

Abstract
Background/Aims:
Metabolic syndrome and type 2 diabetes are associated with some degree of acidosis. Acidosis has also been shown to upregulate renal gluconeogenesis. Whether impaired insulin or insulin-like-growth factor 1 receptor (IGF1) signaling alter this relationship is not known. Our aim was to determine the effects of deletion of insulin and IGF1 receptors (Insr and Igf1r) from renal proximal tubule (PT) on the gluconeogenic response to acidosis.
Methods:
We developed a mouse model with PT-targeted dual knockout (KO) of the Insr/Igf1r by driving Cre-recombinase with the gamma-glutamyl transferase (gGT) promoter. Male and female mice were maintained as control or acidotic by treatment with NH4Cl in the drinking water for 1-week.
Results:
Acidosis in both genotypes increased renal expression of phosphoenolpyruvate carboxykinase (PEPCK) and fructose-1-bisphosphatase (FBP1), but not glucose6-phosphatase catalytic subunit (G6PC), which showed significantly lower expression in the KO regardless of treatment. Several differences between KO and WT suggested a protective role for insulin/IGF1 receptor signaling in maintaining relative euglycemia in the face of acidosis. First, the increase in FBP1 with acid was greater in the KO (significant interactive term). Secondly, proximal-tubule-associated FOXO1 and AKT overall protein levels were suppressed by acid loading in the KO, but not in the WT. Robust intact insulin signaling would be needed to reduce gluconeogenesis in PT. Third, phosphorylated FOXO1 (pS256) levels were markedly reduced by acid loading in the KO PT, but not in the WT. This reduction would support greater gluconeogenesis. Fourth, the sodium-glucose cotransporter (SGLT1) was increased by acid loading in the KO kidney, but not the WT. While this would not necessarily affect gluconeogenesis, it could result in increased circulatory glucose via renal reabsorption. Reduced susceptibility to glucose-homeostatic dysregulation in the WT could potentially relate to the sharp (over 50%) reduction in renal levels of sirtuin-1 (SIRT1), which deacetylates and regulates transcription of a number of genes. This reduction was absent in the KO.
Conclusion:
Insulin resistance of the kidney may increase whole-body glucose instability a major risk factor for morbidity in diabetes. High dietary acid loads provide a dilemma for the kidney, as ammoniagenesis liberates α-ketoglutarate, which is a substrate for gluconeogenesis. We demonstrate an important role for insulin and/or IGF1 receptor signaling in the PT to facilitate this process and reduce excursions in blood glucose. Thus, medications and lifestyle changes that improve renal insulin sensitivity may also provide added benefit in type 2 diabetes especially when coupled with metabolic acidosis.
Keywords: Gluconeogenesis, Type 2 diabetes, Metabolic acidosis, Sirtuin-1, FOXO-1
Introduction
Liver, kidney, and intestine work in concert to regulate blood glucose levels during fed and fasted states; thus impairment in the processes in one of these organs, such as during metabolic acidosis, can offset balance, leading to episodes of hypo and hyperglycemia [1, 2]. For over ½ century it has been known that metabolic acidosis or NH4Cl loading (in animals) increases the capacity of the kidney to produce glucose [3]. In fact, the first reported evidence of renal gluconeogenesis came from human patients with respiratory acidosis [4]. The explanation for this phenomenon partly resides in the fact that the kidney proximal tubule (PT) not only produces glucose, but also plays a major role in hydrogen ion secretion and bicarbonate production during acidosis [5]. The mechanism involves increased glutaminase I activity, thereby converting glutamine to ammonia and glutamate. Glutamate enters the citric acid cycle as α-ketoglutarate, a substrate for gluconeogenesis. Glucose is synthesized in the PT primarily from amino acid precursors via activation of gluconeogenic enzymes. Rate-limiting (unidirectional) enzymes in this cascade include glucose-6-phosphatase (G6P), fructose-bisphosphatase (FBP1), and phosphoenolpyruvate carboxykinase (PEPCK) [2, 6].
Insulin downregulates gluconeogenesis in liver and kidney [2, 7, 8]. In fact, recent studies suggest kidney may be more sensitive than liver to hormonal down-regulation [9]. Insulin and IGF1 receptors (Insr and Igf1r, respectively) are coupled primarily via insulinreceptor substrate(s) (IRS proteins) to phosphoinositide-3-kinase (PI-3K)-protein kinase B (Akt) signaling [10, 11]. In our previous work, we showed that PT-targeted Insr deletion in mice resulted in elevation in fasting blood glucose and increased renal protein and mRNA expression of G6P, at least in males [12]. More recently, in cell culture, we found that siRNA knockdown of Insr (but not Igf1r) abrogated insulin’s inhibitory effects on glucose production [13], suggesting Insr-specific signaling may be necessary for this effect.
Insulin has been demonstrated to reduce the rate of renal gluconeogenesis via phosphorylation of the transcription factor, forkhead box O1 (FOXO1), thereby limiting its nuclear relocation, and upregulation of gluconeogenic enzymes [14–16]. FOXO1 has a co-activator, the proliferator-activated receptor gamma coactivator 1α (PGC1α), which increases gluconeogenic potential of cells [7]. PGC1α expression is enhanced by the NAD-dependent histone deacetylase, Sirtuin 1 (SIRT1). SIRT1 has been reported to have a number of beneficial actions in the kidney by responding to an increased NAD+/NADH ratio in the cell [17]. Thus, it is plausible that reduced SIRT1 activity in CKD or metabolic acidosis could contribute to a reduced ability to produce glucose under fasting conditions. On the other hand, over-activity of FOXO1, due to reduced Insr signaling could inappropriately enhance gluconeogenesis under fed conditions, leading to glucose instability.
Insulin may also regulate gluconeogenesis by affecting substrate bioavailability. Basolateral glutamine uptake in the PT occurs via Slc38a3 (SNAT3) [18]. One recent study showed elevation in renal PEPCK, as well as, SNAT3 protein levels in cafeteria-diet-fed obese mice, suggesting metabolic syndrome activates substrate uptake for ammoniagenesis and gluconeogenesis in PT [19]. Thus, insulin may regulate acid/base secretion by the PT and this may indirectly affect the rate of gluconeogenesis. Insulin has been demonstrated to increase the activity of the sodium bicarbonate cotransporter (Slc4a4) or NBCe1 in renal PT [20].
Finally, kidney PT is distinct from liver in that it not only produces glucose, but it is the major site for glucose reabsorption through sodium-glucose transporters, SGLT1 and SGLT2. SGLT1 and −2 are the high-affinity/low capacity and low-affinity/high capacity transporters, respectively. They are expressed somewhat in series with SGLT2 in the S1 and S2 segments of the PT and SGLT1 in the S3 segment. Current medications of the Gliflozin class target the high-capacity SGLT2 for maximal effectiveness. The role that these transporters or glucose concentrations in the PT per se, play in regulation of gluconeogenesis is under debate [8, 21]. Sasaki et al. conducted an elegant study showing intracellular glucose down-regulated gluconeogenesis through inhibiting SIRT1 (and thus PGC1 α) activity.
Thus, the role of insulin/IGF1 receptor signaling in modulating the homeostatic balance between transporter activities (sodium, chloride, glucose, bicarbonate etc.) and gluconeogenesis in PT remains murky. Therefore, the primary aim of the current study was to elucidate the mechanistic basis of insulin and IGF1 regulation of renal PT gluconeogenesis and acid/base homeostasis using PT-select dual IR/IGF1R knockout mice.
Materials and Methods
Knockout mice
Mice with targeted dual knockout (KO) of the insulin receptor (Insr) and IGF1 receptor (Igf1r) from the proximal tubule (PT) were generated at Georgetown University by crossing mice that were homo-zygously floxed for both genes with mice carrying Cre-recombinase driven by the γ-glutamyltransferase (γGT) promoter [22]. Mice were housed in conventional cages with 12-hour light/dark cycles. KO mice (GGT1cre+/−;Insrflox/flox;Igf1rflox/flox) were positive for Cre-recombinase and homozygous for floxed Insr and Igf1r. WT mice (GGT1cre−/−;Insrflox/flox;Igf1rflox/flox) were negative for Cre-recombinase and homozygous for floxed Insr and Igf1r, as described previously [12].
Study design
Adult male and female WT and KO mice (n = 8–13/group) were offered a “control” drinking solution [0.1% sodium (Na+) saccharin in distilled water] or acid loaded (1.5% NH4Cl/ 0.1% Na+ saccharin in drinking water, ad libitum) for 7 days. Mice were euthanized under thiobutabarbital and blood and kidneys harvested.
Fasting glucose levels
Mice treated as control for 1-week were fasted 6 hours (9:00–15:00) and blood glucose was sampled from the tail vein. Immediately following this, the same mice were acid loaded for a week, and the 6-hr fasting blood glucose determinations repeated. This protocol was repeated in a separate set of mice, with fasting for 18-hours (17:00–11:00) prior to blood glucose determinations.
Immunocytochemistry
To determine regions of the kidney with active Cre-recombinase, LacZ/GGT1-Cre doubly heterozygous mice were perfused transcardially with 1X phosphate-buffered saline (PBS). The kidneys were bisected and immediately stained for the product of β-galactosidase at 37°C for 1 hour using a B-Gal staining kit (Sigma). The kidney pieces were then photographed using a Zeiss dissecting scope and a Canon A590 camera with a fitted objective at 1.2X magnification. Immunohistochemistry was performed to assess expression and location of Cre-recombinase, IR-β and IGF1R-β on kidney from KO and WT mice, as previously described [23]. Briefly, after PBS perfusion, kidneys were bisected, fixed in 4% paraformaldehyde, paraffin-embedded, sliced, and mounted onto glass slides prior to staining with appropriate primary antibodies.
Urine and blood analyses
A 24-hour urine collection was obtained in metabolic cages (Hatteras Instruments) on day 6 of acid or control treatment. Volumes were recorded, and then urine was centrifuged at a slow speed to remove food particles and other small debris. Urinary pH was determined by 5.8–8.0 plastic pH strips (Fisher Scientific). Blood chemistry was performed by an iSTAT (Abbott Laboratories) with EG8+ cartridges. Insulin was measured in plasma by ELISA (Ultra-Sensitive Mouse ELISA kit, Crystal Chem).
Quantitative polymerase-chain reaction (q-PCR)
SIRT1 mRNA in kidney cortex was analyzed by quantitative real-time PCR. Total RNA was extracted using the RNeasy Mini Kit (Qiagen) and cDNA synthesized using the QuantiTect Reverse Transcription Kit (Qiagen). For gene expression analysis, the RT2 qPCR mouse primer assay for Sirtuin 1 (Sirt1) (Qiagen, Assay name: PPM05054A) was used in SYBR Green-based quantitative real-time PCR, which was performed using the RT2 SYBR Green ROX qPCR Mastermix (Qiagen) in the StepOnePlus Real Time PCR System. Gene expression was normalized to GAPDH and relative gene expression calculated using the 2−ΔCT approach.
Proximal tubule (PT) suspensions
In a separate set of mice treated with acid- or control- drinking fluid for 1-week, PT-enriched suspensions were prepared from kidney cortex as described [24]. Briefly minced cortex was digested with collagenase and dexoxyribonuclease in Dulbecco’s modified Eagle’s medium (DMEM F12, Hepes-buffered, Gibco) and mechanical disruption at 37°C for 40 minutes. Suspended tubules were further sorted using a 45% Percoll solution and centrifugation at 25,000g. PT migrated the greatest distance in the Percoll, and were collected by Pasteur pipet. Aliquots of isolated PT were washed 2X with glucose-free DMEM, then pre-treated with one of 5 concentrations of insulin (in 600 μl) encompassing normal to high physiological range (0, 100, 500, 2000, and 5000 pM) for 10 minutes in an incubation medium containing: glucose-free DMEM buffered with HEPES, sodium heptanoate, 2 mM, sodium lactate, 2 mM, L-glutamine, 4 mM, sodium pyruvate, 2 mM, glycerol, 2 mM, and sodium butyrate, 10 mM. Then dexamethasone, 50 nM and cyclic-3’ adenosine monophosphate, 10 μM were added and the incubation was continued for 50 additional minutes at 37°C in a shaking water bath. Aliquots were then cold-centrifuged @ 2,300g to pellet tubules and stop the reaction. Collected supernatants were analyzed for glucose by the Amplex Red Glucose Kit (Invitrogen). The pellets were dissolved in 30 μl isolation solution [25] containing protease/phosphatase inhibitors (HALT, Pierce), measured for protein concentration, and solubilized with Laemmli buffer for dot blotting.
Dot and western blotting
Whole kidney homogenates were prepared into Laemmli sample buffer [25]. Western blotting was performed as previously described [25]. Band densities were normalized to Ponceau Red (ThermoFisher). For PT suspension samples, 5 μl of each sample was dotted onto supported nitrocellulose membrane (BioRad) in a 96-well format and dried. The membrane was then stained with Ponceau Red and then blocked with 5% dry powdered milk in a blot wash solution [25] for 30 minutes prior to probing with primary antibodies overnight at 4°C. The following day, blots were washed and probed with the species-appropriate secondary antibodies and chemiluminescent reagents [25]. Dot blots were first probed with antibodies against the phosphorylated form of the protein, stripped, and then probed with the pan antibodies. Chemiluminescent images were obtained on an Amersham Imager 600 and analyzed for density by ImageJ (NIH).
Primary Antibodies
Antibodies used included: polyclonal rabbit against mouse 1) SGLT1 and 2) SGLT2 from Hermann Koepsell [26, 27]; 3) G6PC (PAS-42541, polyclonal rabbit, Invitrogen); 4) PEPCK (SC-271029, monoclonal mouse, Santa Cruz); 5) FBP1 (109020, monoclonal rabbit, Abcam); 6) SNAT3 (14315, polyclonal rabbit, Proteintech); 7) NBCe1 (11885, polyclonal rabbit, Proteintech); 8) NHE3 (our own polyclonal rabbit); 9) FOXO1 (9454S, polyclonal rabbit, Cell Signaling); 10) SIRT1 (PA517074, polyclonal rabbit, Life Technologies Corporation); 11) PGC1α (NBP1–04676SS, polyclonal rabbit, Novus Biologicals); 12) mTOR (PAS-34663, polyclonal rabbit, Invitrogen); 13) Akt (4691S, monoclonal rabbit, Cell Signaling); 14) pS473-Akt (4060S, monoclonal rabbit, Cell Signaling); 15) pS2448-mTOR (5536S, rabbit monoclonal, Cell Signaling); 16) pS256-FOXO1 (9461, rabbit polyclonal, Cell Signaling); 17) Cre-recombinase (PA5–32245, rabbit polyclonal, Invitrogen); 18) IR-β (20433, rabbit polyclonal, Proteintech); 19) IGF1R-β (3027S, rabbit polyclonal, Cell Signaling).
Statistical analysis
Group sizes were determined a priori using Power Analysis with power set at 0.80 of an alpha of 0.05 with a small sample size using expected variability of glucose measures in PT suspensions. Resulting data are, in general, presented as mean ± sem. Outliers were excluded using the 1.5 Interquartile Rule. GraphPad Prism Software, 8.2.1 was used to analyze data by unpaired t-test (WT versus KO) or two- and three-way analysis of variance followed by Tukey’s multiple comparisons testing (adjusted p-values < 0.05 considered significant). For some analyses, i.e., fasted glucose and PT suspensions, repeated measures ANOVA was used as indicated.
Results
Characterization of the knockout
Some aspects of the Insr/Igf1r PT-select double KO mouse have been previously described [22]. Fig. 1A demonstrates specificity of Cre-recombinase activity to the cortex of the kidney in γGT-Cre positive mice. Immunohistochemistry (IHC) of Cre recombinase protein showed labeling of nuclei in the proximal tubules of KO, but not WT, mice (Fig. 1B). Western blotting of whole kidney homogenates demonstrated an approximate 50% reduction in band density for the mature protein (β-subunits) of IR and IGF1R (Fig. 1C–D). Furthermore, IHC showed less intense basolateral staining for Igf1r (β-subunit, Fig. 1E), and Insr (β-subunit, Fig. 1F) in KO PT, relative to WT mice.
Fig. 1.
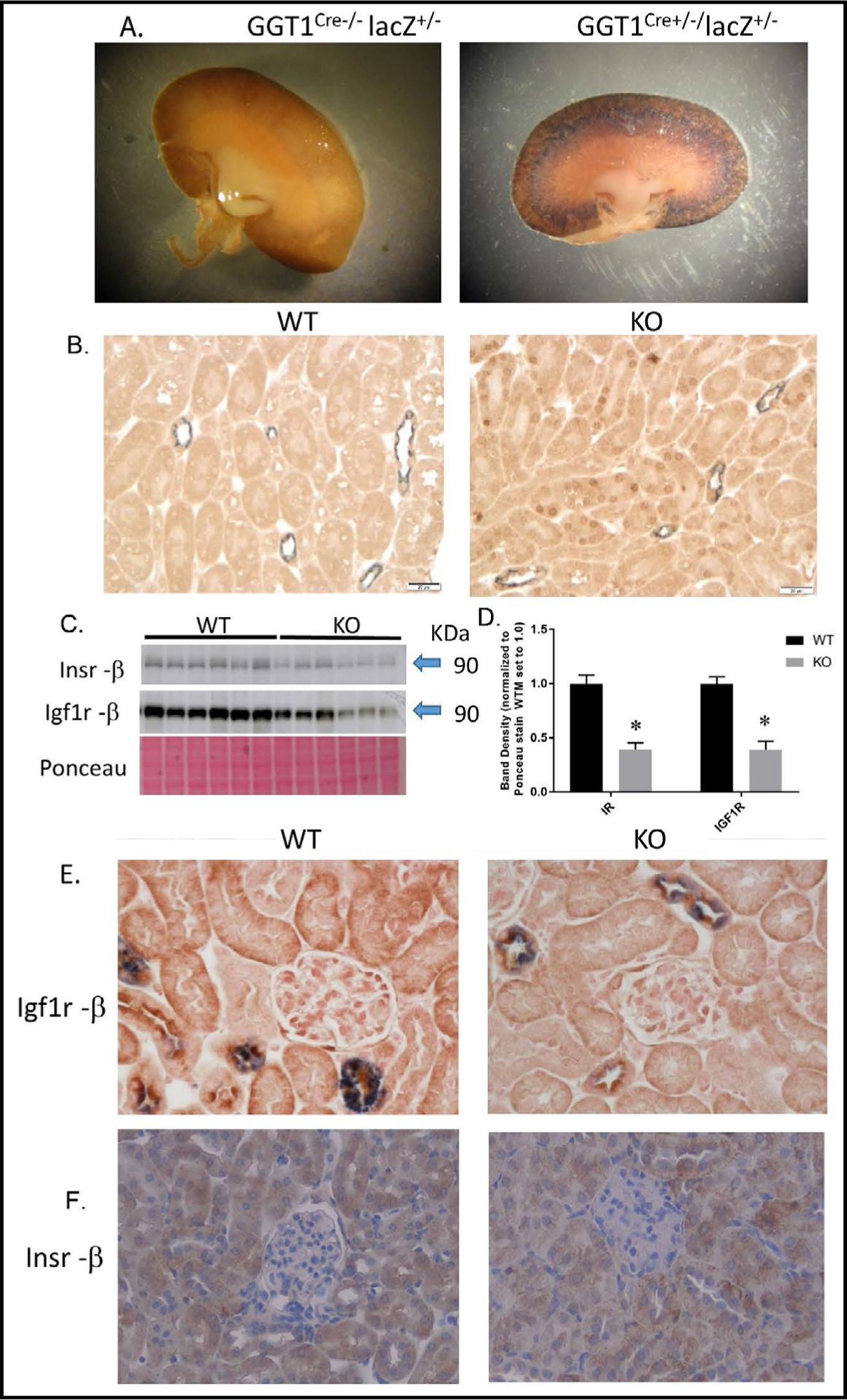
Characterization of Insr/Igf1r PT-targeted dual KO. A. β -galactosidase-generated staining in a Cre-negative (left) and a Cre-positive (right) kidney cross-sections obtained from a mating between transgenic GGT1Cre+/− mice and lacZ reporter mice. B. Representative immunohistochemistry (IHC, brown stain) for nuclear Cre-recombinase in cortex sections from WT (left) and KO (right) mice; (gray stain; aquaporin-2 as a marker for collecting duct/connecting tubule). C. Western blotting for insulin receptor (β-subunit) and IGF1 receptor (β-subunit) in whole-kidney homogenates from WT and KO mice (n = 6/genotype); D. band densities for receptor blots normalized to Ponceau staining; E. IHC for Igf1r (β-subunit, brown) in the cortex of WT (left) and KO (right) mice; gray staining is aquaporin-2; F. IHC for Insr (β-subunit) in WT (left) and KO (right) mice; *indicates a significant (p<0.05) difference between WT and KO by unpaired t-test.
Confirmation of acidosis and general physiology
For Fig. 2 the same groups of mice were fasted first under control, followed by one-week of acid-loading conditions. Separate groups of male and female mice were evaluated in the 6-hour versus 18-hour fasting studies. No significant differences due to genotype, treatment, or their interactions were observed in mean blood glucose in male 6-hr fasted mice (Fig. 2A). However, with 18-hr fasting, acid loading led to a significant interaction (p = 0.046) between genotype and treatment in that the acid loading slightly increased mean fasting glucose in KO, but it remained fairly steady in the WT (Fig. 2B). For females, 6-hour fasting glucose was lower in the KO with acid loading, but not different under control conditions leading to a significant genotype × treatment interactive term. There were no significant differences due to acid loading or genotype in 18-hour fasting glucose in females (Fig. 2D).
Fig. 2.
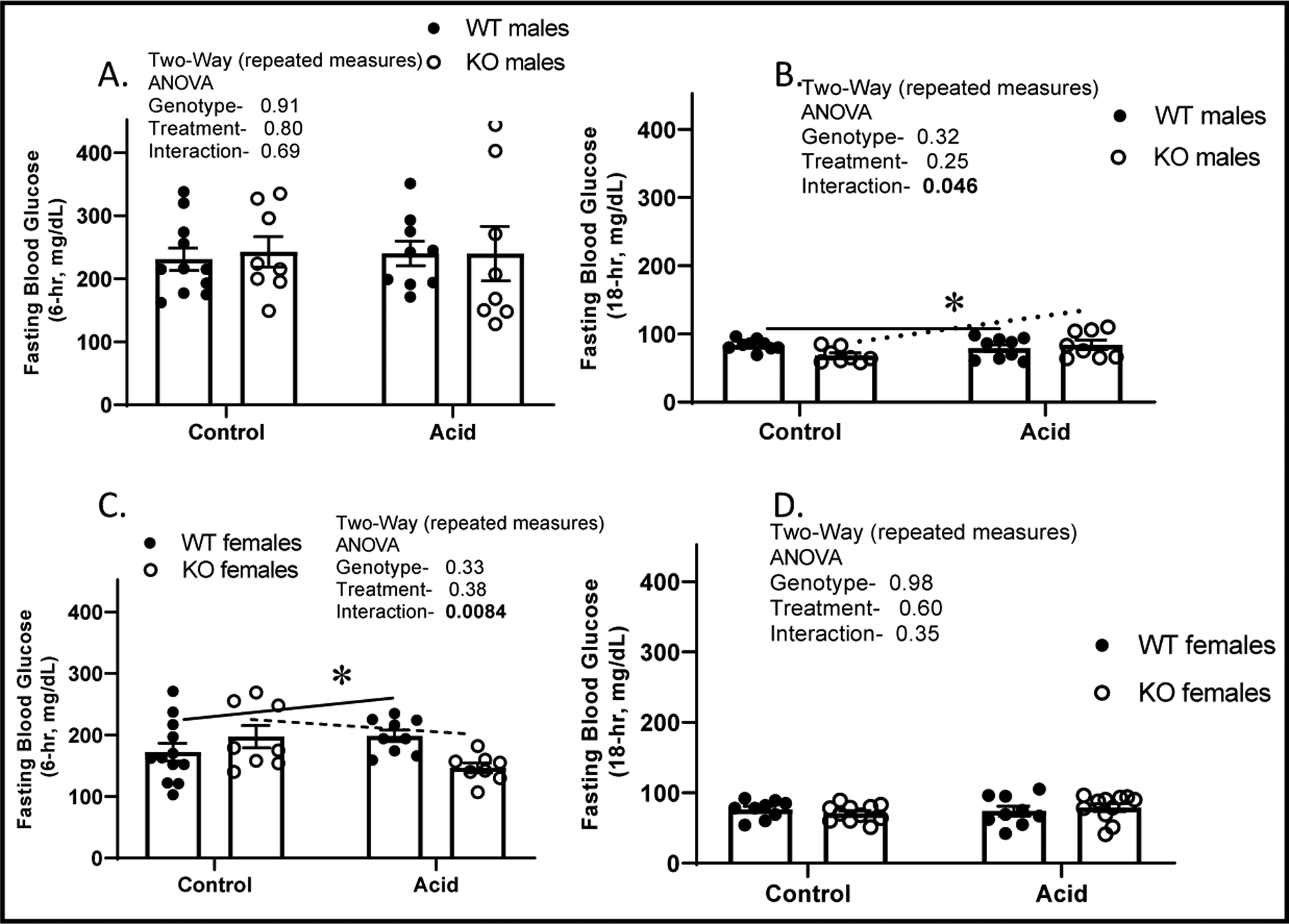
Fasting glucose. Mice sampled under control then 1-week acid loading; blood glucose after A. 6-hour fast in males; B. 18-hour fast in males; C. 6-hour fast in females; and D. 18-hr fast in females; (n = 8–11/genotype, mean ± standard deviation); WT (closed circles) and KO (open circles). *indicates a significant interaction between genotype and treatment by two-way repeated measures ANOVA. P-values shown in the inset with significant values (p<0.05) bolded.
For remaining Figures, mice were maintained as either control or acid-loaded and euthanized after one-week. For Fig. 3 and 4, we show only male results, as females were similar. Neither genotype nor treatment affected final body weight in males or females. For males-final body weight (g): 25 ± 1, 26 ± 2, 26 ± 1, and 26 ± 2 for WTC, KOC, WTA, and KOA, respectively. Kidney weight was increased by acid loading and reduced by KO (Fig. 3A). Acid loading reduced blood HCO3 (Fig. 3B), and blood pH (Fig. 3C), and modestly increased blood Na+ and Cl- concentrations (not shown). There were no differences due to genotype on these factors. There was a trend for plasma insulin to be higher in the KO under control conditions, and lower (than the WT) under acid conditions (Fig. 3D, p = 0.065 for interaction).
Fig. 3.
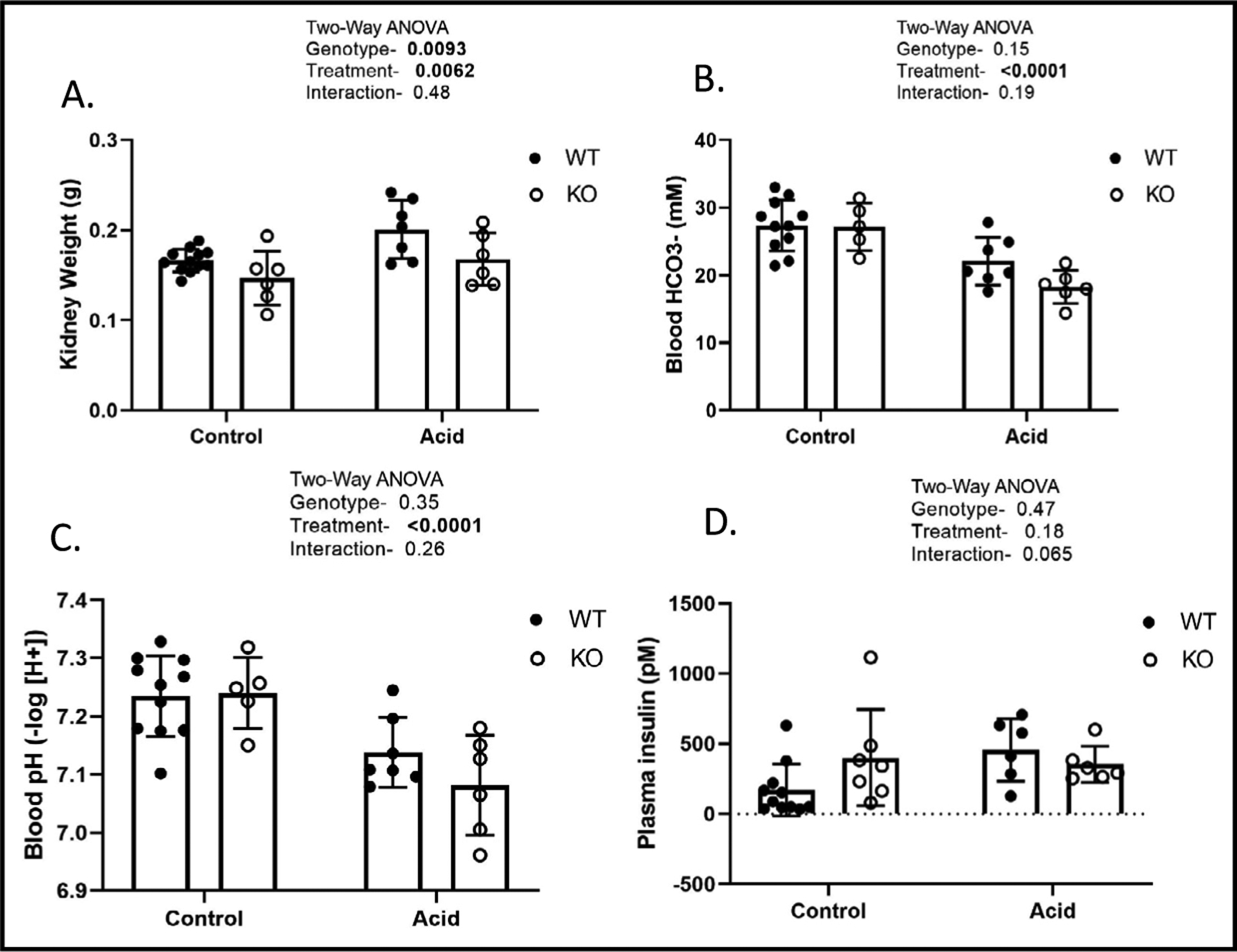
Kidney weight and blood chemistry. A. final kidney weight; B. blood bicarbonate; C. blood pH; and D. plasma insulin (mean ± standard deviation) after one-week of either control or acid-loading conditions; significant differences due to genotype, treatment or their interactions, as determined by two-way ANOVA. P-values shown in the inset with significant values (p<0.05) bolded.
Fig. 4.
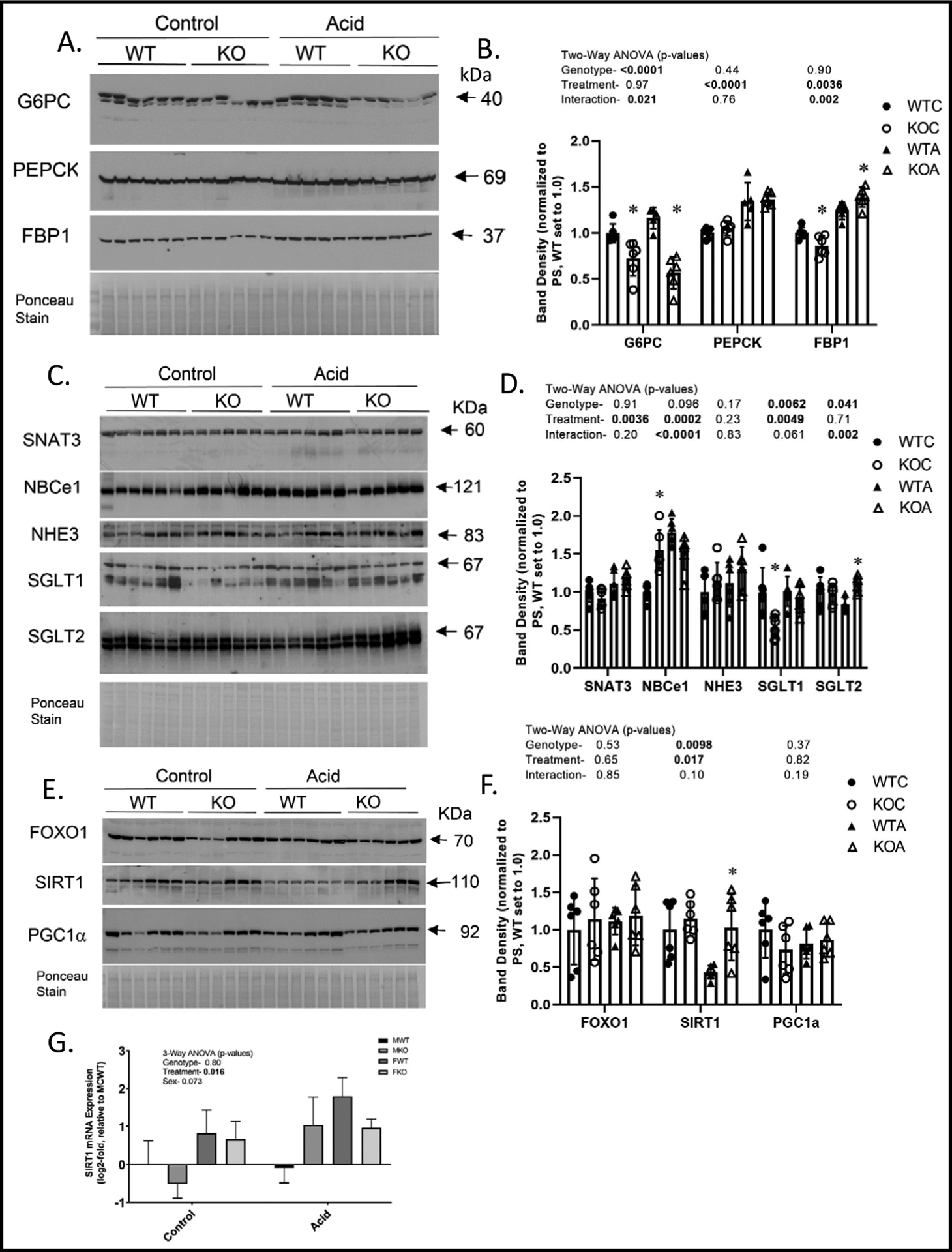
Western blotting of whole kidney homogenates. WT and KO mice were treated under control or acid-loading for 1-week and kidneys harvested (n = 5 or 6/group). Representative western blots and densitometry summary for males- A-B. rate-limiting gluconeogenic enzymes; C-D. PT transporters/exchangers; E-F. transcription factors and a prominent deacetylase. G. SIRT1 mRNA. Western blotting were normalized by Ponceau staining of membranes and the WTC mean was set to 1.0. Two-way ANOVA p-values are provided in graph inserts; *indicates a significant difference (p<0.05) from WT (within a treatment) by Tukey’s multiple comparisons testing following a significant two-way ANOVA. WTC- wildtype control-closed circles; KOC-knockout control- open circles; WTA- wildtype acid- closed triangles; KOA- knockout acid- open triangles.
Gluconeogenic enzymes
Protein regulation of rate-limiting gluconeogenic enzymes in kidney was determined by western blotting of whole-kidney homogenates (Fig. 4A–B). G6PC (glucose-6-phosphatase catalytic subunit), the terminal enzyme in the gluconeogenic cascade, was significantly lower in KO as compared to WT, with no effect of treatment. PEPCK (phosphoenolpyruvate carboxykinase) was increased by acid loading (as has been previously demonstrated) [28, 29] and not affected by genotype. A significant interactive term existed for FBP1 (fructosebisphosphatase-1) in that it was increased by acid loading only in the KO mice.
Transporters/exchangers in PT
Western blots of relevant transport proteins in whole kidney are shown in Fig. 4C–D. SNAT3 (sodium-coupled neutral amino acid transporter, type 3) was increased by acid loading and not affected by genotype. NBCe1 (sodium bicarbonate cotransporter, electroneutral) was increased by acid loading, but was also elevated in the KO under control conditions leading to a significant interaction. NHE3 (sodium hydrogen exchanger) abundance was not affected by genotype or acid treatment. SGLT1 (sodium glucose transporter, type 1) was lower overall in the KO, and increased by acid loading only in the KO. Whereas, SGLT2 (sodium glucose transporter type 2) was similar under control conditions between genotypes, and marginally reduced in the WT, but similar to SGLT1, increased in the KO under acid-loading (significant interaction).
Gluconeogenic transcription factors and SIRT1
Protein levels of transcription factors FOXO1 or PGC1α in whole-kidney homogenates were not affected by genotype or treatment (Fig. 4E–F). KO mice had higher band density for SIRT1 in kidney; especially under acid-loading conditions. Acid loading, overall, caused a 54 and 11% reduction in SIRT1 band density in KO and WT, respectively. We assessed the mRNA levels of SIRT1 in kidney to determine whether this effect was transcriptionally regulated (Fig 4G). We found mRNA to be regulated in the opposite direction of protein (increased by acid loading), suggesting the reduction in SIRT1 protein was post-transcriptionally mediated, and the increase in expression may have been a response to this reduction.
Glucose production in PT suspensions
PT were enriched as a suspension harvested from mice treated as control or acid-loaded for 1-week. PT suspensions were incubated for 60 minutes in a medium with cyclic AMP, glucocorticoid and unlimited substrate. PT obtained from mice treated with acid had significantly lower overall capacity to produce glucose, under these conditions (Fig. 5). Furthermore, there was a strong trend for KO mice PT to produce more glucose.
Fig. 5.
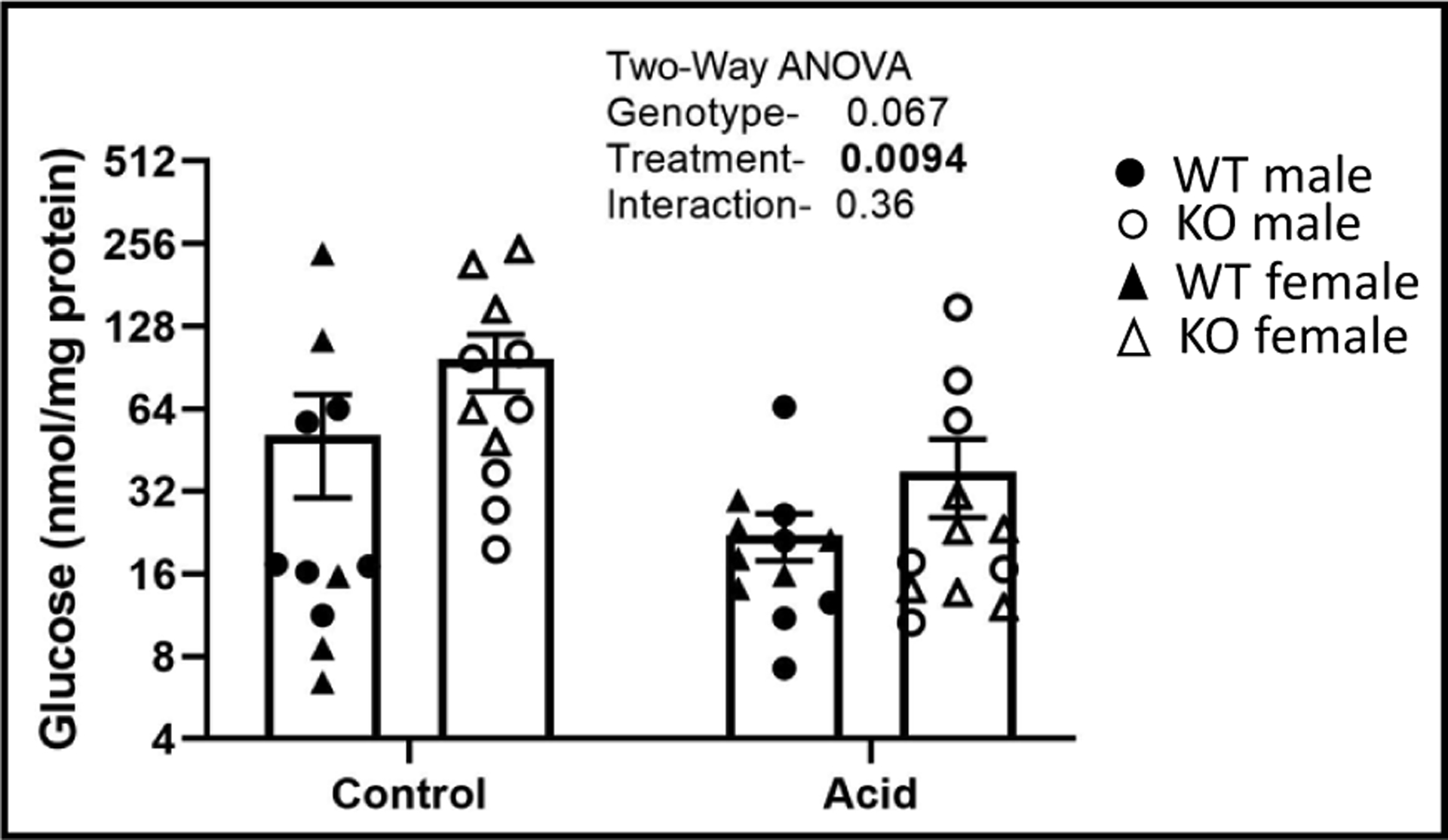
Glucose production in proximal tubule (PT) suspensions. WT and KO mice (n = 5–6/sex/genotype/treatment) were treated under control or acid-loading for 1-week (n = 6/group) and PT suspensions prepared, aliquoted, and stimulated with glucocorticoid and cyclic AMP to produce glucose while in a glucose-free medium for 70 minutes. Glucose in medium was measured and normalized to protein; closed circles- WT male, open circles- KO males; closed triangles- WT females; open triangles- KO females. Two-way ANOVA p-values shown in the inset with significant values (< 0.05) in bold.
Insulin signaling in PT suspensions
Next we tested whether PT proteins involved in key steps in insulin/IGF1 receptor signaling relating to gluconeogenesis were altered in abundance or phosphorylation in the KO or by acidosis (Fig. 6). Insulin was applied ex vivo (physiological to supra-physiological range) to suspension aliquots from acid-loaded and control-treated WT and KO mice (both males and females). Suspensions were incubated for 60 minutes with the insulin prior to stopping the reaction with cold-centrifugation. Proteins were solubilized and dot blotting performed to allow for comparison of the density of 92 dots simultaneously. Representative portions of the blots are shown in the upper right hand corner of each line graph. Phospho forms of the proteins are shown in the right hand column. Pan Akt (protein kinase B), abundance, an early central signaling node for Insr/Igf1r was reduced overall by the increasing doses of insulin (Fig. 6A). Acid loading reduced Akt abundance particularly in the environment of 0 insulin. Surprisingly phosphorylation of Akt on serine 473 (an activating site) was also significantly reduced by insulin dose and not affected by treatment or genotype (Fig. 6B). MTOR, a member of mTORC1 and mTORC2 complexes, also a known target of Insr/Igf1r signaling was likewise, although much more modestly reduced in overall (pan) abundance by insulin dose (Fig. 6C). For mTOR, there was a significant genotype × treatment interaction, primarily in that mTOR was reduced by acid loading in WT, but not in KO. Phospho-mTOR (serine 2448) was increased by insulin dose (Fig. 6D) and the increase was greater in the KO (genotype × insulin dose interaction). Finally, we evaluated FOXO1 PT levels and phosphorylation. FOXO1 pan levels were subject to complex interactions (Fig. 6E). Overall insulin dose reduced FOXO1 only in the KO controls. At 0 insulin dose, there was a large reduction in FOXO with acid in the KO (not apparent in the WT). With regard to p-FOXO (serine 256), which attenuates FOXO1 translocation to the nucleus and thereby reduces its transcriptional activity, there was a strong interaction between genotype and treatment in that acid loading increased levels in the WT, but reduced them markedly in the KO. Insulin dose also markedly reduced phosphorylation of FOXO1 especially in the supraphysiological range between 500 and 5000 pM.
Fig. 6.
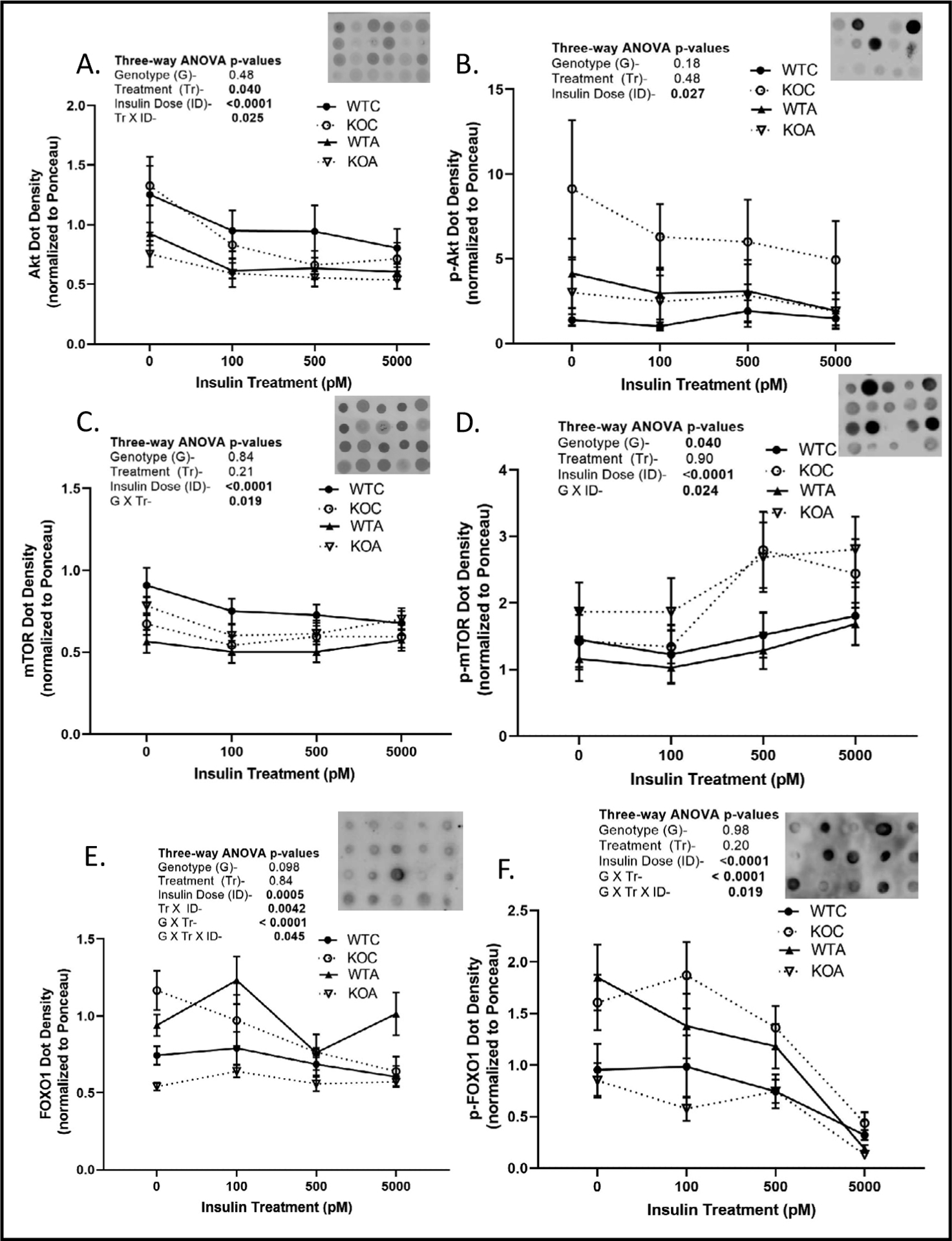
Insulin signaling in proximal tubule (PT) suspensions. WT and KO mice (n = 5–6/sex/genotype/treatment) were treated under control or acid-loading for 1-week and PT suspensions prepared and incubated with different concentrations of insulin for 70 minutes then cold centrifuged. Protein from pellets was solubilized for dot blotting (5 μl) and density was normalized to Ponceau staining. Representative dot blot and density summary for A. Akt; B. pS473-Akt; C. mTOR; D. pS2448-mTOR; E. FOXO1; and F. pS256-FOXO1. Results of 3-way repeated measures ANOVA are shown in inserts; p<0.05 considered significant; closed circles- WT control, open circles- KO control; closed triangles- WT acid; open triangles- KO acid.
Discussion
Through the years, the renal PT has had an under-appreciated role in the regulation of day-to-day blood glucose homeostasis. Early reports suggested it was primarily important during fasting conditions [30]. Re-evaluation in the early 2000’s of prior balance studies revealed that renal gluconeogenesis accounts for about 20% of all endogenous glucose release (considering liver and kidney) and approximately 40% of total body gluconeogenesis (liver also uses glycogenolysis to liberate glucose) in the post-absorptive state [1, 9]. Thus, glucose production in the kidney is substantial, and perturbation in this regulation might be predicted to result in hypo or hyperglycemic episodes.
Insr/Igf1r signaling is known to counter both renal and hepatic gluconeogenesis primarily by phosphorylation or dephosphorylation of FOXO1, retarding its ability to facilitate transcription of the rate-limiting gluconeogenic genes [8]. Metabolic acidosis challenges the kidney in that the PT needs to both produce bicarbonate and NH4+, as well as, control glucose reabsorption and production. Ammonium chloride in the drinking water or diet is a commonly employed model of metabolic acidosis in rodents [5, 31, 32], which in our hands, as expected, successfully resulted in reduced blood pH and bicarbonate after a week of consumption in the mice.
Significant interactive terms, i.e., genotype-by-treatment, were observed for fasting glucose. This suggests that the KO and WT responded differently to acidosis. Our findings are somewhat difficult to interpret because they are not significant in both sexes for the same period of fasting, and they are “opposite” responses with regard to the shorter (6-hr) versus longer (18-hr) fast. We interpret the 18-hour fast as being a substantially fasted state, where one might expect the need for robust renal gluconeogenesis. In this case, we saw what might be interpreted as a relative protection in the male KO. One way in which this may have been accomplished was due to upregulation of FBP1. Not a lot is known about the role of FBP1 in kidney; however, loss of FBP1 has been shown to be associated with tumor progression in clear cell renal cell carcinoma [33], probably by allowing unchecked cell proliferation and glycolytic flux. Second, we found protein levels of the NAD-dependent deacetylase, SIRT1, was down regulated in the WT, but not KO kidneys with acid loading. In addition to an anti-fibrotic and anti-inflammatory role, SIRT1 has a fundamental role in homeostatic control of gluconeogenesis in the fed-and-fasting cycles, through lysine deacetylation and activation of transcription factors, PGC1α and FOXO1 [34]. Thus, this may have allowed relatively elevated glucose production in the KO mice with acid loading. Third, we found that acid loading increased protein levels of both isoforms of SGLT in the KO, but not in the WT. While this would not directly affect glucose production, it could increase glucose reabsorption and attenuate hypoglycemia in the KO with acidosis. Female KO mice were not “as protected” perhaps due to smaller overall kidneys, with resulting less gluconeogenic capacity.
With regard to the shorter (6 hour) fasting, this took place in the day when mice are generally sleeping, and probably relates to a relatively random blood glucose measure. In this case, post-prandial effects may be still somewhat apparent with reabsorption of a large number of substances in the PT. Overall, male blood glucose was still quite high in all groups, and unaffected by the fast. Females, on the other hand, were lower (significantly so, by 3-way ANOVA, p = 0.0001), and more so in the KO with acid loading. We speculate WT females had more effective uptake of substrates needed for glucose production in this case, although additional studies would be warranted to elucidate fully these genetic and sex differences.
In our study, acidosis increased renal protein expression of PEPCK and FBP1, but not G6PC. PEPCK was increased about 50% by acid, consistent with another study [28]. Despite the increase in 2 out of 3 rate-limiting enzymes in gluconeogenesis with acidosis, we did not observe upregulation of ex vivo glucose-producing capacity in the PT with acidosis. In fact, there was a significant reduction in both the WT and KO. The reason for this reduction is unclear. We may speculate that an increase may require a reduced intracellular pH (acidosis), to stimulate glutamine uptake, ammoniagenesis, and the release of α-ketoglutarate (glucose precursor). When the cells were in suspension, they were no longer in an acidotic environment. Perhaps, then the reduced glucose production represents an adaptive down-regulation of glucose producing capacity to try to normalize glucose production in the face of acid loading and the requirement for ammonium production. This adaptation appeared to be independent of Insr/Igf1r expression as there was no interactive term. It is also important to note that the suspension-glucose production was normalized to protein in the suspension. The fact that the kidneys hypertrophied with acid loading would increase the denominator in our equation. Thus, we may be under-estimating total glucose-producing capacity of the kidney per se with acidosis.
There was a trend (p = 0.067) for increased glucose producing capacity of PT due to Insr/Igf1r deletion. This agreed with our previous findings in mice with solely PT IR deletion [35], and supports other work showing insulin down-regulates gluconeogenesis in both liver and kidney [2, 7, 8, 13]. This may be particularly relevant to metabolic syndrome, where insulin receptor expression is reduced in kidney [36] and insulin receptor resistance is common.
A number of genotype differences were noted in transporters of the PT involved in acidosis and/or glucose homeostasis. NBCe1 was increased in the KO under control conditions, but insensitive to the acid load, whereas, the acid loading increase expression in the WT over 70%. This supports insulin/IGF1R regulation of this transporter, which has been shown before [37], and suggests KO may cause modest acidosis in the KO, not present in the WT. Both isoforms of the sodium glucose cotransporter (SGLT1 and 2) were differentially regulated by genotype. SGLT1 has been previously demonstrated to be regulated by a number of factors [38], and our study suggests that KO does cause a modest reduction in expression of this isoform. In contrast, SGLT2 was slightly higher in the KO and regulated in the opposite direction by acidosis. Additional studies will be needed to tease out the cause underlying these genotype differences. It is important to note, however, that while these changes could influence overall PT gluconeogenesis, we could not detect them using our PT-suspension, as luminal glucose would be close to zero with our ”0 added glucose medium”.
Another novel finding was that G6PC was reduced in kidney of KO mice. Moreover, the reduction in total renal G6PC may have been under-estimated because the kidneys were also 7–18% lighter weight in the KO. Our previous study [22] showed smaller-sized cells in the cortex of the KO mice; therefore, in this case, the smaller cells were associated with lower expression of G6PC. These findings are somewhat in contrast to the recent report by Sasaki et al. [8], who found feeding previously fasted mice (rise in circulating insulin) reduced mRNA for both PEPCK and G6P. Thus, acutely, G6PC is down-regulated by insulin, as one would expect to reduce gluconeogenesis; however, constitutive down-regulation of IR and IGF1R appears to reduce its expression. This may explain why these dual KO mice do not have higher fasting glucose levels, as we observed in the single PT-IR KO mice [35]. We can deduce IGF1R deletion is driving the reduction in G6PC and kidney size; however, additional testing of this hypothesis is warranted. It also suggests clear distinction, and possibly opposing roles, for IGF1R and IR signaling in the regulation of gluconeogenesis.
Incubation of the suspensions for 1 hour with increasing doses of insulin, in general, down regulated the protein levels of mTOR, Akt, and FOXO1. Insulin phosphorylation of FOXO1 has been shown to enhance its ubiquitination and target it for proteasomal degradation [39]. Nonetheless, the rapid nature of this down-regulation was surprising, as well as, the fact that the greatest slope occurred between 0 and 100 pM insulin (in the insulin-sensitive physiological range). This opens the door to the possibility for diurnal changes in the levels of these proteins to refine signaling.
Furthermore, phosphorylation of Akt (serine 473) and FOXO1 (serine 256) were also, in general reduced, by increasing doses of insulin. Akt activity is increased by mTORC2 phosphorylation on serine 473 [40]. Activated Akt then phosphorylates (and deactivates) FOXO1 on serine 256. This supports, as predicted, desensitization of established insulin/IGF1 signaling pathways with increasingly supraphysiologic doses of insulin. The circulating final insulin levels (fed state) in our mice were, in general, in the range of 20–500 pM. The highest dose we tested was 5000 pM. The one phosphorylation event that was not desensitized, but rather increased with increasing doses of insulin, was the phosphorylation of mTOR on serine 2448. A recent report suggested that serine 2448 on mTOR, may be an inhibitory site and reduces activity of the complex (at least mTORC1) [41]. Therefore, increased phosphorylation at this site would similarly predict attenuated signaling.
With regard to the effects of acid and genotype on the levels and phosphorylation state of these proteins, we found numerous significant effects, too many to unpack at one sitting. To mention a few that may facilitate our understanding of the phenotype in these mice. First, FOXO1 overall abundance and phosphorylation were reduced substantially by acidosis in the KO, but not in the WT. Under control conditions, in contrast the KO had greater levels of both FOXO and p-FOXO1. In sum, the relative p-FOXO was more stable (between acid and control conditions) in the WT, than in the KO, which would agree with a fine-tuning role for insulin/IGF1 signaling in FOXO1 regulation. Furthermore, p-mTOR levels were higher in the KO, than in the WT, and showed responsivity to insulin. It is unclear what the physiological relevance of this finding is; however, overall protein levels were highest in the WT controls.
Finally, we chose to emphasize the findings in male mice in this study due to greater prevalence of metabolic syndrome in the male mice of our background strain (C57Bl6) [42]; this does not diminish the importance of the findings in the females. Additional studies are required to flesh out sex differences.
Conclusion
Overall, our studies suggest a complex relationship between insr/igf1r signaling in PT and gluconeogenesis in the setting of acidosis. We find increased stability of blood glucose homeostasis under control versus acid loading in the WT animals, suggesting a role for insulin/igf1 buffering (regulated feedback) of production of glucose under these conditions. This may be particularly relevant in type 2 diabetes or insulin resistance, which is associated with reduced expression of insulin receptors in kidney [36].
Acknowledgements
Funding
This work was supported by a Marriott Foundation Cardiovascular Fellowship (CME), as well as, Georgetown University Biomedical Graduate Research Organization (CME). MBF received salary support from the American Diabetes Association in the form of a Minority Fellowship (1-18-PMF-002). AA received funding from the Georgetown University Biochemistry and Biotechnology Master’s Degree Program. BS is supported by NIH K01 K01DK-106400. ST is supported by Sanjay Gandhi Postgraduate Institute of Medical Sciences.
Footnotes
Disclosure Statement
The authors have no conflicts of interest to declare.
Statement of Ethics
All animal experiments and breeding were approved by the Georgetown University Institutional Animal Care and Use Committee (IACUC). Georgetown University animal housing and care facilities have been continuously approved by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC).
References
- 1.Gerich JE, Meyer C, Woerle HJ, Stumvoll M: Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 2001;24:382–391. [DOI] [PubMed] [Google Scholar]
- 2.Swe MT, Pongchaidecha A, Chatsudthipong V, Chattipakorn N, Lungkaphin A: Molecular signaling mechanisms of renal gluconeogenesis in nondiabetic and diabetic conditions. J Cell Physiol 2018;234:8134–8151. [DOI] [PubMed] [Google Scholar]
- 3.Steiner AL, Goodman AD, Treble DH: Effect of metabolic acidosis on renal gluconeogenesis in vivo. Am J Physiol 1968;215:211–217. [DOI] [PubMed] [Google Scholar]
- 4.Aber GM, Morris LO, Housley E: Gluconeogenesis by the human kidney. Nature 1966;212:1589–1590. [DOI] [PubMed] [Google Scholar]
- 5.Curthoys NP, Moe OW: Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 2014;9:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rognstad R: Rate-limiting steps in metabolic pathways. J Biol Chem 1979;254:1875–1878. [PubMed] [Google Scholar]
- 7.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. : Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003;423:550–555. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T, et al. : Dual Regulation of Gluconeogenesis by Insulin and Glucose in the Proximal Tubules of the Kidney. Diabetes 2017;66:2339–2350. [DOI] [PubMed] [Google Scholar]
- 9.Cano N: Inter-relationships between renal metabolism (both in physiology and renal dysfunction) and the liver. Curr Opin Clin Nutr Metab Care 2001;4:279–285. [DOI] [PubMed] [Google Scholar]
- 10.Butlen D, Vadrot S, Roseau S, Morel F: Insulin receptors along the rat nephron: [125I] insulin binding in microdissected glomeruli and tubules. Pflugers Arch 1988;412:604–612. [DOI] [PubMed] [Google Scholar]
- 11.Catena C, Cavarape A, Novello M, Giacchetti G, Sechi LA: Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int 2003;64:2163–2171. [DOI] [PubMed] [Google Scholar]
- 12.Tiwari S, Li J, Tsukerman S, Singh R, Pandey G, Ecelbarger CM: Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. J Am Soc Nephrol 2013;24:1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey G, Shankar K, Makhija E, Gaikwad A, Ecelbarger C, Mandhani A, et al. : Reduced Insulin Receptor Expression Enhances Proximal Tubule Gluconeogenesis. J Cell Biochem 2017;118:276–285. [DOI] [PubMed] [Google Scholar]
- 14.DeFronzo RA, Davidson JA, Del Prato S: The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012;14:5–14. [DOI] [PubMed] [Google Scholar]
- 15.Accili D, Arden KC: FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004;117:421–426. [DOI] [PubMed] [Google Scholar]
- 16.Schmoll D, Walker KS, Alessi DR, Grempler R, Burchell A, Guo S, et al. : Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J Biol Chem 2000;275:36324–36333. [DOI] [PubMed] [Google Scholar]
- 17.Kim MY, Lim JH, Youn HH, Hong YA, Yang KS, Park HS, et al. : Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia 2013;56:204–217. [DOI] [PubMed] [Google Scholar]
- 18.McGivan JD, Pastor-Anglada M: Regulatory and molecular aspects of mammalian amino acid transport. Biochem J 1994;299:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busque SM, Stange G, Wagner CA: Dysregulation of the glutamine transporter Slc38a3 (SNAT3) and ammo-niagenic enzymes in obese, glucose-intolerant mice. Cell Physiol Biochem 2014;34:575–589. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura M, Shirai A, Yamazaki O, Satoh N, Suzuki M, Horita S, et al. : Roles of renal proximal tubule transport in acid/base balance and blood pressure regulation. Biomed Res Int 2014;2014:504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeFronzo RA, Norton L, Abdul-Ghani M: Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol 2017;13:11–26. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Byrd M, Doh K, Dixon PD, Lee H, Tiwari S, et al. : Absence of renal enlargement in fructose-fed proximal-tubule-select insulin receptor (IR), insulin-like-growth factor receptor (IGF1R) double knockout mice. Physiol Rep 2016;4:e13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiwari S, Nordquist L, Halagappa VK, Ecelbarger CA: Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am J Physiol Renal Physiol 2007;293:F178–185. [DOI] [PubMed] [Google Scholar]
- 24.Doctor RB, Chen J, Peters LL, Lux SE, Mandel LJ: Distribution of epithelial ankyrin (Ank3) spliceoforms in renal proximal and distal tubules. Am J Physiol 1998;274:F129–138. [DOI] [PubMed] [Google Scholar]
- 25.Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, et al. : Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 2000;279:F46–53. [DOI] [PubMed] [Google Scholar]
- 26.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, et al. : SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 2011;22:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, et al. : Revised immunolocalization of the Na+−D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 2008;295:C475–489. [DOI] [PubMed] [Google Scholar]
- 28.Curthoys NP, Taylor L, Hoffert JD, Knepper MA: Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol 2007;292:F140–147. [DOI] [PubMed] [Google Scholar]
- 29.Harris AN, Lee HW, Osis G, Fang L, Webster KL, Verlander JW, et al. : Differences in renal ammonia metabolism in male and female kidney. Am J Physiol Renal Physiol 2018;315:F211–F222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr., : Liver and kidney metabolism during prolonged starvation. J Clin Invest 1969;48:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, et al. : Genetic ablation of Rhbg in the mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol 2005;289:F1281–1290. [DOI] [PubMed] [Google Scholar]
- 32.Frische S, Kwon TH, Frokiaer J, Madsen KM, Nielsen S: Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am J Physiol Renal Physiol 2003;284:F584–593. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, et al. : Fructose-1, 6-bisphosphatase opposes renal carcinoma progression. Nature 2014;513:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morigi M, Perico L, Benigni A: Sirtuins in Renal Health and Disease. J Am Soc Nephrol 2018;29:1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiwari S, Singh RS, Li L, Tsukerman S, Godbole M, Pandey G, et al. : Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. J Am Soc Nephrol 2013;24:1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiwari S, Halagappa VK, Riazi S, Hu X, Ecelbarger CA: Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol 2007;18:2661–2671. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura M, Satoh N, Suzuki M, Kume H, Homma Y, Seki G, et al. : Stimulatory effect of insulin on renal proximal tubule sodium transport is preserved in type 2 diabetes with nephropathy. Biochem Biophys Res Commun 2015;461:154–158. [DOI] [PubMed] [Google Scholar]
- 38.Poulsen SB, Fenton RA, Rieg T: Sodium-glucose cotransport. Curr Opin Nephrol Hypertens 2015;24:463469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A: Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A 2003;100:11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore SF, Hunter RW, Hers I: mTORC2 protein complex-mediated Akt (Protein Kinase B) Serine 473 Phosphorylation is not required for Akt1 activity in human platelets [corrected]. J Biol Chem 2011;286:2455324560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Figueiredo VC, Markworth JF, Cameron-Smith D: Considerations on mTOR regulation at serine 2448: implications for muscle metabolism studies. Cell Mol Life Sci 2017;74:2537–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, et al. : Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 2010;18:463–469. [DOI] [PubMed] [Google Scholar]