

Abstract
Alveolar soft part sarcoma (ASPS) is a rare soft tissue tumor of unknown histogenesis generally characterized by the der(17)t(X;17)(p11.2;q25) translocation which results in the ASPSCR1-TFE3 gene fusion. Primary ASPS of the thyroid gland has not yet been reported. During oncology follow-up for breast cancer, a pulmonary nodule and thyroid gland mass were identified in a 71-year-old Korean male. Thyroid ultrasound showed a 5.7 cm left thyroid gland mass. After several fine needle aspirations, a thyroid gland lobectomy was performed after documenting only non-caseating granulomatous inflammation in a biopsy of the lung nodule. A 7.6 cm bulging nodular thyroid gland mass was identified, showing significant destructive invasion. Alveolar nests of large polygonal, eosinophilic, granular neoplastic cells were separated by vascularized stroma. Colloid was absent. Tumor necrosis and increased mitoses were identified. The neoplastic cells were positive with TFE3 and CD68, but negative with pancytokeratin, thyroglobulin, TTF-1, napsin-A, calcitonin, PAX8, CAIX, S100 protein, HMB45, SMA, and desmin. FISH confirmed a TFE3 gene rearrangement. The differential includes several primary thyroid gland epithelial neoplasms, paraganglioma, PEComa, melanoma, crystal storage disease, and metastatic carcinomas, especially Xp11 translocation renal cell carcinoma. The patient has refused additional therapy, but is alive without tumor identified (primary or metastatic).
Keywords: Thyroid gland; Sarcoma, alveolar soft part; Humans; Biopsy, fine-needle; Gene rearrangement
Introduction
Alveolar soft part sarcoma (ASPS) is a rare sarcoma of uncertain histogenesis, accounting for < 1% of soft tissue sarcomas [1]. ASPS is defined by histologically large, eosinophilic cells that are arranged in nests or clusters, frequently exhibiting central dyscohesion that yields an alveolar appearance, while characterized by a specific translocation der(17)t(X;17)(p11.2;q25) that yields the ASPCR1-TFE3 fusion gene [2]. ASPS is most commonly seen in patients aged 15–35 years (median 25 years), with a majority below 30 years of age [1, 3]. There is a predominance in females, although not as well developed in patients > 30 years of age [4]. The most common sites of involvement in adults are deep soft tissue of the thigh/buttock and trunk [4]. In children or infants, the most commons sites of involvement are head and neck region, with the orbit and tongue especially [5, 6]. Primary ASPS has been reported in almost every anatomic site, including: bone [7], breast [8], female genital tract [9], heart [10], larynx [11], liver [12], lung [13], stomach [14], and urinary bladder [15]. However, to the best of our knowledge, we herein report the first case of primary thyroid gland ASPS. Additionally, we discuss the unique differential produced by ASPS presenting as a thyroid gland neoplasm and the immunohistochemistry necessary to parse ASPS from its histologic mimics.
Case Report
Clinical Presentation
The patient was a 71-year-old Korean male with a past medical history significant for breast carcinoma diagnosed 36 years prior to presentation, managed with surgery only (no radiation or chemotherapy). During follow-up imaging studies, a pulmonary nodule was identified, previously followed by serial scans. The patient noted mild thyromegaly. The patient had a previous thyroid gland ultrasound performed outside the country and was told it was abnormal. A repeat thyroid gland ultrasound showed a 5.7 × 4.5 × 4.4 cm mass in the left thyroid gland lobe, with Doppler color flow demonstrating chaotic/turbulent flow (Fig. 1). On physical exam, there was a firm left thyroid gland nodule, which had increased in size recently. The patient denied dysphagia, odynophagia and hoarseness, although reported a 20 pound unintentional weight loss. A thyroid fine needle aspiration (FNA) was performed, and interpreted as Bethesda III (atypia of unknown significance). Before further thyroid gland evaluation, a computed tomography study was performed due to left upper quadrant pain. The study demonstrated both a thyroid gland mass and an 11 mm right lower lung lobe nodule. A PET scan demonstrated left hilar adenopathy and right lower lobe mass with SUV maximum of 17.19, and increased uptake in the left thyroid gland lobe mass. No other areas of abnormal hypermetabolic activity were detected. A computed tomography guided core needle biopsy of the lung demonstrated only non-caseating granulomatous inflammation, with sarcoid considered the most likely candidate. Another thyroid gland FNA was performed, but was non-diagnostic, which was followed by a third thyroid gland FNA, with ThyGeNEXT molecular oncogene panel reporting no mutations detected. The Diff-Quik and Papanicolaou-stained smears were moderately cellular and revealed groups of cohesive cells with abundant granular eosinophilic cytoplasm and round nuclei. There were many “naked” nuclei and clusters arranged around delicate endothelium (Fig. 2). Many of the nuclei were unusually large (in comparison to uninvolved follicular thyroid epithelium). The cells displayed significant anisocytosis and most showed prominent nucleoli (Fig. 2) within nuclei that displayed vesicular chromatin. Colloid was inconspicuous, but there was a hemorrhagic and granular background. Based on these findings a thyroid gland lobectomy was performed. The diagnosis of ASPS prompted further evaluation but failed to identify any soft tissue or organ based primary site. Thus, an alternate primary site was not identified. Given the presentation of two unusual tumors in an adult patient (male breast cancer and adult-onset ASPS), genetic testing was offered and is still under consideration at this time. No additional surgery was performed for the positive tumor margin, and at present the patient has opted to not receive radiotherapy or chemotherapy, but is alive without gross disease at last contact (5 months).
Fig. 1.
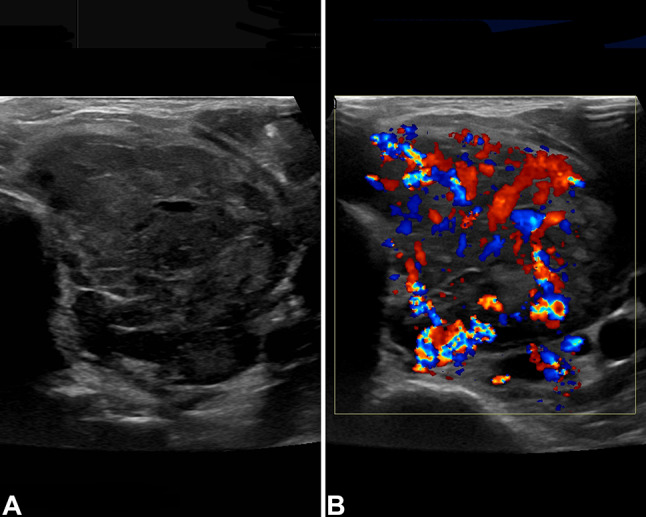
By ultrasound, there was a 5.7 × 4.5 × 4.4 cm mass in the left thyroid gland lobe (a), demonstrating chaotic/turbulent flow with Doppler color flow (b)
Fig. 2.
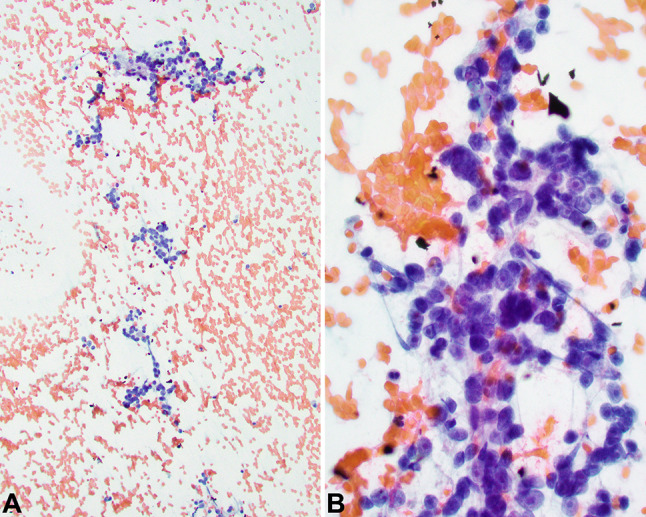
Fine needle aspiration demonstrated moderate cellularity with three-dimensional clusters and single cells in a hemorrhagic and granular background (a). Uni- and binucleated tumor cells showed abundant delicate cytoplasm eccentrically surrounding large nuclei with vesicular chromatin and prominent nucleoli (b)
Pathology
The resected thyroid gland lobectomy weighed 76 g and measured 8.7 × 5.4 × 3.7 cm. Gross examination revealed a mass that closely approximated the entire volume of the lobectomy. Serial sectioning revealed a 7.6 × 5.3 × 3.6 cm firm, tan-yellow, and bulging nodular mass.
Histologic examination revealed a widely infiltrative and destructive neoplasm that nearly replaced the entire lobectomy (Fig. 3). The neoplasm was partially encapsulated, showing significant capsular invasion along with well developed and easily identified lymphovascular invasion. Direct invasion into the parathyroid gland tissue was also observed, with tumor identified on the inked margins of resection. The neoplastic cells were arranged in large sheets and smaller nests which were separated by thick fibrous connective tissue septae. The nests were divided into alveolar spaces and associated with a very richly vascularized stroma (Fig. 4). The neoplastic cells were large and polygonal, with central round to irregular vesicular nuclei containing large, prominent, brightly hypereosinophilic single nucleoli (Fig. 4). The nuclei were surrounded by abundant granular to eosinophilic cytoplasm, although a few cells had vacuolated to cleared cytoplasm. Isolated cells had crystals within the cytoplasm that were arranged in aggregates. Retraction artifacts were seen in the cytoplasm, which created a dilapidated or pseudo-alveolar pattern to the cells (Fig. 4). There were easily identified mitotic figures, including atypical forms, with 18/2 mm2. Tumor central comedonecrosis was easily identified.
Fig. 3.
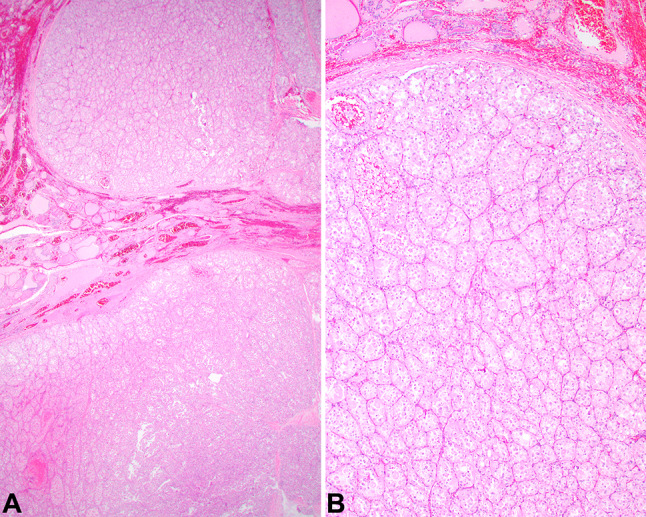
a The neoplasm shows a nodular periphery, surrounded by a thin capsule, separating the tumor from the adjacent thyroid gland parenchyma. b A nested, alveolar pattern of growth was easily identified, with prominent vessels between the tumor nests. Colloid was not seen, although native thyroid gland was present (top of field)
Fig. 4.
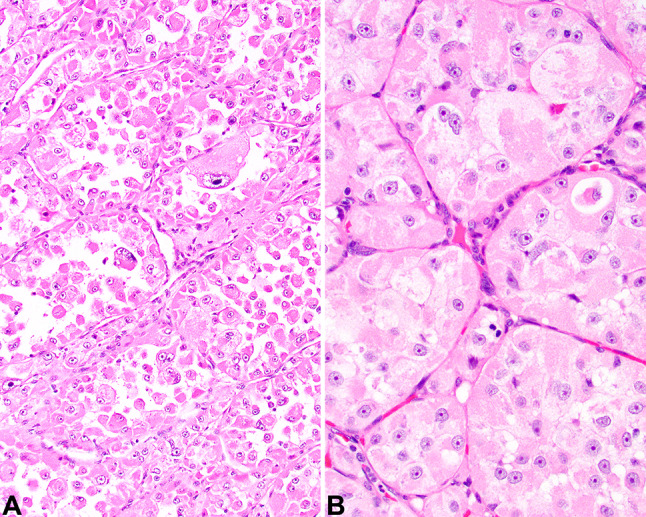
a Sheets and nests of cells separated by a richly vascularized stroma creating alveolar spaces. b The polygonal neoplastic cells were large with a low nuclear to cytoplasmic ratio, with open chromatin and single, prominent macronucleoli
With a working diagnosis of alveolar soft part sarcoma, several additional studies were performed. A PAS-D highlighted PAS positive diastase resistant cytoplasmic rod shaped crystals. The neoplastic cells showed strong, diffuse nuclear reactivity for TFE3 (Fig. 5), along with CD68. There was no reaction in the neoplastic cells for pancytokeratin, thyroglobulin (Fig. 5), TTF-1, napsin-A, calcitonin, PAX8 (Fig. 5), CAIX, S100 protein, SOX10, inhibin, HMB45, SMA, synaptophysin, chromogranin, desmin, and calretinin. These findings supported a diagnosis of alveolar soft part sarcoma. In light of the most unusual presentation clinically and by anatomic site of involvement, a TFE3 gene rearrangement was confirmed by FISH (Fig. 6).
Fig. 5.

Immunohistochemistry studies to exclude thyroid origin as well as prove the histologic diagnosis included negative a thyroglobulin, b pancytokeratin AE1/AE3, and c PAX8, while the neoplastic cells showed a strong and diffuse TFE3 nuclear reaction (d)
Fig. 6.
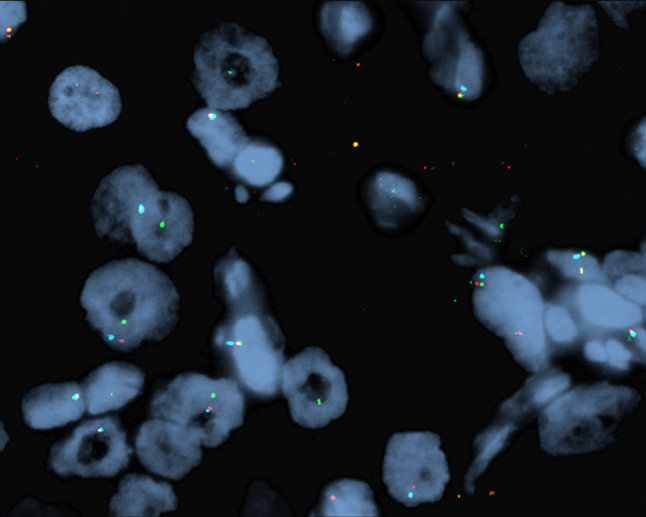
A FISH breakapart was employed to document spatial separation of the green (3′TFE3) and red (5′TFE3) signals, supporting a Xp11.23 rearrangement (yellow signals are intact green/red fusion; the translocation partner is not documented)
Discussion
ASPS is a tumor that affects predominantly adolescents and young adults, and thus considered quite rare in a patient of this age and exceptional in the thyroid gland as a primary tumor. The tumor is usually slow growing, with a relatively high risk (up to 50%) of local recurrence [1], and a high delayed metastatic potential, including up to decades after the primary tumor has been resected [4, 16]. Therefore, long term clinical follow-up with complete, radical surgical excision is usually recommended, as the overall 5-year survival is about 56% [1].
There is a well-known genetic alteration: der(17)t(X;17)(p11;q25). This translocation results in the fusion of the TFE3 transcription factor gene (from Xp11) with the ASPSCR1 at 17q25 [2]. Recently, other fusion partners with TFE3 have been identified, and include HNRNPH3-TFE3, DVL2-TFE3 and PRCC-TFE3 [17], expanding the genetic diversity in ASPS, identifying overlap with translocation-associated renal cell carcinomas and PEComa. The ASPSCR1-TFE3 fusion protein localizes to the nucleus and can be detected by the immunohistochemical stain TFE3, seen irrespective of the fusion partner. TFE3 immunohistochemistry is not specific for ASPS, with nuclear expression identified in a number of neoplasms, including (but not limited to): Xp11 translocation renal cell carcinomas [18], PEComas [19], granular cell tumors [20, 21], paraganglioma [21, 22], solid pseudo-papillary tumors of the pancreas [23], adrenal cortical carcinoma [21, 24], and epithelioid hemangioendotheliomas [25]. From these tumors, a subset of renal cell carcinomas and PEComas have the same ASPSCR1-TFE3 fusion [18, 19, 26].
The morphologic features of this tumor raise a broad differential diagnosis within the thyroid gland, including follicular carcinoma (oncocytic type), medullary carcinoma (oncocytic type), granular cell tumor, metastatic renal cell carcinoma, malignant paraganglioma, PEComa, and metastatic melanoma, while a crystal storage disease was considered unlikely. A pertinent and targeted discussion of the histologic and immunophenotypic characteristics of these differential considerations will be set in the context of the ASPS diagnosis.
A follicular carcinoma tends to show a follicular pattern of growth, although a solid architecture may be seen; shows colloid within the follicles; and would express epithelial and thyroid linage markers (CK-pan, TTF-1, thyroglobulin, PAX8) [27]. Medullary thyroid carcinoma is one of the primary thyroid gland lesions that is a great mimic, with many different architectural patterns and cytologic appearances. However, despite overlap, the neoplastic cells would express epithelial markers and TTF-1, calcitonin, synaptophysin, chromogranin, and PAX8 (variable/weak) [28]. A granular cell tumor generally shows a more solid to packeted architecture, while generally lacking invasion (although malignant granular cell tumors of the thyroid gland are recognized); the cytoplasm would be granular and eosinophilic in appearance and would express a combination of S100 protein, SOX10, inhibin, and CD68 [29]. The lack of epithelial marker reactivity argues against metastatic renal cell carcinoma, however the Xp11 translocation renal cell carcinomas usually underexpress epithelial markers, but they uniformly express positivity for PAX8 [30]. Interestingly, ASPSCR1-TFE3 renal cell carcinomas typically present with metastases and are known to metastasize 20–30 years after diagnosis, much like ASPS [18]. The absence of a PAX8 staining, renal mass, or history of renal cell carcinoma argues for an alternative diagnosis. Head and neck paraganglioma, and especially those within the thyroid gland, are all considered to be malignant by definition, although now classified by a risk stratification approach. There is a smaller nest size (zellballen), with isolated pleomorphic cells usually easily identified; mitoses are usually inconspicuous; the neoplastic cells may express TFE3, but show chromogranin, synaptophysin, and SDHB (the latter may be present or absent depending on possible mutation status, but is positive in most cases), with S100 protein positive sustentacular cells, the latter lacking in ASPS [22, 31]. A perivascular epithelioid cell tumor (PEComa) is a mesenchymal epithelioid neoplasm often associated with blood vessel walls and usually showing myomelanotic differentiation with HMB45 (most sensitive) and smooth muscle actin [32], although not yet documented as a primary tumor in the thyroid gland. Primary thyroid gland melanoma is not reported, but metastatic melanoma to the thyroid gland may show morphologic overlap with ASPS. Melanoma typically shows large polygonal cells that are nested, but not really in a specific pattern, while frequently showing cytoplasmic melanin pigment, prominent nucleoli and intranuclear cytoplasmic inclusions; the neoplastic cells typically express S100 protein, SOX10, HMB45, and Melan-A among other markers [33]. In this patient, the lack of melanoma history argues against such a lesion. Crystal storage disease usually has an increased number of lymphocytes and inflammatory cells, with the histiocytes stuffed with crystals, while showing a strong and diffuse CD68 immunoreactivity [34]; the current case failed to show any significant inflammatory cells. Rhabdomyomas and alveolar rhabdomyosarcoma were considered, but the age of the patient along with the tumor site militated against such a consideration, further supported by a lack of muscle marker immunoexpression [35, 36]. The patient’s known lung lesion was considered, but demonstrated only non-caseating granulomatous inflammation rather than metastatic disease, or as a source of a primary lesion. Given the very vascular nature of the thyroid gland, a metastatic tumor to the thyroid gland must be considered. It is well known that renal cell carcinomas can present with a large single, encapsulated tumor nodule in the thyroid gland [37], and so the presentation of a large, single mass would not preclude the possibility of a metastatic tumor. However, during careful evaluation by imaging studies, no primary tumor has been identified anywhere else, and thus a primary thyroid gland ASPS can be diagnosed.
The major negative prognostic factors influencing overall outcome are older patient age at presentation, tumors > 10 cm, primary trunk location and metastases at presentation. Thus, no histologic features were prognostically significant in this patient [16]. The 5-year disease free survival for localized disease and those that present with metastases is 71% and 20%, respectively [4]. This patient showed a microscopic positive tumor margin, but additional surgery was not accepted and no additional therapy has been performed at this time. Long term follow-up is recommended in light of the significant risk of delayed metastases. Our patient is currently disease free, but will require essentially lifelong follow-up, due to the risk of delayed metastases. Clinical management will evolve as more cases are reported.
Acknowledgements
With sincere appreciation to Robert B. Jenkins, MD (Mayo Clinic Genomics Laboratory, Mayo Clinic, Rochester, MN) for performing the clinical patient sample FISH interpretation. The views expressed are those of the authors solely and do not represent endorsement from Southern California Permanente Medical Group.
Funding
None.
Compliance with Ethical Standards
Conflict of interest
All authors declare that they have no conflict of interest as it relates to this research project.
Ethical Approval
All procedures performed in this case report involving human participants were in accordance with the ethical standards of the institutional review board (IRB #5968), which did not require informed consent.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wang H, Jacobson A, Harmon DC, Choy E, Hornicek FJ, Raskin KA, Chebib IA, DeLaney TF, Chen YL. Prognostic factors in alveolar soft part sarcoma: a SEER analysis. J Surg Oncol. 2016;113:581–586. doi: 10.1002/jso.24183. [DOI] [PubMed] [Google Scholar]
- 2.Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, Healey JH, Ueda T, Yoshikawa H, Meloni-Ehrig A, Sorensen PH, Mertens F, Mandahl N, van den Berghe H, Sciot R, Dal Cin P, Bridge J. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:48–57. doi: 10.1038/sj.onc.1204074. [DOI] [PubMed] [Google Scholar]
- 3.Ordóñez NG, Ladanyi M. Alveolar soft part sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. Lyon: IARC; 2017. [Google Scholar]
- 4.Portera CA, Jr, Ho V, Patel SR, Hunt KK, Feig BW, Respondek PM, Yasko AW, Benjamin RS, Pollock RE, Pisters PW. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91:585–591. doi: 10.1002/1097-0142(20010201)91:3<585::AID-CNCR1038>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Kayton ML, Meyers P, Wexler LH, Gerald WL, LaQuaglia MP. Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg. 2006;41:187–193. doi: 10.1016/j.jpedsurg.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Fanburg-Smith JC, Miettinen M, Folpe AL, Weiss SW, Childers EL. Lingual alveolar soft part sarcoma; 14 cases: novel clinical and morphological observations. Histopathology. 2004;45:526–537. doi: 10.1111/j.1365-2559.2004.01966.x. [DOI] [PubMed] [Google Scholar]
- 7.Park YK, Unni KK, Kim YW, Han CS, Yang MH, Wenger DE, Sim FH, Lucas DR, Ryan JR, Nadim YA, Nojima T, Fletcher CD. Primary alveolar soft part sarcoma of bone. Histopathology. 1999;35:411–417. doi: 10.1046/j.1365-2559.1999.035005411.x. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Brinker DA, Haas M, Montgomery EA, Argani P. Primary alveolar soft part sarcoma (ASPS) of the breast: report of a deceptive case with xanthomatous features confirmed by TFE3 immunohistochemistry and electron microscopy. Int J Surg Pathol. 2005;13:81–85. doi: 10.1177/106689690501300112. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen GP, Oliva E, Young RH, Rosenberg AE, Dickersin GR, Scully RE. Alveolar soft-part sarcoma of the female genital tract: a report of nine cases and review of the literature. Int J Gynecol Pathol. 1995;14:283–292. doi: 10.1097/00004347-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Luo J, Melnick S, Rossi A, Burke RP, Pfeifer JD, Dehner LP. Primary cardiac alveolar soft part sarcoma. A report of the first observed case with molecular diagnostics corroboration. Pediatr Dev Pathol. 2008;11:142–147. doi: 10.2350/06-12-0199.1. [DOI] [PubMed] [Google Scholar]
- 11.Kusafuka K, Muramatsu K, Yabuzaki T, Ishiki H, Asano R, Kamijo T, Iida Y, Ebihara M, Onitsuka T, Kameya T. Alveolar soft part sarcoma of the larynx: a case report of an unusual location with immunohistochemical and ultrastructural analyses. Head Neck. 2008;30:1257–1263. doi: 10.1002/hed.20766. [DOI] [PubMed] [Google Scholar]
- 12.Shaddix KK, Fakhre GP, Nields WW, Steers JL, Hewitt WR, Menke DM. Primary alveolar soft-part sarcoma of the liver: anomalous presentation of a rare disease. Am Surg. 2008;74:43–46. doi: 10.1177/000313480807400109. [DOI] [PubMed] [Google Scholar]
- 13.Sonobe H, Ro JY, Mackay B, Ordóñez NG, Rundell MM, Ayala AG. Primary pulmonary alveolar soft-part sarcoma: report of a case. Int J Surg Pathol. 1994;2:57–61. doi: 10.1177/106689699400200110. [DOI] [Google Scholar]
- 14.Yaziji H, Ranaldi R, Verdolini R, Morroni M, Haggitt R, Bearzi I. Primary alveolar soft part sarcoma of the stomach: a case report and review. Pathol Res Pract. 2000;196:519–525. doi: 10.1016/S0344-0338(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 15.Amin MB, Patel RM, Oliveira P, Cabrera R, Carneiro V, Preto M, Balzer B, Folpe AL. Alveolar soft-part sarcoma of the urinary bladder with urethral recurrence: a unique case with emphasis on differential diagnoses and diagnostic utility of an immunohistochemical panel including TFE3. Am J Surg Pathol. 2006;30:1322–1325. doi: 10.1097/01.pas.0000213298.34520.2b. [DOI] [PubMed] [Google Scholar]
- 16.Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989;63:1–13. doi: 10.1002/1097-0142(19890101)63:1<1::AID-CNCR2820630102>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 17.Dickson BC, Chung CT, Hurlbut DJ, Marrano P, Shago M, Sung YS, Swanson D, Zhang L, Antonescu CR. Genetic diversity in alveolar soft part sarcoma: a subset contain variant fusion genes, highlighting broader molecular kinship with other MiT family tumors. Genes Chromosom Cancer. 2019 doi: 10.1002/gcc.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis CL, Eble JN, Subhawong AP, Martignoni G, Zhong M, Ladanyi M, Epstein JI, Netto GJ, Argani P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: impact of fusion subtype, age, and stage. Mod Pathol. 2014;27:875–886. doi: 10.1038/modpathol.2013.208. [DOI] [PubMed] [Google Scholar]
- 19.Argani P, Aulmann S, Illei PB, Netto GJ, Ro J, Cho HY, Dogan S, Ladanyi M, Martignoni G, Goldblum JR, Weiss SW. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol. 2010;34:1395–1406. doi: 10.1097/PAS.0b013e3181f17ac0. [DOI] [PubMed] [Google Scholar]
- 20.Chamberlain BK, McClain CM, Gonzalez RS, Coffin CM, Cates JM. Alveolar soft part sarcoma and granular cell tumor: an immunohistochemical comparison study. Hum Pathol. 2014;45:1039–1044. doi: 10.1016/j.humpath.2013.12.021. [DOI] [PubMed] [Google Scholar]
- 21.Rekhi B, Ingle A, Agarwal M, Puri A, Laskar S, Jambhekar NA. Alveolar soft part sarcoma ‘revisited’: clinicopathological review of 47 cases from a tertiary cancer referral centre, including immunohistochemical expression of TFE3 in 22 cases and 21 other tumours. Pathology. 2012;44:11–17. doi: 10.1097/PAT.0b013e32834d7ba4. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa K, Nakashima Y, Yamabe H, Hamashima Y. Alveolar soft part sarcoma, granular cell tumor, and paraganglioma. An immunohistochemical comparative study. Acta Pathol Jpn. 1986;36:895–904. doi: 10.1111/j.1440-1827.1986.tb03122.x. [DOI] [PubMed] [Google Scholar]
- 23.Harrison G, Hemmerich A, Guy C, Perkinson K, Fleming D, McCall S, Cardona D, Zhang X. Overexpression of SOX11 and TFE3 in solid-pseudopapillary neoplasms of the pancreas. Am J Clin Pathol. 2017;149:67–75. doi: 10.1093/ajcp/aqx142. [DOI] [PubMed] [Google Scholar]
- 24.Williams A, Bartle G, Sumathi VP, Meis JM, Mangham DC, Grimer RJ, Kindblom LG. Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch. 2011;458:291–300. doi: 10.1007/s00428-010-1039-9. [DOI] [PubMed] [Google Scholar]
- 25.Doyle LA, Fletcher CD, Hornick JL. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol. 2016;40:94–102. doi: 10.1097/PAS.0000000000000511. [DOI] [PubMed] [Google Scholar]
- 26.Argani P, Antonescu CR, Illei PB, Lui MY, Timmons CF, Newbury R, Reuter VE, Garvin AJ, Perez-Atayde AR, Fletcher JA, Beckwith JB, Bridge JA, Ladanyi M. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001;159:179–192. doi: 10.1016/S0002-9440(10)61684-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LiVolsi V, Abdulkader Nallib I, Baloch ZW, Bartolazzi A, Chan JKC, DeLellis RA, El-Naggar AK. Follicular thyroid carcinoma. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs. Lyon: IARC; 2017. [Google Scholar]
- 28.DeLellis RA, Al Ghuzlan A, Albores Saavedra J, Baloch ZW, Basolo F, Elisei R, Kaserer K, LiVolsi V, Matias-Guiu X, Mete O, Moley JF, Nikiforov YE, Nosé V, Pinto AE. Medullary thyroid carcinoma. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs. Lyon: IARC; 2017. [Google Scholar]
- 29.Bowry M, Almeida B, Jeannon JP. Granular cell tumour of the thyroid gland: a case report and review of the literature. Endocr Pathol. 2011;22:1–5. doi: 10.1007/s12022-011-9146-y. [DOI] [PubMed] [Google Scholar]
- 30.Argani P, Hicks J, De Marzo AM, Albadine R, Illei PB, Ladanyi M, Reuter VE, Netto GJ. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol. 2010;34:1295–1303. doi: 10.1097/PAS.0b013e3181e8ce5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelizzo MR, Conti C, Pennelli G, Bellan E, Cook GJ, Wong KK, Colletti PM, Merante Boschin I, Rubello D. Thyroid Paraganglioma: our experience and systematic review of the literature on a rare tumor. Am J Clin Oncol. 2018;41:416–423. doi: 10.1097/COC.0000000000000295. [DOI] [PubMed] [Google Scholar]
- 32.Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1–15. doi: 10.1016/j.humpath.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433–444. doi: 10.1111/j.1600-0560.2007.00891.x. [DOI] [PubMed] [Google Scholar]
- 34.Dogan S, Barnes L, Cruz-Vetrano WP. Crystal-storing histiocytosis: report of a case, review of the literature (80 cases) and a proposed classification. Head Neck Pathol. 2012;6:111–120. doi: 10.1007/s12105-011-0326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sebire NJ, Malone M. Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol. 2003;56:412–416. doi: 10.1136/jcp.56.6.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin B, Saleh H. Pitfalls in the diagnosis of adult rhabdomyoma by fine needle aspiration: report of a case and a brief literature review. Diagn Cytopathol. 2009;37:483–486. doi: 10.1002/dc.21033. [DOI] [PubMed] [Google Scholar]
- 37.Heffess CS, Wenig BM, Thompson LD. Metastatic renal cell carcinoma to the thyroid gland: a clinicopathologic study of 36 cases. Cancer. 2002;95:1869–1878. doi: 10.1002/cncr.10901. [DOI] [PubMed] [Google Scholar]