

Abstract
Tuberculosis (TB) is a chronic inflammatory infectious disease caused by Mycobacterium tuberculosis (Mtb), which induces irreversible pulmonary damage. Oxysophocarpine (OSC) is a natural alkaloid that exhibits multiple pharmacological activities, including anti-inflammation; however, the protective effects of OSC against TB and the mechanisms involved are unknown. Here, we established murine and cellular models of TB with C3HeB/FeJ mice and neutrophils infected with H37Rv to investigate the biological functions of OSC in TB. We found that OSC reduced the mortality, inhibited the pulmonary H37Rv growth, and alleviated the lung pathology injury in the Mtb-infected mice. OSC also repressed neutrophil recruitment to the lesions of the Mtb-infected mice as evidenced by a decrease in the number and percentage of neutrophils in the lungs. OSC hampered the production of proinflammatory cytokines and chemokines, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, macrophage inflammatory protein-2 (MIP-2), granulocyte colony stimulating factor (G-CSF), and keratinocyte chemoattractant (KC) in the lungs of Mtb-infected mice. The results of the in vitro experiments showed that OSC repressed the adhesion and F-actin polymerization of the Mtb-infected neutrophils by inhibiting the toll-like receptor 2/myeloid differentiation primary response gene 88/Src/extracellular signal-regulated kinase 1/2 signaling. Moreover, OSC abolished the Mtb-induced expression and release of TNF-α, IL-1β, IL-6, MIP-2, G-CSF, and KC in neutrophils. Overall, these findings indicate that OSC can treat TB partly by lessening the neutrophilic recruitment and inflammation.
Keywords: Tuberculosis, Mycobacterium tuberculosis, oxysophocarpine, neutrophils, inflammation
Introduction
Tuberculosis (TB), which is caused by Mycobacterium tuberculosis (Mtb), is recognized as a major chronic infectious disease worldwide, with nearly 10.4 million new cases and over 1.7 million deaths annually [1]. Clinically, patients with TB are usually divided into latent infection and active disease [2]. Most infected individuals develop latent TB with no symptoms, but approximately 10% of them can develop active TB and ultimately die [2]. Despite remarkable advances in chemotherapy, drug resistance remains a problem. Therefore, novel treatments for TB should be employed.
Mtb infection leads to severe inflammatory pulmonary disease, specifically the formation of granulomas, which are intended to separate resistant bacteria [3]. Inflammatory tuberculous lesions consist of a central area of Mycobacterium-infected macrophages surrounded by noninfected phagocytes and lymphocytes [4]. Phagocytosis of Mtb by alveolar macrophages can prevent phagosome-lysosome fusion and consequently is eliminated by lysosomes [5]. After infection, Mtb primarily reaches the lungs, where it is sensed and recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), expressed by alveolar macrophages, dendritic cells, and epithelial cells [6]. PRR sensing prompts the mobilization of effector functions, and infected antigen-presenting cells secrete various pro-inflammatory cytokines and chemokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, and macrophage inflammatory protein-2 (MIP-2), to recruit and activate other innate immune cells, including inflammatory monocytes and neutrophils [7,8]. Neutrophils are the most abundant cells in the sputum and bronchoalveolar lavage fluid of patients with active TB and have the highest Mtb load [9]. Neutrophil influx is correlated with increased pathology in genetically susceptible mouse models of Mtb [10,11] and exacerbated inflammatory conditions [12]. Reportedly, the depletion of neutrophils prolongs the survival of TB-prone animals [13,14]. However, molecular events that orchestrate neutrophil recruitment and accumulation at the site of mycobacterial infections, as well as the mechanisms mediating TB inflammation, should be further clarified.
Oxysophocarpine (OSC) is a widely used traditional Chinese herbal medicine isolated from Sophora flavescens Ait. (Kushen), Sophora alopecuroides Linn. (Kudouzi or Kugancao), and other leguminous plants of the genus Robinia [15]. In the literature, OSC reportedly possesses many pharmacological effects, including neuroprotective [16-18], analgesic [19], antinociceptive [20], antiviral [21,22], and antitumor [23,24] properties. OSC elicits remarkable inflammation inhibition and pain relief, accompanied by reduced neutrophil accumulation, TNF-α, IL-1β, and IL-6 production, and extracellular signal-regulated kinase 1/2 (ERK1/2) activation [19]. Nevertheless, the anti-Mtb functions of OSC by abating neutrophil infiltration and inflammation are not certain, and the underlying mechanisms for the alleviation of active TB injury by OSC are unclear.
In this study, we investigated the effects of OSC on the lungs of mice with active TB in vivo and Mtb-infected neutrophils in vitro. OSC extended the survival time of H37Rv-infected mice, limited the growth of Mtb, and ameliorated the pathology of TB in the lungs. OSC also hampered neutrophil infiltration and reduced the inflammatory factors in the lungs of H37Rv-infected mice. In vitro, OSC repressed H37Rv-led neutrophil adhesion and F-actin polymerization by inhibiting the TLR2/myeloid differentiation primary response gene 88 (MyD88)/Src/ERK1/2 pathway. Furthermore, OSC suppressed the release of inflammatory mediators in H37Rv-infected neutrophils. These findings indicate that OSC has anti-inflammatory effects against active TB and can thus be a potential novel therapeutic agent for TB.
Materials and methods
Animals and ethics
Male C3HeB/FeJ mice aged 6-8 weeks were purchased from Jackson Laboratories (Bar Harbor, Maine, USA) and maintained in a specific pathogen-free facility at the Laboratory Animal Center at Henan Provincial Peoples’ Hospital (Zhengzhou, China). The animal experiments were approved by the Institutional Committee for Animal Research and performed in conformity with the guidelines of the Laboratory Animal Center at Henan Provincial Peoples’ Hospital.
The Mtb infection of the mice and the OSC administration
The C3HeB/FeJ mouse model of active TB disease was established as previously described [8]. Briefly, virulent Mtb strain H37Rv (ATCC, Manassas, VA, USA) was cultured in Middlebrook 7H9 broth (BD Difco, Cockeysville, MD, USA) supplemented with 0.2% glycerol, 0.05% Tween 80, and 10% oleic acid-albumin-dextrose-catalase (OADC; BD Difco). In the Mtb group, the mice (n = 36) were infected with 2 × 105 colony forming units (CFU) of H37Rv via the tail vein. In the Mtb + OSC group, OSC (Zi Jin Hua Pharmaceutical Co., Ningxia, China) was dissolved in normal saline and alternatively administered in the infected mice through an intraperitoneal injection with a volume of 0.1 mL/10 g at a dose of 40 mg/kg. The treatment started from the day after the infection (day 1; n = 36) and was conducted every day for 4 weeks. In the normal control (Ctrl) group, the mice (n = 36) received neither the Mtb infection nor the OSC treatment. At the indicated time points after the Mtb infection, the mice were euthanized, and their lungs were immediately removed to analyze the bacillary load, histopathology and histometry, and inflammatory mediators.
Bacillary load counts
For the bacterial load assessment, the lung samples were harvested and homogenized at 1, 2, 3, and 4 weeks after the Mtb infection. The samples were homogenized, and several serial dilutions were plated on Middlebrook 7H10 agar (BD Difco) with 0.5% glycerol and 10% OADC. The plates were then incubated at 37°C for the indicated time duration, and the number of CFU was counted and expressed as log10CFU/lung.
Histopathology scoring and calculating the number of lesions and the affected area in lungs
At 4 weeks after the Mtb infection, the lung tissues were fixed in 10% buffered formalin and embedded in paraffin. Then, 5 μm sections were stained with hematoxylin eosin (HE) and Ziehl-Neelsen for microscopic observation and histometry analysis using the NIS-Elements D version 3.0x software package (Nikon Instruments Inc., Tokyo, Japan). Two experienced pathologists evaluated the results using the Leica system (Leica, Heidelberg, Germany). On the basis of the histopathological changes, such as peribronchiolitis, perivasculitis, alveolitis, and granuloma formation, the lung pathology was scored as 0, 1, 2, 3, 4, or 5 for absent, minimal, slight, moderate, marked, or strong changes, respectively; the maximum sum was 20.
The quantitation of the pathology was conducted using Image J. The image was first adjusted to the color threshold between blue and violet, which eliminated most of the pink stains in each lung section. The adjusted image was converted to an 8-bit image. The tissue section was outlined with the selection tool. The threshold on the gray-scale image was readjusted until mostly only the interested areas were highlighted in black. The threshold was set and was used for all subsequent images. The measurement on the selected lung section is reported as a percent area. Four recuts stained with HE were used to determine the granulomatous area as a percentage of the total lung area. One HE-stained recut per animal was used to count the number of lesions as previously described [8].
Isolation of the lung leukocytes
At 4 weeks after the Mtb infection, the lungs were perfused with 1 × phosphate buffered solution (PBS) to reduce the blood leukocytes. Left lung lobes were dissected and incubated with RPMI 1640 medium (Gibco, BRL, Grand Island, USA) supplemented with 10% heat-inactivated fetal calf serum (FCS; Gibco), glutamine, Na-pyruvate, 2-ME, penicillin, streptomycin, collagenase type VIII (Sigma-Aldrich, St. Louis, MO, USA), and collagenase D (Roche, Basel, Switzerland) at 37°C for 30 min. After the depletion of the red blood cells using a lysis buffer, single-cell suspensions of the lungs were prepared using an iron mesh sieve. Viable cells were counted using a trypan blue exclusion assay. Equal viable cells were collected for culture preparation and subsequent analyses.
Flow cytometry analysis
For the immunophenotyping, the isolated leukocytes were stained with fluorescence-conjugated antibodies, including Ly6G (clone 1A8), CD11c (clone HL3), Ly6C (clone AL-21), CD11b (clone M1/70), CD3e (clone 145-2C11), and CD19 (clone 1D3; BD Pharmingen, San Diego, CA). The propidium iodide-positive cells were excluded by gating prior to collecting at least 10,000 events. The stained cells were acquired using FACSCalibur (BD Biosciences, San Jose, CA, USA) and analyzed with FACS Analyzer software (BD Biosciences). The neutrophils were counted as the CD11b- and Ly6G-positive cells. The neutrophil percentage was calculated from the frequencies among the total leukocytes (n = 1 × 106/each group).
Immunohistochemistry (IHC) assay
The paraffin-embedded sections were rehydrated in graded alcohols and subjected to microwave-antigen-retrieval using a citrate buffer (Beyotime, Haimen, China). The endogenous peroxidase activity was quenched by incubating the sections with 3% H2O2 for 15 min. The sections were probed at 37°C for 1 h with rabbit anti-Ly6G polyclonal antibody (Abcam, Cambridge, UK) and then treated with a biotinylated secondary antibody using the ChemMate EnVision Kit (DAKO, Hamburg, Germany) for 20 min. The visualization was conducted using a diaminobenzidine substrate-chromogen solution (DAKO), resulting in a colored precipitate at the antigen site. The sections were counterstained with hematoxylin, and the slides were examined under a lighted microscope (Carl Zeiss).
Isolation of bone marrow neutrophils, Mtb infection, and OSC administration
The murine bone marrow neutrophils were isolated using Percoll (GE Healthcare, Little Chalfont, UK) density gradient centrifugation and hypotonic lysis of red blood cells. Murine bone marrow cells were layered on top of a 53%/63%/76% three-layer Percoll gradient after the red blood cells were removed. After centrifugation, the mature neutrophils were recovered at the interface of the 63% and 76% fractions and were > 90% pure and > 95% viable, which was determined by using Diff-Quick staining and trypan blue exclusion. The neutrophils were cultured in a RPMI-1640 medium with 5% FCS (Gibco).
Ly6G-positive neutrophils were infected with H37Rv at a multiplicity of infection (MOI) of 5. The cells were then extensively washed with pre-warmed PBS to remove the non-adherent bacteria. OSC (5 μM) was added to the cells immediately after the H37Rv infection. The cells were incubated at 37°C and 5% CO2 for the indicated time duration.
Immunoassays
After the H37Rv infection or OSC treatment, the levels of cytokines and chemokines, such as TNF-α, IL-1β, IL-6, MIP-2, granulocyte colony stimulating factor (G-CSF), and keratinocyte chemoattractant (KC), in the lung homogenates or the neutrophil culture supernatants were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D systems, San Diego, CA, USA) in accordance with the manufacturer’s instructions. The absorbance was recorded at 450 nm using an ELISA microplate reader (Molecular Devices, Sunnyvale, CA, USA).
After the H37Rv infection or OSC treatment, the levels of IL-8 (R&D Systems) in the lung homogenates were evaluated using Luminex (L200, Millipore, Massachusetts, USA). The results were presented in median fluorescence intensity (MFI) units.
Quantitative real-time PCR (qPCR) assay
After the H37Rv infection or OSC treatment, total RNA was isolated from the neutrophils using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s protocols. Complementary DNA was synthesized with SuperScript III Reverse Transcriptase (Invitrogen). The mRNA levels of the mouse TNF-α, IL-1β, IL-6, MIP-2, G-CSF, and KC were measured with qPCR assays using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The amplification was carried out on an ABI PRISM 7900HT thermocycler (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was employed as an internal control. The fold changes were calculated using the 2-ΔΔCt method. The primers used were as follows: for TNF-α, forward 5’-CACAGAAAGCATGATCCGCGAC-3’ and reverse 5’-TGCCACAAGCAGGAATGAGAAGAG-3’; for IL-1β, forward 5’-CAGGATGAGGACATGAGCACC-3’ and reverse 5’-CTCTGCAGACTCAAACTCCAC-3’; for IL-6, forward 5’-GTCCGGAGAGGAGACTTCAC-3’ and reverse 5’-CTGCAAGTGCATCATCGTTGT-3’; for MIP-2, forward 5’-CCAAGGGTTGACTTCAAGAAC-3’ and reverse 5’-AGCGAGGCACATCAGGTACG-3’; for G-CSF, forward 5’-GCAGGCTCTATCGGGTATTTCC-3’ and reverse 5’-GCAACATCCAGCTGAAGCAA-3’; for KC, forward 5’-TGGCTGGGATTCACCTCAA-3’ and reverse 5’-GAGTGTGGCTATGACTTCGGTTT-3’; and for GAPDH, forward 5’-AACTTTGGCATTGTGGAAGG-3’ and reverse 5’-ACACATTGGGGGTAGGAACA-3’.
Adhesion assay
After pretreatment with the Src kinase inhibitor PP1 (5 μM), ERK1/2 inhibitor U0126 (10 μM), p38MAPK inhibitor SB202190 (10 μM), or JNK inhibitor AG490 (25 μM; Biomol, Plymouth Meeting, PA, USA) for 30 min or with siRNA targeting TLR2 (siTLR2; 100 nM; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA) for 24 h, neutrophils (3 × 105 cells/well) were then uninfected or infected with H37Rv (5 MOI) in the presence or absence of 5 μM OSC and allowed to adhere to 48-well plates for 6, 12, 18, and 24 h. The non-adherent cells were washed away, and the adherent neutrophils were quantified using a myeloperoxidase (MPO) assay. The MPO activities were determined through the H2O2-dependent oxidation of 3,3’,5,5’-tetramethylbenzine (Sigma-Aldrich). Adherence was expressed as the ratio of the MPO activity of the adherent neutrophil to that of the total neutrophils, and the percentages of the adherent cells were calculated.
F-actin polymerization
After pretreatment with the Src kinase inhibitor PP1 (5 μM), the ERK1/2 inhibitor U0126 (10 μM), the p38MAPK inhibitor SB202190 (10 μM), or the JNK inhibitor AG490 (25 μM) for 30 min or siRNA targeting TLR2 (siTLR2; 100 nM) for 24 h, the neutrophils were uninfected or infected with H37Rv (5 MOI) in the presence or absence of 5 μM OSC and then cultured on coverslips at 37°C for 24 h. The cells were fixed with 4% paraformaldehyde, permeabilized with BD Perm/Wash Buffer (BD Biosciences), and stained with Alexa568-phalloidin (Molecular Probes, Eugene, Oregon, USA). The MFI was measured according to the fluorescence intensities of the individual cells. More than 20 cells from at least five randomly selected fields from each slide were analyzed under a microscope (Carl Zeiss).
Western blot analysis
The neutrophils were infected with H37Rv (5 MOI) in the presence or absence of OSC (5 μM) and cultured at 37°C for 24 h. The cells were lysed in a RIPA lysis buffer (Beyotime), and the cell debris was pelleted by centrifugation at 12,000 × g and 4°C for 10 min. The protein concentration in each supernatant was determined using a Bradford protein assay (Beyotime). The cell lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk diluted in tris-buffered saline containing 0.5% Tween-20 (TBST) for 1 h and incubated with primary antibodies against TLR2, MyD88, Src, phosphorylated (p)-ERK1/2, ERK1/2, p-p38 MAPK, p38 MAPK, p-JNK, JNK, and β-actin (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight. The membranes were then washed three times with TBST for 1 h at 37°C and incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) for 1 h at room temperature. Immunoblots were developed and visualized through enhanced chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
The data were expressed as the mean ± standard deviation (SD). The statistical comparisons were analyzed using ANOVA followed by Bonferroni’s multiple comparison tests. The survival curves were plotted using Kaplan-Meier method and analyzed using log-rank tests. The statistical analyses were performed using GraphPad Prism 5.0 (San Diego, California, USA). P < 0.05 was considered statistically significant.
Results
OSC alleviates H37Rv-induced lung lesions in mice
To investigate the effects of OSC on Mtb-induced damage, we first determined whether the Mtb infection affected mouse mortality. As shown in Figure 1A, the mortality of the mice infected intravenously with 2 × 105 CFU of H37Rv increased notably compared with the control mice. The OSC treatment substantially prolonged the survival time of the H37Rv-infected mice. Moreover, the bacterial loads in the lungs were enhanced after the H37Rv infection, and the number of bacilli ingested by the OSC-treated mice was remarkably increased compared with the number of mice infected with H37Rv only (Figure 1B). HE staining and pathologic scoring in Figure 1C and 1D revealed that the lung pathology injury, including the formation of granulomas, was intensified in the H37Rv-infected mice, and this aggravation was mitigated by OSC. As shown in Figure 1E, the number of lesions was increased postinfection, and this enhancement was attenuated by OSC. The percentage of the affected area was markedly reduced in the OSC-treated H37Rv-infected mice (Figure 1F). These results indicated that OSC limited Mtb replication in the lungs and decreased lung lesions and the mortality of the Mtb-infected mice.
Figure 1.

OSC mitigated the lung lesions in H37Rv-infected mice. C3HeB/FeJ mice were infected with a dose of 2 × 105 CFU of H37Rv via the tail vein. OSC (40 mg/kg) was intraperitoneally administered starting on the day after the infection (day 1) and employed every day for 4 or 8 weeks. A. The Kaplan-Meier method was used to plot the survival curve and the comparisons were analyzed using log-rank tests (n = 12). B. Intracellular viable bacteria were measured using CFU assays at 7, 14, 21, and 28 days postinfection (n = 6 times per duration). C, D. HE staining was conducted to evaluate the pathology of the lungs at 4 weeks after the H37Rv infection (n = 6). E. Changes in the number of lung lesions (n = 6). F. The affected lung area was measured to determine the histological changes (n = 6). The data are expressed as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 compared with the Ctrl group. #P < 0.05, ##P < 0.01 compared with Mtb group. Ctrl: control; OSC: Oxysophocarpine.
OSC inhibits the active recruitment of neutrophils to the lungs in the H37Rv-infected mice
To probe the effect of OSC on the neutrophil recruitment to the lungs of the mice infected with H37Rv, we performed a series of immunoassays. IL-8 is an important pro-inflammatory mediator that induces chemotaxis, activation, and neutrophil extracellular trap release in human neutrophils [25]. As depicted in Figure 2A, the IL-8 level was enhanced in the TB-infected lungs, which was repressed by the OSC treatment. The neutrophil count, including CD11b+ and Ly6G+, was significantly increased with prolonged Mtb infection, but the OSC treatment evidently reduced the neutrophil accumulation in the lungs (Figure 2B). The IHC results also showed a pronounced elevation in the Ly6G+ cells in the lungs of the H37Rv-infected mice, which was again decreased by the OSC administration (Figure 2C). The neutrophil percentages in the leukocytes was calculated to further confirm the effects of OSC on the accumulation of the recruited neutrophils. As shown in Figure 2D, OSC reduced the abundance of neutrophils in the leukocytes of the mouse lungs with the H37Rv infection. These data demonstrated that the active recruitment of neutrophils was suppressed by OSC in the lungs of the Mtb-infected mice.
Figure 2.

OSC repressed the neutrophil infiltration into the lungs of the H37Rv-infected mice. C3HeB/FeJ mice were infected with a dose of 2 × 105 CFU of H37Rv via the tail vein. OSC (40 mg/kg) was intraperitoneally administered starting on the day after the infection (day 1) and employed every day for 4 weeks. A. The IL-8 levels in the lung homogenates were evaluated using a Luminex model L200 (n = 6). The results are presented in MFI units. B. The number of neutrophils (CD11b+, Ly6G+) was identified in the lungs using flow cytometry. C. The IHC staining of the neutrophils with Ly6G antibody (n = 6). D. A flow cytometry assay was conducted to calculate the recruited leukocytes. The percentages of neutrophils in the various leukocytes were counted (n = 6). The data are expressed as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 compared with the Ctrl group. #P < 0.05, ##P < 0.01 compared with the Mtb group. Ctrl: control; OSC: Oxysophocarpine.
OSC alleviates the inflammatory responses in the lungs of the H37Rv-infected mice
To address the effects of OSC on the H37Rv infection-induced inflammation, we evaluated the production of several inflammatory cytokines and chemokines in the lungs of the Mtb-infected mice at different time points. The levels of TNF-α (Figure 3A), IL-1β (Figure 3B), IL-6 (Figure 3C), MIP-2 (Figure 3D), G-CSF (Figure 3E), and KC (Figure 3F) were increased in the lungs of the H37Rv-infected mice. The OSC treatment reduced the generation of inflammatory mediators (Figure 3A-F). These results suggested that OSC hindered the Mtb-induced inflammation in the mice’s lungs.
Figure 3.
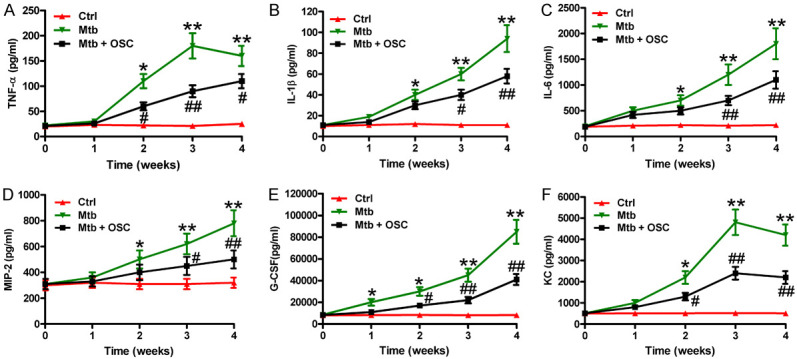
OSC alleviated the inflammation in the lungs of the H37Rv-infected mice. The C3HeB/FeJ mice were infected with a dose of 2 × 105 CFU of H37Rv via the tail vein. OSC (40 mg/kg) was intraperitoneally administered starting on the day after the infection (day 1) and employed every day for 4 weeks. (A-F) The levels of (A) TNF-α, (B) IL-1β, (C) IL-6, (D) MIP-2, (E) G-CSF, and (F) KC in the lung homogenates of the mice at days 7, 14, 21, and 28 postinfection were measured using ELISAs (n = 6 per time duration). The data are expressed as the mean ± SD of the three independent experiments. *P < 0.05, **P < 0.01 compared with the Ctrl group. #P < 0.05, ##P < 0.01 compared with the Mtb group. Ctrl: control; OSC: oxysophocarpine.
OSC reduces the H37Rv-led neutrophil adhesion and F-actin polymerization by inactivating the TLR2/MyD88/Src-dependent ERK1/2 signaling in vitro
Next, we explored whether OSC represses neutrophil adhesion and F-actin polymerization after H37Rv infections. The adhesion of the H37Rv-infected neutrophils was enhanced, but it was reduced by the OSC treatment (Figure 4A). To examine whether the H37Rv-induced neutrophil adhesion was accompanied by F-actin remodeling, we stained H37Rv-infected neutrophils with fluorescently labeled phalloidin. As depicted in Figure 4B, the H37Rv infection promoted F-actin polymerization in neutrophils, which was hampered by OSC. Reportedly, TLR2/MyD88/Src signaling is implicated in Mtb infection in neutrophils, and F-actin remodeling and neutrophil adhesion is regulated by MAPKs [26]. Here, we investigated whether similar pathways are involved in the H37Rv-caused neutrophil adhesion. The results showed that the H37Rv infection increased the levels of TLR2, MyD88, Src, p-ERK1/2 in the neutrophils, but no effect was observed on the p-p38 MAPK or the p-JNK (Figure 4C). Moreover, OSC decreased the expression of TLR2, MyD88, and Src and the phosphorylation of ERK1/2 (Figure 4C). In Figure 4D and 4E, pretreated neutrophils with siTLR2, the Src kinase inhibitor PP1, and the ERK1/2 inhibitor U0126 had a similar action as OSC, which inhibited the H37Rv-induced neutrophil adhesion and the F-actin remodeling. However, pretreatment with the p38 MAPK inhibitor SB202190 or the JAK inhibitor AG490 did not affect the adhesion or the F-actin remodeling in the H37Rv-infected neutrophils. These findings indicate that the inactivation of the TLR2/MyD88/Src/ERK1/2 signaling is essential for the action of OSC in reducing adhesion and F-actin remodeling in H37Rv-infected neutrophils.
Figure 4.

OSC hindered the adhesion and F-actin polymerization in the H37Rv-infected neutrophils by the inhibition of the TLR2/MyD88/Src/ERK1/2 signaling pathway. A-C. The neutrophils were uninfected or infected with H37Rv (5 MOI) in the presence or absence of 5 μM OSC. A. The firmly adherent neutrophils were counted after the H37Rv infection for 6, 12, 18, and 24 h. B. F-actin polymerization was monitored using immunofluorescent microscopy at 24 h after the H37Rv infection. C. A Western blot analysis was performed to assess the expressions of TLR2, MyD88, Src, p-ERK1/2, ERK1/2, p-p38 MAPK, p38 MAPK, p-JNK, and JNK at 24 h after the H37Rv infection. β-actin was used as an endogenous control. D and E. The neutrophils were preincubated with the Src kinase inhibitor PP1 (5 μM), ERK1/2 inhibitor U0126 (10 μM), p38MAPK inhibitor SB202190 (10 μM), or the JNK inhibitor AG490 (25 μM) for 30 min or pretransfected with siTLR2 (100 nM) for 24 h, and then uninfected or infected with H37Rv (5 MOI) in the presence or absence of OSC (5 μM) for 24 h. D. The firmly adherent neutrophils were counted. E. F-actin polymerization was monitored. Data are expressed as the mean ± SD of the three independent experiments. *P < 0.05, **P < 0.01 compared with Ctrl group. #P < 0.05 compared with the Mtb group. Ctrl: control; OSC: Oxysophocarpine; NS: no significance.
OSC restrains H37Rv-induced expression and the release of the inflammatory mediators in neutrophil in vitro
We subsequently addressed the effects of OSC on the H37Rv-induced mRNA expression and release of inflammatory cytokines and chemokines in neutrophils. The OSC administration reduced the mRNA levels of TNF-α (Figure 5A), IL-1β (Figure 5B), IL-6 (Figure 5C), MIP-2 (Figure 5D), G-CSF (Figure 5E), and KC (Figure 5F) in the H37Rv-infected neutrophils. Similarly, the ELISA results showed increased releases of TNF-α (Figure 5G), IL-1β (Figure 5H), IL-6 (Figure 5I), MIP-2 (Figure 5J), G-CSF (Figure 5K), and KC (Figure 5L) from the H37Rv-infected neutrophils, but were abolished by the OSC treatment. These data revealed that OSC resolved the H37Rv inflammation caused by the H37Rv in neutrophils in vitro.
Figure 5.

OSC reduced the generation of cytokines and chemokines in the H37Rv-infected neutrophils. The neutrophils were uninfected or infected with H37Rv (5 MOI) in the presence or absence of OSC (5 μM) for 12 or 24 h. (A-F) The mRNA expressions of (A) TNF-α, (B) IL-1β, (C) IL-6, (D) MIP-2, (E) G-CSF, and (F) KC were determined using qPCR assays at 12 h after the H37Rv infection. (G-L) ELISAs were conducted to measure the release of (G) TNF-α, (H) IL-1β, (I) IL-6, (J) MIP-2, (K) G-CSF, and (L) KC in the media after 24 h of H37Rv infection. The data are expressed as means ± SD of three independent experiments. *P < 0.05 compared with the Ctrl group. #P < 0.05 compared with the Mtb group. Ctrl: control; OSC: Oxysophocarpine.
Discussion
In this study, we elucidated the beneficial roles of OSC in an active TB murine model and with Mtb-infected neutrophils. First, OSC restrained H37Rv growth in the lungs and reduced the Mtb-induced mouse mortality and lung lesions. Second, OSC inhibited the neutrophil recruitment into the H37Rv-infected sites and the production of the pro-inflammatory cytokines and chemokines in the lungs. Third, OSC retarded the adhesion and F-actin polymerization of the H37Rv-infected neutrophils through the inactivation of the TLR2/MyD88/Src/ERK1/2 pathway. Finally, OSC repressed the H37Rv-induced mRNA expression and the release of the inflammatory mediators in the neutrophils. In all, the protective effects of OSC on H37Rv-induced inflammation and neutrophil activation in the lungs may indicate a promising therapeutic feasibility of OSC in Mtb-induced pulmonary TB.
TB is an inflammatory infectious disease caused by Mtb inhalation, which leads to the deaths of almost 2 million individuals annually. An ongoing inflammation of the lungs can remodel their architecture, manifesting as extensive fibrosis, cavitation, bronchostenosis, traction bronchiectasis, or parenchymal lung destruction [27,28]. Mtb enters the airways, reaches the pulmonary alveolus, and infects the resident alveolar macrophages [29]. Granulomatous inflammatory disease occurs when the infected macrophages are unable to contain the replication of Mtb [30]. Mtb-activated alveolar macrophages release pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1α, and IL-1β, which recruit other phagocytes to the infection site [31,32]. Neutrophils are the first phagocytes recruited from the pulmonary vasculature to the lung interstitium [33]. Upon arrival at the infectious sites, the neutrophils phagocytose bacteria by recognizing them either directly or indirectly [34]. In contrast to the description of phagocytosis, the data on the ability of neutrophils to kill Mtb are conflicting [35]. At the early stage of Mtb infection, neutrophils may contribute to protection by favoring the generation of effector T-cells and participating in the formation of granulomas, but the neutrophils become largely detrimental at the later stages [36]. Several clinical and experimental studies have clarified the pathological roles of neutrophils during Mtb infection [8-10]. In humans, neutrophils represent the most abundant cells in the sputum and bronchoalveolar lavage fluid from active TB patients and carry the main mycobacterial load [9]. In previous studies, extensive neutrophilic infiltration was observed in TB-prone mice but not in TB-resistant mice [8,10]. In the literature, after neutrophil depletion by a Ly6G-specific antibody, Mtb-infected I/St mice show reduced weight loss, low mycobacterial counts, mild pulmonary pathology, and increased survival times [37]. In this study, Mtb infection resulted in a reduction of the survival rate, severe pulmonary lesions, massive neutrophil infiltration into the lesion sites, an excessive generation of proinflammatory cytokines and chemokines in the active TB mouse model and promoted the adhesion and F-actin polymerization and release of the proinflammatory factors in neutrophils. OSC reduces the neutrophil influx and decreases TNF-α, IL-1β, and IL-6 production in an inflammatory pain mouse model [19]. Here we demonstrated that OSC mitigated the pathological changes, enhanced the mortality of Mtb-infected mice, in parallel with reduced neutrophil infiltration into the lesion sites, and decreased inflammatory mediators in vivo and in vitro. Overall, the findings indicate that OSC is beneficial to Mtb-induced lung TB by suppressing neutrophilic infiltration and inflammation.
Neutrophil recruitment is closely associated with the overproduction of inflammatory mediators in TB [38]. The increased infiltration of neutrophil-like cells coincides with the production of TNF-α, IL-1β, IL-6, CCL3, CCL4, and other factors in TB mice [39]. Reportedly, alveolar macrophages and immune, endothelial, and epithelial cells secrete pro-inflammatory cytokines and chemokines that influence neutrophil differentiation, mobilization, and recruitment. For instance, IL-1β and G-CSF prolong neutrophil survival; and TNF-α, IL-1β, CXCL1/KC, CXCL2/MIP-2, CXCL8/IL-8, and G-CSF stimulate neutrophil migration, degranulation, oxidative burst, and secretory activity [40-43]. Neutrophils can transcribe a wide range of pro-inflammatory factors, including TNF-α, IL-1α, IL-1β, CXCL1/KC, CXCL8/IL-8, CCL3/MIP-1α, CCL4/MIP-1β, and G-CSF, to attract and activate immune cells and other neutrophils at the infection sites [34,44,45]. After the activation, neutrophil spreading, adhesion, and F-actin polymerization occur. Cell adhesion-associated actin remodeling is regulated by diverse receptor-activated MAPKs [46,47]. Lee et al. [26] reported that activation of Src and ERK1/2 is required for adhesion and F-actin polymerization in trehalose dimycolate-stimulated neutrophils. TLR2 expression is upregulated on the neutrophil surface upon Mtb infection [45], and TLR2 signaling through MyD88 initiates the innate host defenses [6]. In the present study, we demonstrated that Mtb infection results in excessive neutrophil adhesion and F-actin polymerization, coinciding with the enhancement of TLR2 and MyD88 and the activation of Src and ERK1/2. OSC administration reduces adhesion and F-actin polymerization in Mtb-infected neutrophils in parallel with the results of siTLR2 transfection or pretreatment with the Src kinase inhibitor PP1 and the ERK1/2 inhibitor U0126. These results show that OSC abates Mtb-induced neutrophil adhesion and F-actin polymerization by inhibiting theTLR2/MyD88/Src/ERK1/2 pathway.
In conclusion, this study provides evidence that OSC is beneficial against active TB by reducing Mtb-induced neutrophil recruitment and inflammation. OSC significantly prolongs survival, suppresses Mtb replication and neutrophil infiltration in the lungs, alleviates pulmonary lesions, and decreases the production of inflammatory cytokines and chemokines in H37Rv-infected mice. OSC also reduces H37Rv-induced neutrophil adhesion and F-actin polymerization by the inactivation of the TLR2/MyD88/Src/ERK1/2 signaling pathway. Overall, these findings show that OSC hampers Mtb infection-caused neutrophil accumulation and inflammation in mouse lungs. Further studies on the cell lines and animals are needed to clarify whether OSC has potential therapeutic implications in active lung TB.
Acknowledgements
This study is partly supported by National Natural Science Foundation of China (No. 81904050).
Disclosure of conflict of interest
None.
References
- 1.Marimani M, Ahmad A, Duse A. The role of epigenetics, bacterial and host factors in progression of Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2018;113:200–214. doi: 10.1016/j.tube.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, Menzies D, Raviglione M. Tuberculosis. Nat Rev Dis Primers. 2016;2:16076. doi: 10.1038/nrdp.2016.76. [DOI] [PubMed] [Google Scholar]
- 3.Saunders BM, Cooper AM. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol Cell Biol. 2000;78:334–341. doi: 10.1046/j.1440-1711.2000.00933.x. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- 5.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol. 2011;2011:405310. doi: 10.1155/2011/405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawn TR, Matheson AI, Maley SN, Vandal O. Host-directed therapeutics for tuberculosis: can we harness the host? Microbiol Mol Biol Rev. 2013;77:608–627. doi: 10.1128/MMBR.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) 2014;94:55–64. doi: 10.1016/j.tube.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, Cho SN, Via LE, Barry CE 3rd. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, Apt AS. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73:1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74:4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyadova IV, Tsiganov EN, Kapina MA, Shepelkova GS, Sosunov VV, Radaeva TV, Majorov KB, Shmitova NS, van den Ham HJ, Ganusov VV, De Boer RJ, Racine R, Winslow GM. In mice, tuberculosis progression is associated with intensive inflammatory response and the accumulation of Gr-1 cells in the lungs. PLoS One. 2010;5:e10469. doi: 10.1371/journal.pone.0010469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, Gross O, Ruland J, Kaufmann SH. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song JZ, Xu HX, Tian SJ, But PP. Determination of quinolizidine alkaloids in traditional Chinese herbal drugs by nonaqueous capillary electrophoresis. J Chromatogr A. 1999;857:303–311. doi: 10.1016/s0021-9673(99)00758-x. [DOI] [PubMed] [Google Scholar]
- 16.Zhu QL, Li YX, Zhou R, Ma NT, Chang RY, Wang TF, Zhang Y, Chen XP, Hao YJ, Jin SJ, Ma L, Du J, Sun T, Yu JQ. Neuroprotective effects of oxysophocarpine on neonatal rat primary cultured hippocampal neurons injured by oxygen-glucose deprivation and reperfusion. Pharm Biol. 2014;52:1052–1059. doi: 10.3109/13880209.2013.877039. [DOI] [PubMed] [Google Scholar]
- 17.Liu G, Wang J, Deng XH, Ma PS, Li FM, Peng XD, Niu Y, Sun T, Li YX, Yu JQ. The anticonvulsant and neuroprotective effects of oxysophocarpine on pilocarpine-induced convulsions in adult male mice. Cell Mol Neurobiol. 2017;37:339–349. doi: 10.1007/s10571-016-0411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao P, Chang RY, Liu N, Wang J, Zhou R, Qi X, Liu Y, Ma L, Niu Y, Sun T, Li YX, He YP, Yu JQ. Neuroprotective effect of oxysophocarpine by modulation of MAPK pathway in rat hippocampal neurons subject to oxygen-glucose deprivation and reperfusion. Cell Mol Neurobiol. 2018;38:529–540. doi: 10.1007/s10571-017-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Li YX, Wang HL, Jin SJ, Zhou R, Qiao HQ, Du J, Wu J, Zhao CJ, Niu Y, Sun T, Yu JQ. Oxysophocarpine ameliorates carrageenan-induced inflammatory pain via inhibiting expressions of prostaglandin E2 and cytokines in mice. Planta Med. 2015;81:791–797. doi: 10.1055/s-0035-1546153. [DOI] [PubMed] [Google Scholar]
- 20.Xu T, Li Y, Wang H, Xu Y, Ma L, Sun T, Ma H, Yu J. Oxysophocarpine induces anti-nociception and increases the expression of GABAAalpha1 receptors in mice. Mol Med Rep. 2013;7:1819–1825. doi: 10.3892/mmr.2013.1414. [DOI] [PubMed] [Google Scholar]
- 21.Chen JX, Shen HH, Niu M, Guo YM, Liu XQ, Han YZ, Zhang YM, Zhao YL, Bai BK, Zhou WJ, Xiao XH. Anti-hepatitis B virus effect of matrine-type alkaloid and involvement of p38 mitogen-activated protein kinase and tumor necrosis factor receptor-associated factor 6. Virus Res. 2016;215:104–113. doi: 10.1016/j.virusres.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, Li Y, Wang Q, Ma X, Zhang Y. Oxysophocarpine inhibits lung injury induced by respiratory syncytial virus. Am J Transl Res. 2017;9:4083–4093. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Xu J, Li X, Zhang D, Han Y, Zhang X. Comprehensive two-dimensional PC-3 prostate cancer cell membrane chromatography for screening anti-tumor components from radix sophorae flavescentis. J Sep Sci. 2017;40:2688–2693. doi: 10.1002/jssc.201700208. [DOI] [PubMed] [Google Scholar]
- 24.Liu R, Peng J, Wang H, Li L, Wen X, Tan Y, Zhang L, Wan H, Chen F, Nie X. Oxysophocarpine retards the growth and metastasis of oral squamous cell carcinoma by targeting the Nrf2/HO-1 axis. Cell Physiol Biochem. 2018;49:1717–1733. doi: 10.1159/000493615. [DOI] [PubMed] [Google Scholar]
- 25.de Melo MGM, Mesquita EDD, Oliveira MM, da Silva-Monteiro C, Silveira AKA, Malaquias TS, Dutra TCP, Galliez RM, Kritski AL, Silva EC Rede-TB Study Group. Imbalance of NET and alpha-1-antitrypsin in tuberculosis patients is related with hyper inflammation and severe lung tissue damage. Front Immunol. 2018;9:3147. doi: 10.3389/fimmu.2018.03147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY, Cho SN, Kim YJ. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the mincle pathway. PLoS Pathog. 2012;8:e1002614. doi: 10.1371/journal.ppat.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan TS, Spencer EM, Davies P. Tuberculosis, bronchiectasis and chronic airflow obstruction. Respirology. 2010;15:623–628. doi: 10.1111/j.1440-1843.2010.01749.x. [DOI] [PubMed] [Google Scholar]
- 28.Dheda K, Booth H, Huggett JF, Johnson MA, Zumla A, Rook GA. Lung remodeling in pulmonary tuberculosis. J Infect Dis. 2005;192:1201–1209. doi: 10.1086/444545. [DOI] [PubMed] [Google Scholar]
- 29.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 30.Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011;4:271–278. doi: 10.1038/mi.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Law K, Weiden M, Harkin T, Tchou-Wong K, Chi C, Rom WN. Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. Am J Respir Crit Care Med. 1996;153:799–804. doi: 10.1164/ajrccm.153.2.8564135. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 33.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 34.Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. 2000;80:617–653. doi: 10.1038/labinvest.3780067. [DOI] [PubMed] [Google Scholar]
- 35.Denis M. Human neutrophils, activated with cytokines or not, do not kill virulent Mycobacterium tuberculosis. J Infect Dis. 1991;163:919–920. doi: 10.1093/infdis/163.4.919. [DOI] [PubMed] [Google Scholar]
- 36.Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? Mediators Inflamm. 2017;2017:8619307. doi: 10.1155/2017/8619307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeremeev V, Linge I, Kondratieva T, Apt A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb) 2015;95:447–451. doi: 10.1016/j.tube.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Riedel DD, Kaufmann SH. Chemokine secretion by human polymorphonuclear granulocytes after stimulation with Mycobacterium tuberculosis and lipoarabinomannan. Infect Immun. 1997;65:4620–4623. doi: 10.1128/iai.65.11.4620-4623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech. 2015;8:1141–1153. doi: 10.1242/dmm.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silva MT. When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol. 2010;87:93–106. doi: 10.1189/jlb.0809549. [DOI] [PubMed] [Google Scholar]
- 42.Matzer SP, Baumann T, Lukacs NW, Rollinghoff M, Beuscher HU. Constitutive expression of macrophage-inflammatory protein 2 (MIP-2) mRNA in bone marrow gives rise to peripheral neutrophils with preformed MIP-2 protein. J Immunol. 2001;167:4635–4643. doi: 10.4049/jimmunol.167.8.4635. [DOI] [PubMed] [Google Scholar]
- 43.Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunol Today. 1995;16:21–26. doi: 10.1016/0167-5699(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 44.Petrofsky M, Bermudez LE. Neutrophils from Mycobacterium avium-infected mice produce TNF-alpha, IL-12, and IL-1 beta and have a putative role in early host response. Clin Immunol. 1999;91:354–358. doi: 10.1006/clim.1999.4709. [DOI] [PubMed] [Google Scholar]
- 45.Hilda JN, Das SD. TLR stimulation of human neutrophils lead to increased release of MCP-1, MIP-1alpha, IL-1beta, IL-8 and TNF during tuberculosis. Hum Immunol. 2016;77:63–67. doi: 10.1016/j.humimm.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Wang Q, Doerschuk CM. The p38 mitogen-activated protein kinase mediates cytoskeletal remodeling in pulmonary microvascular endothelial cells upon intracellular adhesion molecule-1 ligation. J Immunol. 2001;166:6877–6884. doi: 10.4049/jimmunol.166.11.6877. [DOI] [PubMed] [Google Scholar]
- 47.Pichon S, Bryckaert M, Berrou E. Control of actin dynamics by p38 MAP kinase - Hsp27 distribution in the lamellipodium of smooth muscle cells. J Cell Sci. 2004;117:2569–2577. doi: 10.1242/jcs.01110. [DOI] [PubMed] [Google Scholar]