

Abstract
Objective: Neuronal apoptosis plays an important pathological process in early brain injury (EBI) after subarachnoid hemorrhage (SAH). This pathological process leads to a poor neurological prognosis for patients. This study aimed to investigate whether endoplasmic reticulum (ER) stress mediates cortical neuron apoptosis in EBI after SAH. Methods: Eighty-four male Sprague-Dawley rats were randomly assigned to different groups as follows: the control and the 3, 6, 12, 24, 48, and 72 h groups after SAH. The SAH model was established by injecting 0.3 mL of nonheparinized blood into the prechiasmatic cistern. Hematoxylin-eosin staining, Garcia scoring, Western blotting, transmission electron microscopy, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining were performed. Results: SAH reduced the neurological scores and reached a trough at 24 h after the SAH. The GRP78 expression was significantly upregulated at 6 h after the SAH, peaked at 24 h after the SAH, and then decreased. By comparison, the CHOP, caspase-12, ASK1, and p-c-Jun N-terminal kinase expressions were significantly upregulated at 12 h after the SAH and peaked at 24 h after the SAH. The most serious swelling of the rough ER was observed at 24 h after the SAH and remained notably swollen at 72 h after the SAH. The number of TUNEL-positive cells substantially increased significantly at 12 h after the SAH, and the neuronal apoptosis decreased ratio after reaching a peak at 24 h after the SAH. The apoptosis ratio at 72 h after the SAH was still significantly different from the ratio in the control group. Conclusion: Our study clearly demonstrated that ER stress mediates cortical neuron apoptosis after experimental subarachnoid hemorrhage in rats.
Keywords: Subarachnoid hemorrhage, early brain injury, endoplasmic reticulum stress, neuron apoptosis
Introduction
Subarachnoid hemorrhage (SAH) is a clinical syndrome caused by cerebrovascular rupture that forces blood to enter the subarachnoid space. SAH has high mortality and morbidity rates, accounting for approximately 10% of patients with acute stroke [1]. Early brain injury (EBI) is a series of complex pathological and physiological processes, including brain ischemia, blood-brain barrier destruction, brain edema, the oxidative stress response, the immune inflammatory pathway, and neuronal apoptosis, that occurs within a few minutes of SAH and lasts for several days or longer [2]. Although the control of cerebral vasospasms after SAH can alleviate symptoms, the patients’ neurological prognosis does not drastically improve. In recent years, more and more studies have found that neuronal apoptosis plays an important role in the nerve function prognosis in patients with SAH [3,4].
Endoplasmic reticulum (ER) stress is involved in the pathophysiological process after SAH [5,6]. Unfolded and misfolded proteins during ER stress will activate the unfolded protein response (UPR). After UPR is activated, inositol-requiring enzyme1α (IRE1α), protein kinase R-like ER kinase (PERK), and ATF6, which are anchored on the ER membrane, dissociate from GRP78 and initiate three apoptotic pathways, namely, the IRE1α/c-Jun N-terminal kinase (JNK), PERK/eukaryotic initiation factor-2 (eIF2α), and the ATF6 pathways [7,8].
Moderate UPR promotes normal cell metabolism by eliminating unfolded or misfolded proteins in the ER. However, severe ER stress can induce an excessive UPR to induce apoptosis [9,10]. EBI is an important pathological process that occurs after SAH, and apoptosis is the most common EBI mechanism. Antiapoptotic therapy improves the prognosis of SAH patients. Therefore, determining the molecular mechanism of apoptosis after SAH is important [11,12]. In the present study, we explored whether ER stress is involved in the occurrence of neuronal apoptosis in the EBI process after SAH.
Materials and methods
Animals
Adult male Sprague-Dawley (SD) rats (250 g-300 g) were obtained from the Animal Center of Xinjiang Medical University, China. The SD rats were cultured under the conditions of 24°C on a normal light/dark cycle (12/12 h, light: 7:00 AM to 7:00 PM) and could drink water and eat food freely. The rats were weighed twice a week.
Experimental grouping
Eighty-four rats were randomly divided into the control and the 3 h, 6 h, 12 h, 24 h, 48 h, and 72 h groups after the SAH, with 12 rats in each group. In the SAH group, the subarachnoid space was injected with fresh unheparinized autologous arterial blood, but the control group was not surgically treated.
Prechiasmatic cistern SAH model
The establishment of the SAH model was done under aseptic surgery. The rats were intraperitoneally injected with pentobarbital (40 mg/kg) for the anesthesia and then the rats were fixed on the orientator for the operation. A needle with an oblique edge was tilted 45° on a sagittal plane and inserted 7.5 mm anterior to the bregma at the midline. The needle puncture site was located on the right side of the harnpan. The point of the needle was inserted into the harnpan base and reached 2.5 mm before the chiasma and then the needle was withdrawn by 0.5 mm. Approximately 0.3 mL of fresh arterial blood was removed from the femoral artery and then injected into the prechiasmatic cistern using aseptic manipulation. Hematoxylin-eosin (HE) staining was used to identify the SAH model (Figure 1).
Figure 1.
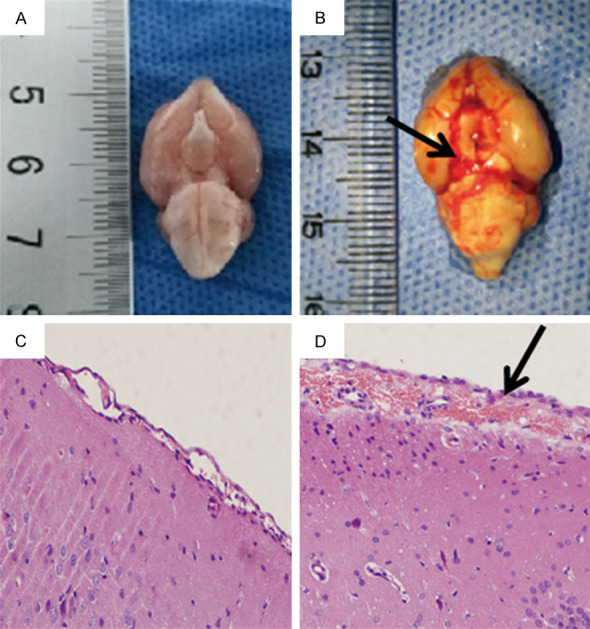
Hematoxylin-eosin staining was used to identify the SAH model (×200). (A and C) represent macroscopic and microscopic observations of the control group, while (B and D) represent the SAH group. No blood clotting was observed in the control group (A), but clots are diffused throughout the brain cisterns in the SAH group (black arrow, B). The subarachnoid space was normal and no red blood cell aggregation was observed in the control group (C), but the SAH group showed a large amount of red blood cell aggregation in the expanded subarachnoid space (black arrow, D).
Neurological scoring
Garcia scoring was performed for the neurological scoring, which includes autonomous movement, spontaneous movements of the limbs, forearm stretching, climbing, proprioception, and sensitivity to the stimulation of the beard. The lowest score is three points, and a low score represents a severe neurological deficit.
Terminal deoxynucleotidyl transferase dUTP nick end labeling stain (TUNEL) staining
The cortical samples obtained were used for the TUNEL staining. Conventional dewaxing hydration for paraffin sectioning was performed. The obtained specimens were digested in 20 µg/ml proteinase K (Merck, Germany) at 37°C for 15 min and rinsed with PBS three times. Next, 50 μL of the TUNEL reaction mixture (A:B = 1:9) was added to the wells and incubated in a humid chamber at 37°C for 60 min. The excess moisture around the samples after they were washed with PBS three times for 5 min was wiped before 50 μL signal transducer (POD) was added. The cells were incubated at 37°C for 30 min in a humid chamber. The microscopic samples were prepared for observation after being washed with PBS three times for 5 min and after the hematoxylin and eosin staining. The cells with dark brown nuclei represented TUNEL positive cells. The TUNEL staining results were observed under a ×200 optical microscope. Finally, the obtained data were averaged.
Transmission electron microscope (TEM) observation
The fresh brain cortical tissues were fixed with 2.5% glutaraldehyde at 25°C for 5 h, washed with 0.1 mol/L PBS buffer at 25°C (3 times, once every 6 min), and fixed with 1% citric acid at 25°C for 1 h. For the second round of embedding, the tissues were washed with 0.1 mol/L PBS buffer at 25°C (three times, once every 6 min) and fixed with ketone gradient dehydration at 25°C. A mixture of Epon812 embedding solution and acetone (ratio = 4:1) was prepared at 35°C for 30 min, placed in the mold, and polymerized at 75°C for 12 h to create a semi-thin sectioning slide. The embedding block was modified to a trapezoidal shape under a stereoscopic microscope. The thickness of the slide varied from 0.5 μm to 5 μm. The tissue was fully exposed, and the area to be observed by the electron microscope was determined. A glass or diamond knife was used to create an ultrathin section after two rounds of embedding. The section thickness ranged from 60 nm to 90 nm. For staining, the sequence was 2% uranyl acetate for 30 min and lead citrate for 20 min, which was labeled and observed under TEM (JEM-1230, JEOL, Japan).
Western blotting (WB) analysis
The cortical frozen samples were homogenized in PBS and clarified by centrifugation for 30 min at 4°C. Then, the lysates were centrifuged at 13,000× g for 15 min. The resulting supernatant (40 μg of protein) was handled with polyacrylamide gel electrophoresis. A transfer apparatus was used to shift the separated proteins into a polyvinylidene difluoride membrane (Millipore, USA) at 300 mA for 40 min. The membrane was then incubated with the following antibodies: anti-GRP78 (1:500, ab108613, Abcam), anti-caspase-12 (1:1000, ab62484, Abcam), anti-CHOP (1:500, ab11419, Abcam), anti-ASK1 (1:1200, ab131506, Abcam), and anti-p-JNK (1:5000, ab76572, Abcam). The membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature after they were washed with Tris-Buffered Saline and Tween 20. The blotted protein bands were observed using enhanced chemiluminescence (Pierce Inc., USA) and then exposed to X-ray film (Kodak; Ximen, China). Densitometry was conducted on the immunoreactive bands using a Gel-Pro Analyzer 4 (Media Cybernetics, USA). After they were incubated with an anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution, Zhongshan Jinqiao Biotechnology Co., Beijing, China), the imprinted protein bands were observed using an enhanced chemiluminescence system.
Statistical analysis
The data are expressed as the mean ± SD and analyzed using a one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls multiple comparison processing. The statistical analyses were performed using SPSS 21.0 (SPSS Inc., Chicago, IL, USA). Significant differences were accepted at P < 0.05.
Results
Morphological observation of the rats in the control and SAH groups
The naked eyes of the rats in the control group had no hemorrhages (Figure 1A). The SAH group had diffusely distributed blood on the surfaces of the basis cranii, and there was a large amount of blood in the circles of Willis, cerebellar medulla pools, and ventral sides of the brain stem (Figure 1B). Compared with the control group (Figure 1C), the HE staining showed that the subarachnoid spaces of the rats in the SAH group were enlarged and accompanied by a large amount of red blood cell deposition (Figure 1D).
The SAH reduced the neurological scores
The SAH caused neurological deficits in the rats. As shown in (Figure 2), the SAH decreased the Garcia scores at 6 h and reached a trough at 24 h after the SAH compared with the control group (P < 0.05). The scores at 72 h after the SAH were still significantly lower than they were in the control group (P < 0.05).
Figure 2.
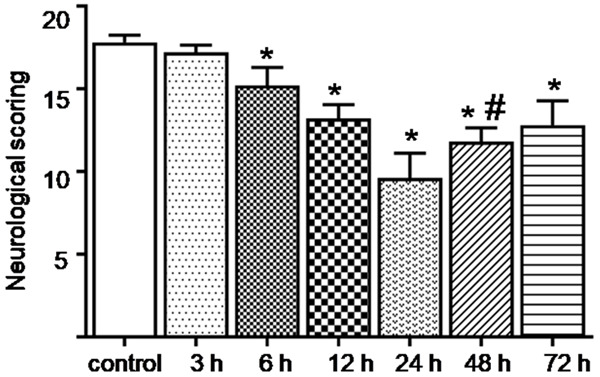
Changes in the Garcia scores at the different time points after the SAH. After the SAH, the neurological function of the rats decreased and then improved after reaching a trough at 24 h. The Garcia scores were still significantly lower than they were in the control group at 72 h after the SAH. *; P < 0.05 versus the control group, #; P < 0.05 versus the 24 h group after SAH.
The SAH increased cortical neuron apoptosis
TUNEL staining was used to determine the degree of neuronal apoptosis in the rat’s cerebral cortexes. As shown in (Figure 3), dark brown staining represents TUNEL-positive cells. The number of apoptotic cells increased significantly at 12 h and peaked at 24 h after the SAH (P < 0.05) (Figure 3A-G). The amount of neuron apoptosis was significantly reduced at 72 h after the SAH, but there was still a significant difference compared with the control group (P < 0.05) (Figure 3H).
Figure 3.
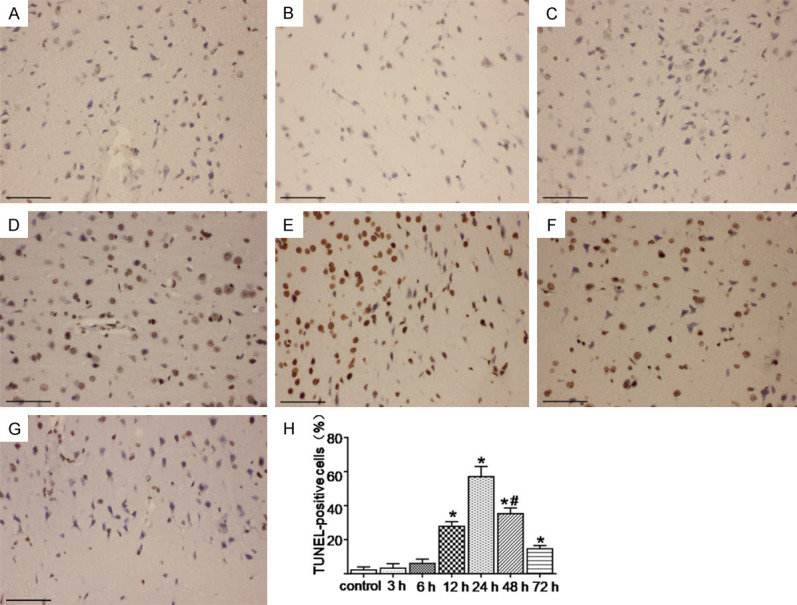
TUNEL staining was used to determine the changes in neuronal apoptosis in the cerebral cortex at various time points after the SAH. (A-G) Represents the control and the 3 h, 6 h, 12 h, 24 h, 48 h, and 72 h groups after the SAH, respectively. (H) Represents the change of neuronal apoptosis rate in each group. The most severe neuronal apoptosis was observed at 24 h after the SAH, and then the level decreased. The number of apoptotic cells was still significantly different from the control group at 72 h after the SAH. *; P < 0.05 versus the control group, #; P < 0.05 versus the 24 h group after SAH. Bar = 100 μm.
The SAH caused swelling of the rough ER
The newly composited proteins were machined and modified in the cisterna of the ER, and the unfolded or misfolded proteins were mainly gathered on the rough ER. The changes in the rough ER in each group were observed using TEM. As shown in (Figure 4), the rough ER was significantly swollen in the SAH groups compared with the control group (P < 0.05) (Figure 4A-G), and was most serious at 24 h after the SAH and burst into vesicles of different sizes. The swelling could be still observed at 72 h after the SAH compared with the control group (P < 0.05) (Figure 4H).
Figure 4.
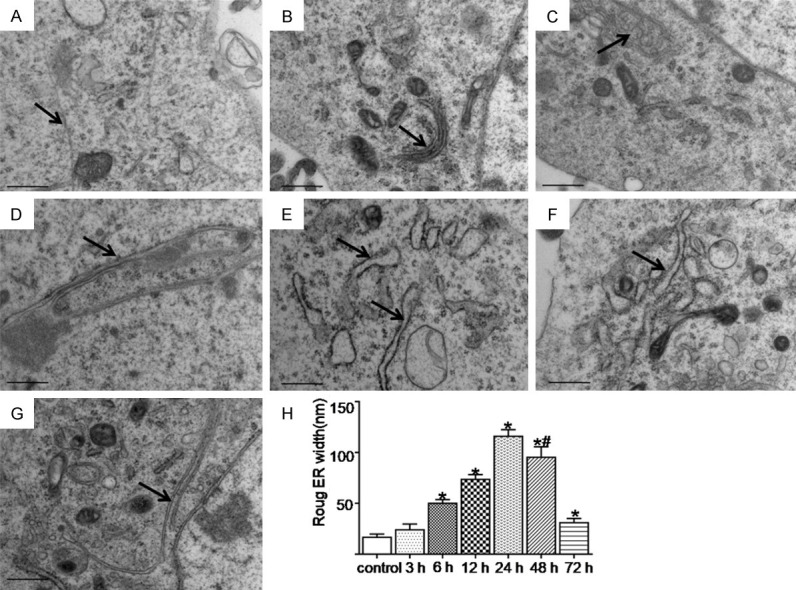
The ultrastructural changes of the rough ER at the different time points were observed using TEM after SAH. The black arrow indicates the rough ER. (A-G) Represents the control and the 3 h, 6 h, 12 h, 24 h, 48 h, and 72 h groups after SAH, respectively. (H) Represents the width variation of rough ER in each group. The rough ER swelling was most serious at 24 h after the SAH. Compared with the control group, rough ER swelling was still observed at 72 h after the SAH. *; P < 0.05 versus the control group, #; P < 0.05 versus the 24 h group after SAH. Bar = 0.5 μm.
The SAH increased the ER stress-related apoptotic proteins expressions
The ER stress-related proteins expressions were analyzed using western blotting. As shown in Figure 5, compared with the control group, the GRP78 expression in the SAH group was upregulated at 6 hours, peaked at 24 h, and then decreased (P < 0.05). Meanwhile, the CHOP, caspase-12, ASK1, and p-JNK expressions increased significantly at 12 h after the SAH, peaked at 24 h, and then decreased significantly (P < 0.05) (Figure 5B-F).
Figure 5.

The expression changes in the ER stress-related proteins at different time points after the SAH were determined using a WB analysis (A). (B-F) Represents the quantitative analysis of the GRP78, CHOP, caspase-12, ASK1, and p-JNK expressions, respectively. *; P < 0.05 versus the control group, #; P < 0.05 versus the 24 h group after the SAH.
Discussion
We investigated the temporal changes in the neurological scores, ER stress-related apoptotic protein expressions, rough ER swelling degrees, and cortical neuron apoptosis in experimental SAH in rats. After the SAH, the neurological scores decreased, but the GRP78, CHOP, caspase-12, ASK1, and p-JNK expressions significantly increased. The rough ER seriously swelled, and numerous apoptotic cortical neurons appeared.
Neuronal apoptosis, cerebral edema, and cerebral vasospasm affect patients’ nerve function. However, neuronal apoptosis is a key link in many pathological factors, and the degree of neuron survival is closely related to prognosis [8,13]. We found that the Garcia scores were the lowest at 24 h after SAH, but the apoptosis was the most active at this time, suggesting that neuronal loss reduced the neurological function in the rats. Although no external intervention was performed, the Garcia scores improved to a certain extent at 48 h after the SAH, probably because of the reduction in neuronal apoptosis and the relief of the acute phase of the cerebral vasospasm [14,15].
Recently, neuronal apoptosis has been the focus of research on EBI after SAH [16]. Apoptosis can increase the permeability of the blood-brain barrier and the degree of brain edema and can reduce cerebral blood flow, thereby decreasing the neurological function [5,17]. Apoptosis is manifested in the cerebral cortex and hippocampus and is related to delayed cognitive and memory dysfunction after SAH [18]. Moreover, neuron survival directly affects patients’ neurological function [13,19]. TUNEL staining revealed that the neuronal apoptosis rate significantly increased at 12 h after SAH and was most severe at 24 h after SAH compared with the rate in the control group. These results are consistent with those of our previous study [20]. The results indicated that apoptosis is an important pathological process of EBI that can lead to the loss of numerous neurons after SAH. Therefore, inhibiting neuronal apoptosis is necessary to improve the prognosis of patients with SAH.
ER is a site for the synthesis and folding of secreted and membrane proteins. Properly folded and modified proteins can be transported into the Golgi apparatus for further processing. The width of rough ER reflects the extent of ER expansion under pressure, which can reflect the severity of ER [21,22]. The morphology of rough ER in the SAH group changed within 6 h after the SAH, and the degree of swelling was the most severe at 24 h after the SAH. However, the swelling of the rough ER at 48 h after SAH was less severe than it was at 24 h after the SAH, consistent with the expression of the ER stress-related apoptosis proteins CHOP, caspase-12, ASK1, and p-JNK. Studies have found that autophagy activated by UPR can relieve ER stress by eliminating unfolded or misfolded proteins [23,24], which may be the reason for the changes in the above results at 48 h after SAH. The GRP78, CHOP, caspase-12, ASK1, and p-JNK expressions at 72 h after the SAH were not significantly different from what they were in the control group. However, the rough ER was still swollen, suggesting that the recovery of the ER morphology lagged behind its functional recovery.
SAH activates multiple pathways of ER stress to induce apoptosis. Intracellular protein synthesis, modification, and folding occur in the ER. ER also stores Ca2+ and regulates Ca2+ metabolism. Stress responses caused by hypoxia, glucose deficiency, inflammation, and oxidative stress lead to an abnormal accumulation of misfolded and unfolded proteins in the ER cavity, which initiates UPR to relieve ER stress [25,26]. Mild ER stress promotes cell survival, but prolonged ER stress upregulates the expression of apoptotic proteins CHOP, caspase-12, ASK1, and JNK in the ATF6, IRE1α/JNK, and PERK/eIF2α pathways. CHOP mediates apoptosis by regulating cell sensitivity to oxidative stress and Bax and Bcl-2 expressions [27,28]. As a proteolytic enzyme unique to ER, caspase-12 specifically binds to the ER membrane, and its activation can be recognized as a marker of ER stress-mediated neuronal apoptosis [29,30]. The downstream molecule JNK of ASK1 is a member of the signal transduction protein family, which regulates the expression of antiapoptotic genes by activating MAPKS, JNKs, and p38MAPKS, consequently inducing apoptosis [31].
At the early stage of ER stress, GRP78 was upregulated to correct the protein misfolding. The GRP78 expression significantly increased at 6 h after the SAH but not at 3 h after the SAH. This result was likely obtained because the glucose stored in cells maintains a normal metabolism in the short term, and the subsequent energy expenditure leads to a widespread activation of the UPR within 6 h after the SAH [32,33]. The Western blot analysis revealed that the CHOP, caspase-12, ASK1, and P-JNK expressions peaked at 24 h after the SAH and significantly decreased at 48 h after the SAH, indicating that the ER stress reached its peak within 48 h after the SAH. This finding is consistent with the neuron apoptosis and the changes in the rough ER width. All the above results indicate that ER stress mediates cortical neuron apoptosis in EBI after SAH.
The persistence of UPR indicates that ER stress is not relieved and homeostasis is not restored. The severity and duration of ER stress are related to neuron survival [34]. The relief of ER stress helps reduce neuronal apoptosis and improves the prognosis of SAH patients. In this study, the neuronal apoptosis rate reached its peak within 48 h after the SAH. This result provides a time reference for the clinical treatment of EBI after SAH. Therefore, relevant measures should be implemented within 48 h after SAH; otherwise, the critical treatment window will be missed. In addition, there may be differences in the pathological progress of the SAH between the experimental animals and human beings, which need to be further explored.
Conclusion
Our study clearly demonstrates that ER stress mediates cortical neuron apoptosis after experimental SAH in rats.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China, (No. 81960222), the International Science and Technology Cooperation Promotion Plan of Shihezi University, (No. GJHZ201704), the Young and Middle-Aged Leading Talents in Scientific and Technological Innovation of XPCC, (No. 2020CB011) and the Doctor Fund Project of The First Affiliated Hospital of Shihezi University, (No. BS201701).
Disclosure of conflict of interest
None.
References
- 1.Feigin VL, Rinkel GJ, Lawes CM, Algra A, Bennett DA, van Gijn J, Anderson CS. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke. 2005;36:2773–2780. doi: 10.1161/01.STR.0000190838.02954.e8. [DOI] [PubMed] [Google Scholar]
- 2.Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1341–1353. doi: 10.1038/sj.jcbfm.9600283. [DOI] [PubMed] [Google Scholar]
- 3.Hasegawa Y, Suzuki H, Sozen T, Altay O, Zhang JH. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;110:43–48. doi: 10.1007/978-3-7091-0353-1_8. [DOI] [PubMed] [Google Scholar]
- 4.Friedrich V, Flores R, Sehba FA. Cell death starts early after subarachnoid hemorrhage. Neurosci Lett. 2012;512:6–11. doi: 10.1016/j.neulet.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 6.Yan F, Cao S, Li J, Dixon B, Yu X, Chen J, Gu C, Lin W, Chen G. Pharmacological inhibition of PERK attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of Akt. Mol Neurobiol. 2017;54:1808–1817. doi: 10.1007/s12035-016-9790-9. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Gao L, Li T, Zheng J, Shao A, Zhang J. Apelin-13 alleviates early brain injury after subarachnoid hemorrhage via suppression of endoplasmic reticulum stress-mediated apoptosis and blood-brain barrier disruption: possible involvement of ATF6/CHOP pathway. Neuroscience. 2018;388:284–296. doi: 10.1016/j.neuroscience.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 8.Takayanagi S, Fukuda R, Takeuchi Y, Tsukada S, Yoshida K. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones. 2013;18:11–23. doi: 10.1007/s12192-012-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Y, Hao Y, Li H, Liu Q, Gao F, Liu W, Duan H. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33:809–816. doi: 10.3892/ijmm.2014.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Q, Che X, Zhang H, Fan P, Tan G, Liu L, Jiang D, Zhao J, Xiang X, Liang Y, Sun X, He Z. Thioredoxin-interacting protein links endoplasmic reticulum stress to inflammatory brain injury and apoptosis after subarachnoid haemorrhage. J Neuroinflammation. 2017;14:104. doi: 10.1186/s12974-017-0878-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J Neurosurg. 2005;102:1046–1054. doi: 10.3171/jns.2005.102.6.1046. [DOI] [PubMed] [Google Scholar]
- 13.Sabri M, Kawashima A, Ai J, Macdonald RL. Neuronal and astrocytic apoptosis after subarachnoid hemorrhage: a possible cause for poor prognosis. Brain Res. 2008;1238:163–171. doi: 10.1016/j.brainres.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, Ley KF, Kassell NF, Lee KS. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003;53:123–133. doi: 10.1227/01.neu.0000068863.37133.9e. discussion 133-125. [DOI] [PubMed] [Google Scholar]
- 15.Sun J, Zhang Y, Lu J, Zhang W, Yan J, Yang L, Zhou C, Liu R, Chen C. Salvinorin A ameliorates cerebral vasospasm through activation of endothelial nitric oxide synthase in a rat model of subarachnoid hemorrhage. Microcirculation. 2018;25:e12442. doi: 10.1111/micc.12442. [DOI] [PubMed] [Google Scholar]
- 16.Duris K, Manaenko A, Suzuki H, Rolland WB, Krafft PR, Zhang JH. α7 nicotinic acetylcholine receptor agonist PNU-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke. 2011;42:3530–3536. doi: 10.1161/STROKEAHA.111.619965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caner B, Hou J, Altay O, Fujii M, Zhang JH. Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem. 2012;123(Suppl 2):12–21. doi: 10.1111/j.1471-4159.2012.07939.x. [DOI] [PubMed] [Google Scholar]
- 18.Kreiter KT, Copeland D, Bernardini GL, Bates JE, Peery S, Claassen J, Du YE, Stern Y, Connolly ES, Mayer SA. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke. 2002;33:200–208. doi: 10.1161/hs0102.101080. [DOI] [PubMed] [Google Scholar]
- 19.Liang Y, Che X, Zhao Q, Darwazeh R, Zhang H, Jiang D, Zhao J, Xiang X, Qin W, Liu L, He Z. Thioredoxin-interacting protein mediates mitochondrion-dependent apoptosis in early brain injury after subarachnoid hemorrhage. Mol Cell Biochem. 2019;450:149–158. doi: 10.1007/s11010-018-3381-1. [DOI] [PubMed] [Google Scholar]
- 20.Zhao D, Liu Q, Ji Y, Wang G, He X, Tian W, Xu H, Lei T, Wang Y. Correlation between nitric oxide and early brain injury after subarachnoid hemorrhage. Int J Neurosci. 2015;125:531–539. doi: 10.3109/00207454.2014.951442. [DOI] [PubMed] [Google Scholar]
- 21.Ling ZQ, Tian Q, Wang L, Fu ZQ, Wang XC, Wang Q, Wang JZ. Constant illumination induces Alzheimer-like damages with endoplasmic reticulum involvement and the protection of melatonin. J Alzheimers Dis. 2009;16:287–300. doi: 10.3233/JAD-2009-0949. [DOI] [PubMed] [Google Scholar]
- 22.Vekich JA, Belmont PJ, Thuerauf DJ, Glembotski CC. Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J Mol Cell Cardiol. 2012;53:259–267. doi: 10.1016/j.yjmcc.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Dai RY, Duan CY, Liu YP, Chen SK, Yan DM, Chen CN, Wei M, Li H. Unfolded protein response suppresses cisplatin-induced apoptosis via autophagy regulation in human hepatocellular carcinoma cells. Folia Biol (Praha) 2011;57:87–95. doi: 10.14712/fb2011057030087. [DOI] [PubMed] [Google Scholar]
- 25.Bánhegyi G, Baumeister P, Benedetti A, Dong D, Fu Y, Lee AS, Li J, Mao C, Margittai E, Ni M, Paschen W, Piccirella S, Senesi S, Sitia R, Wang M, Yang W. Endoplasmic reticulum stress. Ann N Y Acad Sci. 2007;1113:58–71. doi: 10.1196/annals.1391.007. [DOI] [PubMed] [Google Scholar]
- 26.Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22:R622–626. doi: 10.1016/j.cub.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi W, Cao D, Li Y, Peng A, Wang Y, Gao K, Tao C, Wu Y. Atorvastatin ameliorates early brain injury through inhibition of apoptosis and ER stress in a rat model of subarachnoid hemorrhage. Biosci Rep. 2018;38:BSR20171035. doi: 10.1042/BSR20171035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Datta D, Khatri P, Singh A, Saha DR, Verma G, Raman R, Mazumder S. Mycobacterium fortuitum-induced ER-Mitochondrial calcium dynamics promotes calpain/caspase-12/caspase-9 mediated apoptosis in fish macrophages. Cell Death Discov. 2018;4:30. doi: 10.1038/s41420-018-0034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Yu JS, Zhang HS, Yang YQ, Huang LT, Zhang DD, Hang CH. Increased expression of caspase-12 after experimental subarachnoid hemorrhage. Neurochem Res. 2016;41:3407–3416. doi: 10.1007/s11064-016-2076-9. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 32.Shiu RP, Pouyssegur J, Pastan I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin Rous sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci U S A. 1977;74:3840–3844. doi: 10.1073/pnas.74.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park HR, Ryoo IJ, Choo SJ, Hwang JH, Kim JY, Cha MR, Shin-Ya K, Yoo ID. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology. 2007;229:253–261. doi: 10.1016/j.tox.2006.11.049. [DOI] [PubMed] [Google Scholar]
- 34.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]