

Abstract
Aims
Coronavirus disease 2019 (COVID-19) has appeared in Wuhan, China but the fast transmission has led to its widespread prevalence in various countries, which has made it a global concern. Another concern is the lack of definitive treatment for this disease. The researchers tried different treatment options which are not specific. The current study aims to identify potential small molecule inhibitors against the main protease protein of SARS-CoV-2 by the computational approach.
Main methods
In this study, a virtual screening procedure employing docking of the two different datasets from the ZINC database, including 1615 FDA approved drugs and 4266 world approved drugs were used to identify new potential small molecule inhibitors for the newly released crystal structure of main protease protein of SARS-CoV-2. In the following to validate the docking result, molecular dynamics simulations were applied on selected ligands to identify the behavior and stability of them in the binding pocket of the main protease in 150 nanoseconds (ns). Furthermore, binding energy using the MMPBSA approach was also calculated.
Key findings
The result indicates that simeprevir (Hepatitis C virus NS3/4A protease inhibitor) and pyronaridine (antimalarial agent) could fit well to the binding pocket of the main protease and because of some other beneficial features including broad-spectrum antiviral properties and ADME profile, they might be a promising drug candidate for repurposing to the treatment of COVID-19.
Significance
Simeprevir and pyronaridine were selected by the combination of virtual screening and molecular dynamics simulation approaches as a potential candidate for treatment of COVID-19.
Keywords: COVID-19, Simeprevir, Repurpose, Virtual screening, Docking, Molecular dynamics simulation
1. Introduction
Novel coronavirus, designated as SARS-CoV-2, was first identified in December 2019 in Wuhan, China [1]. SARS-CoV-2 belongs to the Coronaviridae (CoV) family, enveloped positive-sense, single-stranded RNA viruses (+ssRNA) that are spread broadly among humans and other mammals which cause a wide range of infections from common cold symptoms to fatal disease like respiratory syndrome. In summery the pathogenesis of SARS-CoV-2 is like this: The envelope spike glycoprotein binds to the ACE2 as a receptor and after membrane fusion, the virus enters to the host cells, RNA genome initiates to translate into structural and non-structural proteins and in the same time the replication of viral genome begins. With the aim of endoplasmic reticulum, the vesicle of the new virus is formed and after fusion to the plasma membrane, the new virus will release. Concurrent the viral entrance to the host cell, its antigen will exposure to antigen presentation cells (APC) and then identified by cytotoxic T lymphocytes (CTLs). But reducing the number of CD4 and CD8 T cells in COVID-19 patients prevents T cell proliferation and activity. The down-regulation in the expression of the APC gene is also possible. Another immunopathological effect is cytokine storm which leads to extensive inflammatory response and multiple organ failure. The IFN-I pathway is also inhibited by this virus [2].
SARS-CoV-2 is considered distinct from two high pathogenic SARS-CoV and MERS-CoV which were responsible for Severe Acute Respiratory Syndrome in 2002 and Middle East Respiratory Syndrome in 2012 respectively [3]. The fatality rate of SARS-CoV-2 seems to be around 2% in China [4], which is much less than the fatality rate of SARS and MERS. It should be considered that most of the fatal cases are vulnerable people with a medical condition such as immunosuppression, diabetes, or heart disease. Apart from the mortality rate, the point that has made it the global concern is the efficient transmission from human-to-human, leading to its widespread outbreaks in many countries around the world [5].
Up to now, there is no FDA-approved or specific treatment for COVID-19. Clinical guidance of the World Health Organization (WHO) and Centers for Disease Control and Prevention (CDC), suggested a lot of guidance for treatment including prompt supportive care, oxygen therapy, boosting the immune system, and empiric antimicrobials (in case of sepsis) and others [6]. However, some investigational medication has been suggested for the treatment of COVID-19, such as remdesivir (adenosine triphosphate analog for the treatment of Ebola) or chloroquine (treatment of malaria) [7] and combined HIV protease inhibitor lopinavir-ritonavir [8] which are previously used to treat SARS and MERS-CoV but their efficacy for treatment of COVID-19 is still unclear and need further evaluation. However, finding a cure can still be a great help to the international community. Fig. 1 , indicates the pathogenicity and the treatment of SARS CoV-2.
Fig. 1.

Pathogenicity and probable treatment approaches of COVID-19.
(Virus picture from the Center for Disease Control and Prevention (CDC))
One way to overcome the virus replication is to considered essential proteins of the virus as an inhibitory target. One of the most critical proteins for transcription and replication is the main protease of the virus (also known as non-structural protein5, Nsp5), which cleaves the polyproteins into smaller fragments [9]. The main protease monomer has three domains, domain I (1–101), II (102–183) and III (200–306), and a long loop between domains II and III (184–199) with total 306 residues. The active site of the main protease is placed between domains I and II with catalytic pair residues Cys145 and His41 with four highly conserved subunits S1-S4.
S1 contains residue Phe 140, Ser 144, Cys 145, His 163, Gln 166, and conserved in all coronaviruses. S2 subunit surrounded by His 41, Ser 46, Met 49, Asn 51, Asn 52. S4 contains Phe 185, Gln 189, and Gln 192 which created an entirely hydrophobic environment in the active site [10,11]. Fig. 2 , represents the main protease of SARS-CoV-2 with colored marked for each domain and the place of the active site.
Fig. 2.

a. Cartoon and surface representation of the main protease of SARS-CoV-2 with colored marked for each domain and the place of the active site which is shown in the rectangle. Domain 1 in green, domain 2 in grey, domain 3 in orange, and long loop in yellow color. b. Closer view of the active site with its important residue and related subunit. S1 in cyan, S2 in sand, and S4 in magenta color. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In the current crisis, to achieve a fast and reliable drug for the treatment of COVID-19, we decided to rely on the repurposing concept [12] and consider available FDA and world approved drugs for use in this disease. In this regard, to find potential small molecule inhibitors virtual screening procedure, employing docking of a total of 5881 FDA and world approved drugs over the binding pocket of the main protease protein of SARS-CoV-2 has been done. Afterward, molecular dynamics simulation was performed to validate the docking result and further investigation of the behavior of two selected ligands in a dynamic environment. The binding energy using the MMPBSA approach was also calculated. The results of this study could be promising and may provide a new approach to combat the coronavirus outbreaks.
2. Methods
2.1. Virtual screening and molecular docking
To identify the compounds with the favorable interaction with the main protease of SARS-CoV-2, 1615 FDA-approved and 4266 world approved drugs were screened with molecular docking simulations over the binding pocket of the main protease of SARS-CoV-2. The newly released crystal structure of SARS-CoV-2 main protease was retrieved from protein data bank (www.rcsb.org) with PDB ID: 6LU7 [13]. AutoDockTools (ADT, Ver.1.5.6) [14] was used for preparing the input files and analyzing the result. For the preparation of protein input files, all water molecules, ligands, and ions were removed from the PDB file. Then polar hydrogens were added and the Kollman-united charge was used to calculate the partial atomic charge and the prepared file was saved in pdbqt format to use in the following steps.
3D structures of FDA and world approved drugs were downloaded from the ZINC database [15] in structure-data file (SDF) format which contains a total of 5881 compounds. Then OpenBabel (version 2.3.1) [16] was used to convert SDF to PDB format. Rotatable bonds and Gasteiger-Marsili charges were assigned to all ligands and saved in pdbqt for further docking process using AutoDock 4.2. A 50 × 50 × 50 Å (x, y, and z) grid box was centered on the protease binding pocket with 0.375 nm spacing for each dimension. AutoGrid 4.2 was used to prepare grid maps. Docking parameters were set as follows: the number of Lamarckian job = 40, initial population = 150, the maximum number of energy evaluation = 2.5 × 105, other parameters were set in their default value, and finally, docking was performed by AutoDock 4.2.
All docking results were sorted from the lowest to highest of the docking score. Docking procedures were done automatically by scripts written in-house. Also, docking validation was carried out using previously published methods [17] with re-docking of the co-crystal structure as an inhibitor in the main protease of SARS-CoV-2 with the above-mentioned parameters and values. Visualization of docking results has been done by Discovery Studio visualizer version 17.2 [18] and PyMol version 1.1evel [19]. The best complexes with the lowest docking score were used for further investigation as input files for molecular dynamics simulation.
2.2. Classical Molecular dynamics simulation
GROMACS package Version 2020.1 [20] was run on a high-performance Linux cluster to determine the behavior of selected ligands, simeprevir, and pyronaridine in complex with main protease protein of SARS-CoV-2 during the 150 ns. CGenFF web server [21] was used to generate the topology file of the two ligands. From the docking study, the best complex of ligand-protein with the lowest docking score and favorable interactions was selected as an input file for MD simulations. CHARMM36 (March 2019) was used as a force field. Dodecahedron shape box of water surrounded the complex with the TIP3P water model. To neutralize the net charge of the system, Na+, and Cl− counter ions substituted by water molecules. The steepest-descent algorithm with a tolerance of 1000 kJ/mol/nm was used for the energy minimization of the system.
The Van der Waals cutoff was 12 Å and periodic boundary conditions were assigned in all directions. After convergence, NVT ensemble MD simulation in 100 ps, and then the system went through NPT in 100 ps in a periodic boundary condition. Berendsen barostat and thermostat were used to keep the temperature and pressure constant at 300 K and 1 bar with a coupling time of τT = 0.1 ps, τp = 2 ps respectively.
The Particle mesh Ewald (PME) method was applied to calculate the long-range electrostatic interactions. The bond lengths were constrained using the LINCS algorithm. MD simulation run was repeated twice for 150 ns in constant temperature and pressure for both ligands and the results are reported on average. Finally, root mean square deviation (RMSD) of protein and ligand, root mean square fluctuation (RMSF), and the number of hydrogen bonds were analyzed from the final production run. The binding energy of the simeprevir and pyronaridine in complex with the main protease were calculated based on Molecular Mechanics – Poisson Boltzmann Surface Area (MM-PBSA) method using g_mmpbsa v2020.1 package by taking snapshots at every 100 ps from 150 ns MDs [22].
3. Result and discussion
3.1. Virtual screening and molecular docking
In an attempt to find potential treating for COVID-19 from available drugs, molecular docking simulation was performed over 1615 FDA approved and 4266 worlds approved drugs on the binding pocket of the main protease of the virus. Through the docking method, all compounds were compared with each other and the results were sorted from lowest to highest docking score. Each dataset was investigated separately. In FDA approved dataset, the top 25 compounds with lowest docking score were chosen for further investigation and all compounds were evaluated for their clinical applications. According to the reported by WHO and CDC, SARS-CoV-2 mostly affects the respiratory system and leading to symptoms such as fever, cough, and shortness of breath. Therefore, the compounds with specific effects on other systems like the nervous system and skin were omitted (perampanel, thiothixene, and ergotamine). Corticosteroid compounds were omitted because of the special alert by CDC about using them for treatment of viral pneumonia which has no effectiveness and possible harm. Some drugs were ignored because of their side effect (conivaptan and daunorubicin). As a result, after applying these filters there were 10 compounds left which are listed in Table 1 .
Table 1.
Potential compounds to treat the COVID-19 from FDA-approves dataset.
| No. | Drug IUPAC name | Structure | Docking score | Usage |
|---|---|---|---|---|
| 1 | Paclitaxel [(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-diacetyloxy-15-[(2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl]oxy-1,9-dihydroxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate |
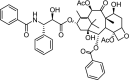 |
−12.31 | Kaposi's sarcoma, cancer of the lung, ovarian, and breast. |
| 2 | Simeprevir (1R,4R,6S,7Z,15R,17R)-N-cyclopropylsulfonyl-17-[7-methoxy-8-methyl-2-(4-propan-2-yl-1,3-thiazol-2-yl)quinolin-4-yl]oxy-13-methyl-2,14-dioxo-3,13-diazatricyclo[13.3.0.04,6]octadec-7-ene-4-carboxamide |
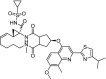 |
−11.33 | Hepatitis C virus (HCV) NS3/4A protease inhibitor |
| 3 | Docetaxel [(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4-acetyloxy-1,9,12-trihydroxy-15-[(2R,3S)-2-hydroxy-3-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]oxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate |
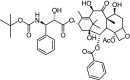 |
−10.64 | Breast, ovarian, and non-small cell lung cancer |
| 4 | Palbociclib 6-Acetyl-8-cyclopentyl-5-methyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrido[2,3-d]pyrimidin-7-one |
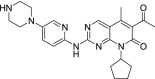 |
−10.62 | Breast cancer |
| 5 | Cabazitaxel [(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4-acetyloxy-1-hydroxy-15-[(2R,3S)-2-hydroxy-3-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]oxy-9,12-dimethoxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate |
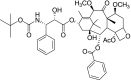 |
−10.53 | Prostate cancer |
| 6 | Alectinib 9-Ethyl-6,6-dimethyl-8-(4-morpholin-4-ylpiperidin-1-yl)-11-oxo-5H-benzo[b]carbazole-3‑carbonitrile |
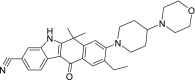 |
−10.49 | Non-small cell lung cancer |
| 7 | Imatinib 4-[(4-Methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide |
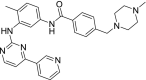 |
−10.36 | Chronic myelogenous leukemia (CML) |
| 8 | Plerixafor 1-[[4-(1,4,8,11-Tetrazacyclotetradec-1-ylmethyl)phenyl]methyl]-1,4,8,11-tetrazacyclotetradecane |
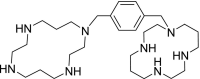 |
−10.15 | Non-Hodgkin lymphoma |
| 9 | Azelastine 4-[(4-Chlorophenyl)methyl]-2-(1-methylazepan-4-yl)phthalazin-1-one |
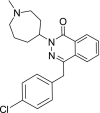 |
−9.98 | Allergic and vasomotor rhinitis |
| 10 | Dasabuvir N-[6-[3-tert-butyl-5-(2,4-dioxopyrimidin-1-yl)-2-methoxyphenyl]naphthalen-2-yl]methanesulfonamide |
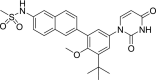 |
−9.76 | Chronic Hepatitis C |
All data retrieved from Drug Bank Databases www.drugbank.ca and https://pubchem.ncbi.nlm.nih.gov/.
It should be mentioned that the ideal compounds are those that could fit well to the binding site with the lowest docking score and favorable interactions. Base on Table 1 provided, paclitaxel indicates the significant affinity to the binding pocket of the main protease protein but the side effect of bone marrow suppression leads to a worsening of the immune system and the condition of the patient with COVID-19. The same can be said for the rest anticancer such as docetaxel, palbociclib, cabazitaxel, imatinib, alectinib, and plerixafor. Azelastine that is an antihistamine agent may useful for symptomatic treatment of shortness of breath, and if it can reach the main protease of the virus could inhibit the protease activity. Dasabuvir, used in combination therapy to treat chronic Hepatitis C, since it prevents polymerase to elongate viral RNA [23] therefore it may through the cell membrane and reach to the binding pocket of the main protease, also, it has no particular side effect and may effective. And the last but not the least, simeprevir, a directly acting antiviral agent (DAA) of the hepatitis C virus (HCV) as NS3/4A protease inhibitor, interacts with the main protease with a low docking score −11.33 which shows its significant affinity to interact with the receptor. The docking conformation of simeprevir indicates three hydrogen bonds with Asn 119, His 163, and Thr 26 and three sigma and pi interactions with the key binding site residues His 41, Cys 145, and Met 49 (Fig. 3 b–c).
Fig. 3.

a) Superimpose of simeprevir (magenta) and pyronaridine (cyan) in the binding pocket of the main protease. b) 3D and c) 2D display of the interactions of simeprevir in the binding pocket. d) 3D and e) 2D display of the interactions of pyronaridine in the binding pocket. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Interestingly, simeprevir identified in several other virtual screening. The screening results of Alamri et al. on the same receptor at the same time, but with different docking software, identified simeprevir as one of the potential inhibitors of the main protease [24]. A pharmacophore modeling and virtual screening on the drug bank dataset with maestro software also introduced the simeprevir as a potent inhibitor of the main protease of SARS-CoV-2 [25]. A comparative computational study of SARS-CoV-2 receptors antagonists by Oliveira et al. was described simeprevir as one of the most effective inhibitors, not only for the main protease but also, 3CL protease and NSP12 RNA polymerase [26]. A drug-target interaction (DTI) deep learning model to predicting available antiviral drugs against SARS-CoV-2 by beck et al., described simeprevir as a potential inhibitor on 3C-like proteinase, helicase and 3′-to-5′ exonuclease and endoRNAse [27]. Simeprevir is a broad-spectrum antiviral agent beyond hepatitis C. It can be used in patients with AIDS (Acquired immunodeficiency syndrome) [28] and other virus infections like HSV-1 (Herpes simplex type 1), ZIKV (Zika virus) and EV-A71 (Enterovirus A71) [29]. The HIV protease inhibitors lopinavir-ritonavir and darunavir, which have suggested as highly effective therapeutic agents against SARS-CoV-2 [8,25,[30], [31], [32]], showed less docking score −5.36, −5.04 and −7.49 respectively than simeprevir (Table 2 ).
Table 2.
Investigational treatment of COVID-19.
| No. | Drug IUPAC name |
Structure | Docking score | Usage |
|---|---|---|---|---|
| 1 | Lopinavir (2S)-N-[(2S,4S,5S)-5-[[2-(2,6-dimethylphenoxy)acetyl]amino]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl)butanamide |
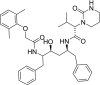 |
−5.36 | HIV protease inhibitor |
| 2 | Ritonavir 1,3-Thiazol-5-ylmethyl N-[(2S,3S,5S)-3-hydroxy-5-[[(2S)-3-methyl-2-[[methyl-[(2-propan-2-yl-1,3-thiazol-4-yl)methyl]carbamoyl]amino]butanoyl]amino]-1,6-diphenylhexan-2-yl]carbamate |
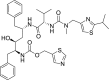 |
−5.04 | HIV protease inhibitor |
| 3 | Darunavir [(3aS,4R,6aR)-2,3,3a,4,5,6a-hexahydrofuro[2,3-b]furan-4-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate Create Date: 2005-06-24 |
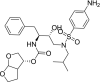 |
−7.49 | HIV protease inhibitor |
| 4 | Chloroquine 4-N-(7-chloroquinolin-4-yl)-1-N,1-N-diethylpentane-1,4-diamine |
 |
−7.5 | Antimalarial agent |
| 5 | Hydroxychloroquine 2-[4-[(7-Chloroquinolin-4-yl)amino]pentyl-ethylamino]ethanol |
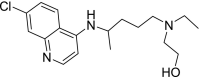 |
−6.7 | Rheumatoid arthritis |
| 6 | Captopril (2S)-1-[(2S)-2-methyl-3-sulfanylpropanoyl]pyrrolidine-2-carboxylic acid |
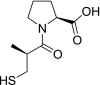 |
−4.22 | Hypertension |
| 7 | Enalapril (2S)-1-[(2S)-2-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]propanoyl]pyrrolidine-2-carboxylic acid |
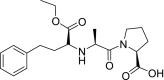 |
−5.6 | Hypertension |
| 8 | Remdesivir 2-Ethylbutyl (2S)-2-[[[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate |
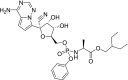 |
−5.8 | Antiviral |
All data retrieved from Drug Bank Databases www.drugbank.ca and https://pubchem.ncbi.nlm.nih.gov/.
Chloroquine and hydroxychloroquine used to treat malaria, systemic lupus erythematosus, rheumatoid arthritis, and Q fever. And through increasing the endosomal pH effects on the SARS-CoV-2 and disrupt virus/cell fusion [7,33]. But, the docking score of these drugs −7.5 and −6.7 respectively indicates that they do not interact effectively with the main protease. Captopril and enalapril can inhibit the binding between the SARS-CoV-2 and human ACE2, and reduces symptoms of severe pneumonia [34] but based on the obtained docking score −4.22 and −5.6 respectively, these two drugs are not able to interact appropriately with the mentioned target.
Subsequently, the top 25 compounds of virtual screening of world approved drug datasets, were investigated. The results were analyzed with the same logic in excluding the FDA-approved drugs, like the affected system and side effects. For example, thioproperazine, N-desmethyleletriptan, ergoloid, sulforidazine, and other compounds, were excluded from the study because of their particular effects on the nervous system. Testosterone, nandrolone, and abiraterone also omitted. The rest of the compounds listed in Table 3 .
Table 3.
Potential compounds to treat the COVID-19 from world approves dataset.
| No. | Drug IUPAC name |
Structure | Docking score | Usage |
|---|---|---|---|---|
| 1 | Pyronaridine 4-[(7-Chloro-2-methoxybenzo[b][1,5]naphthyridin-10-yl)amino]-2,6-bis(pyrrolidin-1-ylmethyl)phenol |
 |
−10.9 | Antimalaria |
| 2 | Rupatadine 13-Chloro-2-[1-[(5-methylpyridin-3-yl)methyl]piperidin-4-ylidene]-4-azatricyclo[9.4.0.03,8]pentadeca-1(11),3(8),4,6,12,14-hexaene |
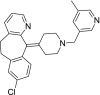 |
−10.06 | Antihistamine |
| 3 | Clofazimine N,5-bis(4-chlorophenyl)-3-propan-2-yliminophenazin-2-amine |
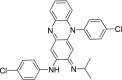 |
−9.66 | antileprotic |
| 4 | Tasosartan 2,4-Dimethyl-8-[[4-[2-(2H-tetrazol-5-yl)phenyl]phenyl]methyl]-5,6-dihydropyrido[2,3-d]pyrimidin-7-one |
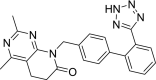 |
−9.65 | Antihypertensive |
| 5 | Encorafenib Methyl N-[(2S)-1-[[4-[3-[5-chloro-2-fluoro-3-(methanesulfonamido)phenyl]-1-propan-2-ylpyrazol-4-yl]pyrimidin-2-yl]amino]propan-2-yl]carbamate |
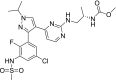 |
−9.47 | Anticancer |
| 6 |
N-Desmethyl imatinib N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]-4-(piperazin-1-ylmethyl)benzamide |
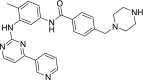 |
−9.42 | Anticancer |
| 7 | Mizolastine 2-[[1-[1-[(4-Fluorophenyl)methyl]benzimidazol-2-yl]piperidin-4-yl]-methylamino]-1H-pyrimidin-6-one |
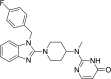 |
−9.37 |
Antihistamine |
| 8 | Desmethylazelastine 2-(Azepan-4-yl)-4-[(4-chlorophenyl)methyl]phthalazin-1-one |
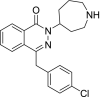 |
−9.34 | Antihistamine |
All data retrieved from Drug Bank Databases www.drugbank.ca and https://pubchem.ncbi.nlm.nih.gov/.
The kinase inhibitors to treat cancer, encorafenib and N-Desmethyl imatinib indicate high binding affinity to the active site, but due to their unintended side effects they are not an ideal option for treatment of COVID-19.
Regarding tasosartan, probably because of its effect on the angiotensin II (Ang II) receptor and blood pressure, it couldn't be the right choice. The antihistamine agents, rupatadine, mizolastine, and desmethylazelastine could be helpful as symptomatic treatment of shortness of breath. Clofazimine, which used as anti-mycobacterium, anti-trypanosomal [35], and anti-cryptosporidium parvum agent [36] may reach the target because of its solubility in fat, and show its inhibitory effect. Pyronaridine, which indicates a high affinity with the lowest docking score −10.9 to the main protease. Fig. 3d–e shows that pyronaridine involve in two hydrogen bonds with Ser 144 and Cys 145 and halogen bond with Phe 140. Also three pi interactions were formed with His 41 which plays an important role in the catalytic activity of the enzyme, and one pi interaction was formed with Met 165. Because of these interactions, it can play its inhibitory effect well.
Pyronaridine was synthesized as an antimalarial agent; it has a similar structure to chloroquine but indicates superiority in potency [37], pharmacokinetic properties, and also less toxicity [38]. Pyronaridine is effective against acute Chagas disease [39] and the Ebola virus (EBOV). Also, its immunomodulation effect during the EBOV infection may synergistically increase its antiviral activity [40].
As long as the Ebola drug like remdesivir and anti-malaria agent like chloroquine can work against coronavirus, therefore pyronaridine which affects both diseases can be promising for the treatment of COVID-19 and could be the ideal compound of this dataset.
Finally, remdesivir a prodrug of adenosine nucleotide analog for the treatment of Ebola has entered into clinical phases for COVID-19 [41]. This drug has recently been considered for the treatment of COVID-19, with its mechanism of action on viral RNA polymerase and making a mistake in proofreading by viral exoribonuclease (ExoN), which causes a decrease in viral RNA production [42]. The implication stated for the other compounds is also correct here, and docking score −5.8 indicates the inability of this compound to interact well with the main protease binding pocket. However, a promising study has been published that simeprevir suppresses the replication of SARS-CoV-2 in vitro and revealed synergizes with the remdesivir in that way [43].
We can infer that simeprevir and pyronaridine are potential drugs for repurposing in treating COVID-19, due to their favorable interactions with the main protease and also their broad-spectrum antiviral activity.
Fig. 3a illustrates the binding mode of these two drugs in the binding pocket of the main protease. As it clear, the simeprevir placed well in the binding pocket due to its long and flexible structure.
3.2. Molecular dynamics simulation
To determine the stability and behavior of selected ligands, simeprevir, and pyronaridine in complex with the main protease, molecular dynamics simulations were performed during 150 ns and the analysis was carried out on its output as follows. To ensure the reliability of the results, the simulation of both systems repeated twice. The results of each run presented separately in the supplementary material (Figs. S1–S5).
The backbone root mean square deviation (RMSD) of both complexes was plotted on simulation time. Fig. 4a indicates that both complexes converged to an equilibration state in the last 30 ns of simulation. However, the main protease in complex with pyronaridine reached to an equilibration state much faster and remained almost constant until the end of the simulation. But the main protease in complex with simeprevir undergoes significant conformational changes during the simulation time and reaches the steady-states more slowly. The macrocyclic structure of simeprevir and the resulting flexibility could be the reason for more fluctuation in the RMSD plot in comparison with pyronaridine. The same phenomenon can be seen in the RMSD plot of the active site in Fig. 4b that the active site undergoes conformational changes to interact properly with simeprevir.
Fig. 4.

a) Backbone RMSD plots of the entire main protease and b) backbone RMSD plots of the active site of the main protease in complex with simeprevir (magenta) and pyronaridine (cyan). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Further analysis of ligands RMSD plot Fig. 5 showed that both ligands reach the steady-state in the last 50 ns and the more fluctuation of simeprevir is related to its macrocyclic and flexible structure.
Fig. 5.

Backbone RMSD plots of the ligands simeprevir (magenta) and pyronaridine (cyan). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 6 shows the conformational changes and interactions of both ligands during the simulation in three different frames from the beginning, middle, and end. As seen in Fig. 6a, simeprevir rotated along with the active site and three hydrogen bonds that hold the ligand for up to 75 ns of simulation disappear at the end. However, pi interaction with key residue His 41 has remained. Although pyronaridine initially provides hydrogen bond with Cys 145 and pi cation interaction with His 41 and has the one hydrogen bond at the end but due to improper rotation and small size, it has come out of the active site and lost its effective interactions.
Fig. 6.

a) 2D and 3D display of the interactions of simeprevir during simulation b) 2D and 3D and display of the interactions of pyronaridine during the simulation.
Detailed analysis of the root-mean-square fluctuation (RMSF) Fig. 7 versus each residue number was also applied to determine the residue fluctuations and flexibility through the simulation period. Both complexes present similar RMSF distributions with slight differences. Simeprevir indicates higher fluctuations for the amino acids around the active site which means simeprevir is capable of disrupting the active site conformation and transfigure the catalytic site. Residues in the catalytic site which contain His 41 and Cys 145 didn't show the high fluctuation means being stabilized and dysfunction.
Fig. 7.

a) RMSF plot of the main protease in complex with simeprevir (magenta) and pyronaridine (cyan). Closer view of RMSF plot of key residue b) His 41 and c) Cys 145. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Analysis of the number of hydrogen bonds between two ligands and main protease Fig. 8 showed that Simeprevir formed more hydrogen bonds number with binding pocket during simulation in comparison with pyronaridine.
Fig. 8.

Number of the hydrogen bond between main protease and (a) simeprevir (magenta) (b) pyronaridine (cyan). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
MM-PBSA calculation was performed to account for the binding free energy from three energetic terms. The first term combines the covalent energy, van der Waals and electrostatic interactions of both bonded and non-bonded terms, the second term is solvation free energy and the third is entropy term [44].
Table 4 shows the results of the prediction of binding free energy. MM-PBSA calculations confirmed the results obtained with AutoDock and between the two selected ligands, simeprevir shows less binding energy −252.54 ± 85.69 kcal/mol in compare with pyronaridine with −171.56 ± 59.56 kcal/mol which means that the simeprevir is more stable in the binding pocket. Regarding van der Waals energies, both ligands indicate negative values which mean that both have appropriate hydrophobic interactions; however, the effect of the macrocyclic and flexible structure of simeprevir causes a more negative value. The contribution of electrostatic energies is much less than the other energies.
Table 4.
results of the binding free energy calculation for simeprevir and pyronaridine in complex with the main protease.
| Compound | ∆G binding energy | ∆G Vdw | ∆G elec | ∆G polar |
|---|---|---|---|---|
| Simeprevir | −252.54 ± 85.69 | −181.16 ± 62.17 | −27.68 ± 17.90 | −21.85 ± 6.35 |
| Pyronaridine | −171.56 ± 59.56 | −130.61 ± 40.92 | −14.74 ± 20.33 | −13.10 ± 4.48 |
All energies are in kcal/mol.
The results of the MD, such as stability, the number of hydrogen bonds, and the binding free energy confirmed that simeprevir not only have better docking score but it also retains its interaction and stability over the time. Then it may have more inhibitory effects on the main protease of the virus.
4. Conclusion
COVID-19 has become a global concern, due to widespread outbreaks and uncertainty in treatment. In this study, we rely on the effectiveness of virtual screening [45] and the repurposing concept to identify new potential inhibitors for the main protease protein of SARS-CoV-2. Virtual screening procedure employing docking of 5881 FDA and world approved drugs were performed over the main protease and subsequently, MDs were applied to determine the stability of selected ligands in the binding pocket in 150 ns. The virtual screening result consisted of two drugs, simeprevir, and pyronaridine. Both drugs indicate proper interaction with the main protease. MDs results were also in good agreement with docking results and indicate the suitable stability along with proper interaction during the time, for both ligands.
However, simeprevir indicate superiority in many terms, such as interaction with both catalytic residue Cys 145 and His 41 during the time, approved by FDA which confirms its pharmacokinetic or ADME properties, identified as a potential inhibitor by different software for different proteins of the virus, broad-spectrum antiviral activity, low free binding energy, common receptor, the more number of hydrogen bonds and stability during the time and finally its suppression the replication of the virus in in-vitro which makes it a reasonable option for repurposing to the treatment of COVID-19. However, pyronaridine is also a potential choice for the treatment of COVID-19 because of its proper interaction with the receptor and its effectiveness against both malaria and EBOV. Taking together both drugs can be promising for the treatment of COVID-19. Nevertheless, a significant need for clinical approval remains.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgment
This study was financially supported by the Research Council of Tehran University of Medical Sciences and Health Services, Tehran, Iran, and Iran National Science Foundation (INSF-99008187).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.lfs.2020.118205.
Appendix A. Supplementary data
Supplementary figures
References
- 1.Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S., Lau E.H., Wong J.Y., Xing X. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X., Geng M., Peng Y., Meng L., Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020;10(2):102–108. doi: 10.1016/j.jpha.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taherizadeh M., Tabibzadeh A., Panahi M., TAMESHKEL F.S., Golahdooz M., NIYA M.H. An introduction to SARS coronavirus 2; comparative analysis with MERS and SARS coronaviruses: a brief review. Iran. J. Public Health. 2020;49:30–37. doi: 10.18502/ijph.v49iS1.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Coronavirus Disease 2019 (COVID-19): Situation Report, 24.
- 5.Munster V.J., Koopmans M., van Doremalen N., van Riel D., de Wit E. A novel coronavirus emerging in China—key questions for impact assessment. N. Engl. J. Med. 2020;382:692–694. doi: 10.1056/NEJMp2000929. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization World Health Organization; 2020. Clinical management of severe acute respiratory infection when novel coronavirus (2019-nCoV) infection is suspected: interim guidance. https://www.who.int/
- 7.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020:1–3. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim J., Jeon S., Shin H.Y., Kim M.J., Seong Y.M., Lee W.J., Choe K.W., Kang Y.M., Lee B., Park S.J. Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: the application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J. Korean Med. Sci. 2020;35(6):e79. doi: 10.3346/jkms.2020.35.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goetz D.H., Choe Y., Hansell E., Chen Y.T., McDowell M., Jonsson C.B., Roush W.R., McKerrow J., Craik C.S. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46(30):8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- 10.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10(5):766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirza M.U., Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020 doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polamreddy P., Gattu N. The drug repurposing landscape from 2012 to 2017: evolution, challenges, and possible solutions. Drug Discov. Today. 2019;24(3):789–795. doi: 10.1016/j.drudis.2018.11.022. [DOI] [PubMed] [Google Scholar]
- 13.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 14.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sterling T., Irwin J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015;5(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminformatics. 2011;3(1):33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bagherzadeh K., Shirgahi Talari F., Sharifi A., Ganjali M.R., Saboury A.A., Amanlou M. A new insight into mushroom tyrosinase inhibitors: docking, pharmacophore-based virtual screening, and molecular modeling studies. J. Biomol. Struct. Dyn. 2015;33(3):487–501. doi: 10.1080/07391102.2014.893203. [DOI] [PubMed] [Google Scholar]
- 18.Dassault Systèmes BIOVIA . Dassault Systèmes. Version 17.2 [software]; San Diego: 2016. Discovery Studio Modeling Environment, Release 2017.https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio Available from. [Google Scholar]
- 19.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminformatics. 2011;33 doi: 10.1186/1758-2946-3-33. https://pymol.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 21.Yu W., He X., Vanommeslaeghe K., MacKerell A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumari R., Kumar R., Open Source Drug Discovery, C, Lynn A. g_mmpbsa—a GROMACS tool for highthroughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 23.Gentile I., Buonomo A.R., Borgia G. Dasabuvir: a non-nucleoside inhibitor of NS5B for the treatment of hepatitis C virus infection. Rev. Recent Clin. Trials. 2014;9(2):115–123. doi: 10.2174/1574887109666140529222602. [DOI] [PubMed] [Google Scholar]
- 24.Alamri M.A., Tahir ul Qamar M., Alqahtani S.M. 2020. Pharmacoinformatics and Molecular Dynamic Simulation Studies Reveal Potential Inhibitors of SARS-CoV-2 Main Protease 3CLpro. Preprints. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francis D., CS S., Variyar J. 2020. Repurposing Simeprevir, Calpain Inhibitor IV and a Cathepsin F Inhibitor Against SARS-CoV-2: A Study Using In Silico Pharmacophore Modeling and Docking Methods. [Google Scholar]
- 26.de Oliveira L., Davi M., de Oliveira T., Mota K. Comparative computational study of SARS-CoV-2 receptors antagonists from already approved drugs. ChemRxiv. Preprint. 2020 doi: 10.26434/chemrxiv.12044538.v3. [DOI] [Google Scholar]
- 27.Beck B.R., Shin B., Choi Y., Park S., Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagan S., Crawford-Jones A., Orkin C. Simeprevir for the treatment of hepatitis C and HIV/hepatitis C co-infection. Expert. Rev. Clin. Pharmacol. 2014;7(6):691–704. doi: 10.1586/17512433.2014.956091. [DOI] [PubMed] [Google Scholar]
- 29.Li Z., Yao F., Xue G., Xu Y., Niu J., Cui M., Wang H., Wu S., Lu A., Zhong J., Meng G. Antiviral effects of simeprevir on multiple viruses. Antivir. Res. Dec 1, 2019;172 doi: 10.1016/j.antiviral.2019.104607. [DOI] [PubMed] [Google Scholar]
- 30.Chang Y.C., Tung Y.A., Lee K.H., Chen T.F., Hsiao Y.C., Chang H.C., Hsieh T.T., Su C.H., Wang S.S., Yu J.Y., Shih S.S. 2020. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. Preprints. [DOI] [Google Scholar]
- 31.Contini A. 2020. Virtual Screening of an FDA Approved Drugs Database on Two COVID-19 Coronavirus Proteins. Chemrxiv. [DOI] [Google Scholar]
- 32.Lin S., Shen R., He J., Li X., Guo X. 2020. Molecular Modeling Evaluation of the Binding Effect of Ritonavir, Lopinavir and Darunavir to Severe Acute Respiratory Syndrome Coronavirus 2 Proteases. bioRxiv. [Google Scholar]
- 33.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. BioSci. Trends. 2020 doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 34.Sun M.L., Yang J.M., Sun Y.P., Su G.H. Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43(0):E014. doi: 10.3760/cma.j.issn.1001-0939.2020.0014. [DOI] [PubMed] [Google Scholar]
- 35.Love M.S., Beasley F.C., Jumani R.S., Wright T.M., Chatterjee A.K., Huston C.D., Schultz P.G., McNamara C.W. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl. Trop. Dis. 2017;11(2) doi: 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lombardo F.C., Perissutti B., Keiser J. Activity and pharmacokinetics of a praziquantel crystalline polymorph in the Schistosoma mansoni mouse model. Eur. J. Pharm. Biopharm. 2019;142:240–246. doi: 10.1016/j.ejpb.2019.06.029. [DOI] [PubMed] [Google Scholar]
- 37.Okoth W.A., Dukes E.J., Sullivan D.J. Superior pyronaridine single-dose pharmacodynamics compared to artesunate, chloroquine, and amodiaquine in a murine malaria luciferase model. Antimicrob. Agents Chemother. 2018;62(9) doi: 10.1128/AAC.00394-18. (e00394-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naisbitt D.J., Williams D.P., O’Neill P.M., Maggs J.L., Willock D.J., Pirmohamed M., Park B.K. Metabolism-dependent neutrophil cytotoxicity of amodiaquine: a comparison with pyronaridine and related antimalarial drugs. Chem. Res. Toxicol. 1998;11(12):1586–1595. doi: 10.1021/tx980148k. [DOI] [PubMed] [Google Scholar]
- 39.Ekins S., de Siqueira-Neto J.L., McCall L.I., Sarker M., Yadav M., Ponder E.L., Kallel E.A., Kellar D., Chen S., Arkin M., Bunin B.A. Machine learning models and pathway genome data base for Trypanosoma cruzi drug discovery. PLoS Negl. Trop. Dis. 2015;9(6) doi: 10.1371/journal.pntd.0003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lane T.R., Massey C., Comer J.E., Anantpadma M., Freundlich J.S., Davey R.A., Madrid P.B., Ekins S. Repurposing the antimalarial pyronaridine tetraphosphate to protect against Ebola virus infection. PLoS Negl. Trop. Dis. 2019;13(11) doi: 10.1371/journal.pntd.0007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilead Sciences initiates two phase 3 studies of investigational antiviral remdesivir for the treatment of COVID-19. 2020. https://www.gilead.com/
- 42.Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., Smith E.C., Case J.B., Feng J.Y., Jordan R., Ray A.S. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2) doi: 10.1128/mBio.00221-18. (e00221-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lo H.S., Hui K.P., Lai H.M., Khan K.S., Kaur S., Li Z., Chan A.K., Cheung H.H., Ng K.C., Ho J.C., Che Y.W. 2020. Simeprevir Suppresses SARS-CoV-2 Replication and Synergizes With Remdesivir. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C., Greene D.A., Xiao L., Qi R., Luo R. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 2018;4:87. doi: 10.3389/fmolb.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavecchia A., Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 2013;20(23):2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures