

Abstract
Water deficit has a significant impact on growth, development and yield of fava bean (Vicia fava L.) in arid and semi-arid climates. The aim of this study was to identify differentially expressed genes in the Qinghai 13 genotype under soil drought through leaf transcriptome analysis. A total of 256.95 M clean reads were obtained and assembled into 176334 unigenes, with an average length of 766 bp. A total of 9126 (4439 upregulated and 4687 downregulated) differentially expressed genes (DEGs) were identified in faba bean leaves under soil drought. In total, 324 putative transcription factors were identified and classified as belonging to different transcription factor families. According to GO and KEGG analysis, the soil drought stress-inducible DEGs encoded proteins mainly involved in regulating photosynthesis, osmotic adjustment, detoxification, autophagy and other functions. In addition, a large portion of DEGs appeared to be novel because they could not be annotated in any functional databases, therefore, suggesting a specific response to soil drought in faba bean. Finally, RNA-seq analysis was validated by quantitative reverse-transcription PCR analysis. This work provides comprehensive and valuable information for understanding the molecular mechanisms which faba bean uses to respond to soil drought.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02374-3) contains supplementary material, which is available to authorized users.
Keywords: Faba bean, Transcriptome, Soil drought, Differentially expressed genes, Function
Introduction
Faba bean (Vicia faba L.) is the most widely cultivated cool-season legume reported by the FAOSTAT (http://www.fao.org/faostat/en/). It is consumed worldwide as a plant protein source for humans and animals due to its high protein content (Abdelmula et al. 1999; Amede et al. 1999; Maalouf et al. 2019). In addition, faba bean is a globally important nitrogen-fixing legume (Sosulski and McCurdy 1987; Doyle and Luckow 2003; Cazzato et al. 2012; Webb et al. 2016). Thus, faba bean cultivation is widespread in the temperate and subtropical regions of the world (Torres et al. 2006).
The Qinghai province, which is located in the northwest area of the Qinghai-Tibet Plateau has an average altitude above 3000 m and is one of the main faba bean producing areas in China. Faba bean is grown in Qinghai because this area has intense sunlight, large diurnal temperature variation, and low pest pressure compared to other areas in China (Li et al. 2018). To adjust to the agricultural policy of local government in recent years, the major producing areas of faba bean have been extended from irrigated agricultural areas to rain-fed lands (dry areas or semi-arid areas), but the planting area of faba bean in rain-fed land still only accounts for 20% of the total area in Qinghai (Li et al. 2018). Water deficit is one of the most severe abiotic stresses that significantly impacts plant growth, development and yield (Chaves et al. 2003; Farooq et al. 2009; Hossain et al. 2016; Hong et al. 2020). Faba bean is known to have a low tolerance to water deficit compared with other grain legumes (McDonald and Paulsen. 1997; Amede et al. 2003; Khan et al. 2010), which limits its cultivation in rain-fed areas. Farmers have been forced to select faba beans with better drought tolerance when growing in rain-fed lands.
In recent years, physiological and molecular mechanisms underlying plant drought resistance have been studied in several leguminous crops, including Glycine max (Kron et al. 2008; Du et al. 2009), Medicago lupulina (Küchenmeister et al. 2013), Cicer arietinum (Varshney et al. 2014), Vigna radiata (Sengupta et al. 2011) and Cajanus cajan (Varshney et al. 2013). Though the morphological changes and physiological responses to drought stress have been studied in faba bean (Khan et al. 2007; Abid et al. 2016; Ammar et al. 2015, 2017), the molecular mechanisms underlying drought tolerance still need to be investigated. This is especially important in Qinghai, which often experiences drought. Some genetic and genomic studies in faba bean have been carried out (Abid et al. 2015; Webb et al. 2016; Yang et al. 2019), though the genome of faba bean (approximated 13 Gb) is unknown so far. Identifying candidate genes is fundamental to unraveling the molecular mechanism of plant drought stress survival. There are several studies which have attempted transcriptomic profiling of faba bean recently (Ammar et al. 2015; Ray et al. 2015; Siddiqui et al. 2015; O’Sullivan et al. 2016; Braich et al. 2017; Cooper et al. 2017; Alghamdi et al. 2018). RNA-Seq, has frequently been applied for identifying stress-inducible transcripts and determining the complex networks that underly plant stress biology (Kolev et al. 2010; Mizuno et al. 2010; Siegel et al. 2010; Zhai et al. 2013). In the previous study, researchers attempted to identify and evaluate the drought-responsive genes of faba bean genotypes by RNA-Seq with a treatment of 15% polyethylene glycol (PEG) 6000 (Khan et al. 2019). PEG is widely used to simulate drought stress artificially (Skriver and Mundy. 1990), but the actual genes expression profile of plants responding to soil drought has been shown to significantly differ compared with PEG or mannitol treatment (Bray 2004; Forner-Giner et al. 2011). Little is known about the actual molecular responses of faba bean under soil drought stress.
In our previous work, we found Qinghai 13 was able to cope with water deficit better than other local varieties (Zhang et al. 2015). For this work, Qinghai 13 was cultivated in soil and then subjected to water deficit to simulate a natural drought. Although the leaves and roots have distinct developmental trajectories, plants also evolved highly concerted biological processes to combat drought conditions by fine-tuning energy production in leaves and nutrients in roots (Khan et al. 2012; Zhu 2016). It has been reported lots of DEGs showed tissue-specific expression and leaves are more sensitive to drought stress than roots when analyzing the drought resistance transcriptome in vetch (Vicia sativa L) (Min et al. 2020). In previous study, drought is linked to changes in leaves of Qinghai 13 including shrinkage in the size of leaves, chlorophyll content (Chl), superoxide dismutase activity (SOD), leaf relative water content (RWC) and increasement in soluble sugar content (SSC)(Zhang et al. 2015), which revealed obvious morphological and physiological changes in leaves of faba bean under drought stress. Therefore, the leaves are chosen for transcriptome analysis in faba bean under soil drought in this study. We aimed to investigate the drought stress transcriptome profile of faba bean under these more natural drought conditions, which could provide a comprehensive reference for the drought tolerance mechanism in faba bean.
Materials and methods
Plant materials, growth conditions and stress treatments
Qinghai 13, cultivated by Qinghai Academy of Agricultural and Forestry Science was used in this work because of the well adaptation to drought tolerance in the previous study (Zhang et al. 2015). The seeds were surface-sterilized in 5% (v/v) sodium hypochlorite solution for 10 min followed by three sterile distilled water washes and soaked for 7 days. After that, seedlings were sowed in pots containing a mixture of soil and vermiculite (2: 1, w/w) in an controlled growth chambers (temperature of 25/18 °C (day/night), the relative humidity of 70 ± 5%, and a 16 h/8 h (day/night) photoperiod (250 μmol/m2/s light intensity)) (Li et al. 2019). Seedlings were equally irrigated with half-strength Hoagland solution for 10 days, then the pots were divided into two batches (control and water stress). After that, the control plants were irrigated regularly and the water-stressed plants were subjected to water deficit for 7 days after a sufficient irrigation. Leaves were collected and instantly frozen in liquid nitrogen, and stored at − 80 °C.
RNA isolation and sequencing
Total RNA was extracted from 100 mg leaf using the total RNA extraction kit (TIANGEN, China) according to the manufacturer’s instructions. Total RNA of each sample was quantified and qualified by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), NanoDrop (Thermo Fisher, USA) and 1% agarose gel. 1 μg total RNA with RIN value above 7 was used for following library preparation. Next-generation sequencing library preparations were constructed according to the manufacturer’s protocol (NEBNext® Ultra™ RNA Library Prep Kit for Illumina®).
The poly(A) mRNA isolation was performed using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, USA) or Ribo-Zero™ rRNA removal Kit (Illumina, USA). First-strand cDNA was synthesized using ProtoScript II Reverse Transcriptase and the second strand cDNA was synthesized using Second Strand Synthesis Enzyme Mix. The purified double-stranded cDNA was then treated with End Prep Enzyme Mix to repair both ends and add a dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends.
Size selection of Adaptor-ligated DNA was then performed using AxyPrep Mag PCR Clean-up (Axygen, USA), and fragments of ~ 360 bp (with the approximate insert size of 300 bp) were recovered. Each sample was then amplified by PCR for 11 cycles using P5 and P7 primers, with both primers carrying sequences which can anneal with flow cell to perform bridge PCR and P7 primer carrying a six-base index allowing for multiplexing. The PCR products were cleaned up using AxyPrep Mag PCR Clean-up (Axygen, USA), validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified by Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). Then libraries with different indices were multiplexed and loaded on an Illumina HiSeq X ten instrument according to manufacturer’s instructions (Illumina, San Diego, CA, USA). The sequences were processed and analyzed by GENEWIZ (China).
Data analysis
Quality Control: To remove technical sequences, including adapters, polymerase chain reaction (PCR) primers, or fragments thereof and quality of bases lower than 20, pass filter data of fasta format were processed by Cutadapt (version 1.9.1) to be high-quality clean data.
Assembly: First, assembled by Trinity, which represents a novel method for the efficient and robust de novo reconstruction of transcriptomes from RNA-Seq data. Trinity combines three independent software modules: Inchworm, Chrysalis, and Butterfly were applied sequentially to process large volumes of RNA-seq reads. Second, remove the duplicated contigs by cd-hit, then get the unigene sequence file.
Expression analysis: With the unigene sequence file as a reference gene file, RSEMestimated gene and isoform expression levels from the pair-end clean data (Li and Dewey 2011).
Differential expression analysis: Differential expression analysis used the DESeq 2 Bioconductor package, a model based on the negative binomial distribution, and calculated by the fragments per kilobase of transcript per million mapped transcript (FPKM) method for each sample. After adjusted by Benjamini and Hochberg’s approach for controlling the false discovery rate, differentially expressed genes (DEGs) were determined by setting the thresholds for false discovery rate (FDR) < 0.05 and the |log2ration| ≥ 1 by performing pairwise comparisons for the treatment and control samples.
GO and KEGG enrichment analysis: GO-TermFinder was used to identify Gene Ontology (GO) terms that annotate a list of enriched genes with a p value that less than 0.05. KEGG (Kyoto Encyclopedia of Genes and Genomes) is a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances (http://en.wikipedia.org/wiki/KEGG).
Annotation: Use blast software to annotate the unigene sequence. All the database includes NCBI nonredundant protein (Nr), clusters of orthologous groups (COG), Swissprot, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO).
Quantitative qRT-PCR analysis
RNA-Seq results were verified by qRT-PCR with 12 randomly selected DEGs. RNA exacted from the stored sample at -80 °C which mentioned in the plant material above. The experiment was conducted using 2 × SYBR Premix Ex Taq™ II (TaKaRa) on a LightCycle®480II (Roche) Real-time Detection System. The primers were designed with Primer 5. The ELF1A gene of in faba bean was used as an internal control (Gutierrez et al. 2011). Three independent biological replicates were used in this assay and the relative mRNA expression level was calculated as 2−∆∆Ct.
Results
Analysis of transcriptome sequencing data
The leaves of Qinghai 13 that were drought-stressed for 7 days were selected because of significant differences observed in soil water content (SWC), relative water content (RWC) and physiological indices compared with that in the control leaves (Zhang et al. 2015). Six cDNA libraries were prepared from mRNA, that was extracted from the leaves of the control and drought-stressed plants of Qinghai 13. The libraries were termed as C-1, C-2, C-3 (three replications of the control group) and D-1, D-2, D-3 (three replications of the stress group). These libraries were sequenced by Illumina deep-sequencing, and a filtering process was performed on the raw sequencing reads.
The transcriptome sequencing and assembly are shown in Table 1. The cDNA libraries from C-1, C-2 and C-3 (control samples) produced a total of 42.5 M, 39.26 M and 40.48 M raw reads, with 42.45 M, 39.22 M and 40.43 M total clean reads. Clean reads had a Q20 of 97.69%, 97.85% and 97.35%, respectively (Table 1). The cDNA libraries from D-1, D-2 and D-3 (drought samples) produced total raw reads of 39.86 M, 47.77 M and 47.35 M, with total clean reads of 39.82 M, 47.73 M and 47.30 M. Clean reads had a Q20 of 97.76%, 97.46% and 97.33%, respectively (Table 1). The Q20 clean reads were consistently greater than 97%, indicating high-quality sequencing (Table 1).
Table 1.
Summary of transcriptome sequencing
| Sample no. | Total raw reads (M) | Total clean reads (M) | Bases | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|
| C-1 | 42504992 (42.5 M) | 42450700 (42.45 M) | 6298313539 | 97.69 | 93.38 | 43.36 |
| C-2 | 39266402 (39.26 M) | 39225018 (39.22 M) | 5822251138 | 97.85 | 93.72 | 43.30 |
| C-3 | 40480440 (40.48 M) | 40438798 (40.43 M) | 6002963474 | 97.35 | 92.64 | 43.26 |
| D-1 | 39865584 (39.86 M) | 39825266 (39.82 M) | 5912995882 | 97.76 | 93.47 | 43.28 |
| D-2 | 47778828 (47.77 M) | 47730534 (47.73 M) | 7091246545 | 97.46 | 92.89 | 43.17 |
| D-3 | 47358562 (47.35 M) | 47308692 (47.30 M) | 7025370995 | 97.33 | 92.56 | 43.21 |
C-1, C-2, C-3: three replications of the control group leaves; D-1, D-2, D-3: three replications of the stress group leaves; Total raw reads: the number of reads before filtering; Total clean reads: the number of reads after filtering; Bases: total number of bases after filtration; Q: Q scores are used to measure base calling accuracy; GC: GC content
All libraries were assembled with high stringency and the results are summarized in Table 2. 14042026 contigs consisting of 664050651 bases were assembled into 176334 unigenes with an average length of 766 bp and N50 length of 1239 bp (Table 2). The distribution of unigene length is shown in Table S1 and Fig.S1. A total of 97302 (55.18%) unigenes ranged from 200 to 500 bp in length. A total of 38346 (21.75%) unigenes ranged from 500 to 1000 bp in length and a total of 31369 (23.05%) unigenes were longer than 1500 bp. The proportions of A + T and G + C were 61.52% and 38.48%, respectively (Table S1). The transcriptome data of the six faba bean libraries were deposited in the NCBI-SRA database with the following accessions: SRX7873340, SRX7873341, SRX7873342, SRX7873343, SRX7873344 and SRX7873345.
Table 2.
Assembly statistics of unigenes
| Type | Sequences | Bases | Min | Max | Average | N50 | (A + T) % | (C + G) % |
|---|---|---|---|---|---|---|---|---|
| All_Contig | 14042026 | 664050651 | 25 | 18087 | 47.29 | 47 | 58.91 | 41.09 |
| All_Unigene | 176334 | 135066464 | 201 | 20142 | 765.97 | 1239 | 61.52 | 38.48 |
Annotation statistic of unigenes in different databases
A sequence similarity search for all assembled unigenes was executed against the Nr, COG, Swissprot, KEGG and GO databases (Table 3 and Fig.S2). Of the 176334 unigenes, 85057 (48.23%) shares homology with members of the Nr database and 85206 unigenes had hits in the GO database (48.32%). Additionally, 37597 (21.32%) unigenes were annotated in COG, 51948 (29.46%) in SwissProt and 18047 (10.23%) in KEGG (Table 3 and Fig.S2).
Table 3.
Annotation statistic of unigenes in different database
| Database categories | Number | Percentage |
|---|---|---|
| All assembled unigenes | 176334 | 100% |
| Nr | 85057 | 48.23% |
| KOG | 37597 | 21.32% |
| Swissprot | 51948 | 29.46% |
| KEGG | 18047 | 10.23% |
| GO | 85206 | 48.32% |
| All annotated unigenes | 88593 | 50.24% |
Characterization of all unigenes
The distribution of the top annotated unigenes against the Nr database is shown in Fig.S3. Approximately 35540 (41.78%) unigene sequences were similar to the model legume Medicago truncatula, 19696 unigenes (23.15%) were similar to Cicer arietinum, 4599 unigenes (5.4%) were similar to Cajanus cajan and 4013 unigenes (4.71%) were similar to Glycine max. An additional 8.03% the sequences showed similarities to other organisms, including Pisum sativum, Trifolium subterraneum, Glycine soga, Mus musculus, Vigna radiata var. radiata and Phaseolus vulgaris (Fig. S3). Overall, nearly 70% of the assembled unigenes had high similarity with the leguminous plants.
To predict and classify the putative functions of these unigenes, all unigenes were compared against the COG database. A total of 37579 COG -annotated putative proteins were classified into 25 families (Fig. S4). The largest category among these families was the “general function prediction only” (5098 unigenes) followed by “signal transduction mechanisms” (4953 unigenes) and “posttranslational modification, protein turnover, chaperones” (4252 unigenes). In addition, there were a large number of unigenes belonging to “carbohydrate transport and metabolism” (2385 unigenes), “translation, ribosomal structure and biogenesis” (2271), “transcription” (2153), “intracellular trafficking, secretion, and vesicular transport” (1976), “RNA processing and modification” (1833), “lipid transport and metabolism” (1829), “energy production and conversion” (1795) and “amino acid transport and metabolism” (1795). There were also 2521 unigenes belonging to the category of “function unknown”.
For GO terms, a total of 85206 (48.32%) unigenes were assigned to GO ontologies based on their sequence similarity with genes that have known functions. These hits were categorized into 57 functional groups in the three main categories of molecular function, biological process and cellular components (Fig. 1, Table S2). In the molecular function category, the most represented GO terms were related to binding (15776) and catalytic activity (15433). In addition, the most frequent terms in the molecular function category were transporter activity (1576), structural molecule activity (712), and electron carrier activity (499). In the cellular component category, cell part (4250), membrane part (3144) and organelle (3052) were the most highly represented categories. Additionally, macromolecular complex (2306), organelle part (1938) and membrane (1836) were also highly represented in the cellular component category. In the biological process category, the most represented terms were associated with the metabolic process (11359) and cellular process (9029). In addition, single-organism process (4706), biological regulation (3140), response to a stimulus (1386) and localization (1360) were the most frequent groups in the biological process category (Fig. 1, Table S2).
Fig. 1.

Histogram of GO terms assigned to all assembled unigenes. X axis represents corresponding number of unigenes in certain category. Y axis represents unigenes are categorized into three main groups: molecular function, cellular components and biological process
Next, the metabolic pathways that annotated unigenes participated in were investigated by comparing to the KEGG database. A total of 18047 annotated unigenes were mapped to 148 KEGG reference pathways. These can be classified into five different functional groups including metabolism, genetic information processes, environmental information processing, organismal system and cellular processes (Fig. 2). In the metabolism functional groups, unigenes were predominantly involved in “global and overview maps” (13592), “carbohydrate metabolism” (4190), “amino acid metabolism” (2179), “lipid metabolism” (2088), “energy metabolism” (1473) and “nucleotide matabolism” (1063). Among the genetic information processing functional groups, the majority of the unigenes were involved in “translation” (3673), “folding, sorting, and degradation” (2195), “transcription” (1098) and “replication and repair (912)”. In the environmental information processing functional groups, “signal transduction” (932) and “membrane transport” (255) were most frequent. In the organismal systems functional groups, unigenes only involved in “environmental adaptation” (682) were found (Fig. 2). Meanwhile, in cellular processes functional groups, unigenes involved in only “transport and catabolism” (1694) were found (Fig. 2). Detailed information about functional pathways found by comparing against the KEGG database is shown in Table S3.
Fig. 2.

Functional distribution of KEGG terms assigned to all assemble unigenes. X axis represents the number of unigenes in a certain category. Y axis represents the KEGG functional category
Identification of differentially expressed genes
The expression values of the unigenes were analyzed via the FPKM method (Table S4). Genes which were differentially expressed between the control group and the stress group were identified in pairwise comparisons with the following criteria: log2FoldChange ≥ 1 or ≤ − 1, FDR ≤ 0.05. As shown in Fig.S5, a total of 4439 upregulated genes and 4687 downregulated genes were identified when comparing the drought stress group against the control group. The overall differential expression pattern was also visualized with a volcano plot (Fig. S6). The results above revealed a specific expression profile in response to soil drought stress in faba bean leaf. The genes with the strongest drought induction included a ribulose bisphosphate carboxylase small chain member, SLOW GREEN1 (chloroplastic), delta-1-pyrroline-5-carboxylate synthase, nuclear poly(A) polymerase and protein translocase subunit SECA2 (Table S4). Genes which were most repressed by drought included glutaredoxin, phosphatase 2C and cyclic nucleotide-binding and lysine-specific demethylase JMJ30 (Table S4).
In addition to the above genes, a total of 324 putative transcription factors (TFs) were found to be significantly regulated during drought stress. As shown in Fig. 3, the top 11 transcription factor families included 54 NAC, 36 bHLH, 26 MYB, 23 C3H, 18 GRAS, 17 WRKY, 9 FAR1, 6 HSF, 9 AP-BREBP, 4 Trihelix and 4 GRF. As is known, these transcription factor families play important roles during abiotic stress including salt, drought, cold, heat and so on (Udvardi et al. 2007). Finally, a hierarchical clustering heat map of DEGs was constructed to show control and soil drought transcriptomes (Fig. 4).
Fig. 3.
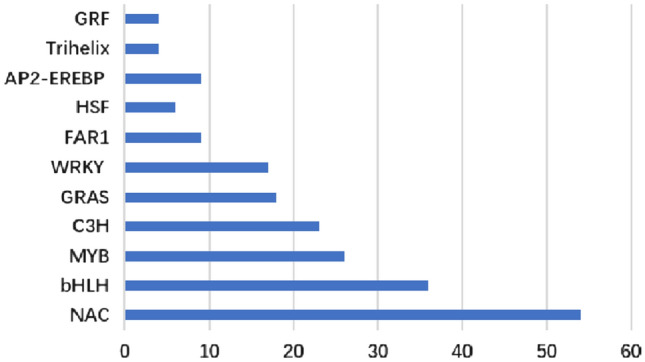
Top 11 families of differentially expressed transcription factors response to soil drought stress in faba bean leaves. X axis represents the corresponding number of differentially expressed unigenes in certain transcription factor family. Y axis represents the transcription factor family classification
Fig. 4.

A hierarchical clustering heat map showing DEGs identified in the transcriptomes of control samples (three replications:C-1, C-2, C-3) and soil drought-stressed samples (three replications:D-1, D-2, D-3). Relative gene expression fold-changes (log2FPKM) are color coded (red upregulated and green downregulated, the deeper color representing greater difference in relative gene expression)
A total of 12 DEGs were randomly selected to conduct qRT-PCR for validation of the RNA-Seq data. Detail description and the primers of the 12 selected DEGs are listed in Table 4. Six upregulated DEGs, including D-TRINITY_DN33510 (ribulose-1,5-bisphosphate carboxylase small subunit), D-TRINITY_DN24736 (chitinase family protein), D-TRINITY_DN43139 (Rab GTPase activator), D-TRINITY_DN68230 (NAC transcription factor) and 2 genes of unknown function (D-TRINITY_DN47859 and D-TRINITY_DN47873), were assayed with qRT-PCR. Six downregulated DEGs, including C-TRINITY_DN38456 (myb transcription factor), C-TRINITY_DN32684 (transmembrane protein), C-TRINITY_DN37999 (ethylene response factor), D-TRINITY_DN46507 (auxilin-related protein) and 2 genes of unknown function (C-TRINITY_DN32373 and C-TRINITY_DN24815), were also screened for qRT-PCR. The expression levels of selected DEGs that were measured by qRT-PCR were quantified with log2foldchange value and all the DEGs showed an approximately similar expression compared with the transcriptome analysis (Table 4). These qRT-PCR results demonstrated the high quality and reliability of the RNA-seq data.
Table 4.
Detail of the primers of 12 randomly selected DEGs for qRT-PCR
| Gene no. | Gene ID | Description | Primers sequence (5′–3′) | Production (bp) |
log2Foldchange (RNA-Seq) |
log2Foldchange (qRT-PCR) |
|---|---|---|---|---|---|---|
| 1 |
D-TRINITY_ DN33510_c1_g1_i1 |
ribulose-1,5-bisphosphate carboxylase small subunit |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
177 | 7.68 | 6.5 |
| 2 |
D-TRINITY_ DN47859_c3_g5_i1 |
Unknown |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
169 | 6.39 | 5.4 |
| 3 |
D-TRINITY_ DN24736_c1_g1_i1 |
chitinase family protein |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
167 | 6.1 | 5.9 |
| 4 |
D-TRINITY_ DN47873_c3_g7_i6 |
Unknown |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
158 | 4.9 | 3.7 |
| 5 |
D-TRINITY_ DN43139_c2_g1_i13 |
Rab GTPase activator |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
146 | 3.2 | 2.7 |
| 6 |
D-TRINITY_ DN68230_c0_g1_i1 |
NAC transcription factor |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
166 | 3.03 | 1.8 |
| 7 |
D-TRINITY_ DN32373_c1_g2_i1 |
Unknown |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
150 | − 6.15 | − 4.6 |
| 8 |
D-TRINITY_ DN38456_c0_g1_i3 |
myb transcription factor |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
148 | − 5.34 | − 5.4 |
| 9 |
D-TRINITY_ DN32684_c0_g1_i1 |
hitinase family protein |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
175 | − 5 | − 4.6 |
| 10 |
D-TRINITY_ DN37999_c1_g1_i6 |
ethylene response factor |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
165 | − 3 | − 1.9 |
| 11 |
D-TRINITY_ DN46507_c0_g1_i2 |
auxilin-related protein |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
151 | − 3.9 | − 4.3 |
| 12 |
D-TRINITY_ DN24815_c0_g1_i1 |
Unknown |
Forward:CTTTGAGTTGAAGCATGAAGCA Reverse:ATTGTTGGCCTTCTTGGTAACCG |
148 | − 2.9 | − 2 |
Functional annotation of DEGs
There were a significant amount of DEGs in response to soil drought. To classify the functions of the DEGs, we compared all the DEGs to the GO database to search for significantly enriched GO terms. This analysis revealed enrichment in major biological processes, molecular functions and cellular components (Fig. 5). In the molecular function category, the most significant GO terms were catalytic activity, binding and transport activity, electron carrier activity, nucleic binding transcription factor activity, structural molecule activity, enzyme regulator activity, antioxidant activity, molecular transducer activity, nutrient reservoir activity and protein binding transcription factor activity. The most enriched terms in the cellular component category were cell part, membrane part, organelle, membrane, macromolecular complex, organelle part, extracellular region, membrane-enclosed lumen and extracellular region part. In the biological process category, DEGs were enriched in metabolic process, cellular process, single-organism process, biological regulation, response to stimulus, localization, multi-organism process, development process, cellular component organization biogenesis, cell killing, immune system process and growth.
Fig. 5.

GO enrichment distribution of differentially expressed genes. X axis represents the corresponding number of DEGs. Y axis indicates DEGs are categorized into three main groups: molecular function, cellular components and biological process
KEGG pathway enrichment of DEGs
As shown in Table 5, DEGs were mainly enriched in three major KEGG pathways, including “metabolism”, “Environmental Information Processing” and “Genetic Information Processing”. The largest number of unigene-containing pathways in the “metabolism” category were assigned to the metabolic pathway (ko01100; 486, 46.15%) and biosynthesis of secondary metabolites (ko01110; 281, 26.69%). The largest number of unigene-containing pathways in the “Genetic Information Processing” were assigned to DNA replication (ko03030; 20, 1.9%). There were also two pathways belonging to the “Environmental Information Processing” category, including MAPK signaling pathway-plant (ko04016; 38, 3.61%) and plant hormone signal transduction (ko04075; 74, 7.03%).
Table 5.
KEGG pathway annotation and number of DEGs
| Pathway category | Pathway ID |
DEGs with pathway annotation (1053) | All genes with pathway annotation (18047) | Qvalue |
|---|---|---|---|---|
| Pentose and glucuronate interconversions | ko00040 | 20 (1.90%) | 158 (0.88%) | 4.30E − 03 |
| Flavonoid biosynthesis | ko00941 | 11 (1.04%) | 64 (0.35%) | 4.01E − 03 |
| Linoleic acid metabolism | ko00591 | 16 (1.52%) | 103 (0.57%) | 1.44E − 03 |
| Photosynthesis—antenna proteins | ko00196 | 16 (1.52%) | 100 (0.55%) | 1.31E − 03 |
| MAPK signaling pathway-plant | ko04016 | 38 (3.61%) | 306 (1.70%) | 1.55E − 04 |
| Plant hormone signal transduction | ko04075 | 74 (7.03%) | 707 (3.92%) | 2.19E − 05 |
| Cutin, suberine and wax biosynthesis | ko00073 | 9 (0.85%) | 48 (0.27%) | 4.30E − 03 |
| Ubiquinone and other terpenoid-quinone biosynthesis | ko00130 | 17 (1.61%) | 146 (0.81%) | 1.58E − 02 |
| Isoquinoline alkaloid biosynthesis | ko00950 | 12 (1.14%) | 65 (0.36%) | 1.44E − 03 |
| Tropane, piperidine and pyridine alkaloid biosynthesis | ko00960 | 13 (1.23%) | 89 (0.49%) | 5.95E − 03 |
| Tyrosine metabolism | ko00350 | 18 (1.71%) | 149 (0.83%) | 9.15E − 03 |
| Biosynthesis of secondary metabolites | ko01110 | 281 (26.69%) | 3670 (20.23%) | 1.62E − 05 |
| Phenylpropanoid biosynthesis | ko00940 | 47 (4.46%) | 451 (2.5%) | 1.05E − 03 |
| Metabolic pathways | ko01100 | 486 (46.15%) | 7274 (40.31%) | 1.02E − 03 |
| Monoterpenoid biosynthesis | ko00902 | 6 (0.57%) | 31 (0.17%) | 1.36E − 02 |
| Zeatin biosynthesis | ko00908 | 6 (0.57%) | 25 (0.14%) | 4.50E − 03 |
| Sesquiterpenoid and triterpenoid biosynthesis | ko00909 | 4 (0.38%) | 21 (0.12%) | 4.36E − 02 |
| DNA replication | ko03030 | 20 (1.90%) | 166 (0.92%) | 6.12E − 03 |
In addition to the pathways mentioned above, there were some pathways assigned to pentose and glucuronate interconversions (ko00040; 20, 1.9%), flavonoid biosynthesis (ko00941; 11, 1.04%), linoleic acid metabolism (ko00591; 16, 1.52%), photosynthesis-antenna proteins (ko00197; 16, 1.52%), cutin, suberine and wax biosynthesis (ko00073; 9, 0.85%), ubiquinone and other terpenoid-quinone biosynthesis (ko00130; 17, 1.61%), isoquinoline alkaloid biosynthesis (ko00950; 12, 1.14%), tropane, piperidine and pyridine alkaloid biosynthesis (ko00960; 13, 1.23%), tyrosine metabolism (ko00350; 18, 1.71%), phenylpropanoid biosynthesis (ko00940; 47, 4.46%), monoterpenoid biosynthesis (ko00902; 6, 0.57%), zeatin biosynthesis (ko00908; 6, 0.57%) and sesquiterpenoid and triterpenoid biosynthesis (ko00909; 4, 0.38%). As a result, a total of 1053 DEGs were identified as being associated with 18 most strongly represented KEGG pathways through mapping DGEs to the KEGG database (Fig. 6). These results are visualized in Fig. 6 and full information is available in Table S5.
Fig. 6.

Scatter diagram of enriched KEGG pathways for DEGs in soil drought-stressed samples (D) vs. control samples (C). The 18 most strongly representative pathways are displayed. X axis represents RichFactor, a result of the ratio of DEG number to all annotated genes in certain pathway. Y axis represents KEGG term. The colour of the dots represents the range of the -log10 (QValue). The area of black dot means the number of DEGs in certain pathway
Discussion and conclusion
Water scarcity greatly influences the yield of faba bean (Vicia faba L.) in the Qinghai province, but the mechanisms behind faba bean drought tolerance are not well-understood. Therefore, the main objective of this study was to determine how the drought-tolerant faba bean genotype Qinghai 13 alters its transcriptome in response to water deficit.
The leaves of Qinghai 13 were sequenced with an Illumina HiSeq X ten instrument, which resulted in 257.22 M raw reads and 256.95 M clean reads from six cDNA libraries. This is greater than the depth obtained by some previous studies 65.8 M (Arun-Chinnappa and McCurdy. 2015), 33.023 M (Ocaña et al. 2015), 304680 (Kaur et al. 2012), but is smaller than others (606.35 M, Khan et al. 2019). However, the high Q20 percentage (97%) indicated that the sequencing was extremely high quality. In this study, we obtained a total of 176, 334 unigenes, which was larger than 164679 reported by Khan (Khan et al. 2019). Among the assemble unigenes, 88593 (50.24%) were annotated, while 87751 (49.76%) unigenes were not. The annotated unigenes showed high similarity with the model legumes Medicago truncatula (41.78%) and Cicer arietinum (23.15%), which is consistent with the result reported by Khan (Khan et al. 2019). GO terms of the annotated unigenes mainly involved categories associated with molecular function and biological process, which may indicate a common role in plant growth and development, and stress tolerance mechanisms.
DEGs were identified through transcriptome comparison between the control and drought-stressed leaf samples. A total of 9126 unigenes with significant differential expression were identified with the following criteria: log2FoldChange ≥ 1 or ≤ − 1, FDR ≤ 0.05. The number of the DEGs in this work is far smaller than the number found in leaves of different developmental stages under PEG6000 stress, which shows that a different spectrum of drought-responsive genes in faba bean is present when using different water-deficit-stress experimental systems. Similar work in wheat has also found that its proteome exhibited obvious differences under soil drought compared to PEG stress (Cui et al. 2019). We concluded that the transcriptome response to drought in faba bean may be different due to a variety of factors, including stress time, osmotic pressure and others.
Meanwhile, GO enrichment analysis of DEGs showed enrichment in “catalytic activity” and “binding” in the molecular function category, as well as “cellular process” and “metabolic process” in the biological process category. This indicated a common response mechanism in both PEG stress and soil drought, which has also been reported in peanut (Arachis hypogaea L.) and Glycine max (Brasileiro et al. 2015; Tripathi et al. 2016; Yang et al. 2017).
There were many pathways (133) enriched under PEG in a previous study (Khan et al. 2019), but only 18 of them overlapped with our results. This lack of overlap could have several possible explanations. For example, carbon fixation in photosynthetic organisms and glycolysis/gluconeogenesis were significantly increased by PEG6000 stress in faba bean and other crops (Valluru and Van den Ende 2008; Yang et al. 2017; Fan et al. 2018; Wu et al. 2018). Additionally, arginine and proline metabolism were also significantly increased under PEG6000 stress in faba bean and other species (Armengaud et al. 2004; Wu et al. 2018; Cui et al. 2019). Genes involved in ribosome biogenesis have also been shown to be differentially expressed in PEG6000 stressed plants (Cui et al. 2019; Khan et al. 2019), but were unchanged in faba bean under soil drought stress. In a previous study, genes involved in the citrate cycle, glyoxylate and dicarboxylate metabolism and pyruvate metabolism were differentially expressed under PEG6000 stress, but they remained largely unchanged under soil drought in Jute (Yang et al. 2017).
Although drought and PEG stress elicit some similar response (such as phytohormonal balance and leaf water content), the osmotic stress caused by PEG causes the two to diverge for several reasons, including the different length of possible treatment (Cui et al. 2019). Undoubtedly, there are several common pathways that are shared by PEG stress and soil drought, because majority of the responsive pathways in soil drought can be annotated from the PEG stress. For example, phenylpropanoid biosynthesis, biosynthesis of secondary metabolites, photosynthesis-antenna proteins, MAPK signaling pathway-plant, plant hormone signal transduction, cutin, suberin and wax biosynthesis, ubiquinone and other terpenoid-quinone biosynthesis, phenylpropanoid biosynthesis are all regulated in both stresses. Considering these results, PEG stress may be redundant compared to simply testing soil drought directly. Soil drought could undoubtedly be better understood by the addition of experiment which uses multiple water-deficit-stress experimental systems, or studies which combine transcriptome, proteome and metabolome data (Hamanishi et al. 2015; Kosová et al. 2016; Shen et al. 2016; Savoi et al. 2017). Such work would enable a better understanding of the physiological or biochemical processes associated with drought stress and provide insight into the possible molecular mechanisms behind these responses in faba bean.
In this work, the drought-induced genes were mainly classified into two major categories: regulatory proteins and functional proteins. Regulatory proteins mainly included transcription factors, protein kinases, protein phosphatases and regulators in signal transduction. Differentially expressed transcription factors, including NAC, bHLH, MYB, WRKY, AP2-EREBP were identified under soil drought and also found to some extent in PEG stress (Khan et al. 2019). This overlap indicated a common function of the transcription factor families of NAC, bHLH, WRKY, MYB, AP2-EREBP during water deficit (Reddy et al. 2008; de Zélicourt et al. 2012; Bhatnagar-Mathur et al. 2014;Sosa-Valencia et al. 2017). Among the transcription factors, the expression of putative MYB59 was found to be substantially reduced, which fits with its known role in calcium signal regulation in Arabidopsis thaliana during stress (Fasani et al. 2019). Members of NAC family are involved in abiotic stress response and symbiotic nodule senescence (de Zélicourt et al. 2012), and also represent the family with the most differentially expressed members in faba bean under soil drought. Transcription factors of the bHLH family were also differentially expressed in faba bean under drought stress. It has been reported that the bHLH member AtMYC2 can function as a transcriptional activator in ABA-inducible gene expression under drought stress in plants (Abe et al. 2003). DEGs also included members of AP2/EREBP family, which is known to function in mediating cuticular permeability, sensitivity to abscisic acid (ABA), and drought resistance by regulating wax biosynthesis (Zhang et al. 2019).
DEGs encoding receptor-like kinases and LRR receptor-like kinases were found in our differential testing. DEGS encoding protein kinases including calcium-dependent protein kinases, serine/threonine protein kinases, light-sensor protein kinases, cysteine-rich receptor-kinase-like protein and mitogen-activated protein kinases were also differently expressed in faba bean leaves under soil drought. The protein kinase is known to function as sensor response genes for initiating phosphorylation cascades (Singh et al. 2015). Besides, DEGs involved in the regulation of signal transduction such as plant hormone regulation, signaling molecule regulation, redox reaction, and carbohydrate and sugar metabolism were differentially expressed in this work.
Additionally, functional genes encoding proteins involved in various functional processes were differently regulated under soil drought in faba bean. Several DEGs identified in this study belong to the aquaporin family, whose members facilitate water uptake across cell membranes in maintaining cellular water homeostasis (Javot and Maurel 2002). That would be accountable for the low water uptake and consequent reduction in relative water content (RWC) of leaves in Qinghai 13 faba bean under soil drought (Zhang et al. 2015). In addition, DEGs encoding soluble sugars synthetases and sugar transporter were also differentially expressed. The soluble sugar is also an osmolyte and signaling molecule expressed under drought stress. Furthermore, DEGs encoding functional proteins also included enzymatic compounds triggered by reactive oxygen species (ROS), ABC-transporter proteins, chloride channel, Na+/K+ transporter, late embryogenesis abundant protein (LEA) and other drought-induced proteins.
In this study, the most drought-induced gene encodes a ribulose-1,5-bisphosphate carboxylase small subunit. It is reported that severe drought limits the quantum efficiency of PS II during photosynthesis by reducing the activity of ribulose-1,5-bisphosphate carboxylase (Rubisco) (Carmo-Silva et al. 2012). Additionally, a group of genes encoding chloroplastic were identified, which fits with its known role as a major player in photosynthesis (Carmo-Silva et al. 2012). As a result, chlorophyll content showed a visible reduction in faba bean leaves under soil drought (Zhang et al. 2015). All of the above data indicated that drought-induced differential expression results in major changes to photosynthetic machinery.
In this study, we also identified genes encoding Δ1-pyrroline-5-carboxylate synthetase (P5CS1), which is a major component of proline biosynthesis, and proline dehydrogenase 1 (PDH1), which is related to proline catabolism. Proline is thought to contribute to osmotic adjustment and the stabilization of subcellular structures under stress conditions (Ashraf and Foolad 2007). The Arabidopsis p5cs1 and pdh1 are deficient in stress-induced proline synthesis, which lead to a reduction in proline catabolism (Sharma et al. 2011). This implies that P5CS1 and PDH1 play important roles in osmotic adjustment during water stress through regulating proline biosynthesis and catabolism (Khan et al. 2019). A homolog of the BRCA1 gene 1(NBR1), was the most down-regulated DEG in faba bean during soil water deficit and the Arabidopsis nbr1 mutant was previously shown to have reduced drought tolerance (Zhou et al. 2013). In faba bean, NBR1 also play an important role in adapting to drought stress through mediating autophagy.
The production of reactive oxygen species (ROS) is an early consequence of plant defense response to water stress and acts as a secondary messenger to trigger subsequent adaptive responses (Miller et al. 2010). A large number of genes encoding detoxification enzymes such as glutathione S transferase (GST), ascorbate peroxidase (APX), mono dehydroascorbate reductase (MDAR), glutathione peroxidase (GPX) and glutathione reductase (GR) were differentially expressed to execute cell protection in faba bean under soil drought. ROS signaling under drought is linked to abscisic acid (ABA) and Ca2+ changes (Kaur and Asthir 2017). It is well known that abscisic acid (ABA) is a very important signaling molecule during drought stress. In this study, a group of 2C-type protein phosphatases, which affect the ABA pathway, were differentially expressed. In addition, 9-cis-epoxycarotenoid dioxygenases (NCED1 and NCED3), key enzymes in ABA biosynthesis, were also differentially expressed. Overexpression of several genes involved in ABA biosynthesis has been shown to result in improved drought tolerance in petunia plants (Iuchi et al. 2000).
Lastly, many non-annotated DEGs were identified from the assembled unigenes (Table S4). Although the function of these genes is still unknown, many of them were differentially expressed during drought stress. Elucidating the function of these drought-regulated unannotated genes may prove useful for improving drought tolerance in the future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1 Pie chart for length distribution of all assembled unigenes
Fig. S2 Venn diagram between Nr, Swissport, KEGG and COG. The number of unique and shared unigenes are shown
Fig. S3 Analysis of the distribution of annotated species through mapping annotated unigenes against NR database. X axis shows top ten annotated species which shared a high similarity with faba bean. Y axis represents annotated unigenes number in certain annotated species
Fig. S4 Clusters of orthologous groups (COG) function classification of all assembled unigenes. X axis shows the unigenes were assigned to 25 categories and the categories were listed on the right of the histogram. Y axis indicates the corresponding number of unigenes in certain COG categories
Fig. S5 Differentially expressed genes in soil drought stressed samples vs. control samples. X axis represents two groups: C, control group; D, soil drought stressed group. Y axis represents the number of upregulated (red) and downregulated (blue) genes
Fig. S6 Volcano plot (log2 (fold change) vs -log10(FDR)) of DEGs in faba bean leaves under soil drought stress. C, control group; D, soil drought stressed group; red dots, upregulated genes; blue dots, downregulated genes; black dots, non-differentially expressed genes
Supplementary material 10 (XLSX 3041 kb)
Acknowledgements
This work was supported by The Project of Qinghai Science & Technology Department (2018-ZJ-940Q), The Open Project of State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University (2017-ZZ-12, 2016-ZZ-04), The China Agriculture Research System (CARS-09), Open Fund of Qinghai Province Key Laboratory of Physical Geography and Environmental Process (2018-QZH-K06) and National Natural Science Foundation of China (31960339).
Author contributions
YL developed the experimental design; XW and YF prepared the samples for RNA-seq, conducted physiological experiments and RT-qPCR; YL and LL supervised all experiments; XW and LL wrote the article.
Availability of data and material
All the sequencing data have been deposited in the NCBI-SRA database with the following accessions: SRX7873340, SRX7873341, SRX7873342, SRX7873343, SRX7873344 and SRX7873345.
Compliance with ethical standards
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Abdelmula AA, Link W, Kittlitz EV, Stelling D. Heterosis and inheritance of drought tolerance in faba bean, Vicia faba L. Plant Breed. 1999;118:485–490. doi: 10.1046/j.1439-0523.1999.00411.x. [DOI] [Google Scholar]
- Abe H, Urao T, Ito T, et al. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003;15:63–78. doi: 10.1105/tpc.006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abid G, Muhovski Y, Mingeot D, et al. Identification and characterization of drought stress responsive genes in faba bean (Vicia faba L.) by suppression subtractive hybridization. Plant Cell Tiss Organ Cult. 2015;121:367–379. doi: 10.1007/s11240-014-0707-x. [DOI] [Google Scholar]
- Abid G, M’hamdi M, Mingeot D, et al. Effect of drought stress on chlorophyll fluorescence, antioxidant enzyme activities and gene expression patterns in faba bean (Vicia faba L.) Arch Agron Soil Sci. 2016;63(4):536–552. doi: 10.1080/03650340.2016.1224857. [DOI] [Google Scholar]
- Alghamdi SS, Khan MA, Ammar MH, et al. Characterization of drought stress-responsive root transcriptome of faba bean (Vicia faba L.) using RNA sequencing. 3 Biotech. 2018;8:1–19. doi: 10.1007/s13205-018-1518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amede T, Kittlitz EV, Schubert S. Differential drought responses of faba bean (Vicia faba L.) inbred lines. J Agron Crop Sci. 1999;183:35–45. doi: 10.1046/j.1439-037x.1999.00310.x. [DOI] [Google Scholar]
- Amede T, Schubert S, Stahr K. Mechanisms of drought resistance in grain legumes I: osmotic adjustment. SINET Ethiop J Sci. 2003;26:37–46. [Google Scholar]
- Ammar MH, Anwar F, El-Harty EH, et al. Physiological and yield responses of faba bean (Vicia faba L.) to drought stress in managed and open field environments. J Agron Crop Sci. 2015;201:280–287. doi: 10.1111/jac.12112. [DOI] [Google Scholar]
- Ammar MH, Khan AM, Migdadi HM, et al. Faba bean drought responsive gene identification and validation. Saudi J Biol Sci. 2017;24:80. doi: 10.1016/j.sjbs.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengaud P, Thiery L, Buhot N, et al. Transcriptional regulation of proline biosynthesis in Medicago truncatula reveals developmental and environmental specific features. Physiol Plant. 2004;120:442–450. doi: 10.1111/j.0031-9317.2004.00251.x. [DOI] [PubMed] [Google Scholar]
- Arun-Chinnappa KS, McCurdy DW. De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front Plant Sci. 2015 doi: 10.3389/fpls.2015.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf M, Foolad MR. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ Exp Bot. 2007;59:206–216. doi: 10.1016/j.envexpbot.2005.12.006. [DOI] [Google Scholar]
- Bhatnagar-Mathur P, Rao JS, Vadez V, et al. Transgenic peanut overexpressing the DREB1A transcription factor has higher yields under drought stress. Mol Breed. 2014;33:327–340. doi: 10.1007/s11032-013-9952-7. [DOI] [Google Scholar]
- Braich S, Sudheesh S, Forster JW, Kaur S. Characterisation of Faba Bean (Vicia faba L) transcriptome using RNA-Seq: sequencing, de novo assembly, annotation, and expression analysis. Agronomy. 2017;7:53. doi: 10.3390/agronomy7030053. [DOI] [Google Scholar]
- Brasileiro ACM, Morgante CV, Araujo ACG, et al. Transcriptome profiling of wild arachis from water-limited environments uncovers drought tolerance candidate genes. Plant Mol Biol Rep. 2015;33:1876–1892. doi: 10.1007/s11105-015-0882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray EA. Genes commonly regulated by water-deficit stress in Arabidopsis thaliana. J Exp Bot. 2004;55(407):2331–2341. doi: 10.1093/jxb/erh270. [DOI] [PubMed] [Google Scholar]
- Carmo-Silva AE, Gore MA, Andrade-Sanchez P, et al. Decreased CO2 availability and inactivation of Rubisco limit photosynthesis in cotton plants under heat and drought stress in the field. Environ Exp Bot. 2012;83:1–11. doi: 10.1016/j.envexpbot.2012.04.001. [DOI] [Google Scholar]
- Cazzato E, Tufarelli V, Ceci E, et al. Quality, yield and nitrogen fixation of faba bean seeds as affected by sulphur fertilization. Acta Agric Scand Sect B Soil Plant Sci. 2012;62(8):732–738. [Google Scholar]
- Chaves MM, Maroco JP, Pereira JS. Understanding plant responses to drought—from genes to the whole plant. Funct Plant Biol. 2003;30:239–264. doi: 10.1071/fp02076. [DOI] [PubMed] [Google Scholar]
- Cooper JW, Wilson MH, Derks MFL, et al. Enhancing faba bean (Vicia faba L.) genome resources. J Exp Bot. 2017;68:1941–1953. doi: 10.1093/jxb/erx117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Zhao Y, Zhang J, et al. Proteomic analysis of the similarities and differences of soil drought and polyethylene glycol stress responses in wheat (Triticum aestivum L.) Plant Mol Biol. 2019;100:391–410. doi: 10.1007/s11103-019-00866-2. [DOI] [PubMed] [Google Scholar]
- de Zélicourt A, Diet A, Marion J, et al. Dual involvement of a Medicago truncatula NAC transcription factor in root abiotic stress response and symbiotic nodule senescence. Plant J. 2012;70:220–230. doi: 10.1111/j.1365-313X.2011.04859.x. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Luckow MA. The rest of the Iceberg. Legume diversity and evolution in a phylogenetic context. Plant Physiol. 2003;131:900–910. doi: 10.1104/pp.102.018150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Wang M, Fu S, Yu D. Mapping QTLs for seed yield and drought susceptibility index in soybean (Glycine max L.) across different environments. J Genet Genom. 2009;12:721–731. doi: 10.1016/S1673-8527(08)60165-4. [DOI] [PubMed] [Google Scholar]
- Fan L, Wang G, Hu W, et al. Transcriptomic view of survival during early seedling growth of the extremophyte Haloxylon ammodendron. Plant Physiol Biochem. 2018;132:475–489. doi: 10.1016/j.plaphy.2018.09.024. [DOI] [PubMed] [Google Scholar]
- Farooq M, Wahid A, Kobayashi N, et al. Plant drought stress: effects, mechanisms and management. Sustain Agric. 2009 doi: 10.1007/978-90-481-2666-8_12. [DOI] [Google Scholar]
- Fasani E, DalCorso G, Costa A, et al. The Arabidopsis thaliana transcription factor MYB59 regulates calcium signalling during plant growth and stress response. Plant Mol Biol. 2019;99:517–534. doi: 10.1007/s11103-019-00833-x. [DOI] [PubMed] [Google Scholar]
- Forner-Giner MÁ, Rodríguez-Gamir J, Primo-Millo E, Iglesias DJ. Hydraulic and chemical responses of citrus seedlings to drought and osmotic stress. J Plant Growth Regul. 2011;30:353–366. doi: 10.1007/s00344-011-9197-9. [DOI] [Google Scholar]
- Gutierrez N, Giménez MJ, Palomino C, Avila CM. Assessment of candidate reference genes for expression studies in Vicia faba L. by real-time quantitative PCR. Mol Breed. 2011;28:13–24. doi: 10.1007/s11032-010-9456-7. [DOI] [Google Scholar]
- Hamanishi ET, Barchet GL, Dauwe R, et al. Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner. BMC Genom. 2015;16:329. doi: 10.1186/s12864-015-1535-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Wang Z, Liu X, et al. Two chloroplast proteins negatively regulate plant drought resistance through separate pathways. Plant Physiol. 2020;182:1007–1021. doi: 10.1104/pp.19.01106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Wani SH, Bhattacharjee S, et al., editors. Drought stress tolerance in plants. Cham: Springer; 2016. [Google Scholar]
- Iuchi S, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K. A stress-inducible gene for 9-cis-epoxycarotenoid dioxygenase involved in abscisic acid biosynthesis under water stress in drought-tolerant cowpea. Plant Physiol. 2000;123:553–562. doi: 10.1104/pp.123.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javot H, Maurel C. The role of aquaporins in root water uptake. Ann Bot. 2002;90:301–313. doi: 10.1093/aob/mcf199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Asthir B. Molecular responses to drought stress in plants. Biol Plant. 2017;61:201–209. doi: 10.1007/s10535-016-0700-9. [DOI] [Google Scholar]
- Kaur S, Pembleton LW, Cogan NO, et al. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics. 2012;13:1–12. doi: 10.1186/1471-2164-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan HR, Link W, Hocking TJ, Stoddard FL. Evaluation of physiological traits for improving drought tolerance in faba bean (Vicia faba L.) Plant Soil. 2007;292:205–217. doi: 10.1007/s11104-007-9217-5. [DOI] [Google Scholar]
- Khan HR, Paull JG, Siddique KHM, Stoddard FL. Faba bean breeding for drought-affected environments: a physiological and agronomic perspective. Field Crops Res. 2010;115:279–286. doi: 10.1016/j.fcr.2009.09.003. [DOI] [Google Scholar]
- Khan NA, Nazar R, Iqbal N, Anjum NA, editors. Phytohormones and abiotic stress tolerance in plants. Berlin: Springer; 2012. [Google Scholar]
- Khan MA, Alghamdi SS, Ammar MH, et al. Transcriptome profiling of faba bean (Vicia faba L.) drought-tolerant variety hassawi-2 under drought stress using RNA sequencing. Electron J Biotechnol. 2019;39:15–29. doi: 10.1016/j.ejbt.2019.02.004. [DOI] [Google Scholar]
- Kolev NG, Franklin JB, Carmi S, et al. The transcriptome of the human pathogen trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010 doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosová K, Urban MO, Vítámvás P, Prášil IT. Drought stress response in common wheat, durum wheat, and barley: transcriptomics, proteomics, metabolomics, physiology, and breeding for an enhanced drought tolerance. Drought Stress Toler Plants. 2016;2:277–314. doi: 10.1007/978-3-319-32423-4_11. [DOI] [Google Scholar]
- Kron AP, Souza GM, Ribeiro RV. Water deficiency at different developmental stages of Glycine max can improve drought tolerance. Bragantia. 2008;67:43–49. doi: 10.1590/S0006-87052008000100005. [DOI] [Google Scholar]
- Kuchenmeister K, Kuchenmeister F, Kayser M, et al. Influence of drought stress on nutritive value of perennial forage legumes. International Journal of Plant Production. 2013;7:693–710. [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Zhang Y, Wu X, Liu Y. Drought stress impact on leaf proteome variations of faba bean (Vicia faba L.) in the Qinghai-Tibet Plateau of China. 3 Biotech. 2018;8:1–12. doi: 10.1007/s13205-018-1088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Hou W-W, Liu Y-J. Proteomic analysis of drought stress response on drought resistance for Vicia faba L. variety ‘Qinghai 13’ in Qinghai Plateau of China. Acta Agron Sin. 2019;45:267. doi: 10.3724/SP.J.1006.2019.84075. [DOI] [Google Scholar]
- Maalouf F, Hu J, O’Sullivan DM, et al. Breeding and genomics status in faba bean (Vicia faba) Plant Breed. 2019;138:465–473. doi: 10.1111/pbr.12644. [DOI] [Google Scholar]
- McDonald GK, Paulsen GM. High temperature effects on photosynthesis and water relations of grain legumes. Plant Soil. 1997;196:47–58. doi: 10.1023/A:1004249200050. [DOI] [Google Scholar]
- Miller G, Suzuki N, Ciftci-Yilmaz S, Mittler R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant, Cell Environ. 2010;33:453–467. doi: 10.1111/j.1365-3040.2009.02041.x. [DOI] [PubMed] [Google Scholar]
- Min X, Lin X, Ndayambaza B, et al. Coordinated mechanisms of leaves and roots in response to drought stress underlying full-length transcriptome profiling in Vicia sativa L. BMC Plant Biol. 2020;20:165. doi: 10.1186/s12870-020-02358-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Kawahara Y, Sakai H, et al. Massive parallel sequencing of mRNA in identification of unannotated salinity stress-inducible transcripts in rice (Oryza sativa L.) BMC Genomics. 2010;11:1–13. doi: 10.1186/1471-2164-11-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan DM, Angra D. Advances in faba bean genetics and genomics. Front Genet. 2016 doi: 10.3389/fgene.2016.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaña S, Seoane P, Bautista R, et al. Large-scale transcriptome analysis in Faba Bean (Vicia faba L) under ascochyta fabae infection. PLoS ONE. 2015 doi: 10.1371/journal.pone.0135143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray H, Bock C, Georges F. Faba bean: transcriptome analysis from etiolated seedling and developing seed coat of key cultivars for synthesis of proanthocyanidins, phytate, raffinose family oligosaccharides, vicine, and convicine. Plant Genome. 2015 doi: 10.3835/plantgenome2014.07.0028. [DOI] [PubMed] [Google Scholar]
- Reddy PCO, Sairanganayakulu G, Thippeswamy M, et al. Identification of stress-induced genes from the drought tolerant semi-arid legume crop horsegram (Macrotyloma uniflorum (Lam.) Verdc.) through analysis of subtracted expressed sequence tags. Plant Sci. 2008;3:372–384. doi: 10.1016/j.plantsci.2008.05.012. [DOI] [Google Scholar]
- Savoi S, Wong DCJ, Degu A, et al. Multi-omics and integrated network analyses reveal new insights into the systems relationships between metabolites, structural genes, and transcriptional regulators in developing grape berries (Vitis vinifera L.) exposed to water deficit. Front Plant Sci. 2017 doi: 10.3389/fpls.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D, Kannan M, Reddy AR. A root proteomics-based insight reveals dynamic regulation of root proteins under progressive drought stress and recovery in Vigna radiata (L.) Wilczek. Planta. 2011;233:1111–1127. doi: 10.1007/s00425-011-1365-4. [DOI] [PubMed] [Google Scholar]
- Sharma S, Villamor JG, Verslues PE. Essential role of tissue-specific proline synthesis and catabolism in growth and redox balance at low water potential. Plant Physiol. 2011;157:292–304. doi: 10.1104/pp.111.183210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Fu L, Dai F, et al. Multi-omics analysis reveals molecular mechanisms of shoot adaption to salt stress in Tibetan wild barley. BMC Genomics. 2016;17:1–15. doi: 10.1186/s12864-016-3242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui MH, Al-Khaishany MY, Al-Qutami MA, et al. Response of different genotypes of faba bean plant to drought stress. Int J Mol Sci. 2015;16:10214–10227. doi: 10.3390/ijms160510214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Wang X, et al. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010;38:4946–4957. doi: 10.1093/nar/gkq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Laxmi A. Transcriptional regulation of drought response: a tortuous network of transcriptional factors. Front Plant Sci. 2015 doi: 10.3389/fpls.2015.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skriver K, Mundy J. Gene expression in response to abscisic acid and osmotic stress. Plant Cell. 1990;2:503. doi: 10.1105/tpc.2.6.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Valencia G, Palomar M, Covarrubias AA, Reyes JL. The legume miR1514a modulates a NAC transcription factor transcript to trigger phasiRNA formation in response to drought. J Exp Bot. 2017;68:2013–2026. doi: 10.1093/jxb/erw380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosulski FW, Ar McCURDY. Functionality of flours, protein fractions and isolates from field peas and faba bean. J Food Sci. 1987;52:1010–1014. doi: 10.1111/j.1365-2621.1987.tb14263.x. [DOI] [Google Scholar]
- Torres AM, Román B, Avila CM, et al. Faba bean breeding for resistance against biotic stresses: Towards application of marker technology. Euphytica. 2006;147:67–80. doi: 10.1007/s10681-006-4057-6. [DOI] [Google Scholar]
- Tripathi P, Rabara RC, Reese RN, et al. A toolbox of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genomics. 2016;17:1–22. doi: 10.1186/s12864-016-2420-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udvardi MK, Kakar K, Wandrey M, et al. Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol. 2007;144:538–549. doi: 10.1104/pp.107.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valluru R, Van den Ende W. Plant fructans in stress environments: emerging concepts and future prospects. J Exp Bot. 2008;59:2905–2916. doi: 10.1093/jxb/ern164. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Mohan SM, Gaur PM, et al. Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol Adv. 2013;31:1120–1134. doi: 10.1016/j.biotechadv.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Thudi M, Nayak SN, et al. Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.) Theor Appl Genet. 2014;127:445–462. doi: 10.1007/s00122-013-2230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Cottage A, Wood T, et al. A SNP-based consensus genetic map for synteny-based trait targeting in faba bean (Vicia faba L.) Plant Biotechnol J. 2016;14:177–185. doi: 10.1111/pbi.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhao Q, Sun D, et al. Transcriptome analysis of flax (Linum usitatissimum L.) undergoing osmotic stress. Ind Crops Prod. 2018;116:215–223. doi: 10.1016/j.indcrop.2018.02.035. [DOI] [Google Scholar]
- Yang Z, Dai Z, Lu R, et al. Transcriptome analysis of two species of jute in response to polyethylene glycol (PEG)- induced drought stress. Sci Rep. 2017;7:1–11. doi: 10.1038/s41598-017-16812-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Jiang J, Zhang H, et al. Density enhancement of a faba bean genetic linkage map (Vicia faba) based on simple sequence repeats markers. Plant Breed. 2019;138:207–215. doi: 10.1111/pbr.12679. [DOI] [Google Scholar]
- Zhai R, Feng Y, Wang H, et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics. 2013;14:1–14. doi: 10.1186/1471-2164-14-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li P, Liu Y. Study on phenotypic diversity and drought resistance of northwest broad bean seedling under drought stress. J Northeast Agric Univ. 2015;46:30–37. [Google Scholar]
- Zhang Y-L, Zhang C-L, Wang G-L, et al. Apple AP2/EREBP transcription factor MdSHINE2 confers drought resistance by regulating wax biosynthesis. Planta. 2019;249:1627–1643. doi: 10.1007/s00425-019-03115-4. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wang J, Cheng Y, et al. NBR1-mediated selective autophagy targets insoluble ubiquitinated protein aggregates in plant stress responses. PLoS Genet. 2013;9:e1003196. doi: 10.1371/journal.pgen.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J-K. Abiotic Stress Signaling and Responses in Plants. Cell. 2016;167:313–324. doi: 10.1016/j.cell.2016.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Pie chart for length distribution of all assembled unigenes
Fig. S2 Venn diagram between Nr, Swissport, KEGG and COG. The number of unique and shared unigenes are shown
Fig. S3 Analysis of the distribution of annotated species through mapping annotated unigenes against NR database. X axis shows top ten annotated species which shared a high similarity with faba bean. Y axis represents annotated unigenes number in certain annotated species
Fig. S4 Clusters of orthologous groups (COG) function classification of all assembled unigenes. X axis shows the unigenes were assigned to 25 categories and the categories were listed on the right of the histogram. Y axis indicates the corresponding number of unigenes in certain COG categories
Fig. S5 Differentially expressed genes in soil drought stressed samples vs. control samples. X axis represents two groups: C, control group; D, soil drought stressed group. Y axis represents the number of upregulated (red) and downregulated (blue) genes
Fig. S6 Volcano plot (log2 (fold change) vs -log10(FDR)) of DEGs in faba bean leaves under soil drought stress. C, control group; D, soil drought stressed group; red dots, upregulated genes; blue dots, downregulated genes; black dots, non-differentially expressed genes
Supplementary material 10 (XLSX 3041 kb)
Data Availability Statement
All the sequencing data have been deposited in the NCBI-SRA database with the following accessions: SRX7873340, SRX7873341, SRX7873342, SRX7873343, SRX7873344 and SRX7873345.