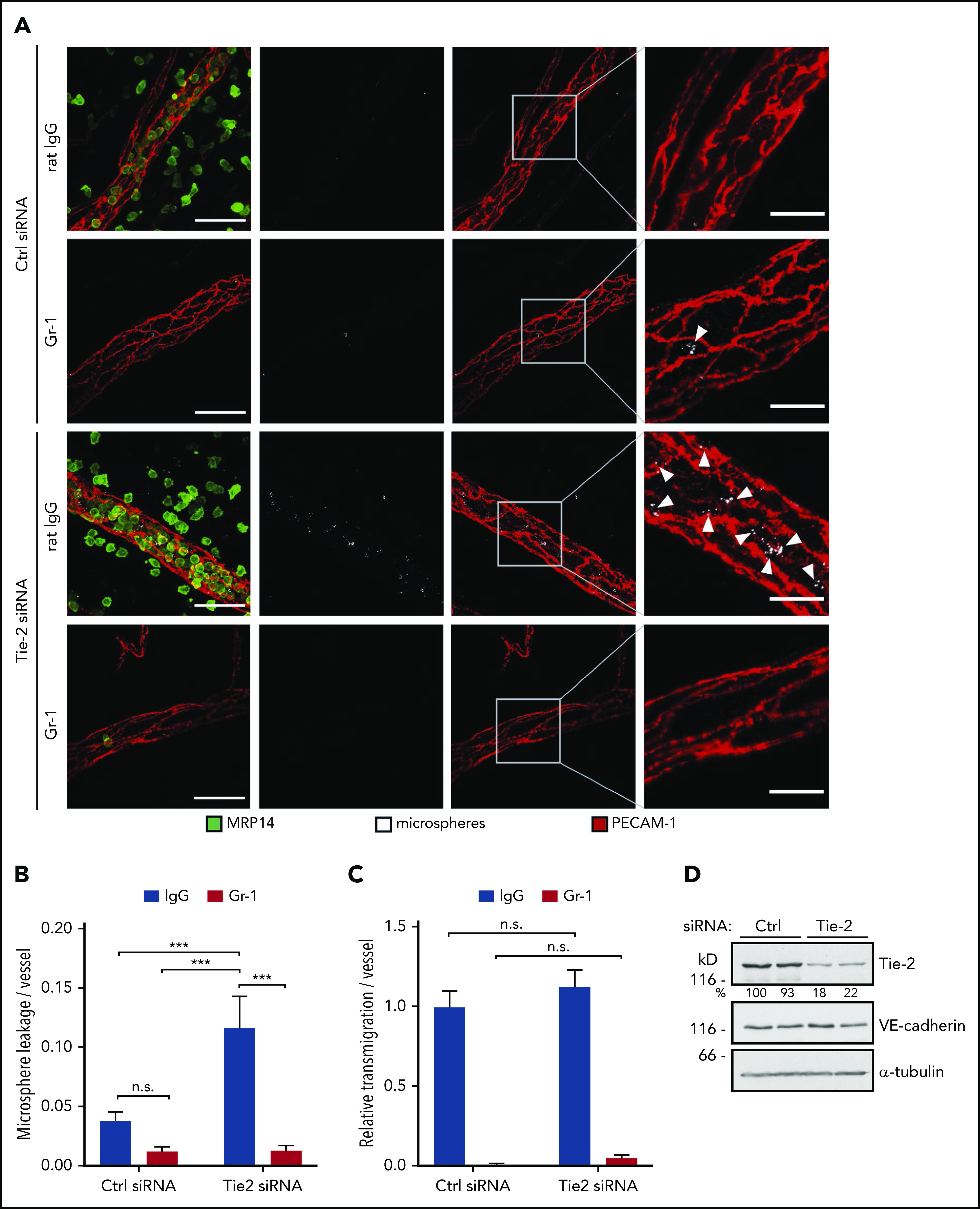
Tie-2 siRNA promotes neutrophil-dependent microsphere leakage from inflamed cremaster vessels. (A) Mice received control (Ctrl) siRNA or Tie-2 siRNA intrascrotally, 24 hours later they received intraperitoneal anti–Gr-1 antibodies or control IgG, and 24 hours thereafter they received IL-1β intrascrotally, followed by IV injection of fluorescent microspheres 3 hours later; mice were euthanized 5 minutes later. Whole mounts of the cremaster muscle were stained for PECAM-1 and MRP14. Arrowheads indicate microphere leakage. Scale bars, 40 µm (left panels), 15 µm (right panels). (B) Microsphere leakage and (C) relative neutrophil extravasation per vessel from experiments as in (A). (D) Total lung lysates from Ctrl siRNA–treated or Tie-2 siRNA–treated mice were analyzed by immunoblot for expression of Tie-2, VE-cadherin, and α-tubulin. Results are representative of (A,D) or pooled from (B-C) 3 independent experiments, with a total of 30 vessels analyzed per condition. Data are mean ± SEM. ***P < .001, 2-way ANOVA. n.s., not significant.
Therefore, we decided to analyze Tie-2–inducible endothelial-specific KO mice (TekiECKO) in similar assays. Intraperitoneal injections of tamoxifen on 5 consecutive days induced efficient inactivation of Tek in lung, heart, and skin, as estimated by western blot analysis of whole-organ lysates 3 days after the last dose (Figure 3A). In addition, Tie-2 staining was largely undetectable in cremasteric venules, confirming high KO efficiency in various tissues (Figure 3B).
Figure 3.
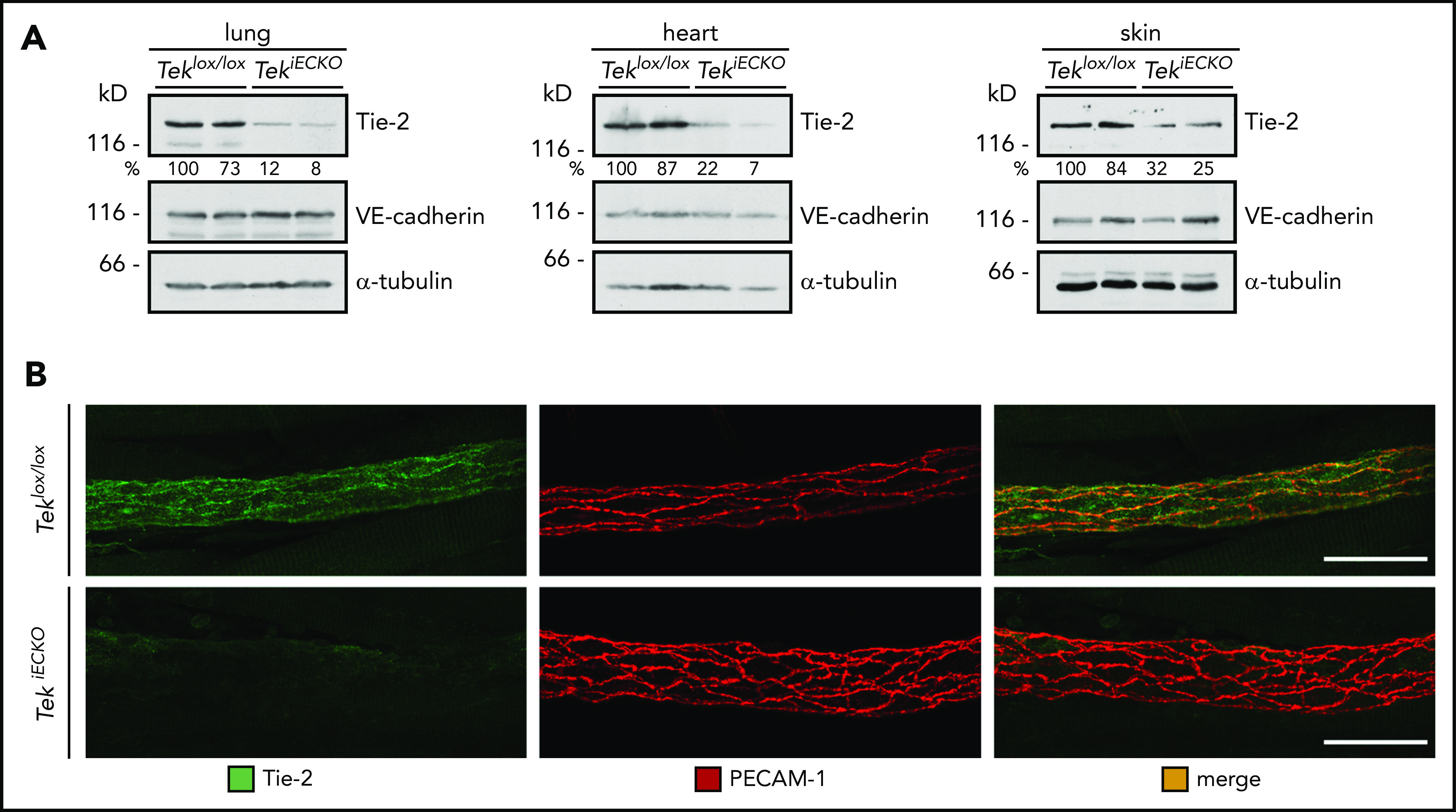
Conditional gene inactivation of Tek in mice. (A-B) Teklox/lox and TekiECKO mice were injected intraperitoneally with tamoxifen on 5 consecutive days and analyzed 3 days later. (A) Total lysates of lung, heart and skin were immunoblotted for Tie-2, VE-cadherin and α-tubulin. (B) Cremaster whole mounts were stained for Tie-2 and PECAM-1. Scale bars, 50 µm.
In line with the results obtained in Tie-2 siRNA–treated animals, leakage of microspheres from venules of the IL-1β–inflamed cremaster muscle was strongly induced in TekiECKO mice compared with Teklox/lox littermates, and it was completely blocked by depletion of neutrophils via anti–Gr-1 antibodies (Figure 4A-B). In fact, TekiECKO mice exhibited a fivefold greater vessel leakiness compared with Teklox/lox control mice, unambiguously demonstrating the relevance of Tie-2 for the maintenance of microvessel stability during neutrophil extravasation. Interestingly, the extravasation process itself did not seem to be affected by genetic inactivation of Tie-2, because no difference in the number of extravasated neutrophils around the vessels was detected between TekiECKO and Teklox/lox littermates (Figure 4C). Efficient inhibition of Tie-2 expression was confirmed by western blot analysis of lung lysates (Figure 4D).
Figure 4.
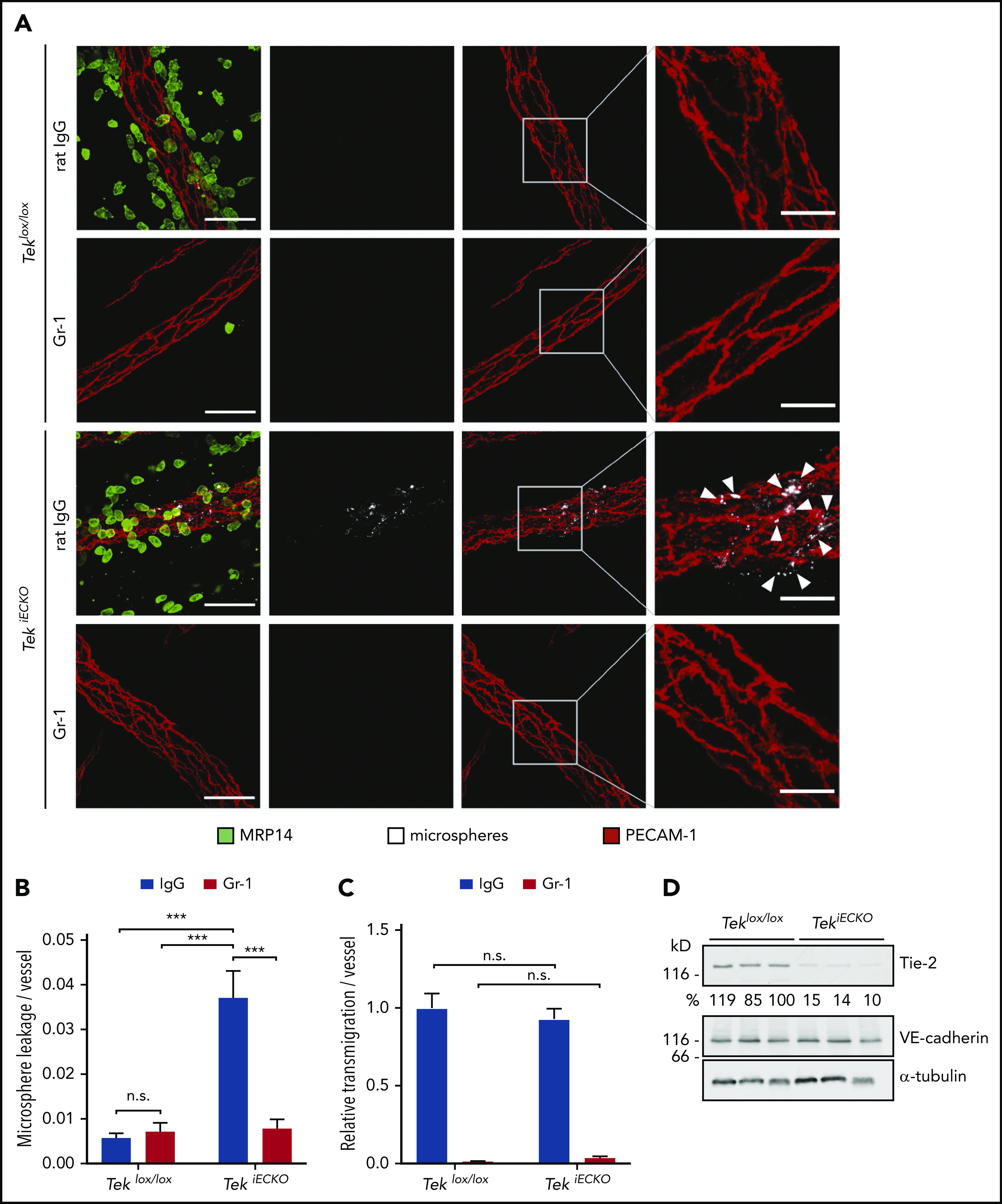
Conditional inactivation of Tie-2 in endothelial cells evokes neutrophil-dependent leakiness of microspheres. (A) Teklox/lox and TekiECKO mice received intraperitoneal injections of anti–Gr-1 or control IgG antibodies and were stimulated 24 hours later for 3 hours with IL-1β. Fluorescent microspheres were injected IV for 5 minutes, and cremaster whole mounts were stained for PECAM-1 and MRP14. Arrowheads indicate microsphere leakage. Scale bars, 40 µm (left panels), 15 µm (right panels). Quantification of microsphere leakage (B) and relative neutrophil extravasation (C) per vessel. (D) Total lung lysates of Teklox/lox and TekiECKO mice were analyzed by immunoblot for the indicated antigens. Results are representative of (A,D) or pooled from (B-C) 4 independent experiments, with 40 or 41 vessels analyzed. Data are mean ± SEM. ***P < .001, 2-way ANOVA. n.s., not significant.
Platelet-derived Angpt1 is required to prevent neutrophil-induced leakiness
The major known agonist of Tie-2 is Angpt1, which is provided by different cell types, among them pericytes and platelets.50-52 Because platelets are known to be present at sites of leukocyte exit and support the leukocyte-extravasation process,26-28,31 we decided to test whether platelet-specific gene inactivation of Angpt1 would impair leak protection in neutrophil extravasation. To this end, we mated Angpt1lox/lox mice with PF4-Cre mice to obtain transgenic animals specifically lacking Angpt1 in platelets (Angpt1PltKO mice). These mice or Cre− littermates were subjected to the microsphere leakage assay in the inflamed cremaster, as described above. Although neutrophil accumulation around venules was not altered in Angpt1PltKO mice vs Angpt1lox/lox mice (Figure 5A,C), the leakage of microspheres was markedly increased in KO animals and only occurred if neutrophils were not depleted (Figure 5A-B). Similar results as for microspheres were found for leakage of 40 kDa FITC-dextran (supplemental Figure 2). Successful depletion of Angpt1 in platelets isolated from Angpt1PltKO mice was controlled by an enzyme-linked immunosorbent assay (Figure 5D). Thus, platelet-released Angpt1 is specifically required for Tie-2 activation during inflammation to prevent neutrophil-induced leakiness. In agreement with this, we found that platelet depletion with anti-GP1bα antibodies also increased microsphere leaks during neutrophil extravasation to a similar extent as gene deletion of Angpt1 in platelets (supplemental Figure 3).
Figure 5.
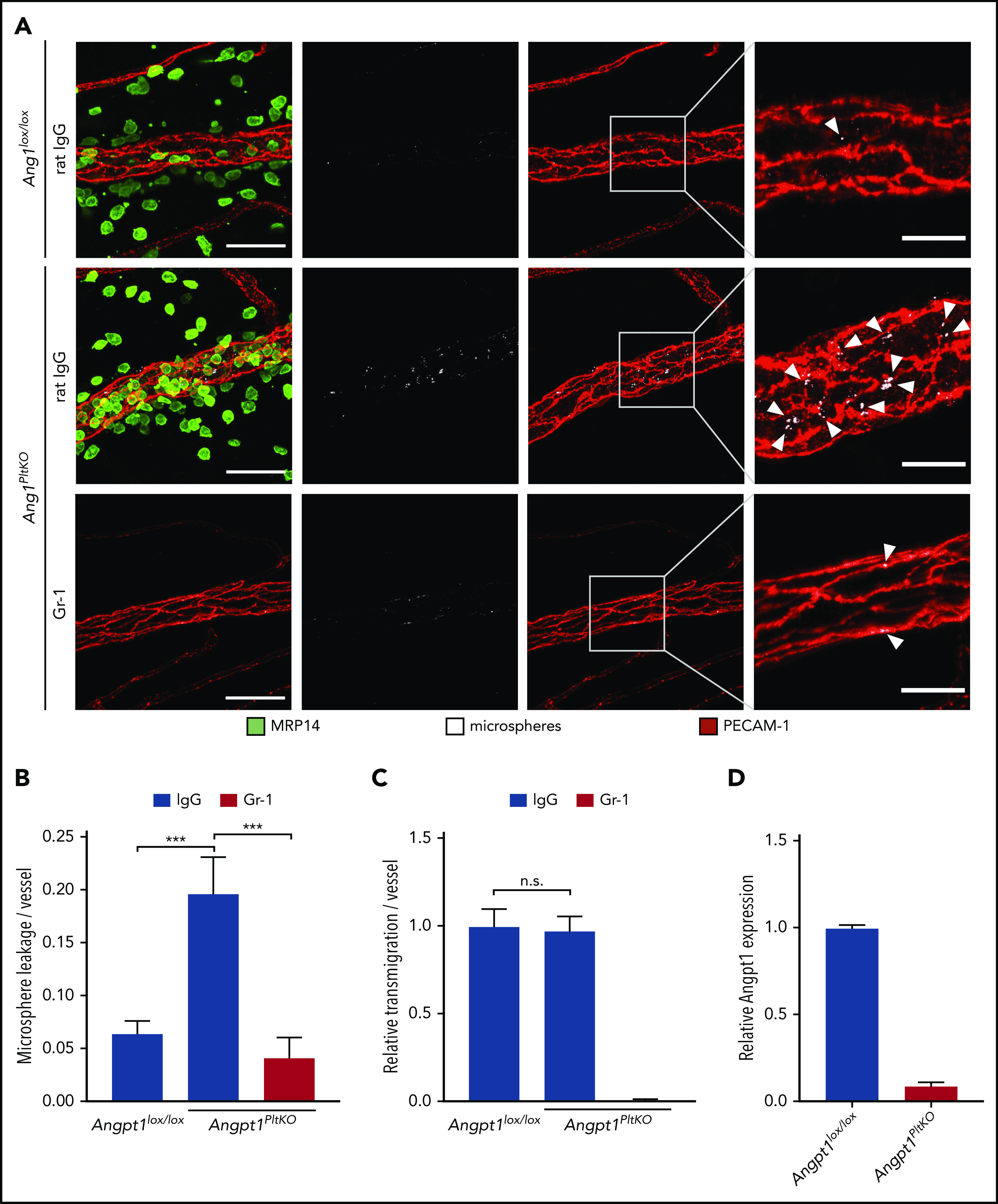
Platelet-derived Angpt1 prevents neutrophil-induced plasma leakage. (A-C) Angpt1lox/lox and Angpt1PltKO mice were injected intraperitoneally with anti–Gr-1 or control IgG antibodies to deplete neutrophils and were stimulated intrascrotally 24 hours later with IL-1β. Four hours later, fluorescent microspheres were injected IV, and cremaster whole mounts were prepared and stained for PECAM-1 and MRP14 5 minutes later. Arrowheads indicate microsphere leakage. Scale bars, 40 µm (left panels), 15 µm (right panels). Microsphere leakage (B) and transmigrated neutrophils (C) per vessel were quantified. (D) Lysates of platelets isolated from Angpt1lox/lox or Angpt1PltKO mice were submitted to an Angpt1 enzyme-linked immunosorbent assay (Bosterbio), according to the manufacturer’s instructions, to measure Angpt1 content. Results are representative of 5 (A) or are pooled from 3 (D) or 5 (B-C) independent experiments, with 50 vessels analyzed. Data are mean ± SEM. ***P < .001, 1-way ANOVA. n.s., not significant.
Blocking VWF prevents protection against diapedesis-induced leaks
It was reported that platelets contribute to systemic Angpt1 levels in the circulation.38 On the other hand, platelets are recruited to extravasation sites. Depending on the inflammatory stimulus, platelets are binding to neutrophils25,26 or to VWF on endothelial cells.27,31 We found that platelet binding to endothelium correlated with leukocyte extravasation (single timeframe in Figure 6A; supplemental Video 1). Eighty percent of all extravasating neutrophils transmigrated at sites of platelet binding (Figure 6B), and the 2 events correlated well (Figure 6C). To determine whether these locally recruited platelets, and not the circulating ones, provide Angpt1 to prevent leaks, we injected VWF-blocking antibodies IV 5 minutes prior to the induction of local inflammation in the cremaster by IL-1β stimulation, followed by IV administration of fluorescent microspheres after 4 hours and preparation of the cremaster muscle of euthanized mice 5 minutes later. Strikingly, VWF blockade increased the leakage of microspheres from the inflamed vessels by more than fourfold (Figure 7A-B). At the same time, neutrophil transmigration was reduced by 33% (Figure 7C), similar to what we have found previously.27 This indicates that, although less transmigration takes place, which should clearly reduce the challenge for endothelial junctions, the lack of VWF-bound platelets strongly impairs sealing of the diapedesis pore, leading to substantial leakiness.
Figure 6.
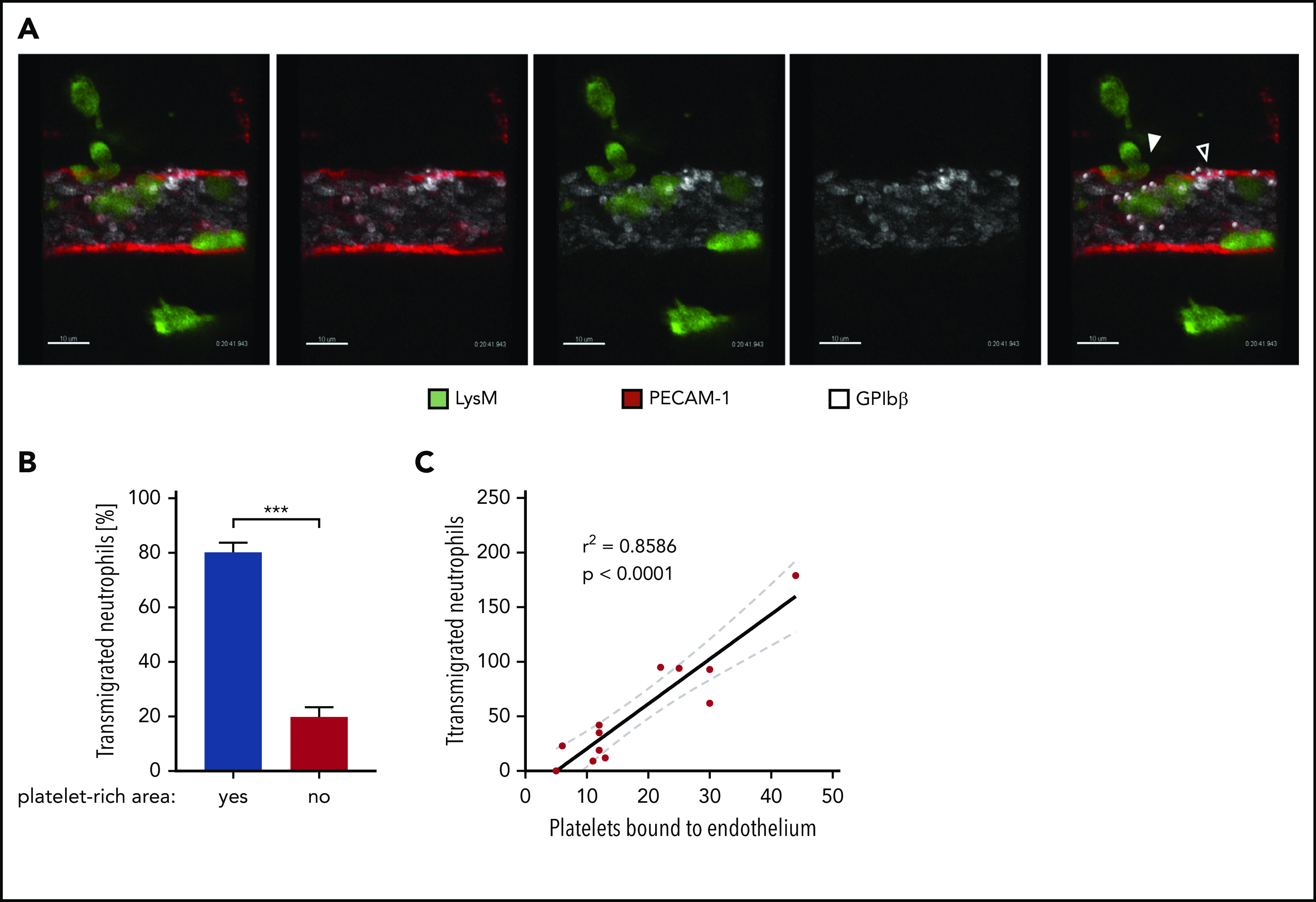
Preferential extravasation of neutrophils in platelet-rich areas of cremasteric venules. (A) Confocal intravital microscopy of cremasteric venules in LysM-eGFP mice that were injected intrascrotally with IL-1β. Endothelial cells and platelets were labeled through IV injection of fluorophore-coupled anti–PECAM-1 and anti–GPIbβ antibodies, respectively. Mice were anaesthetized, the cremaster muscle was prepared for intravital imaging, and videos were taken for 1 to 1.5 hours. White dots mark platelets bound to the endothelium, the filled arrowhead marks areas of neutrophil extravasation, and the open arrowhead marks areas of extravasation (supplemental Video 1). Scale bars, 10 µm. The timestamp of the video is 0:20:41:943. (B) Quantification of neutrophil transmigration in platelet-rich and platelet-poor areas of cremasteric venules from 10 videos shown as the percentage of total transmigrated neutrophils. Data are mean ± SEM. (C) Correlation analysis (Pearson) and linear regression of transmigrated neutrophils and endothelium-bound platelets in videos, as described in (A). Results are representative of (A) or pooled from (B-C) 10 videos taken of 9 mice. ***P < .001, Mann-Whitney rank test.
Figure 7.

Blockade of endothelial VWF prevents microsphere leakage during neutrophil transmigration. (A) C57BL/6 mice received anti–Gr-1 antibodies or control (Ctrl) IgG and were injected IV with VWF-blocking or control antibodies 24 hours later. Five minutes later, local inflammation was induced by intrascrotal IL-1β injection. Fluorescent microspheres were injected IV 4 hours later, and cremaster whole mounts were prepared and stained for PECAM-1 and MRP14 5 minutes later. Arrowheads indicate microsphere leakage. Scale bars, 40 µm (left panels), 15 µm (right panels). Microsphere leakage (B) and neutrophil extravasation (C) in experiments as depicted in (A) were quantified and normalized to vessel surface. Results are representative of (A) or pooled from (B-C) 5 independent experiments, with 50 vessels analyzed. Data are mean ± SEM. *P < .05, ***P < .001, 1-way ANOVA.
Discussion
Leukocyte extravasation requires the opening of large gaps between endothelial cells, yet vascular leaks of plasma content are prevented. This is because an actomyosin-based contractile system confines the diapedesis pore, which prevents the passage of macromolecules.9 The present study establishes the stimulus and the receptor system that trigger this protective mechanism. Platelets are recruited to endothelial VWF released at sites of leukocyte extravasation and provide Angpt1 to activate Tie-2 and its downstream target FGD5 in endothelial cells. Together with our recent findings that Tie-2 and FGD5 reinforce endothelial cortical actin and junction stability,21 our results establish an essential receptor signaling mechanism for the stimulation of a contractile actin ring around the diapedesis pore that was described previously.9
Based on platelet-specific gene inactivation of Angpt1, we could identify platelets as the source of Angpt1 needed for the protection of the endothelial leukocyte diapedesis pore against leakage. To distinguish whether the platelet-derived systemic level of Angpt1 in the circulation or locally secreted Angpt1 at the leukocyte extravasation site was responsible, we blocked the docking of platelets to endothelium at such sites. We and other investigators have shown that platelets are recruited by binding to endothelial VWF, which becomes exposed to the apical endothelial surface at sites of inflammation.27,31 Blocking of this interaction with antibodies was shown to prevent platelets from contributing to the efficiency of the diapedesis process.27,31 Here, we show that anti-VWF antibodies also prevent platelets from sealing the leukocyte diapedesis pore. Collectively, these results indicate that Angpt1 molecules that trigger diapedesis pore confinement are provided by locally recruited platelets.
We believe that this novel function of platelets, diapedesis pore confinement, is different from the well-described protective function against inflammatory bleeding. It is known that thrombocytopenia in patients and in platelet-depleted mice causes hemorrhage at sites of neutrophil extravasation.33,34,53 Thus, platelets somehow prevent the exit of erythrocytes at such sites. Platelet receptors mediating this function are the collagen receptor GPVI and the podoplanin receptor CLEC2.53 It was excluded that platelet secretion contributes to this effect, clearly arguing against a role for Angpt1 in protecting against inflammatory bleeding.36 Instead, single platelets were suggested to plug sites of neutrophil extravasation by a mechanism that required GPVI.37 Clearly, this is an interaction that only occurs if binding sites, such as collagen, become accessible to platelets. This is unlikely to occur while a neutrophil is still transmigrating; it probably happens when complete closure of the diapedesis pore fails to occur or is delayed while the transmigrating neutrophil has already largely disappeared, allowing platelets access to the vascular basement membrane.
Stimulation of Tie-2 is well known to protect against permeability induction by soluble inflammatory mediators.14,17 Mechanistically, it was shown that p190RhoGAP and Rac1 were involved in these junction-stabilizing effects.54,55 All of these studies investigated junction-destabilizing factors, independent of the leukocyte-extravasation process. We showed recently that stimulation of Tie-2 can even stabilize junctions when VE-cadherin has been removed by gene inactivation.20 This was a strong indication that Tie-2 can stabilize junctions in vivo by mechanisms that are not primarily based on the regulation of VE-cadherin, but rather on the regulation of indirect mechanisms, such as cortical actin. In agreement with this, we then found that the Cdc42 GEF FGD5 was an essential downstream signaling target by which Tie-2 stabilizes endothelial junctions in vivo.21 FGD5 was needed for Tie-2–stimulated Cdc42 and Rac1 activation, which supported reinforcement of cortical actin and dampening of radial stress fibers.21 Thus, Tie-2 signaling is highly suitable for the tightening of junctions that are devoid of homophilic adhesion mechanisms because of the presence of a transmigrating leukocyte en route through the diapedesis pore. Formation and confinement of a cortical actin ring are the bases for leak prevention during neutrophil diapedesis, as was reported by Heemskerk et al.9 They found that RhoA, as well as the GEFs Ect2 and LARG, are required to minimize passive plasma leakage during neutrophil transmigration.9 How Tie-2/FGD5–stimulated mechanisms relate to the components identified in that study is not clear and will be interesting to determine in the future. It is possible that both GTPase regulatory mechanisms act independently, while each is necessary. The 2 mechanisms may even be required during different stages of the transmigration process, such as restriction of the pore size during early and mid-diapedesis, or during the actual closure of the pore at a late stage when the largest part of the neutrophil body has already moved through the endothelial barrier.
In summary, we provide evidence for how sites of neutrophil extravasation are protected from passive plasma leaks: platelets recruited to endothelial VWF release Angpt1 to activate endothelial Tie-2, which reinforces cortical actin via FGD5. Our results imply that 2 factors already recognized for their ability to modulate vascular barrier function, endothelial Tie-2 signaling and platelets, are orchestrating the decoupling of neutrophil extravasation and passive fluid leakage.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Sean Morrison for generously providing Angpt1lox/lox mice.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB1009, A1; KFO342, A2) (D.V.).
Footnotes
Contact the corresponding author for original data.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.V. and L.J.B. designed the study and wrote and revised the manuscript; L.J.B., R.I.S., K.S., B.K., S.M.C., and A.F.N. performed experiments; S.V. developed essential tools for microscopy; and L.J.B. analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dietmar Vestweber, Max Planck Institute for Molecular Biomedicine, Röntgenstr. 20, D-48149 Münster, Germany; e-mail: vestweb@mpi-muenster.mpg.de.
REFERENCES
- 1.Carden DL, Smith JK, Korthuis RJ. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ Res. 1990;66(5):1436-1444. [DOI] [PubMed] [Google Scholar]
- 2.Kubes P, Suzuki M, Granger DN. Modulation of PAF-induced leukocyte adherence and increased microvascular permeability. Am J Physiol. 1990;259(5 Pt 1):G859-G864. [DOI] [PubMed] [Google Scholar]
- 3.Sumagin R, Lomakina E, Sarelius IH. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circ Physiol. 2008;295(3):H969-H977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finsterbusch M, Voisin MB, Beyrau M, Williams TJ, Nourshargh S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J Exp Med. 2014;211(7):1307-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baluk P, Bolton P, Hirata A, Thurston G, McDonald DM. Endothelial gaps and adherent leukocytes in allergen-induced early- and late-phase plasma leakage in rat airways. Am J Pathol. 1998;152(6):1463-1476. [PMC free article] [PubMed] [Google Scholar]
- 7.Kim MH, Curry FR, Simon SI. Dynamics of neutrophil extravasation and vascular permeability are uncoupled during aseptic cutaneous wounding. Am J Physiol Cell Physiol. 2009;296(4):C848-C856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wessel F, Winderlich M, Holm M, et al. . Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15(3):223-230. [DOI] [PubMed] [Google Scholar]
- 9.Heemskerk N, Schimmel L, Oort C, et al. . F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7(1):10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumont DJ, Gradwohl G, Fong GH, et al. . Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8(16):1897-1909. [DOI] [PubMed] [Google Scholar]
- 11.Sato TN, Tozawa Y, Deutsch U, et al. . Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376(6535):70-74. [DOI] [PubMed] [Google Scholar]
- 12.Fukuhara S, Sako K, Minami T, et al. . Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol. 2008;10(5):513-526. [DOI] [PubMed] [Google Scholar]
- 13.Saharinen P, Eklund L, Miettinen J, et al. . Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol. 2008;10(5):527-537. [DOI] [PubMed] [Google Scholar]
- 14.Gamble JR, Drew J, Trezise L, et al. . Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. 2000;87(7):603-607. [DOI] [PubMed] [Google Scholar]
- 15.Pizurki L, Zhou Z, Glynos K, Roussos C, Papapetropoulos A. Angiopoietin-1 inhibits endothelial permeability, neutrophil adherence and IL-8 production. Br J Pharmacol. 2003;139(2):329-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baffert F, Le T, Thurston G, McDonald DM. Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol. 2006;290(1):H107-H118. [DOI] [PubMed] [Google Scholar]
- 17.Thurston G, Rudge JS, Ioffe E, et al. . Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460-463. [DOI] [PubMed] [Google Scholar]
- 18.Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation. 2005;111(1):97-105. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, Frye M, Lee BL, et al. . Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. 2014;124(10):4564-4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frye M, Dierkes M, Küppers V, et al. . Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. 2015;212(13):2267-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun LJ, Zinnhardt M, Vockel M, Drexler HC, Peters K, Vestweber D. VE-PTP inhibition stabilizes endothelial junctions by activating FGD5. EMBO Rep. 2019;20(7):e47046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114(3):449-458. [DOI] [PubMed] [Google Scholar]
- 23.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123(18):2759-2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gros A, Ollivier V, Ho-Tin-Noé B. Platelets in inflammation: regulation of leukocyte activities and vascular repair. Front Immunol. 2015;5:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116(12):3211-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hara T, Shimizu K, Ogawa F, et al. . Platelets control leukocyte recruitment in a murine model of cutaneous arthus reaction. Am J Pathol. 2010;176(1):259-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petri B, Broermann A, Li H, et al. . von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116(22):4712-4719. [DOI] [PubMed] [Google Scholar]
- 28.Tamagawa-Mineoka R, Katoh N, Ueda E, Takenaka H, Kita M, Kishimoto S. The role of platelets in leukocyte recruitment in chronic contact hypersensitivity induced by repeated elicitation. Am J Pathol. 2007;170(6):2019-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562-570. [DOI] [PubMed] [Google Scholar]
- 30.Goerge T, Kleinerüschkamp F, Barg A, et al. . Microfluidic reveals generation of platelet-strings on tumor-activated endothelium. Thromb Haemost. 2007;98(2):283-286. [PubMed] [Google Scholar]
- 31.Zuchtriegel G, Uhl B, Puhr-Westerheide D, et al. . Platelets guide leukocytes to their sites of extravasation. PLoS Biol. 2016;14(5):e1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cloutier N, Paré A, Farndale RW, et al. . Platelets can enhance vascular permeability. Blood. 2012;120(6):1334-1343. [DOI] [PubMed] [Google Scholar]
- 33.Goerge T, Ho-Tin-Noe B, Carbo C, et al. . Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111(10):4958-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hillgruber C, Pöppelmann B, Weishaupt C, et al. . Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med. 2015;212(8):1255-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho-Tin-Noé B, Boulaftali Y, Camerer E. Platelets and vascular integrity: how platelets prevent bleeding in inflammation. Blood. 2018;131(3):277-288. [DOI] [PubMed] [Google Scholar]
- 36.Deppermann C, Kraft P, Volz J, et al. . Platelet secretion is crucial to prevent bleeding in the ischemic brain but not in the inflamed skin or lung in mice. Blood. 2017;129(12):1702-1706. [DOI] [PubMed] [Google Scholar]
- 37.Gros A, Syvannarath V, Lamrani L, et al. . Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood. 2015;126(8):1017-1026. [DOI] [PubMed] [Google Scholar]
- 38.Ho-Tin-Noé B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68(16):6851-6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins SJ, Purcell LA, Silver KL, et al. . Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med. 2016;8(358):358ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46(2):74-80. [DOI] [PubMed] [Google Scholar]
- 41.Zhou BO, Ding L, Morrison SJ. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting angiopoietin-1. eLife. 2015;4:e05521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109(4):1503-1506. [DOI] [PubMed] [Google Scholar]
- 43.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96(2):719-726. [PubMed] [Google Scholar]
- 44.Nawroth R, Poell G, Ranft A, et al. . VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 2002;21(18):4885-4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broermann A, Winderlich M, Block H, et al. . Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med. 2011;208(12):2393-2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wegmann F, Petri B, Khandoga AG, et al. . ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J Exp Med. 2006;203(7):1671-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gotsch U, Borges E, Bosse R, et al. . VE-cadherin antibody accelerates neutrophil recruitment in vivo. J Cell Sci. 1997;110(Pt 5):583-588. [DOI] [PubMed] [Google Scholar]
- 48.Koblizek TI, Runting AS, Stacker SA, Wilks AF, Risau W, Deutsch U. Tie2 receptor expression and phosphorylation in cultured cells and mouse tissues. Eur J Biochem. 1997;244(3):774-779. [DOI] [PubMed] [Google Scholar]
- 49.Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275(36):27979-27988. [DOI] [PubMed] [Google Scholar]
- 50.Davis S, Aldrich TH, Jones PF, et al. . Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87(7):1161-1169. [DOI] [PubMed] [Google Scholar]
- 51.Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest. 2002;82(4):387-401. [DOI] [PubMed] [Google Scholar]
- 52.Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost. 2001;85(2):204-206. [PubMed] [Google Scholar]
- 53.Boulaftali Y, Hess PR, Getz TM, et al. . Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mammoto T, Parikh SM, Mammoto A, et al. . Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282(33):23910-23918. [DOI] [PubMed] [Google Scholar]
- 55.David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 requires IQ domain GTPase-activating protein 1 to activate Rac1 and promote endothelial barrier defense. Arterioscler Thromb Vasc Biol. 2011;31(11):2643-2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.